

Abstract
Crystal structures of the bacterial multidrug transporter AcrB in R32 and C2 space groups showing both symmetric and asymmetric trimeric assemblies respectively, supplemented with biochemical investigations, have provided most of the structural basis for a molecular level understanding of the protein structure and mechanisms for substrate uptake and translocation carried out by this 114 kDa inner membrane protein. They suggest that AcrB captures ligands primarily from the periplasm. Substrates can also enter the inner cavity of the transporter from the cytoplasm, but the exact mechanism of this remains undefined.
Analysis of the amino acids sequences of AcrB and its homologs revealed the presence of conserved residues at the N-terminus including two phenylalanines which may be exposed to the cytoplasm. Any potential role that these conserved residues may play in function has not been addressed by existing biochemical or structural studies. Since phenylalanine residues elsewhere in the protein have been implicated in ligand binding, we explored the structure of this N-terminal region to investigate structural determinants near the cytoplasmic opening that may mediate drug uptake. Our structure of AcrB in R32 space group reveals an N-terminus loop, reducing the diameter of the central opening to ∼15 Å as opposed to the previously reported value of ∼30 Å for crystal structures in this space group with disordered N-terminus. Recent structures of the AcrB in C2 space group have revealed a helical conformation of this N-terminus but have not discussed its possible implications. We present the crystal structure of AcrB that reveals the structure of the N-terminus containing the conserved residues. We hope that the structural information provides a structural basis for others to design further biochemical investigation of the role of this portion of AcrB in mediating cytoplasmic ligand discrimination and uptake.
1. Introduction
There are five families of bacterial multidrug exporters (Nikaido 1996; van Veen and Konings 1997; Putman, van Veen et al. 2000) which include H+/drug antiporters from the Major Facilitator Superfamily (MFS), Small Multidrug Resistance (SMR) and Resistance Nodulation Division (RND) transporters, Na+/drug antiporter exemplified by Multidrug and Toxic Compound Extrusion (MATE), and ATP hydrolysis driven transporters which form the ATP-Binding Cassette (ABC) family. These transporters are the key proteins involved in the ejection of antibiotics and chemotherapeutic agents to the external environment of the cell. The major multidrug efflux proteins in mammalian cells are ABC transporters (Dean, Rzhetsky et al. 2000) which are not as prevalent in bacteria (van Veen, Callaghan et al. 1998; Kobayashi, Nishino et al. 2001).
The Acriflavine resistance protein B, AcrB, belongs to the RND family of efflux proteins. Its size of ∼114 kDa comprised of 1049 amino acid residues makes it one of the largest efflux proteins. AcrB is responsible for the primary resistance mechanism in bacteria due to its ability to recognize a large number of toxic compounds including drugs, detergents, dyes and bile acids and has the ability to export from the periplasm and the cytoplasm. The RND family is evolutionarily conserved in humans and includes the Niemann-Pick type C disease protein (Tseng, Gratwick et al. 1999) that is believed to transport fatty acids and cholesterol out of the cell.
Numerous crystal structures of the unliganded and ligand bound AcrB in R32 (symmetric AcrB assembly) and C2 space groups (asymmetric assembly) have been reported (Murakami, Nakashima et al. 2002; Yu, McDermott et al. 2003; Pos, Schiefner et al. 2004; Yu, Aires et al. 2005; Murakami, Nakashima et al. 2006; Seeger, Schiefner et al. 2006). The AcrB monomers associate into a trimeric assembly that is inserted perpendicular to the inner membrane with 12 transmembrane helices (TMs) in each molecule. The periplasmic portions of the molecule span about 70 Å, taper toward each other at the top and form the TolC docking region. This interacts with the TolC outer membrane protein (Koronakis, Sharff et al. 2000) via the adapter protein MexA/AcrA homolog (Akama, Matsuura et al. 2004; Higgins, Bokma et al. 2004; Mikolosko, Bobyk et al. 2006) to complete the AcrB-AcrA-TolC efflux assembly (Eswaran, Koronakis et al. 2004) to eject toxic compounds from the cell.
Taken together, the biochemical and crystallographic studies on AcrB overwhelmingly support the proposed models of drug capture through the vestibules linking the central cavity with the periplasm. It has been more difficult to explain the exact mechanism of cytosolic drug capture (Yu, R. et al. 2003) which remains one of the unresolved questions in multidrug efflux by AcrB. One proposed mechanism for the cytoplasmic uptake is that molecules may laterally diffuse in the inner membrane and slide up the transmembrane grooves on the outside of the trimer and enter through the vestibule. This hypothesis does not exclude the possibility of drug uptake directly from the cytoplasm through the central cytoplasmic opening of the transporter (Murakami, Nakashima et al. 2002).
