

Abstract
Lipopolysaccharide (LPS) is recognized by Toll-like receptor (TLR) 4 and activates NF-κB and a set of MAP kinases. Here we have investigated proteins associated with the cytoplasmic domain of mouse TLR4 by yeast two-hybrid screening and identified JNK-interacting protein 3 (JIP3), a scaffold protein for JNK, as a TLR4-associated protein. In mammalian cells, JIP3, through its N-terminal region, constitutively associates with TLR4. The association is specific to JIP3, as the two other JIPs, JIP1 and JIP2, failed to bind TLR4. In HEK 293 cells exogenously expressing TLR4, MD2 and CD14, co-expression of JIP3 significantly increased the complex formation of TLR4–JNK and LPS-mediated JNK activation. In contrast, expression of C-terminally truncated forms of JIP3 impaired LPS-induced JNK activation in a mouse macrophage cell line, RAW264.7. Moreover, RNA interference of JIP3 inhibited LPS-mediated JNK activation. In RAW264.7 cells, JIP3 associates MEKK-1, but not with TAK-1. Finally, JIP3 also associates with TLR2 and TLR9, but not with TLR1 or TLR6. Altogether, our data indicate the involvement of JIP3 in JNK activation in downstream signals of some TLRs.
Keywords: JIP3/JNK/LPS/MEKK1/Toll-like receptor
Introduction
Toll-like receptors (TLRs) play important roles in host defense mechanisms by pathogen recognition. Ten members of the TLRs (TLR1–10) have so far been reported. TLR4 mediates lipopolysaccharide (LPS) signals in collaboration with other molecules, such as CD14, MD-2, myeloid differentiation factor 88 (MyD88) and Toll receptor–IL-1 receptor domain containing adapter protein (TIRAP)/MyD88-adapter-like (Mal). On the other hand, TLR2 is considered to be an essential receptor for lipoprotein, peptidoglycan, zymosans and lipoteichoic acid presumably by forming heteromers with TLR1 or TLR6. TLR3, TLR5 and TLR9 have recently been shown to mediate signals from double-stranded RNA, flagella and bacterial DNA, respectively (Akira, 2001).
Stimulation of TLRs by specific ligands induces nuclear transport of NF-κB and the activation of a set of mitogen-activated protein kinases (MAPKs): extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs) and p38 kinases. Activation mechanisms of MAPKs are generally through phosphorylation of threonine and tyrosine residues within the signature sequence of T-X-Y by dual specificity MAPK kinases (MKKs). MKKs are in turn phosphorylated and activated by a family of serine/threonine MKK kinases (MKKKs) (Robinson and Cobb, 1997). MKKKs integrate signals mediated by upstream signaling molecules.
The distinct classes of MAPKs play important roles in various cellular events including cell proliferation, differentiation and apoptosis. JNKs are activated by diverse stimuli including DNA damage, heat-shock, bacterial components, inflammatory cytokines and Fas (Leppa and Bohmann, 1999). Activated JNKs play an essential role in the activation of transcriptional factors, such as c-Jun, ATF-2, Elk-1 and ets-2 (Smith et al., 2000). In macrophages, activated JNKs mediate the expression of inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), chemokines and cytokines, all of which potently activate host defense mechanisms. The critical roles of JNK signaling in immuno-regulatory cells are evident as jnk1–/– and jnk2–/– mice exhibit various defective immune responses (Dong et al., 1998; Yang et al., 1998).
MAPK activation may be facilitated by the formation of signaling modules, and it has been well established in both yeast and mammalian cells that ‘scaffold proteins’ play important roles by interacting with MAPKs and their upstream kinases (Whitmarsh and Davis, 1998). For example, in yeast, a scaffold protein, Ste5p, mediates the mating Fus3 MAPK pathway (Choi et al., 1994) and Pbs2p plays a part in the osmoregulatory MAPK pathway (Posas and Saito, 1997). Mammalian scaffold proteins, MP1 and KSR were found to function in the ERK activation pathway (Schaeffer et al., 1998; Cacace et al., 1999). β-arrestin2, which binds to β2-adrenergic receptors, has recently been reported to function as a scaffold protein for JNK activation (McDonald et al., 2000). Also in the JNK signaling cascade, three other putative scaffold proteins have been reported: JNK-interacting protein 1 (JIP1; Whitmarsh et al., 1998), JIP2 (Yasuda et al., 1999) and JIP3 (also termed JSAP1) (Ito et al., 1999; Kelkar et al., 2000). They promote JNK activation by interacting with JNKs and the upstream kinases in vitro. A recent study of jip1 gene-disrupted mice has indicated that JIP1 functions as an JNK activator in vivo (Whitmarsh et al., 2001). The role of JIP2 is somewhat controversial, as it has recently been reported to bind and activate p38 kinase, but not JNK (Buchsbaum et al., 2002). JIPs may also participate in vesicular transport by binding a motor protein, kinesin-1 (Verhey et al., 2001). JIP3 is ubiquitously expressed in mouse tissues, with notably higher expression detected in brain, heart and lung (Kelkar et al., 2000). The homologue of JIP3 has been reported in Drosophila and Caenorhabiditis elegans (Bowman et al., 2000; Byrd et al., 2001), indicating JIP3 is evolutionally well conserved.
Although TLRs activate three groups of MAPKs, the possible involvement of scaffold proteins in the activating processes is not known. LPS, which signals through TLR4, activates two MKKKs, TGFβ-activated kinase (TAK) 1 and MEK kinase (MEKK) 1, presumably through TRAF6 (Kopp et al., 1999; Ninomiya-Tsuji et al., 1999). Activ ation of these two kinases has been indicated to involve interacting proteins, TAK1-binding protein (TAB) 1/2 and ECSIT (evolutionarily conserved signaling intermediate in Toll pathways), respectively.
Here we report that TLR4 constitutively associates with JIP3, but not with JIP1 or JIP2. JIP3 also associates with TLR2 and TLR9, but not with TLR1 or TLR6. In a mouse macrophage cell line, JIP3 associates with both full-length and processed forms of MEKK1. Moreover, expression of C-terminally deleted JIP3 mutants and RNA interference of JIP3 inhibited LPS-mediated JNK activation. These data indicate that JIP3, as a scaffold protein, plays a role in mediating JNK activation by LPS through the association with TLR4.
Results
Identification of JIP3 as a TLR4-interacting protein by yeast two-hybrid screening
In an attempt to identify novel proteins interacting with the cytoplasmic domain of TLR4, we screened a mouse embryo cDNA library by yeast two-hybrid method using the mouse TLR4 cytoplasmic domain as the bait. We isolated 34 independent clones, two of which encode MyD88, a previously known TLR4-interacting protein, confirming validation of the screening. By nucleotide sequencing, we found that two of the isolated clones encode JIP3. These two clones contained different lengths of JIP3 cDNA (encoding amino acids 154–1337 and 172–1337), and were confirmed to associate with TLR4 cytoplasmic domain by re-transfection in yeast with bait plasmid (Figure 1A).

Fig. 1. Interaction of JIP3 and TLR4. (A) The cytoplasmic portion of mouse TLR4 cloned in frame with GAL4 DNA binding domain in pDBLeu vector was cotransformed with pPC86 empty vector containing GAL4 activating domain, pPC86-MyD88 or pPC86-JIP3 into MaV203 yeast strain carrying the three reporter genes: HIS3, URA3 and lacZ. The transformed colonies were grown on either SC/-Leu or SC/-His-Trp-His+3AT selection. (B) HEK 293 cells were transiently transfected with pcDNA3.1(+)-CD14, pcDNA3.1(+)-MD2 and p3XFlagCMV14-TLR4 in combination with either pEFBOSMyc (EV: empty vector) or pEFBOSMyc-JIP3. At 48 h after transfection, cells were either left untreated or treated with 1 µg/ml LPS for 20 min. Anti-Flag and anti-Myc immunoprecipitates were separated by SDS–PAGE and immunoblotting was performed with anti-Flag (upper) and anti-Myc (lower) antibodies. (C) HEK 293 cells were transiently transfected with pEFBOSFlag-JIP3 in combination with either pcDNA3.1(+)-GCSFR/TLR2 or pcDNA3.1(+)-GCSFR/TLR4. At 48 h after transfection, cell lysates were prepared. Anti-Flag and control antibody immunoprecipitates were separated by SDS–PAGE, and immunoblotting was performed with anti-GCSFR antibody (top panel). Cell lysates were also probed with anti-Flag or anti-GCSFR antibody. (D) RAW264.7 cells were stimulated with 1 µg/ml LPS for 20 min or left untreated. Cell lysates were prepared and anti-TLR4 and control antibody immunoprecipitates were separated by SDS–PAGE, followed by the analysis with anti-JIP3 antibody. All the experiments in this paper were repeated at least twice, typically 3–5 times, with reproducible results.
