

Abstract
Individuals with spinal cord injury (SCI) are highly susceptible to infection. This post-traumatic immune suppression is thought to occur via alterations in sympathetic nervous system (SNS) or hypothalamic-pituitary-adrenal (HPA) axis function. Normally, the HPA axis and SNS help coordinate proper immune function. After SCI, the HPA axis becomes activated and descending input to sympathetic preganglionic neurons (SPNs) is impaired. Because lymphoid organs are innervated by SPNs distributed throughout the thoracolumbar spinal cord, we predicted level-dependent immune suppression after SCI due to activation of the HPA axis and loss of descending input to SPNs. We tested this hypothesis by measuring indices of HPA (circulating corticosterone; CORT) and SNS function (norepinephrine (NE) in spleen) as well as antigen-specific antibody synthesis against an exogenous non-self protein following high or low level SCI. Using a mid-thoracic (T9) spinal contusion injury model, we found that CORT was elevated after SCI with aberrant patterns of diurnal CORT synthesis evident through at least the first 24 hours post-injury. However, splenic NE and antibody synthesis were similar to uninjured controls. Injury severity did not change these parameters. Indeed, CORT, NE and antibody synthesis were similar after T9 contusion or transection SCI. In contrast, high level SCI (T3) caused sustained increases in CORT and splenic NE along with impaired antibody synthesis and elevated splenocyte apoptosis. The immunosuppressive effects of T3 SCI were caused by NE acting at β2-adrenergic receptors (β2AR) and could be reversed using β2AR blockers. Interestingly, impaired antibody after T3 SCI could be mimicked after T9 SCI with a β2AR agonist. These data illustrate the immunosuppressive effects of the SNS after high-level SCI and indicate that immune deficits may be overcome using β-blockers.
Keywords: CNS injury, sympathetic nervous system, hypothalamic-pituitary-adrenal axis, antibodies, apoptosis, vaccines
Introduction
Clinical data show that human spinal cord injury (SCI) is accompanied by profound immunological impairment (Cruse et al., 1993; Nash, 2000). Undoubtedly, immune dysfunction after SCI contributes to the significant increase in mortality caused by septicemia, diseases of the lung (e.g., pneumonia), gastrointestinal tract or urinary system (DeVivo et al., 1989). SCI-induced deficiencies in supraspinal control of the sympathetic nervous system (SNS) or hypothalamic-pituitary-adrenal (HPA) axis have long been suspected, but never proven, as mechanisms of post-traumatic immune suppression (Cruse et al., 1996; Nash, 2000).
Activation of the HPA axis causes release of cortisol (humans) or corticosterone (CORT; rodents) from the adrenal cortex into the bloodstream. High or sustained levels of CORT suppress antibody production, cytokine synthesis and leukocyte proliferation (Munck et al., 1984; Barnes, 1998; Moraska et al., 2000). In humans, urinary cortisol remains elevated for months after SCI (Campagnolo et al., 1999) suggesting prolonged dysregulation of HPA function. Activation of the SNS causes the release of norepinephrine (NE). In spleen and lymph nodes, noradrenergic nerves synapse on T and B cells (Felten et al., 1987; Felten and Olschowka, 1987). This “hardwiring” between the spinal cord and lymphoid tissue ensures proper immune function. Indeed, depletion of noradrenergic neurons suppresses antibody synthesis (Kohm and Sanders, 1999). This deficit is overcome by activating B cells in the presence of β2-adrenergic receptor (β2AR) agonists (e.g., terbutaline) (Podojil and Sanders, 2003). However, repeated or prolonged exposure of B cells to NE or other β2AR agonists is immunosuppressive (Melmon et al., 1974; Keller et al., 1983; Maisel, 1994; Harris et al., 1995; Woiciechowsky et al., 1998; Prass et al., 2003).
HPA/SNS responses are coordinated in the spinal cord via supraspinal projections and by afferent feedback from the periphery to sympathetic preganglionic neurons (SPNs) (Hayes et al., 1991; Taylor and Weaver, 1993). SPNs found throughout the thoracic spinal cord (T3-13; (Strack et al., 1989; Cano et al., 2001) influence immune function through post-ganglionic noradrenergic projections to spleen (Wan et al., 1993) and adrenal cortex (Bloom et al., 1988; Engeland, 1998). Because SPNs are influenced by descending input from the brain, we predicted that high level SCI would cause greater dysfunction of the HPA axis and SNS and subsequently, greater immunological impairment than lower level SCI. To test this, mice were subjected to high (T3) vs. mid-thoracic (T9) SCI; after which circulating CORT, splenic NE and antigen-specific antibody responses were measured as indices of HPA activation and SNS and immune competence, respectively. The data show that only after T3 SCI is splenic NE elevated and immune function suppressed. Moreover, the immunological impairment that occurs in T3 SCI mice can be overcome by pharmacological blockade of β2ARs, implicating NE in post-traumatic immune suppression. These studies are of potential clinical significance and could dramatically influence the design of prophylactic or therapeutic vaccines. Indeed, the ability to eradicate extracellular pathogens (Luster et al., 1993; Robbins et al., 1995) or mount an immune response against CNS proteins (e.g., myelin inhibitory proteins) (Huang et al., 1999; Hauben et al., 2001) requires the coordinated activity of T and B cells, dendritic cells and their associated cytokine networks. Here we show that each of these parameters is affected by SCI in a level-dependent fashion.