An analysis of the sequences of AcrB and its homologs revealed the presence of conserved amino acids including two consecutive phenylalanines at the N-terminus of the protein which should be exposed to the cytoplasm. As conserved phenylalanines elsewhere in the protein have been implicated in ligand binding, we have solved the crystal structure of the intact endogenous AcrB at 3.1 Å resolution to reveal the structure of the N-terminus to better understand any potential structural determinants near the cytoplasmic opening which may mediate cytoplasmic drug capture. All previously reported structures of AcrB in R32 space group lacked the first six N-terminal amino acids in the crystal structures due to disorder. Recent crystal structures in C2 space group have this N-terminus present, but do not discuss any potential effects of its presence.
Our structure reveals that the presence of these amino acids results in a central opening of only ∼15 Å that is comparable to the size of most substrates (as opposed to the previously reported value of ∼30 Å in R32 space group and ∼24 Å in C2 space group, mostly due to the disordered N-terminal structures), while still maintaining the inner membrane cavity diameter of ∼30Å. Conserved phenylalanines F4 and F5 face the inside of the channel just like residues F386, F388, F458 and F459 further up the cavity. We suggest that these N-terminal residues may play an important role in the capture of ligands from the cytoplasm and present our structure to aid others in future biochemical experiments to study the effects of these conserved amino acid residues in function.
2. Experimental Procedures
2. 1 Protein expression and purification
Native and seleno-methionine derivatized endogenous AcrB protein was produced in E. coli BL21(DE3)Star and B834(DE3)Star strains in Luria Broth and M9 and PASM media by the method of Studier (Studier 2005). The cell paste was resuspended in lysis buffer comprised of 20 mM Tris-HCl pH 8.0, 0.1 M NaCl, 10 mM βME, 1 mM PMSF, 10 μg/ml DNAseI and Roche Protease Inhibitor Cocktail, EDTA free (Roche Diagnostics, Indianapolis, IN) and lysed in a microfludizer. The lysed cells were centrifuged in a Sorvall Centrifuge at 25000 x g for 25 minutes at 4°C to eliminate inclusion bodies and cell debris. The supernatant was carefully decanted and spun at 100,000 x g in a Beckman ultracentrifuge for 2 hours at 4°C to isolate the membrane fraction. The membrane fraction was extracted by solubilization with 20 mM Tris-HCl pH 8.0, 0.1 M NaCl and 1% DDM (w/v) with stirring for 2 hours at 4°C. After extraction, the sample was centrifuged for 60 minutes at 100,000 x g to obtain solubilized membrane proteins. The sample was diluted 1:1 in 20 mM Tris-HCl pH 8.0, 0.5 M NaCl (to obtain a final salt concentration of 0.3M NaCl) and purified though a 5 ml HisTrap HP column (GE Healthcare, Piscataway, NJ). The sample was purified through a second metal affinity column or by ion-exchange chromatography. AcrB co-purified with another membrane protein but preferentially crystallized.
2.2 Crystallization and x-ray diffraction data collection
Crystallization screens were set up using several commercially available high-throughput crystallization screening kits. Native and selenomethionine derivatized crystals of endogenous AcrB appeared in 3 to 8 weeks at room temperature in several crystal morphologies including pyramids, bipyramids and rhomboids. All of these crystals ranged in size from 30 μ −80 μ in each dimension with the average size of about 50 μ x 50 μ x 50 μ and in several cases up to 200 μ x 200 μ x 200 μ. They appeared in several conditions containing either 0.1 M HEPES pH 7.5, 0.1 M MOPS pH 7.0, or 0.1 M MES pH 6.5; 0.1 M NaCl, 0.1 M Li2SO4 and 12% PEG4000 (w/v) in 96-well sitting drop crystallization screens (Molecular Dimensions Limited, Cambridgeshire, UK). Crystals were harvested from the 96 well plates and soaked in the crystallization solution with the addition of 25% (v/v) glycerol for cryoprotection prior to flash freezing in liquid nitrogen. A native data set to 3.1 Å resolution from a crystal grown in 0.1 M HEPES pH 7.5, 0.3 M Li2SO4 and 12% PEG4000 (w/v), a seleno-methionine derivative data set to 3.2 Å and a 6.0 Å data set on a Ta6Br14 soaked crystal were collected at synchrotron beamlines 5.0.2 and 8.2.2 at the Advanced Light Source, Lawrence Berkeley National Laboratory. Data statistics for the 3.1 Å native data set are reported in Table 1. The crystals belong to space group R32 (Hexagonal setting, H32) with unit cell dimensions of a=b=146.577Å, c=515.118 Å, α=β=90°, γ=120°. The Matthews coefficient (4.53) and solvent content analysis (∼73%) indicated the presence of one AcrB molecule in the asymmetric unit.
Table I.