In order to test the JIP3–TLR4 association in mammalian cells, we expressed JIP3 N-terminally tagged with Myc and TLR4 C-terminally tagged with Flag in HEK 293 cells in combination with mouse CD14 and MD-2. We found that JIP3 was coprecipitated with TLR4 in the absence of LPS stimulation of the cells (Figure 1B). No increase of binding was detected when the cells were stimulated with LPS. JIP3–TLR4 interaction was also detected in the absence of CD14 or MD-2 (data not shown), which were co-expressed in order to increase the LPS-responsiveness of the cells. We next examined the association by using a chimera protein of the extracellular domain of human G-CSF receptor fused to the cytoplasmic domain of TLR4 (Figure 1C). The G-CSFR–TLR4 fusion protein was transiently expressed with Flag-tagged JIP3 in HEK 293 cells. The fusion protein was coprecipitated with JIP3, indicating that the extracellular domain of TLR4 does not contribute to the TLR4–JIP3 association. We further examined the association of endogenous TLR4 and JIP3. Using a mouse macrophage cell line, RAW264.7, we could detect endogenous JIP3 in the TLR4 immunoprecipitates with or without LPS stimulation, but not in the control immunoprecipitates (Figure 1D).
The C-terminal domain of TLR4 is necessary for binding JIP3
In an attempt to identify the region of TLR4 required for JIP3 binding, we utilized two C-terminal deletion mutants: TLR4del13 and TLR4 del130, deleting C-terminal 13 and 130 amino acids of TLR4, respectively. Unlike the full-length TLR4, neither mutant was coprecipitated with JIP3 (Figure 2A). Thus the C-terminal 13 amino acids of TLR4 are essential for binding JIP3.

Fig. 2. Identification of TLR4 region essential for interaction with JIP3. (A) Schematic presentation of the mouse TLR4 mutants used in the assay. Black boxes represent transmembrane domains (TM). (B) C-terminal region of TLR4 is essential for the interaction with JIP3. HEK 293 cells were transiently transfected with expression plasmids of a Myc-tagged JIP3 (wild type or JNK-binding mutant) and an indicated Flag-tagged TLR4. At 48 h after transfection, JIP3 was precipitated with anti-Myc antibody and coprecipitation of TLR4 was detected with anti-Flag antibody. Lysates were also run as controls for the input. (C) Pro712 of TLR4 is not essential for JIP3 binding. HEK 293 cells were transiently transfected with expression plasmids of a Myc-tagged wild-type JIP3 construct and the TLR4 wild-type or Pro712His point mutant. At 48 h after transfection, lysates were prepared and anti-Myc and control immunoprecipitates were analyzed for the coprecipitation of TLR4 with anti-Flag antibody.
Although not included in the C-terminal 13 amino acids, Pro712 is considered essential for the TLR4 signaling. Mutation of the proline to histidine, which is found in the C3H/HeJ mouse strain, severely impaired LPS-stimulated NF-κB and JNK activation presumably by inhibiting TLR4–MyD88 binding (data not shown) (Rhee and Hwang, 2000). We examined if this Pro712His mutation affects TLR4–JIP3 binding and found that the TLR4 Pro712His mutant is capable of binding JIP3 to the same extent as the wild-type TLR4 (Figure 2B), indicating that the TLR4–JIP3 interaction alone is not sufficient to induce JNK activation.
JIP3 increases LPS-stimulated JNK activity
As TLR4 associates with JIP3, which is a known scaffold for a JNK signaling module, we speculated that the expression of JIP3 may enhance JNK activation by LPS. To test this hypothesis, we performed transfection assays using COS7 cells, which do not express endogenous JIP3 protein (Kelkar et al., 2000) (Figure 3). Expression of JIP3 in combination with TLR4, CD14 and MD-2 did not cause the activation of the cotransfected haemagglutinin (HA)-JNK1. However, JIP3 expression significantly increased LPS-induced JNK activity. In contrast, JIP3 expression did not affect JNK activation by anisomycin, a potent activator of MKK4. Thus JIP3 is specifically involved in LPS-mediated JNK activation. Also, the effect of JIP3 is specific to JNK, as activation of p38 kinase by LPS was not affected by JIP3 expression (Figure 3).
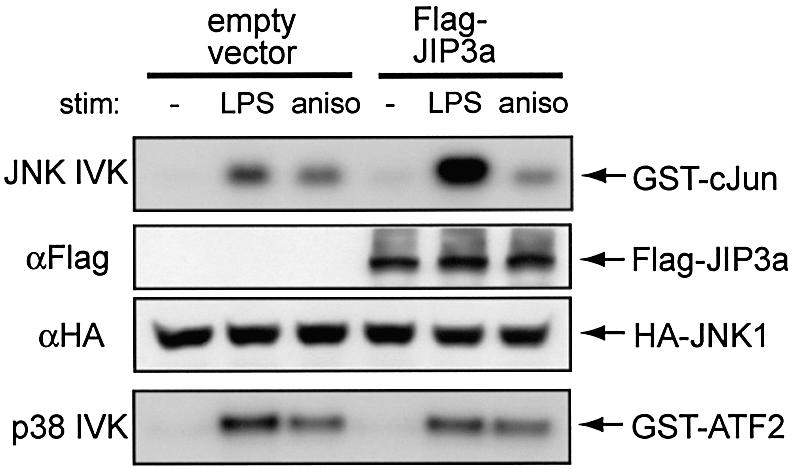
Fig. 3. JIP3 overexpression enhances LPS-mediated JNK activation. COS7 cells were transiently transfected with p3XFlag-CMV14-TLR4, pcDNA3.1(+)-mCD14, p3XFlag-CMV10-mMD-2 and pcDNA3.1(+)-HA-JNK1. At 48 h after the transfection, cells were left untreated or stimulated with 1 µg/ml LPS or 10 µg/ml anisomycin for 20 min, and lysed. Anti-HA immunoprecipitates were tested for in vitro kinase assay on GST–cJun5-89 as the substrate. Cell lysates were also analyzed for p38 kinase activity by anti-p38 immunoprecipitation using GST–ATF-2 as the substrate.
Identification of TLR4 binding domain of JIP3
Two independent partial cDNA clones of JIP3 isolated by the two-hybrid screening both contain parts of the N-terminal region of JIP3. We did not isolate any short cDNA clones encoding only the C-terminal regions of JIP3, hinting that the N-terminal region of JIP3 may be responsible for TLR4 binding. In order to define the TLR4 binding domain of JIP3, we expressed several C-terminal deletion mutants of JIP3 in combination with the wild-type TLR4 for coprecipitation assays. As shown in Figure 4, Flag-tagged TLR4 was coprecipitated with JIP3 C-terminal deletion mutants: 1–221, 1–355, 1–677, 1–1015, at similar levels to the full JIP3 protein. However, no coprecipitation was detected for the JIP3 1–163 mutant, suggesting that the residues 163 to 221 are essential for JIP3 to bind TLR4.