Materials and Methods
Mice
Adult pathogen-free C57BL/6 female mice (6-8 weeks old; 16-21g) were purchased from Taconic Laboratories (Germantown, NY). All experimental procedures described below were approved by the Animal Review Committee at Ohio State University and are in accord with the US Department of Health, Education, and Welfare.
Spinal cord injury
A total of 82 mice received a spinal cord injury (SCI) as described below. Mice were anesthetized (i.p.) with a cocktail of ketamine (80 mg/kg)/xylazine (40 mg/kg), then given prophylactic antibiotics (Gentocin; 1 mg/kg). Using aseptic technique, a partial laminectomy was performed at vertebral level T3 or T9 to expose the dorsal spinal cord. Sham injured animals received a laminectomy (LAM) but no SCI. For contusion injuries (T9; n=46), an electromagnetic spinal contusion device was used to displace the exposed spinal cord a calibrated vertical distance of 0.5 mm over a period of 30 msec (Jakeman et al., 2000). For spinal transection injuries (T3; n=26 and T9; n=10), the periosteum and dura mater were opened with forceps and microscissors. Iridectomy scissors, together with gentle aspiration, were used to cut the spinal cord until a clear separation was noted between the rostral and caudal stumps of the spinal cord. To ensure complete transection, the rostral and caudal stumps were gently lifted from the spinal canal using a blunt probe. Gel foam was used to fill the cavity created by the retracted ends of the spinal cord. After injury, muscles and skin were sutured separately and mice were injected with sterile saline (2 mL, s.q.) then were placed individually into warmed HEPA-filtered cages. Post-operative care included manual bladder expression 2-3 x/day and daily antibiotics for the first 7dpi (Gentocin, 50 mg/kg; s.q.).
Sera collection
Blood was collected from awake, unrestrained mice via retro-orbital puncture. Unless otherwise noted, all bleeds were performed beginning at 8:00am. Individual mice were bled within 30 sec of being removed from their cage and were then placed into a separate “recovery cage” isolated from their remaining cage-mates. These precautions eliminate stress in cage-mates. Serum was collected by centrifugation of whole blood (7000xg for 15 mins), transferred into sterile tubes and stored at -80°C until time of CORT or ELISA analysis (see below).
Measurement of serum corticosterone (CORT)
Pre-injury and post-injury concentrations of CORT were obtained for each mouse, allowing precise analysis of SCI-induced changes in HPA function. Sera were assayed for CORT by (I125) radioimmunoassay (ICN Biomedical, Costa Mesa, CA). The minimum detectable limit for this assay was 12.5ng/mL.
Measurement of splenic norepinephrine (NE)
After the final blood collection (see above), mice were anesthetized (i.p.) with an overdose of ketamine (160 mg/kg) and xylazine (80 mg/kg). Spleens were removed and immediately placed on dry ice to halt degradation of catecholamines. Tissue was stored at -80°C until NE was extracted. For NE analysis, frozen spleens were cut in half before homogenization. One half was used for western blot analysis (see below) while the remaining half was weighed and then homogenized on ice in 1mL of 0.2N acetic acid buffer (1000μL glacial acetic acid, 0.05% EDTA, 0.1% sodium bisulfite in 99mL of distilled water). Spleen homogenate was centrifuged at 160xg for 5 mins. 500μL of NE-containing supernatant was extracted and diluted 1:3 in 0.2N acetic acid. Quantitative analysis of NE was determined via HPLC using a Waters System mobile phase and electrochemical detector (Waters™, division of Millipore, Milford, MA). 3,4-dihydroxybenzylamine was used as an internal standard for the determination of extraction efficiency. The minimum detectable limit for this assay was 20pg/mL.
Ovalbumin immunizations
Mice were immunized (i.p. @ 3dpi) with 100μg of ovalbumin (OVA; Sigma-Aldrich, St. Louis, MO) emulsified in an equal volume of TiterMax adjuvant (Sigma). TiterMax was used as an adjuvant due to its ability to stimulate high antibody titers without inducing non-specific inflammatory responses (Bennett et al., 1992).
β2-adrenergic receptor agonists/antagonists
Mice receiving β2AR-specific agonists or antagonists (terbutaline or butoxamine respectively; Sigma) were injected (i.p. @ 3dpi) with 5mg/kg of drug prepared in PBS. Control injections consisted of sterile PBS only (i.p.).