X-ray diffraction data
| Data set | Native |
| Space group | R32 |
| Wavelength (Å) | 0.9918 |
| Resolution (Å) | 3.1 |
| Reflections (Total/Unique) | 358101/39489 |
| Redundancya | 9.1(8.9) |
| I/σa | 20.4(2.3) |
| Completenessa (%) | 99.9 (100.0) |
| Rsyma,b (%) | 6.9 (75.6) |
| Overall B-factor from Wilson Plot | 66.7 Å2 |
Numbers in parentheses are related to the highest resolution shell (3.21–3.10 Å)
Rsym = Σhkl Σi|Ihkl, i −〈I〉hkl| / Σ |〈I〉hkl|
2.3 Structure determination
We initially resorted to experimental phasing methods for structure determination since there was a possibility of a contaminating protein in the sample preparation having crystallized. Initial phases were obtained from a single tantalum cluster (located using HySS (Grosse-Kunstleve and Adams 2003) in the PHENIX (Adams, Grosse-Kunstleve et al. 2002) package) at 6.0Å from a Ta6Br14 derivatized crystal that were then used to locate some selenium atom positions in the SeMet derivative data set at 4.5Å using difference Fourier methods implemented in SOLVE (Terwilliger and Berendzen 1999). Phases from these two derivatives were then combined (CAD and SIGMAA in CCP4 (1994)) and extended to 3.0Å using RESOLVE (Terwilliger 2000). The electron density map allowed the tracing of a partial model including the transmembrane helices and the β-sheet portion of the molecule in the pore domains of the protein. At this point, it was clear that the molecule was indeed that of AcrB through a DALI (Holm and Sander 1998) search. The 3.08 Å crystal structure of AcrB (1T9X.pdb) was then used as a molecular replacement model in PHASER (Adams, Grosse-Kunstleve et al. 2002) and calculations revealed the presence of the N-terminus region in the electron density maps. Structure refinement was completed using CNS (Brunger, Adams et al. 1998) resulting in final R/Rfree values of 0.305/0.341 (Table II). The higher than expected R/Rfree values are attributed to radiation damage to the crystals in our attempt to collect usable data to 3.1 Å. This was corroborated by splitting up the diffraction data set into two portions and analyzing a model-phased (F_late – F_early) electron density map. The sequence of the protein in the electron density was also verified as there are several copies of AcrB homologs in E. coli. The final model has 1019 amino acids and lacks residues 500-510 and 1031–1049. The overall temperature factor of the model is 69.7 Å2 and that of the N-terminal segment exceeds 100 Å2 which is not inconsistent with the data resolution and the high solvent content of the crystals. The structure of the N-terminus was verified by a composite omit map calculation in CNS (Brunger, Adams et al. 1998) as well as a fo-fc map after removing the first nine N-terminal amino acids from the model (Figure 3).
Table II.
Crystallographic refinement statistics
| Space group | R32 |
| Cell dimensions | a=146.30 Å, b=146.30 Å,c=514.30 Å, α=β=90°, γ=120° |
| Volume fraction of protein | 73% |
| Vm (Å3/Dalton) | 4.53 |
| Total number residues | 1019 /asu |
| Overall temperature factor | 69.7 Å2 |
| Resolution range of reflections used | 50.0–3.1 Å |
| R factor a | 0.305 (0.420) |
| Free R factora | 0.341 (0.476) |
| Reflections in working seta | 36947 (6113) |
| Reflections in test seta | 1952 (289) |
| Total data completeness (working set + test set)a | 38899/99.6 % (6402/100%) |
| R.M.S. deviations: bond | 0.009 |
| angle | 1.6 |
Numbers in parentheses are related to the highest resolution shell (3.29–3.10 Å)
Figure 3.
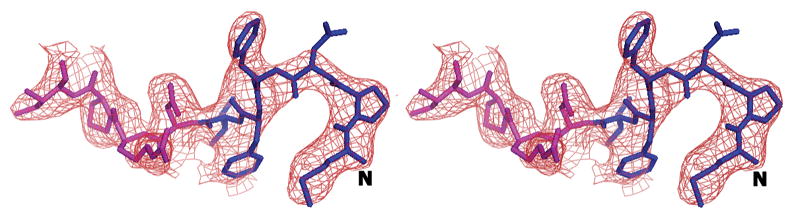
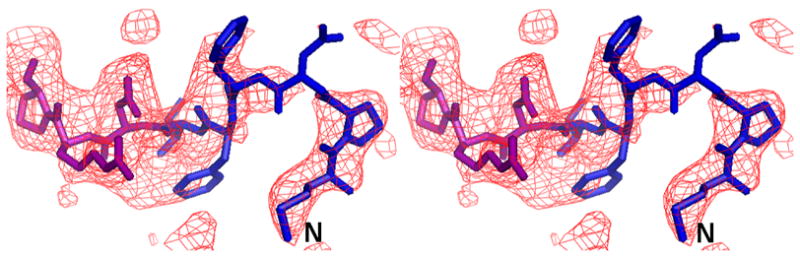
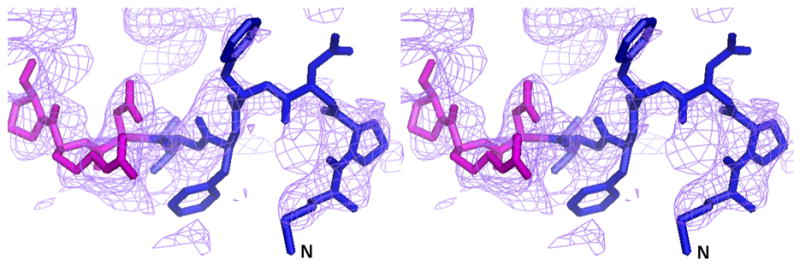
The complete N-terminus of the AcrB structure in the (a) final 2Fo-Fc electron density map at 1.0σ. (b) Fo-Fc difference map calculated by omission of the first nine N-terminal residues at 2.0σ and (c) final composite omit map at 1.0σ. Amino acids 1–6 are in blue and residues 7–10 are in magenta.