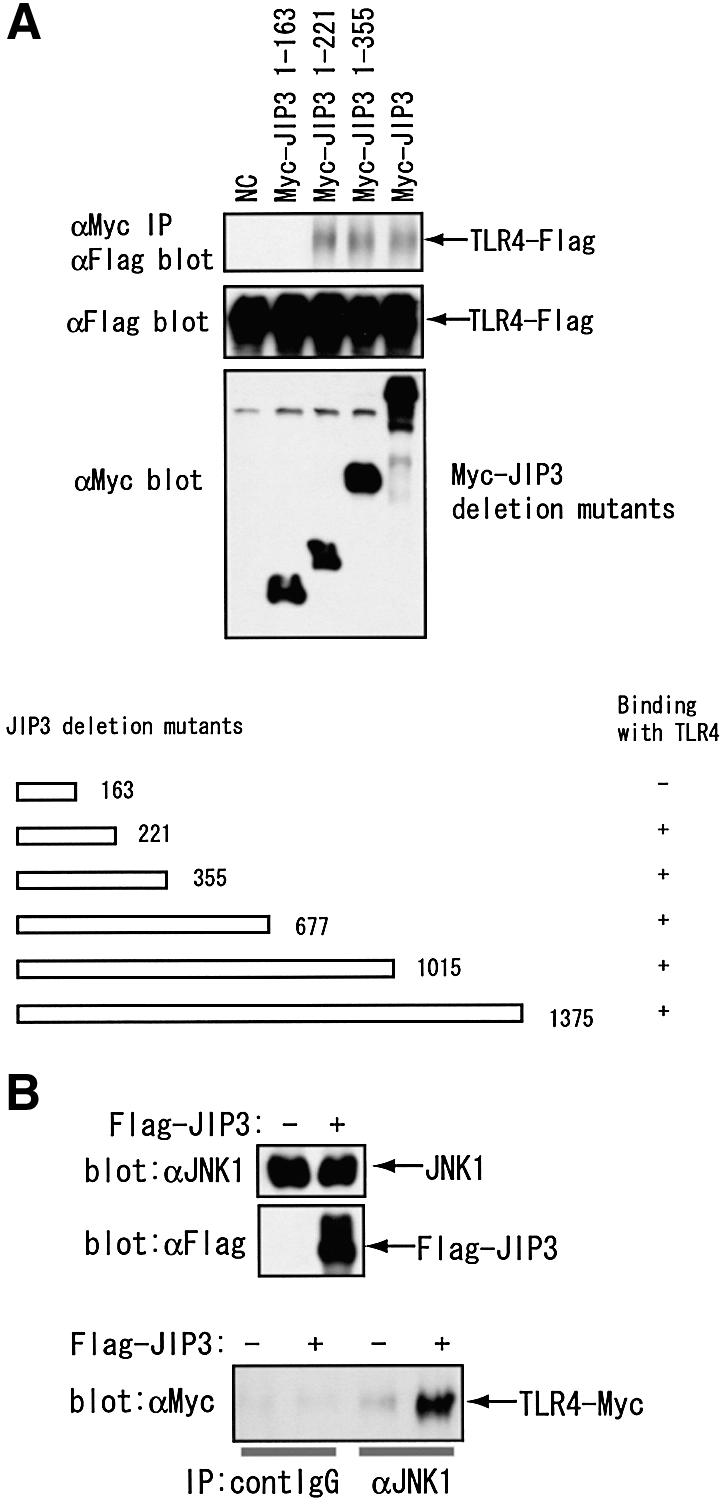
Fig. 4. JIP3 associates with TLR4 through its N-terminal region and induces TLR4–JIP3–JNK complex formation. (A) JIP3 N-terminal region is sufficient for the interaction with TLR4. HEK 293 cells were transiently transfected with p3XFlag-CMV14-mTLR4 in combination with a series of Myc-tagged C-terminal deletion mutants of JIP3 (see the scheme). Cell lysates were prepared at 48 h after transfection, and the anti-Myc immunoprecipitates were tested for the coprecipitation of TLR4 using anti-Flag antibody. The results are summarized in the scheme. The immunoblots for some constructs are shown. (B) JIP3 induces the complex formation by TLR4 and JNK. HEK 293 cells were transiently transfected with pEFBOSMyc-mTLR4 in combination with either pEFBOSFlag (empty vector) or pEFBOSFlag-mJIP3. Cell lysates were prepared at 48 h after transfection, and the protein expression of endogenous JNK1 and Flag-JIP3 was confirmed by immunoblot (top and middle panel). The control or anti-JNK1 antibody immunoprecipitates were separated by SDS–PAGE, and the coprecipitation of TLR4 was tested using anti-Myc antibody (bottom panel).
It has previously been reported that residues 207 to 216 are required for JIP3–JNK binding (Kelkar et al., 2000). To rule out the possibility that TLR4 and JNK compete for the same binding site in JIP3, we examined if JIP3 induces TLR4–JIP3–JNK complex formation. We transiently transfected HEK 293 cells with Myc-tagged TLR4 with or without JIP3, immunoprecipitated endogenous JNK1 and detected coprecipitated TLR4 by anti-Myc antibodies. As shown in Figure 4B, JIP3 expression significantly increased TLR4 coprecipitating with JNK1, indicating that there is TLR4–JIP3–JNK1 complex formation. We also introduced two amino acid substitutions into the JNK-binding motif of JIP3 (Kelkar et al., 2000). This mutant (JIP3-GG) was significantly less potent for JNK binding (data not shown), but was capable of binding TLR4 with similar affinity to the wild-type JIP3 (Figure 2). Thus, JNK- and TLR4-binding is not mutually exclusive for JIP3.
Expression of JIP3 C-terminal deletion mutants and JIP3 RNA interference inhibit LPS-mediated JNK activation
It has previously been reported that JIP3 binds, in addition to JNK, upstream kinases: MKK7 and MLK3, through the central region of the JIP3 molecule (Kelkar et al., 2000). In another report, JIP3 has been reported to bind MKK4 and MEKK1 through the C-terminal and the central region, respectively (Ito et al., 1999). These reports indicated that the expression of N-terminal region of JIP3 alone may function in a dominant negative fashion. We tested this hypothesis with RAW264.7 cells using both transient and stable expression systems. First we confirmed that RAW264.7 cells constitutively express endogenous JIP3 protein, using two antibodies recognizing the N- and C-terminal domains of JIP3 (Figure 5A). We also easily detected JIP3 protein in mouse peritoneal macrophages induced by thioglycollate (data not shown), indicating that JIP3 protein is generally expressed in mouse macrophages. We then transiently transfected RAW264.7 cells with the expression plasmid for C-terminal deletion mutants of JIP3, in combination with the expression plasmid for HA-tagged JNK1, and compared JNK activity of the anti-HA immunoprecipitates after LPS stimulation. We found that transient expression of either JIP3 1–221 or 1–355 significantly decreased JNK activity in response to LPS but not to TNF-α (Figure 5B).