3. Results and Discussion
3. 1 Overall architecture of AcrB
In R32 space group, three AcrB protomers are arranged symmetrically around the crystallographic three-fold axis to form a tripartite complex (Figure 1) as was observed in earlier crystallographic studies (Murakami, Nakashima et al. 2002; Yu, McDermott et al. 2003). Each AcrB molecule, can be categorized into three structural domains. The transmembrane segment spanning ∼50 Å is composed of 36 transmembrane (TM) helices with each molecule contributing 12 helices. Portions of this region also protrude into the cytoplasmic side and the first transmembrane helix starts at residue number 9. This allows the first few N-terminal residues to be exposed up to a distance of 6.0 Å into the cytoplasm. In previously reported crystal structures of AcrB where this portion is not visible, this arrangement of protomers resulted in an opening of ∼30 Å in diameter on the cytoplasmic face and this opening continues upward through the whole of the inner membrane to occupy an inner cavity volume of ∼5000 Å3 in the periplasmic portion.
Figure 1.
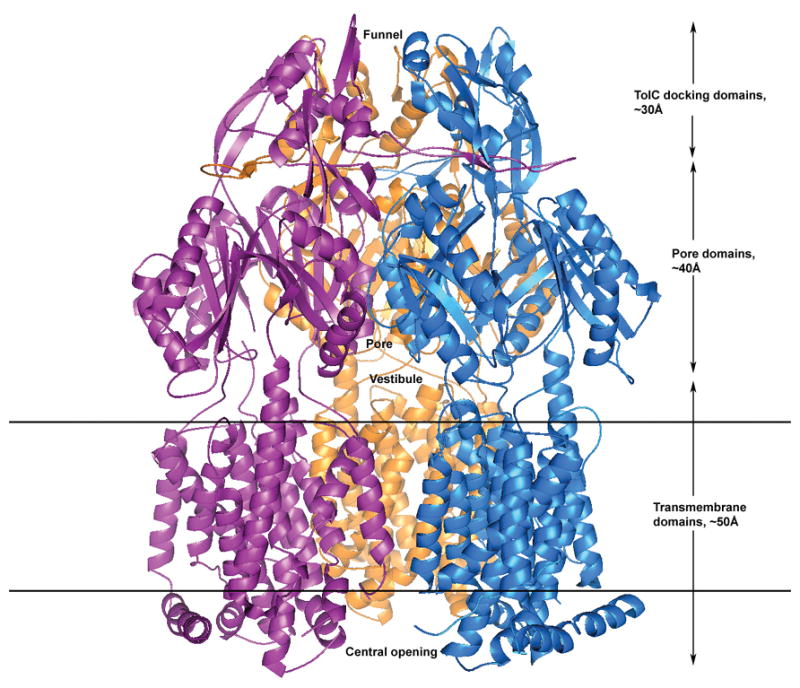
A ribbon representation of the crystal structure of AcrB showing three AcrB protomers that constitute the symmetric trimer of the transporter in R32 space group. The vestibular opening is located in the center just above the outer leaflet of the inner membrane in the periplasm that allows ligands from the periplasm to enter the inner central cavity. The top of the inner central cavity leads to the pore in the pore domain which then opens out into a funnel in the TolC docking domain for conveying the captured molecules to the TolC protein spanning the outer membrane and then ejects the substrates to the external medium. The bottom of the tripartite AcrB assembly extends into the cytoplasm which may allow the direct capture of toxic substrates from the cytosol through the central opening of the trimer cytoplasmic face.
In the periplasm, the protein can be divided into the pore domain and the TolC docking domain, spanning ∼40 Å and ∼30 Å, respectively. The top of the inner central cavity opens up into the inner pore in the pore domain, at the end of which it spreads into a funnel in the TolC docking domain. The periplasmic portions of the proteins have vestibules which can allow the transfer of ligands from the periplasm into the inner cavity of the transporter. In addition, each protomer also has a ∼35 Å lateral projection that inserts into the neighboring molecule and helps to lock the tripartite assembly together.