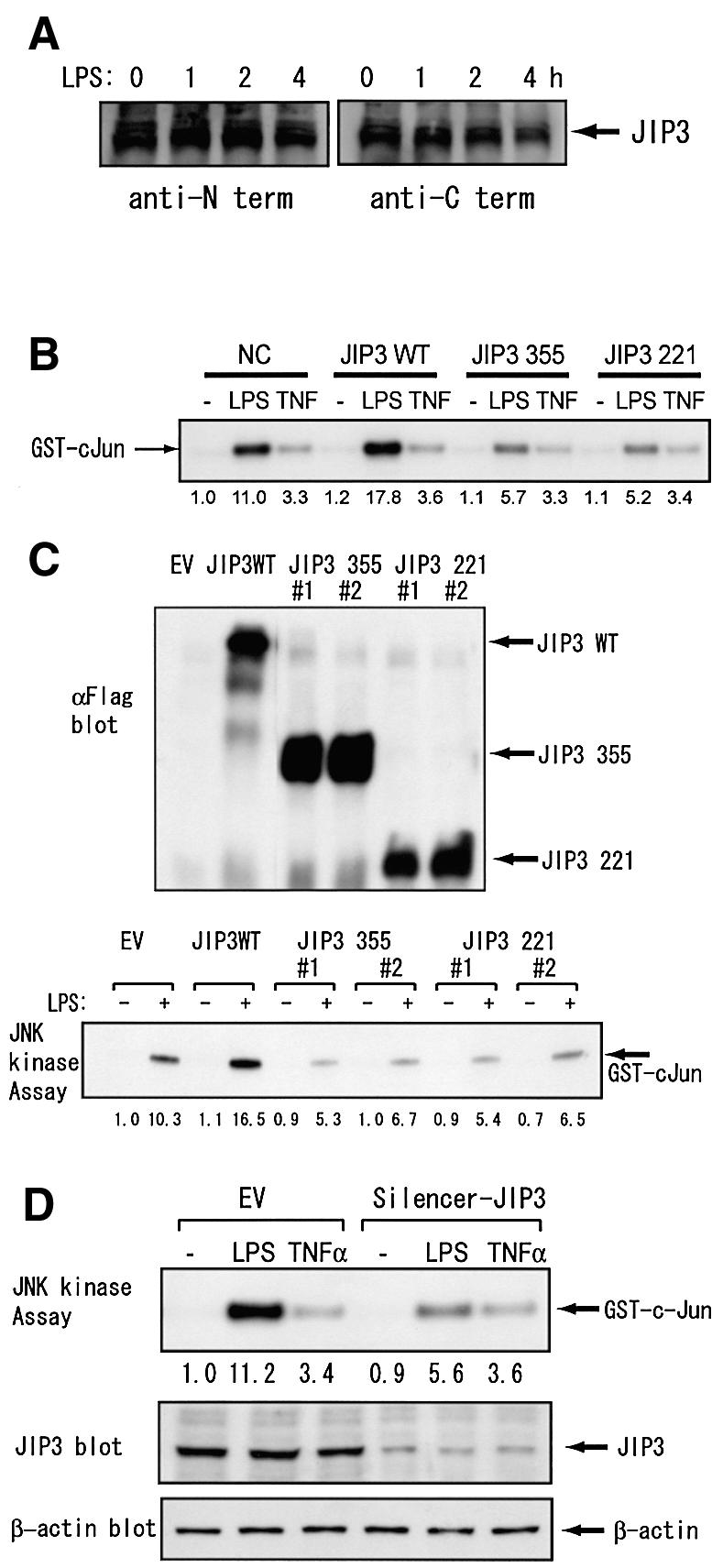
Fig. 5. JIP3 is involved in JNK activation by LPS in a mouse macrophage cell line. (A) RAW264.7 cells were stimulated with 1 µg/ml LPS for the indicated times. Cell lysates were prepared and JIP3 protein contents were analyzed by antibodies specific to the N- and C-terminal domains of JIP3. (B) RAW264.7 cells were transiently transfected with the empty vector, the expression plasmid of the wild-type JIP3 or a JIP3 C-terminal mutant, in combination with the expression plasmid for HA-tagged JNK1. Cells were untreated, treated with 1 µg/ml LPS or 10 ng/ml TNF-α for 20 min, and anti-HA antibody immunoprecipitates were examined for their kinase activity on GST–cJun5-89. (C) RAW264.7 cells stably transfected with the empty vector, the expression plasmid of the wild-type JIP3, or a JIP3 C-terminal mutant were either untreated or treated with 1 µg/ml LPS for 20 min, and anti-JNK1 immunoprecipitates were examined for their kinase activity on GST–cJun5-89. Cell lysates were also examined for the exogenous JIP3 expression with anti-Flag antibody. (D) RAW264.7 cells were stably transfected with either the empty vector or the pSilencer-JIP3 plasmid. Cells were untreated, treated with 1 µg/ml LPS or 10 ng/ml TNF-α for 20 min. JIP3 and β-actin protein contents were examined by their specific antibodies. Subsequently, JNK1 was immunoprecipitated and in vitro kinase assay was performed on GST–cJun5-89.
We next isolated multiple RAW264.7 clones stably expressing full-length or C-terminally deleted JIP3s and analyzed their JNK activity of anti-JNK1 immunoprecipitates after LPS stimulation (Figure 5C). Consistently with the transient transfection experiment described above, more than three independent clones for each of JIP3 1–221 and JIP3 1–355 showed decreased JNK1 activity compared with the empty vector clones (results from two typical clones are shown for each deletion construct in Figure 5B). These results indicate that both JIP3 1–221 and 1–355 function as dominant negative mutants and suggest that JIP3 is physiologically involved in LPS-mediated JNK activation.
Furthermore, we introduced small inhibitory RNA (siRNA) of JIP3 into RAW264.7 cells. It reduced the endogenous protein level of JIP3, but not that of β-actin, which was analyzed as a control (Figure 5D). Introduction of JIP3 siRNA efficiently decreased JNK activation by LPS but not that by TNF-α (Figure 5D), further confirming the specific involvement of JIP3 in LPS signaling.
JIP3 associates with MEKK1, but not TAK1, in RAW264.7 cells
LPS induces activation of several MKKKs. In particular, both MEKK1 and TAK1 have been indicated as essential upstream kinases for JNK activation by LPS. JIP3 has been reported to bind MEKK1 (Ito et al., 1999), whereas the association of TAK1 and JIP3 has not been reported. To examine the possible interaction between JIP3 and the two MKKKs, we used RAW264.7 cells stably expressing Flag-tagged JIP3, and MKKKs coprecipitated with JIP3 were detected with their specific antibodies in the anti-Flag immunoprecipitates. As shown in Figure 6, although no coprecipitation of TAK1 and JIP3 was detected, we found that the full-length form of MEKK1 constitutively associated with JIP3. Furthermore, LPS stimulation rapidly induced the temporary coprecipitation of the processed 72 kDa form of MEKK1 with JIP3. As the ratio of processed/full-length MEKK1 did not change in response to LPS in this cell line (Figure 6A, left panel), it apparently indicated that only a portion of MEKK1 protein that interacts with JIP3 becomes processed on the scaffold protein in response to LPS stimulation.

Fig. 6. Complex formation of JIP3 and MEKK1. RAW264.7 cells stably expressing Flag-tagged JIP3 were stimulated with LPS for the indicated times. (A) The anti-MEKK1 immunoblot of the cell lysates is shown in the left panel. For the immunoprecipitation experiments, anti-Flag or control antibody was cross-linked to protein A beads and incubated with the cell lysates. The immunoprecipitates were separated by SDS–PAGE and probed with anti-MEKK1 antibody (two right panels). (B) Anti-Flag and control antibody immunoprecipitates were separated by SDS–PAGE and probed with anti-TAK1 antibody.
Interaction of other JIP and TLR proteins
Two other JIP family proteins (JIP1 and JIP2), both of which function as JNK scaffold proteins, have been reported (Whitmarsh et al., 1998; Yasuda et al., 1999). Although JIP1 and JIP2 are homologous, JIP3 does not share significant amino acid sequence homology with JIP1 or JIP2. In order to test the possible association of JIP1 and JIP2 with TLR4, we transiently transfected Flag-tagged TLR4 in HEK 293 cells, and analyzed coprecipitated JIP proteins in the anti-Flag immunoprecipitates. HEK 293 cells constitutively express endogenous JIP1, 2 and 3 as shown by the immunoblot using their specific antibodies (Figure 7A). In contrast to JIP3, neither JIP1 nor JIP2 was coprecipitated with TLR4, indicating TLR4 specifically interacts with JIP3. Consistently, expression of the functionally defective mutants of JIP1 (JIP1 281) (Whitmarsh et al., 1998) and JIP2 (JIP2 232) (Yasuda et al., 1999) did not show any significant effects on LPS-induced JNK activation (Figure 7B).

Fig. 7. Involvement of JIP3 in other TLR signals. (A) HEK 293 cells were transiently transfected with p3XFlag-CMV14-TLR4, pcDNA3.1(+)-mCD14, and pcDNA3.1(+)-mMD-2. At 48 h after the transfection, cells were left untreated or stimulated with 1 µg/ml LPS for 20 min and lysed. At 48 h after transfection, cells were lysed and immunoprecipitates of anti-Flag and control antibodies were separated by SDS–PAGE. Immunoblotting was performed with anti-JIP1, anti-JIP2, anti-JIP3 or anti-Flag antibody. (B) HEK 293 cells were transiently transfected with pEEBOS-Flag-JIP3 221, pEFBOS-Flag-JIP1 281 or pEFBOS-Flag-JIP2 232, along with p3XFlag-CMV14-TLR4, pcDNA3.1(+)-mCD14, p3cDNA3.1(+)-mMD-2 and pcDNA3.1(+)-HA-JNK1. At 48 h after the transfection, cells were left untreated or stimulated with 1 µg/ml LPS for 20 min, and lysed. Immunoblotting results using anti-Flag antibody are shown in the upper panel. Anti-HA immunoprecipitates were tested for in vitro kinase assay on GST–cJun5-89 as the substrate. (C) 293T cells were transiently transfected with an expression plasmid of Myc-tagged JIP3 in combination with the indicated expression plasmid of Flag-tagged TLR. At 48 h after transfection, cells were lysed and protein expression was examined using anti-Flag and anti-Myc antibodies (upper two panels). JIP3 was immunoprecipitated with anti-Myc antibody. Coprecipitated TLRs were detected by anti-Flag antibody. (D) RAW264.7 cells stably transfected with the empty vector or the expression plasmid of JIP3 1–355 were treated with 1 µg/ml LPS, 10 μg/ml synthetic lipoprotein, or 1 µM CpG ODN for 20 min and anti-JNK1 antibody immunoprecipitates were examined for their kinase activity on GST–cJun5-89.
We next examined the association of JIP3 with several members of TLR proteins. Flag-tagged TLR1, 2, 4, 6 and 9 were transiently expressed with Myc-JIP3 in HEK 293 cells, and the interaction was analyzed by co-immunoprecipitation assays. As shown in Figure 7C, JIP3 associated with TLR2, 4 and 9, but not with TLR1 or 6. The association was also detected between JIP3 and G-CSFR–TLR2 fusion protein, indicating the TLR2 cytoplasmic domain is sufficient to bind JIP3 (Figure 1C). Furthermore, we stimulated RAW264.7 cells stably expressing a C-terminal deletion mutant of JIP3, JIP3 1–355, with synthetic lipoprotein, which signals through TLR2/TLR6, and CpG bacterial DNA, which signals through TLR9, in addition to LPS (Figure 7D). Consistently, JNK activation was reduced in this cell line after lipoprotein and CpG stimulation as well as LPS treatment compared with that in the control cell line, confirming that JIP3 is also involved in TLR2 and TLR9 signaling.
Discussion
We report that a scaffold protein, JIP3, interacts with TLR4 and appears to enhance JNK activation by LPS. The efficiency of MAPK activation is generally enhanced by the presence of scaffold proteins in both mammals and yeasts. A scaffold protein provides a site for the formation of a signaling module, which contains the elements of an MKKK, an MKK and a MAPK. In yeast, for example, a scaffold protein, Ste5p, is required for the mating Fus3 MAPK activation (Choi et al., 1994). In response to pheromone, a heterotrimeric G protein associates with the N-terminus of Ste5 and transmits the pheromone response pathway (Whiteway et al., 1995). Also, Pbs2p bound to the Sho1p osmosensor, the MKKK Ste11p and the MAPK Hog1p mediate Hog1p activation in response to high extracellular osmolarity (Posas and Saito, 1997). In mammals, the β-arrestin-2 scaffold protein has been reported to be engaged by G protein-coupled receptors and activate the JNK pathway when these receptors bind ligands (McDonald et al., 2000). These examples clearly indicate that at least some scaffold proteins participate in MAPK activation downstream of signaling receptors. However, for most of the many receptors that activate MAPKs, the roles of scaffold proteins in MAPK activation have not been clearly elucidated.
LPS, a cell wall component of Gram-negative bacteria, is a complex glycolipid composed of a hydrophilic polysaccharide region and a hydrophobic domain known as lipid A that is responsible for most of the biological functions of LPS (Schletter et al., 1995). LPS stimulates host cells such as macrophages to produce endogenous mediators including prostaglandins, NO and cytokines. LPS activates MAPKs including ERKs, JNKs, p38 MAPKs and ERK5 (Zhu et al., 2000). Especially, JNK plays an essential role in LPS responses of macrophages by phosphorylating transcription factors including ATF-2 and c-Jun, which are responsible for the transcriptional activation of iNOS and COX-2, and various inflammatory cytokines.
It has recently been shown that most of the LPS signals are mediated by TLR4 (Poltorak et al., 1998; Qureshi et al., 1999). TLRs are a recently identified family of proteins, which recognize molecular patterns of invading pathogens. TLR4 is essential for LPS-mediated JNK activation, as JNK activation is abrogated in mice with TLR4 null-functional mutation, and conversely, ectopic expression of TLR4 causes activation of JNK (Muzio et al., 1998). MyD88, which directly associates with the cytoplasmic domain of TLRs, is required for the early activation of JNK by LPS, because JNK activation is delayed in macrophages of MyD88-deficient mice (Kawai et al., 1999). In contrast, MyD88 is absolutely required for JNK activation by lipoprotein and bacterial DNA, which signal through TLR2 and TLR9, respectively. Another adaptor molecule, TIRAP/Mal, is also essential for the early JNK activation by LPS and lipoprotein, but not by bacterial DNA (Yamamoto et al., 2002). IRAK and IRAK4, serine threonine kinases interacting with MyD88, are also important in LPS-mediated JNK activation, as JNK activation is impaired, but not abrogated, in either IRAK–/– or IRAK4–/– mice (Kanakaraj et al., 1998; Suzuki et al., 2002).
The role of TRAF6, with which IRAKs interact, in LPS-induced JNK activation has remained somewhat controversial. Overexpression of TRAF6 activates JNK (Song et al., 1997). However, an N-terminal deletion mutant of TRAF6, which inhibits NF-κB activation by LPS, failed to inhibit JNK activation in 293T cells ectopically expressing TLR4 (Muzio et al., 1998). In a recent report, however, a similar TRAF6 mutant moderately inhibited JNK activity in an LPS-treated human microvascular endothelial cell line (Hull et al., 2002). The discrepancy may be due to the different involvement of TRAF6-independent induction of IRF-3 transcription factor by LPS, which is also mediated by TLR4. IRF-3 induces the secretion of IFN-β, which may cause TRAF6-independent late JNK activation by an autocrine mechanism. If so, it may be safe to say that TRAF6 is essential for at least the early activation of JNK by LPS.
Two MKKKs, TAK1 and MEKK1, have been indicated as putative JNK activating kinases in association with TRAF6. TAK1 is an ubiquitin-dependent kinase implicated in NF-κB and JNK activation by IL-1 and LPS (Ninomiya-Tsuji et al., 1999; Lee et al., 2000; Wang et al., 2001). TAK1 constitutively binds its activator, TAB1 (Ninomiya-Tsuji et al., 1999). In IL-1-stimulated cells, another adaptor protein, TAB2, links TRAF6 and TAK1, thereby inducing TAK1 activation (Takaesu et al., 2000, 2001). So far, the association of TAK1 and JIP3 has not been reported, and we could not detect the coprecipitation of these two proteins in RAW264.7 cells (Figure 6), indicating that JIP3 may not be involved in TAK1-mediated JNK activation.
In contrast to TAK1, we found that JIP3 constitutively associated with MEKK1 in RAW264.7 cells (Figure 6). TRAF6 interacts with ECSIT, a 435 amino acid ubiquitously expressed protein (Kopp et al., 1999). ECSIT is essential for processing and activation of MEKK1 and is indicated to be a mediator of JNK activation by IL-1 receptor and TLRs. The Drosophila homologue of ECSIT (dECSIT) also associates with Drosophila homologue of TRAF6 (dTRAF6), and expression of dECSIT in Schneider cells induces the production of two antibacterial peptides, defensin and attacin, as efficiently as that of a dominant active mutant of Drosophila Toll (dToll) (Kopp et al., 1999). Thus, ECSIT is an evolutionally conserved mediator of innate immunity responses against pathogens. MEKK1 is a 195 kDa protein that is activated on proteolytic cleavage by DEVD-specific caspases (Cardone et al., 1997; Deak et al., 1998). Notably, we could detect the processed form of MEKK1 temporarily coprecipitated with JIP3 in LPS-treated RAW264.7 cells. As LPS did not seem to increase the whole content of this form of MEKK1, it apparently indicates that LPS specifically process MEKK1 molecules that is associated with JIP3. Interestingly, both caspase 3 and 9 are activated in mouse macrophages by ingestion of Escherichia coli (Hacker et al., 2002), while TLR2 signal activates caspase 1 and 8 (Aliprantis et al., 2000).