An analysis of the electrostatic potential energy surface of the transmembrane portion facing the inside of the trimeric assembly (data not shown) indicates that it is composed primarily of hydrophobic residues and is thus likely, but not necessarily, filled with phospholipids of the inner membrane. The view from the bottom of the transporter (Figure 2, viewed upward from the cytoplasm to the periplasm across the inner membrane, 90° viewing angle from Figure 1), reveals a strikingly different picture from prior reports as described below.
Figure 2.
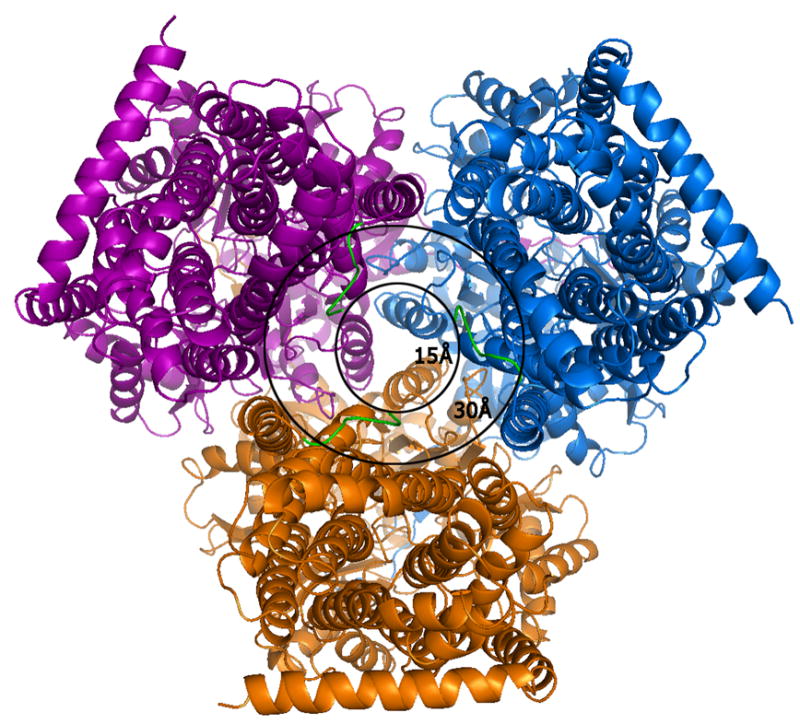
A view of the AcrB trimer from the bottom, i.e.,the cytoplasmic face at a 90° view from that of Figure 1. The outer circle with a diameter of 30Å represents the transporter entry point size as reported in all previous crystal structures of AcrB in R32 space group lacking the first six amino acids. The extensions of the N-terminus (green) represent our model which completes the N-terminal model and reveals the actual cavity entry size of only ∼15Å. The smaller cavity opening and presence of conserved residues in the N-terminus implies a possible role of these initial residues in ligand capture and discrimination during cytosolic drug uptake.
3.2 Structure of the N-terminus of AcrB
The sequence of the N-terminus of AcrB is highly conserved. In previous structures in R32 space group, the N-terminus of the molecule started at residue Asp 7. This resulted in a Cα-Cα distance between the terminal Asp 7 residues of each protomer of ∼29 Å and a channel opening of ∼34 Å (diameter of a circle inscribed in an equilateral triangle of side 29 Å). Recently, the structure of AcrB was determined in C2 space group showing an asymmetric arrangement of the trimers (Murakami, Nakashima et al. 2006; Seeger, Schiefner et al. 2006). This also allowed for the completion of the model in this region but resulted in an opening of ∼24 Å. However, the conformation of the N-terminus in that structure is helical and different from that seen in this structure. In our case, these first six N-terminal amino acids (shown as a green N-terminal extension in each protomer in Figure 2) reduce the real pore size to ∼15 Å in the symmetric trimeric assembly, resulting from a Cα–Cα distance of ∼14 Å between the first residue of each protomer and the corresponding amino acid from a neighboring molecule. A stereo view of the model with the complete N-terminus is shown superimposed in the final 2Fo-Fc electron density map (Figure 3a), a Fo-Fc difference map calculated by removal of the first nine N-terminal residues (Figure 3b) and a final composite omit map (Figure 3c). Though these N-terminal residues extend into the transporter opening and adopt a loop structure, they appear to be held in place due to van der Waal’s interactions with amino acids T431 and R432 in the cytoplasmic portion, and Q439, V443, L483, L486 and I487 of the neighboring TM helices, TM5 and TM6. Amino acid F4 also constitutes part of a hydrophobic patch together with residues M435, L483, L486, I487, and P490 in TM5 and TM6.
The inside of the periplasmic portion of the cavity formed by the trimers is surrounded by 18 conserved phenylalanine residues contributed by the three protomers (Figure 4). These are very well conserved and include F386 and F388 (red), and F458 and F459 (purple) that have been implicated in drug binding through hydrophobic and π-π interactions. There are also two phenylalanines within the first six conserved N-terminal residues, F4 and F5, which may indicate a functional role of these residues in drug binding. All these conserved phenylalanines line the inside of the cylindrical central cavity from the cytoplasm to the periplasm.