MEKK1 is a strong activator of JNK in many cell types. Especially, using MEKK1-deficient embryonic stem cells, Xia and coworkers recently found that, in addition to its function in JNK activation by growth factors, MEKK1 is required for JNK activation by diverse proinflammatory stimuli, including TNF-α, IL-1, double-stranded RNA and LPS, whereas MEKK1 is not required for activation of other MAPK or the IκB kinase signaling cascade (Xia et al., 2000). Thus, based on our current data, we would like to propose a model in which JIP3 is involved in ECSIT–MEKK1-dependent JNK activity in response to LPS (Figure 8), although we can not completely rule out the possibility that TAK1 also binds JIP3 with low affinity. Consistently with our data, association of JIP3 with MEKK1 has previously been reported using COS7 overexpression system (Ito et al., 1999). Quite recently, TAB2-deficient mice have been generated (Sanjo et al., 2003). Notably, in TAB2–/– mouse embryonic fibroblasts, JNK and NF-κB activation occurred normally in response to IL-1 and LPS stimulation, whereas TAK1 phosphorylation was abrogated. These findings indicate that TAK1 activation is at least not essential for JNK activation by LPS or IL-1.

Fig. 8. Schematic demonstration of the role of JIP3 in LPS signaling.
Other than TAK1 and MEKK1, two other MKKKs, ASK1 and NIK, have been shown to interact directly with TRAFs. Although its Drosophila homologue was indicated in association with JNK activation (Su et al., 1998), NIK has not been clearly shown to function upstream of JNK in mammals. On the other hand, ASK1 activates both JNK and p38 kinase pathways in response to various stress signals (Ichijo et al., 1997). Notably, ASK1 has recently been shown to interact with JIP3 protein (Matsuura et al., 2002). Although the role of ASK1 has never been clearly shown in LPS responses, the possible involvement of ASK1 in LPS-mediated JNK activation need to be carefully examined.
Our findings presented here provide some insight into how JIP3 is involved in LPS signaling. Although JIP3 binds the cytoplasmic domain of TLR4, the binding itself appears insufficient to initiate the downstream signals. Firstly, the binding is detected in the absence of LPS stimulation without causing an increase of JNK activity (Figure 3). Secondly, JIP3 is able to associate with the Pro712His mutant of TLR4, which is defective in JNK activation. Thus, although JIP3 increases JNK activation efficiency by providing a scaffold for the kinase module formation near the TLR4 receptor, LPS-mediated JNK activation needs an initiation signal, which seems to be provided by the MyD88/TIRAP–TRAF6–ECSIT–MEKK1 pathway (Figure 8).
Our current data have shown that JIP3, one of the JNK scaffold proteins, interacts with several members of TLRs and promotes JNK activation on receptor engagement. Study of the possible involvement of other scaffold proteins in various immune receptor systems is warranted.
Materials and methods
Antibodies and reagents
Goat polyclonal antibodies against JIP1and JIP2, N- and C-terminal regions of JIP3, an anti-HA mouse monoclonal antibody (12CA5), rabbit polyclonal antibodies against human G-CSF receptor (H-176), JNK1, p38 kinase, TLR4 (H-80), C-terminus of MEKK1 (C-22) and TAK1 were purchased from Santa Cruz Biotech (Santa Cruz, CA). The anti-FLAG M2 mouse monoclonal antibody was from Sigma Chemical Co. (St Louis, MO). A phospho-specific anti-p38 rabbit polyclonal antibody was purchased from New England Biolabs (Beverly, MA). LPS from E.coli serotype B6: 026 was obtained from Sigma Chemical Co. Phosphorothioate-stabilized CpG oligodeoxynucleotide (ODN) (TCCATGACGTTCCTGATGCT) was purchased from Rikaken Co. (Nagoya, Japan). Synthetic lipoprotein [Palmitoyl-Cys{(RS)-2,3-di(palmitoyloxy)- propyl}-Ala-Gly-OH] was obtained from Bachem AG (Bubendorf, Switzerland).
Cells
A mouse macrophage cell line, RAW264.7, a monkey kidney cell line, COS7, and a human embryonic kidney cell line, HEK 293, were obtained from RIKEN cell bank (Tsukuba, Japan), and maintained in DMEM with 10% FCS (Sigma Chemical Co.).
Yeast two-hybrid screening
A mouse fetal (day 10.5) cDNA library (Gibco-BRL, Rockville, MD) was screened using the PROQUEST™ Yeast Two-hybrid System (Gibco-BRL) according to the manufacturer’s instructions. The bait was constructed by fusing the cytoplasmic portion of mouse TLR4 in frame with GAL4 DNA binding domain in pDBLeu vector. The cytoplasmic portion of TLR4 (174 amino acids) was PCR amplified from BALB/c mouse spleen cDNA using CTAGCTAGCTGTAAAAGTACAGCAGAGGA and GTTGCGGCCGCTCAGGTCCAAGTTGCCGTT as primers, and ligated into NheI/NotI sites of pDBLeu. The partial sequences of the pPC86cDNA plasmids purified from the positive yeast colonies were searched for homology to known sequences with the BLAST program (http://www.ncbi.nlm.nih.gov/BLAST).
Mammalian expression plasmids
The expression plasmid for the Flag-tagged mouse JIP3a (pcDNA3-Flag-mJIP3wt) was a generous gift from Dr Roger J.Davis (University of Massachusetts Medical School, Worcester, MA). A series of mJIP3a C-terminal deletion mutants were prepared by PCR from pdDNA3-Flag-mJIP3wt as the template using the 5′-end primer: CGACGCCGTATGATGGAGATCCAGATGGACG, and 3′-end primers: CGACGCGTTCACTCAGGGGTGTAGGACACC (full); CGACGCGTTCATACCA GCACTCGGCCTTTG (1–1015); CGACGCGTTCAGCTCAGCTGCTTGTACTTG (1–677); CGACGCGTTCAACACACATCCAGCTCTGGG (1–355); CGACGCGTTCATGCACGTACCATGCCATCA (1–221); CGACGCGTTCAGGCATTGTATTCCTTCTTC (1–163), and inserted into the MluI site of the expression vector pEFBOS-FLAG or pEFBOS-Myc (Matsuguchi and Kraft, 1998) to yield pEFBOS-FLAG or Myc/JIP3 full, 1–355, 1–221, 1–163, respectively.
The expression plasmid for JNK binding domain mutant of JIP3 (mutated from Arg205Pro206 to Gly205Gly206) was constructed by recombinant PCR (ishizaka-Ikeda et al., 1993) using four primers: CGACGCGTATGGCCCATGAGATGATTGGAA, CGACGCGTCTATTTTTTGTGAACAGGAAAC, GTGCTTATCCACAGCTTA and GATCCCAGCTAAGCTGTG. The PCR product was cloned into the MluI site of pEFBOS-FLAG or Myc to yield pEFBOS-FLAG or Myc/JIP3-GG. The three-nucleotide substitution (from CGTCCC to GGTGGC) was confirmed by nucleotide sequencing.
The expression plasmids for mouse JIP1 and JIP2 C-terminal deletion mutants, pEFBOS-Flag/JIP1 1–281 and pEFBOS-Flag/JIP2 1–232 respectively, were prepared by RT–PCR using 5′-end primer: CGACGCCGTATGGCGGAGCGAGAGAGCGGCC (JIP1) or CGACGCCGTATGGCGGATCGCGCGGAGATGT (JIP2), and 3′-end primer CGA CGCCGTTCATGGGGTCAGGTAGATCTCCTC (JIP1) or CGACGCCGTTCACTCAATACCAGGATCCGAGGA (JIP2) from BALB/c mouse spleen total RNA. The PCR products were ligated into the MluI site of pEFBOS-Flag vector. The orientation of the inserts was determined by restriction enzyme mapping.