Figure 4.
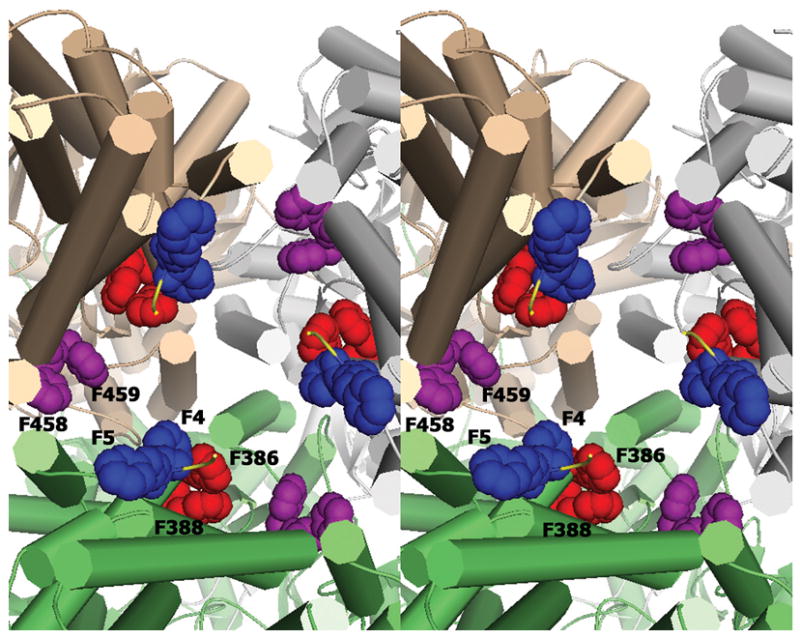
Stereo view of the inner cavity from the cytoplasmic side up into the cavity of the trimer showing the conserved phenylalanine residues lining the cavity. Amino acids F386 and F388 (red), F458 and F459 (purple) have been previously reported to bind substrates. F4 and F5 (blue) show conserved residues in the N-terminus that may be involved in substrate discrimination and uptake from the cytosol.
3.3 Conservation of the N-terminus in other AcrBs and homologs
A PSI-BLAST (Altschul, Madden et al. 1997) search against the non-redundant sequencedatabase shows that AcrBs and its homologs from different organisms have sequence identities ranging from 51–99%, with the majority of sequences showing conservation of the N-terminal residues (data not shown) even in cases of low overall sequence identity. A multiple sequence alignment (Figure 5) of 10 representative members of the RND family of transporters of very similar size (Borges-Walmsley, McKeegan et al. 2003) including AcrB from E. coli, Salmonella typhimurium and Yersinia pestis, MexB and MexD from Pseudomonas aeruginosa, SmeE from Stenotrophomonas maltophila, ArpB from Pseudomonas putida, MtrD from Nisseria gonorrhoeae and Bordotella pertussi, and TbtB from Pseudomonas stutzeri reveal the conservation of the N-terminal residues and presence of F4 and F5 (blue rectangle) as identical amino acids over all the family members, indicating that these residues may play a role in biological function. It is interesting to note that these N-terminal phenyalanines are identical across all these family members compared to the other phenylalanines elsewhere in the sequence that are implicated in ligand binding (F386 and F388 as green circles; F458, F459 as purple circles).
Figure 5.

A multiple sequence alignment of ten representative members of the RND family of transporters including AcrB from E. coli, Salmonella typhimurium and Yersinia pestis, MexB and MexD from Pseudomonas aeruginosa, SmeE from Stenotrophomonas maltophila, ArpB from Pseudomonas putida, MtrD from Nisseria gonorrhoeae and Bordotella pertussi, and TbtB from Pseudomonas stutzeri reveal the conservation of the N-terminal residues and presence of F4 and F5 (blue rectangle) as identical amino acids over all the family members indicating that these residues may play a role in biological function. Other residues implicated in ligand binding in AcrB are F386 and F388 (green circles); and F458 and F459 (purple circles). The figure was made using CLUSTALW (Thompson, Higgins et al. 1994) and ESPRIPT (Gouet, Courcelle et al. 1999).
3.4 Implications for cytosolic drug uptake
The AcrB transporter has a wide range of substrate specificity and has the ability to export from the periplasm as well as the cytoplasm (Yu, R. et al. 2003). Several models have been proposed for the mechanism of drug efflux by this RND-family protein based on crystal structures and biochemical experiments. The drug molecules can be taken up directly into the central cavity of the trimeric assembly from the periplasm through the vestibules on the side of the trimer. Substrates present in the outer leaflet of the inner membrane can translate laterally in the membrane and can also negotiate their way into the central cavity via these vestibules. This model supports the role of AcrB as a “periplasmic vacuum cleaner” (Elkins and Nikaido 2003).