The coding region of the mouse TLR cDNAs were amplified from BALB/c mouse spleen cDNA by PCR using pairs of primers. Additionally, the coding region of the mal-functional mouse TLR4 (TLR4 Pro712His) cDNA was amplified from C3H/HeJ mouse spleen cDNA. The amplified PCR products were cloned into p3X Flag-CMV14 vector (Sigma Chemical Co.). For the C-terminal deletion mutants of TLR4, 3′-end primers: CGGGATCCCTCAGGATTCGAGGCTTTTCCA (TLR4 d13); CGGGATCCGCAGAGGTGAAAGCGGGGCACT (TLR4 d130), were used in combination with the TLR4 5′-end primer for PCR amplification from BALB/c mouse spleen cDNA. The PCR products were digested with NotI/BamHI and inserted into p3X Flag-CMV14.
The expression plasmids of chimeric protein of human G-CSF receptor extracellular domain/mouse TLR2 or TLR4 cytoplasmic domain was prepared by first cloning cDNA coding human G-CSF receptor extracellular domain by RT–PCR and cloned into pcDNA3.1(+) producing a plasmid pcDNA3.1(+)-GCSFR. Cytoplasmic domain cDNAs for mouse TLR2 and TLR4 were prepared by PCR and cloned into pcDNA3.1(+)-GCSFR, producing pcDNA3.1(+)-GCSFR/TLR2 and pcDNA3.1(+)-GCSFR/TLR4.
The coding regions of mouse JNK1 was amplified by RT–PCR and inserted with the N-terminal HA tag into a mammalian expression vector, pcDNA3.1(+) (Invitrogen, Carlsbad, CA) to yield pcDNA3.1-HA/JNK1. The expression plasmids, pcDNA3.1(+)-mMD2 and p3XFlag-CMV10-mMD2, were prepared by cloning the PCR product into pdDNA3.1(+) and p3XFlag-CMV10 vectors, respectively. The pcDNA3.1(+)-mCD14 has previously been described (Matsuguchi et al., 2000). The structure of all plasmid constructs was verified by restriction enzyme mapping and nucleotide sequencing.
Transient transfection
Cells were plated onto 60 mm plates at 1 × 106 cells/plate on the day before transfection. Combinations of expression plasmid DNAs (6 µg total/plate) were dissolved in 300 µl of OptiMEM™ (Invitrogen) and mixed well with 15 µl of LipofectAMINE™ (Invitrogen) dissolved in 300 μl of OptiMEM™. The mixture was incubated at room temperature for 30 min. The mixture was added to 2.4 ml of OptiMEM™ and poured onto cells after the removal of the culture medium. After the incubation at 37°C for 12 h, the DNA–LipofectAMINE™ mixture was replaced with 4 ml of regular medium. The cells were harvested with PBS after a further 36 h incubation at 37°C and used for analyses.
Immunoblotting, immunoprecipitation and immune complex kinase assay
Cell lysate preparation and immunoblotting were performed as previously described (Matsuguchi et al., 2001). Immunoprecipitation and immune complex kinase assays were carried out as previously described (Matsuguchi et al., 2001) except for MEKK1 coprecipitation experiment, in which antibodies were cross-linked to protein A beads by dimethylpimelimidate prior to the incubation with cell lysates.
RNA interference
For the plasmid construct of JIP3 small inhibitory RNA (siRNA), the insert was prepared by annealing two oligonucleotides: sense CAGGCCGAGGAGAAATTCATTCAAGAGATGAATTTCTCCTCG GCCTGTTTTTT; anti-sense AATTAAAAAATGAATTTCTCCTCGG CCTGTCTCTTGAACAGGCCGAGGAGAAATTCAGGCC. The annealed insert was cloned into pSilencer™ 1.0-U6 (Ambion Inc., Austin, TX) digested with ApaI–EcoRI. The correct structure of the positive clone was confirmed by nucleotide sequencing. The resultant plasmid, pSilencer-JIP3, was cotransfected with pcDNA3.1(+) into RAW264.7 cells, and G418-resistant cell clones were isolated. The interference of JIP3 protein expression was confirmed by immunoblot using anti-JIP3 C-terminus antibody.
Acknowledgments
Acknowledgements
We thank Ms K.Itano and Ms A.Nishikawa for their technical assistance. This work was supported in part by grants from Ono Pharmaceutical Company, Ministry of Education, Science and Culture of the Japanese Government and the Yakult Bioscience Foundation.
References
- Akira S. (2001) Toll-like receptors and innate immunity. Adv. Immunol., 78, 1–56. [DOI] [PubMed] [Google Scholar]
- Aliprantis A.O., Yang,R.B., Weiss,D.S., Godowski,P. and Zychlinsky,A. (2000) The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J., 19, 3325–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman A.B., Kamal,A., Ritchings,B.W., Philp,A.V., McGrail,M., Gindhart,J.G. and Goldstein,L.S. (2000) Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell, 103, 583–594. [DOI] [PubMed] [Google Scholar]
- Buchsbaum R.J., Connolly,B.A. and Feig,L.A. (2002) Interaction of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. Mol. Cell. Biol., 22, 4073–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd D.T., Kawasaki,M., Walcoff,M., Hisamoto,N., Matsumoto,K. and Jin,Y. (2001) UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C.elegans. Neuron, 32, 787–800. [DOI] [PubMed] [Google Scholar]
- Cacace A.M., Michaud,N.R., Therrien,M., Mathes,K., Copeland,T., Rubin,G.M. and Morrison,D.K. (1999) Identification of constitutive and ras-inducible phosphorylation sites of KSR: implications for 14-3-3 binding, mitogen-activated protein kinase binding and KSR overexpression. Mol. Cell. Biol., 19, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone M.H., Salvesen,G.S., Widmann,C., Johnson,G. and Frisch,S.M. (1997) The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell, 90, 315–323. [DOI] [PubMed] [Google Scholar]
- Choi K.Y., Satterberg,B., Lyons,D.M. and Elion,E.A. (1994) Ste5 tethers multiple protein kinases in the MAP kinase cascade required for mating in S.cerevisiae. Cell, 78, 499–512. [DOI] [PubMed] [Google Scholar]
- Deak J.C., Cross,J.V., Lewis,M., Qian,Y., Parrott,L.A., Distelhorst,C.W. and Templeton,D.J. (1998) Fas-induced proteolytic activation and intracellular redistribution of the stress-signaling kinase MEKK1. Proc. Natl Acad. Sci. USA, 95, 5595–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C., Yang,D.D., Wysk,M., Whitmarsh,A.J., Davis,R.J. and Flavell,R.A. (1998) Defective T cell differentiation in the absence of Jnk1. Science, 282, 2092–2095. [DOI] [PubMed] [Google Scholar]
- Hacker H., Furmann,C., Wagner,H. and Hacker,G. (2002) Caspase-9/-3 activation and apoptosis are induced in mouse macrophages upon ingestion and digestion of Escherichia coli bacteria. J. Immunol., 169, 3172–3179. [DOI] [PubMed] [Google Scholar]
- Hull C., McLean,G., Wong,F., Duriez,P.J. and Karsan,A. (2002) Lipopolysaccharide signals an endothelial apoptosis pathway through TNF receptor-associated factor 6-mediated activation of c-Jun NH2-terminal kinase. J. Immunol., 169, 2611–2618. [DOI] [PubMed] [Google Scholar]
- Ichijo H. et al. (1997) Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science, 275, 90–94. [DOI] [PubMed] [Google Scholar]
- ishizaka-Ikeda E., Fukunaga,R., Wood,W.I., Goeddel,D.V. and Nagata,S. (1993) Signal transduction mediated by growth hormone receptor and its chimeric molecules with the granulocyte colony-stimulating factor receptor. Proc. Natl Acad. Sci. USA, 90, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. et al. (1999) JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol. Cell. Biol., 19, 7539–7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanakaraj P. et al. (1998) Interleukin (IL)-1 receptor-associated kinase (IRAK) requirement for optimal induction of multiple IL-1 signaling pathways and IL-6 production. J. Exp. Med., 187, 2073–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., Adachi,O., Ogawa,T., Takeda,K. and Akira,S. (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity, 11, 115–122. [DOI] [PubMed] [Google Scholar]