While periplasmic uptake in AcrB supports the role that it may play in the efflux of external materials that may enter the cell from the environment, studies have also shown that this protein may play a role in the excretion of excess toxic materials that may build up in the cytoplasm or to remove other external toxic materials that may escape the periplasmic vacuuming and enter the cytoplasm (Helling, Janes et al. 2002) but the exact mechanism of this remains largely undefined (Yu, R. et al. 2003). Structural information suggests that compounds in the inner leaflet of the inner membrane can pass the membrane along the groove between TM7 and TM8 by a flipping and then merge with the periplasmic-vestibule pathway for entry into the inner cavity (Murakami and Yamaguchi 2003). None of the proposed models negate the possibility of the central trimer opening facing the cytoplasm as a possible entry point for substrates (Murakami, Nakashima et al. 2002).
This crystal structure of AcrB in R32 space group including the complete N-terminus represents a further step in the structural characterization of the RND-type multidrug efflux protein. Our structure reveals that the central hole is smaller that the previously reported size and includes residues that are exposed to the cytoplasm. The size of this opening may actually change in different functional states with the N-terminal arm extending in or out or changing conformations, acting as an aperture control. This hypothesis is supported by the recently reported crystal structures of AcrB in C2 space group (Murakami, Nakashima et al. 2006; Seeger, Schiefner et al. 2006) revealing an aysmmetic arrangement of the protomers that also reveals the N-terminal residues in a different conformation and resulting in another size of the central opening. The size of ∼15 Å of this cytoplasm facing opening in our structure is similar to the size of the vestibular opening in the periplasm and is large enough to accommodate the commonly known drugs that are ejected by the transporter. The N-terminal conserved amino acids, positioned at the opening of the channel, may play a role in the discrimination, uptake and transport of cytosolic drug molecules through the central opening or the space between adjacent N-terminal tails, upward into the channel cavity for eventual extrusion from the cell. The potential role of these amino acids is independent of the determination of the crystal structure of AcrB in R32 or C2 space groups.
This may occur in the following manner. The substrates align near the cytoplasmic opening where they are allowed to pass if they meet specific recognition criteria including size and chemical properties. The molecules then diffuse into and traverse the inner membrane by a slow and spontaneous flipping which may be aided by hydrophobic interactions with amino acid residues lining the inner cavity and reach the periplasmic space of the inner cavity. This mode of molecule capture via the central opening of the assembly would require a similar mechanism of movement as the previously suggested mode of ligand movement from the cytosol in which the ligands diffuse in the inner membrane and enter the periplasmic cavity from the periplasmic vestibule after traveling along the transmembrane groove.
Although speculative, this structural information, whose implications are independent of the space group of the crystal structure, may aid in the design of further biochemical experiments by other researchers including mutational studies and reconstitution of the AcrB into membranes, which is beyond the scope of this current work, to explore the role of these first few residues in molecular uptake. The structure has been deposited in the Protein Data Bank with accession number 2I6W.pdb. Figures 1, 2, 3 and 4 have been made using PyMOL (DeLano 2002).
Acknowledgments
We would like to thank Bruno Martinez and Marlene Henriquez for protein expression, Paul Adams for helpful discussions and computational crystallography support, Christine Trame for user support at beamline 5.0.2 and Corie Ralston at beamline 8.2.2 at the Advanced Light Source. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences Division, of the U.S. Department of Energy under Contract No. DE-AC03–76SF00098 at Lawrence Berkeley National Laboratory. The work described here was supported by a National Institutes of Health grant GM 62412.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Debanu Das, Department of Chemistry, University of California, Berkeley, Calvin Lab, Berkeley, CA 94720.
Qian Steven Xu, Berkeley Structural Genomics Center, Physical Biosciences Division, Lawrence Berkeley National Laboratory, University of California, Berkeley, Calvin Lab, Berkeley, CA 94720.
Jonas Y. Lee, Department of Chemistry, University of California, Berkeley, Calvin Lab, Berkeley, CA 94720
Irina Ankoudinova, Berkeley Structural Genomics Center, Physical Biosciences Division, Lawrence Berkeley National Laboratory, University of California, Berkeley, Calvin Lab, Berkeley, CA 94720.
Candice Huang, Bio-Rad Laboratories, Hercules, CA 94547.
Yun Lou, Berkeley Structural Genomics Center, Physical Biosciences Division, Lawrence Berkeley National Laboratory, University of California, Berkeley, Calvin Lab, Berkeley, CA 94720.
Andy DeGiovanni, Berkeley Structural Genomics Center, Physical Biosciences Division, Lawrence Berkeley National Laboratory, University of California, Berkeley, Calvin Lab, Berkeley, CA 94720.
Rosalind Kim, Berkeley Structural Genomics Center, Physical Biosciences Division, Lawrence Berkeley National Laboratory, University of California, Berkeley, Calvin Lab, Berkeley, CA 94720.
Sung-Hou Kim, Department of Chemistry, University of California, Berkeley, Calvin Lab, Berkeley, CA 94720.
References
- Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Adams PD, Grosse-Kunstleve RW, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 11):1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- Akama H, Matsuura T, et al. Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. J Biol Chem. 2004;279(25):25939–42. doi: 10.1074/jbc.C400164200. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges-Walmsley MI, McKeegan KS, et al. Structure and function of efflux pumps that confer resistance to drugs. Biochem J. 2003;376(Pt 2):313–38. doi: 10.1042/BJ20020957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Cryst. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Dean M, Rzhetsky A, et al. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2000;11:1156–66. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System 2002 [Google Scholar]
- Elkins CA, Nikaido H. 3D structure of AcrB: the archetypal multidrug efflux transporter of Escherichia coli likely captures substrates from periplasm. Drug Resist Updat. 2003;6(1):9–13. doi: 10.1016/s1368-7646(03)00004-9. [DOI] [PubMed] [Google Scholar]
- Eswaran J, Koronakis E, et al. Three's company: component structures bring a closer view of tripartite drug efflux pumps. Curr Opin Struct Biol. 2004;14(6):741–7. doi: 10.1016/j.sbi.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Gouet P, Courcelle E, et al. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15(4):305–8. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- Grosse-Kunstleve RW, Adams PD. Substructure search procedures for macromolecular structures. Acta Cryst. 2003;D59:1966–1973. doi: 10.1107/s0907444903018043. [DOI] [PubMed] [Google Scholar]
- Helling RB, Janes BK, et al. Toxic waste disposal in Escherichia coli. J Bacteriol. 2002;184(13):3699–703. doi: 10.1128/JB.184.13.3699-3703.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins MK, Bokma E, et al. Structure of the periplasmic component of a bacterial drug efflux pump. Proc Natl Acad Sci U S A. 2004;101(27):9994–9. doi: 10.1073/pnas.0400375101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Sander C. Touring protein fold space with Dali/FSSP. Nucleic Acids Res. 1998;26:316–19. doi: 10.1093/nar/26.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Nishino K, et al. Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J Bacteriol. 2001;183(19):5639–44. doi: 10.1128/JB.183.19.5639-5644.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronakis V, Sharff A, et al. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405(6789):914–9. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- Mikolosko J, Bobyk K, et al. Conformational flexibility in the multidrug efflux system protein AcrA. Structure. 2006;14(3):577–87. doi: 10.1016/j.str.2005.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, et al. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006 doi: 10.1038/nature05076. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, et al. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature. 2002;419(6907):587–93. doi: 10.1038/nature01050. [DOI] [PubMed] [Google Scholar]
- Murakami S, Yamaguchi A. Multidrug-exporting secondary transporters. Curr Opin Struct Biol. 2003;13(4):443–52. doi: 10.1016/s0959-440x(03)00109-x. [DOI] [PubMed] [Google Scholar]
- Nikaido H. Multidrug efflux pumps of gram-negative bacteria. J Bacteriol. 1996;178(20):5853–9. doi: 10.1128/jb.178.20.5853-5859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pos KM, Schiefner A, et al. Crystallographic analysis of AcrB. FEBS Lett. 2004;564(3):333–9. doi: 10.1016/S0014-5793(04)00272-8. [DOI] [PubMed] [Google Scholar]
- Putman M, van Veen HW, et al. Molecular properties of bacterial multidrug transporters. Microbiol Mol Biol Rev. 2000;64(4):672–93. doi: 10.1128/mmbr.64.4.672-693.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger MA, Schiefner A, et al. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science. 2006;313(5791):1295–8. doi: 10.1126/science.1131542. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41(1):207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Terwilliger T, Berendzen J. Automated MAD and MIR structure solution. Acta Cryst. 1999;D55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56(Pt 8):965–72. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, et al. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng TT, Gratwick KS, et al. The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J Mol Microbiol Biotechnol. 1999;1(1):107–25. [PubMed] [Google Scholar]
- van Veen HW, Callaghan R, et al. A bacterial antibiotic-resistance gene that complements the human multidrug-resistance P-glycoprotein gene. Nature. 1998;391(6664):291–5. doi: 10.1038/34669. [DOI] [PubMed] [Google Scholar]
- van Veen HW, Konings WN. Drug efflux proteins in multidrug resistant bacteria. Biol Chem. 1997;378(8):769–77. [PubMed] [Google Scholar]
- Yu EW, Aires JR, et al. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J Bacteriol. 2005;187(19):6804–15. doi: 10.1128/JB.187.19.6804-6815.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu EW, McDermott G, et al. Structural basis of multiple drug-binding apacity of the AcrB multidrug efflux pump. Science. 2003;300(5621):976–80. doi: 10.1126/science.1083137. [DOI] [PubMed] [Google Scholar]
- Yu EWAR, et al. AcrB multidrug efflux pump of Escherichia coli: Composite substrate-binding cavity of exceptional flexibility generates its extremely wide substrate specificity. J Bacteriol. 2003;185(19):5657–64. doi: 10.1128/JB.185.19.5657-5664.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]