- Kelkar N., Gupta,S., Dickens,M. and Davis,R.J. (2000) Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol. Cell. Biol., 20, 1030–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp E., Medzhitov,R., Carothers,J., Xiao,C., Douglas,I., Janeway,C.A. and Ghosh,S. (1999) ECSIT is an evolutionarily conserved intermediate in the Toll/IL-1 signal transduction pathway. Genes Dev., 13, 2059–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Mira-Arbibe,L. and Ulevitch,R.J. (2000) TAK1 regulates multiple protein kinase cascades activated by bacterial lipopoly saccharide. J. Leukoc. Biol., 68, 909–915. [PubMed] [Google Scholar]
- Leppa S. and Bohmann,D. (1999) Diverse functions of JNK signaling and c-Jun in stress response and apoptosis. Oncogene, 18, 6158–6162. [DOI] [PubMed] [Google Scholar]
- Matsuguchi T. and Kraft,A.S. (1998) Regulation of myeloid cell growth by distinct effectors of Ras. Oncogene, 17, 2701–2709. [DOI] [PubMed] [Google Scholar]
- Matsuguchi T., Takagi,K., Musikacharoen,T. and Yoshikai,Y. (2000) Gene expressions of lipopolysaccharide receptors, toll-like receptors 2 and 4, are differently regulated in mouse T lymphocytes. Blood, 95, 1378–1385. [PubMed] [Google Scholar]
- Matsuguchi T., Musikacharoen,T., Johnson,T.R., Kraft,A.S. and Yoshikai,Y. (2001) A novel mitogen-activated protein kinase phosphatase is an important negative regulator of lipopoly saccharide-mediated c-Jun N-terminal kinase activation in mouse macrophage cell lines. Mol. Cell. Biol., 21, 6999–7009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura H., Nishitoh,H., Takeda,K., Matsuzawa,A., Amagasa,T., Ito,M., Yoshioka,K. and Ichijo,H. (2002) Phosphorylation-dependent scaffolding role of JSAP1/JIP3 in the ASK1–JNK signaling pathway. A new mode of regulation of the MAP kinase cascade. J. Biol. Chem., 277, 40703–40709. [DOI] [PubMed] [Google Scholar]
- McDonald P.H., Chow,C.W., Miller,W.E., Laporte,S.A., Field,M.E., Lin,F.T., Davis,R.J. and Lefkowitz,R.J. (2000) β-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science, 290, 1574–1577. [DOI] [PubMed] [Google Scholar]
- Muzio M., Natoli,G., Saccani,S., Levrero,M. and Mantovani,A. (1998) The human toll signaling pathway: divergence of nuclear factor κB and JNK/SAPK activation upstream of tumor necrosis factor receptor-associated factor 6 (TRAF6). J. Exp. Med., 187, 2097–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya-Tsuji J., Kishimoto,K., Hiyama,A., Inoue,J., Cao,Z. and Matsumoto,K. (1999) The kinase TAK1 can activate the NIK-I κB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature, 398, 252–256. [DOI] [PubMed] [Google Scholar]
- Poltorak A. et al. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science, 282, 2085–2088. [DOI] [PubMed] [Google Scholar]
- Posas F. and Saito,H. (1997) Osmotic activation of the HOG MAPK pathway via Ste11p MAPKKK: scaffold role of Pbs2p MAPKK. Science, 276, 1702–1705. [DOI] [PubMed] [Google Scholar]
- Qureshi S.T., Lariviere,L., Leveque,G., Clermont,S., Moore,K.J., Gros,P. and Malo,D. (1999) Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J. Exp. Med., 189, 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee S.H. and Hwang,D. (2000) Murine TOLL-like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NF-κB and expression of the inducible cyclooxygenase. J. Biol. Chem., 275, 34035–34040. [DOI] [PubMed] [Google Scholar]
- Robinson M.J. and Cobb,M.H. (1997) Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol., 9, 180–186. [DOI] [PubMed] [Google Scholar]
- Sanjo H., Takeda,K., Tsujimura,T., Ninomiya-Tsuji,J., Matsumoto,K. and Akira,S. (2003) TAB2 is essential for prevention of apoptosis in fetal liver but not for interleukin-1 signaling. Mol. Cell. Biol., 23, 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer H.J., Catling,A.D., Eblen,S.T., Collier,L.S., Krauss,A. and Weber,M.J. (1998) MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science, 281, 1668–1671. [DOI] [PubMed] [Google Scholar]
- Schletter J., Heine,H., Ulmer,A.J. and Rietschel,E.T. (1995) Molecular mechanisms of endotoxin activity. Arch. Microbiol., 164, 383–389. [DOI] [PubMed] [Google Scholar]
- Smith J.L., Schaffner,A.E., Hofmeister,J.K., Hartman,M., Wei,G., Forsthoefel,D., Hume,D.A. and Ostrowski,M.C. (2000) ets-2 is a target for an akt (protein kinase B)/Jun N-terminal kinase signaling pathway in macrophages of motheaten-viable mutant mice. Mol. Cell. Biol., 20, 8026–8034.11027273 [Google Scholar]
- Song H.Y., Regnier,C.H., Kirschning,C.J., Goeddel,D.V. and Rothe,M. (1997) Tumor necrosis factor (TNF)-mediated kinase cascades: bifurcation of nuclear factor-κB and c-jun N-terminal kinase (JNK/SAPK) pathways at TNF receptor-associated factor 2. Proc. Natl Acad. Sci. USA, 94, 9792–9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y.C., Treisman,J.E. and Skolnik,E.Y. (1998) The Drosophila Ste20-related kinase misshapen is required for embryonic dorsal closure and acts through a JNK MAPK module on an evolutionarily conserved signaling pathway. Genes Dev., 12, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N. et al. (2002) Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature, 416, 750–756. [DOI] [PubMed] [Google Scholar]
- Takaesu G., Kishida,S., Hiyama,A., Yamaguchi,K., Shibuya,H., Irie,K., Ninomiya-Tsuji,J. and Matsumoto,K. (2000) TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell, 5, 649–658. [DOI] [PubMed] [Google Scholar]
- Takaesu G., Ninomiya-Tsuji,J., Kishida,S., Li,X., Stark,G.R. and Matsumoto,K. (2001) Interleukin-1 (IL-1) receptor-associated kinase leads to activation of TAK1 by inducing TAB2 translocation in the IL-1 signaling pathway. Mol. Cell. Biol., 21, 2475–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey K.J., Meyer,D., Deehan,R., Blenis,J., Schnapp,B.J., Rapoport,T.A. and Margolis,B. (2001) Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol., 152, 959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C., Deng,L., Hong,M., Akkaraju,G.R., Inoue,J. and Chen,Z.J. (2001) TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature, 412, 346–351. [DOI] [PubMed] [Google Scholar]
- Whiteway M.S., Wu,C., Leeuw,T., Clark,K., Fourest-Lieuvin,A., Thomas,D.Y. and Leberer,E. (1995) Association of the yeast pheromone response G protein βγ subunits with the MAP kinase scaffold Ste5p. Science, 269, 1572–1575. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J. and Davis,R.J. (1998) Structural organization of MAP-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem. Sci., 23, 481–485. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J., Cavanagh,J., Tournier,C., Yasuda,J. and Davis,R.J. (1998) A mammalian scaffold complex that selectively mediates MAP kinase activation. Science, 281, 1671–1674. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J. et al. (2001) Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev., 15, 2421–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y., Makris,C., Su,B., Li,E., Yang,J., Nemerow,G.R. and Karin,M. (2000) MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc. Natl Acad. Sci. USA, 97, 5243–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M. et al. (2002) Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature, 420, 324–329. [DOI] [PubMed] [Google Scholar]
- Yang D.D., Conze,D., Whitmarsh,A.J., Barrett,T., Davis,R.J., Rincon,M. and Flavell,R.A. (1998) Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity, 9, 575–585. [DOI] [PubMed] [Google Scholar]
- Yasuda J., Whitmarsh,A.J., Cavanagh,J., Sharma,M. and Davis,R.J. (1999) The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol., 19, 7245–7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W., Downey,J.S., Gu,J., Di Padova,F., Gram,H. and Han,J. (2000) Regulation of TNF expression by multiple mitogen-activated protein kinase pathways. J. Immunol., 164, 6349–6358. [DOI] [PubMed] [Google Scholar]