ELISA
Sera from immunized mice were assayed for OVA-specific Abs using ELISA. Round bottom 96-well plates (Costar, Cambridge, MA) were coated with 100μg/mL of OVA in PBS + 0.02% sodium azide. Plates were incubated for 2 hrs at 37°C, washed with PBS, and blocked overnight at 4°C with PBS + 1% BSA. Plates were washed with PBS + 0.05% Tween. After the wash, 20μL of diluted serum (to create a dilution curve) was incubated in the appropriate well for 2 hrs at 37°C. Plates were then washed with PBS + 0.05% Tween and incubated with alkaline phosphatase-conjugated goat anti-mouse IgG1 antibody (diluted 1:1000 in PBS + 1% BSA) for 2 hrs at 37°C. Plates were washed with PBS + 0.05% Tween followed by PBS then bound antibodies were visualized by incubating with 1μg/mL p-nitrophenyl phosphate in 10mM diethanolamine-0.5 mM MgCl2 buffer. Spectrophotometric readings were performed on a Spectramax kinetic microplate reader (Molecular Devices, Sunnyvale, CA; minimum detection limit of 0.006 OD ±1.0%) at a wavelength of 405nm.
Western blot
Spleens were homogenized in 500μL Tissue Protein Extraction Reagent (T-PER™; Pierce, Rockford, IL) and 5μL Halt™ Protease Inhibitor cocktail (Pierce). The resulting homogenate was centrifuged for 5 mins at 4500xg and supernatants were transferred into fresh tubes for protein quantification. Protein concentration was determined using Coomassie Plus™ Protein Assay Kit (Pierce). 20μg of protein was added to 6.25μL NuPAGE™ LDS sample buffer with 5% beta-mercaptoethanol and brought up to a volume of 25μL with H2O. Samples were heated at 100°C for 5 minutes. Samples were loaded and run on NuPAGE™ 4-12% Bis-Tris gels. Gels were run at 200V for ∼50 min after which proteins were transferred to immobilon-P membranes (Millipore, Medford, MA) at 30V for 90 mins. Membranes were blocked with 5% milk and 0.5% Tween for 1 hour at RT then hybridized with antibodies against active caspase-3 (1:1000 @ 4°C overnight; R&D systems, Minneapolis, MN) to assess the magnitude of apoptosis. Antibodies against α-Tubulin (1:2500; Sigma) were used to ensure consistent protein loading. Goat anti-mouse HRP and goat anti-rabbit HRP (Sigma; 1:50,000; 1:4000) were used as secondary antibodies for α-tubulin and caspase-3, respectively. Blots were developed using Pierce WestPico chemiluminescent substrate kit and visualized on Kodax Biomax film (Rochester, NY).
Splenocyte isolation
Mice were anesthesized and a laparatomy was performed under aseptic conditions to expose the spleen. After excision, a single cell suspension was prepared from individual spleens by mashing through 40μm nylon mesh strainers. Strainers were washed 3x with 5mL RPMI + 5%FBS. Red blood cells were lysed with 0.8% ammonia chloride. Splenocytes were washed with RPMI + 5% FBS, resuspended in cRPMI (RPMI 1640, 10% FBS (Atlas Biologicals, Ft. Collins, CO), 0.01M Hepes, 0.1nM non-essential amino acids, 1mM sodium pyruvate, 5×10-5M β-mercaptoethanol (Sigma), 0.01% penicillin-streptomycin, and 2nM L-glutamine), then counted on a hemacytometer. Unless specified, all media ingredients were obtained from Invitrogen, Inc. (Carlsbad, CA).
Cytofluorometric analysis
Flow cytometry (FACScalibur; Becton-Dickinson, San Jose, CA) was used to quantify specific cell numbers in spleen and measure SCI-induced apoptosis. Whole splenocyte suspensions (1×106 cells/sample) were analyzed separately using immunofluorescent labeling. Cells were first washed with PBS and resuspended in Annexin V binding buffer (10mM Hepes, 140mM NaCl, 2.5mM CaCl2; pH 7.4). All samples were incubated for 15 minutes with 0.25 μg Fc-block (anti-mouse-CD16/CD32), then labeled with APC or PE-conjugated Abs specific for either B cells (CD19), T cells (CD4 or CD8), or dendritic cells (CD11c). All samples were labeled with Annexin V-FITC to quantify apoptosis within specific cell populations. Whenever possible, samples stained with isotype-matched control antibodies (at identical concentrations) were used to discriminate positively labeled from non-specifically labeled cells. At least 10,000 events were collected per sample. Specific cell counts within spleen were obtained by multiplying the percent at which they were observed in splenocyte suspensions by the total splenocyte count (as determined via hemacytometer). Fc-block and Annexin V were obtained from BD Pharmingen (Franklin Lakes, NJ). All labeling Abs for cellular surface markers, and their corresponding isotype controls, were obtained from eBioscience (San Diego, CA).
Statistical analyses
All results are expressed as mean ± SEM. A Kolmogorov-Smirnov normality test confirmed that representative data exhibited Gaussian distribution. Therefore, Gaussian distribution was assumed for all data and parametric analyses were used. Group means were compared using one-way ANOVA with Tukey’s post-test or a two-way ANOVA when comparing multiple variables. Significance for all analyses was set at p<0.05. All statistical tests were performed using GraphPad prism version 4.03 (GraphPad Software, San Diego, CA, USA). For clarity of presentation in some Figures (Fig. 3), measures of statistical significance (e.g., *) are described in the text or figure legends, but not on the graph.
Fig. 3.
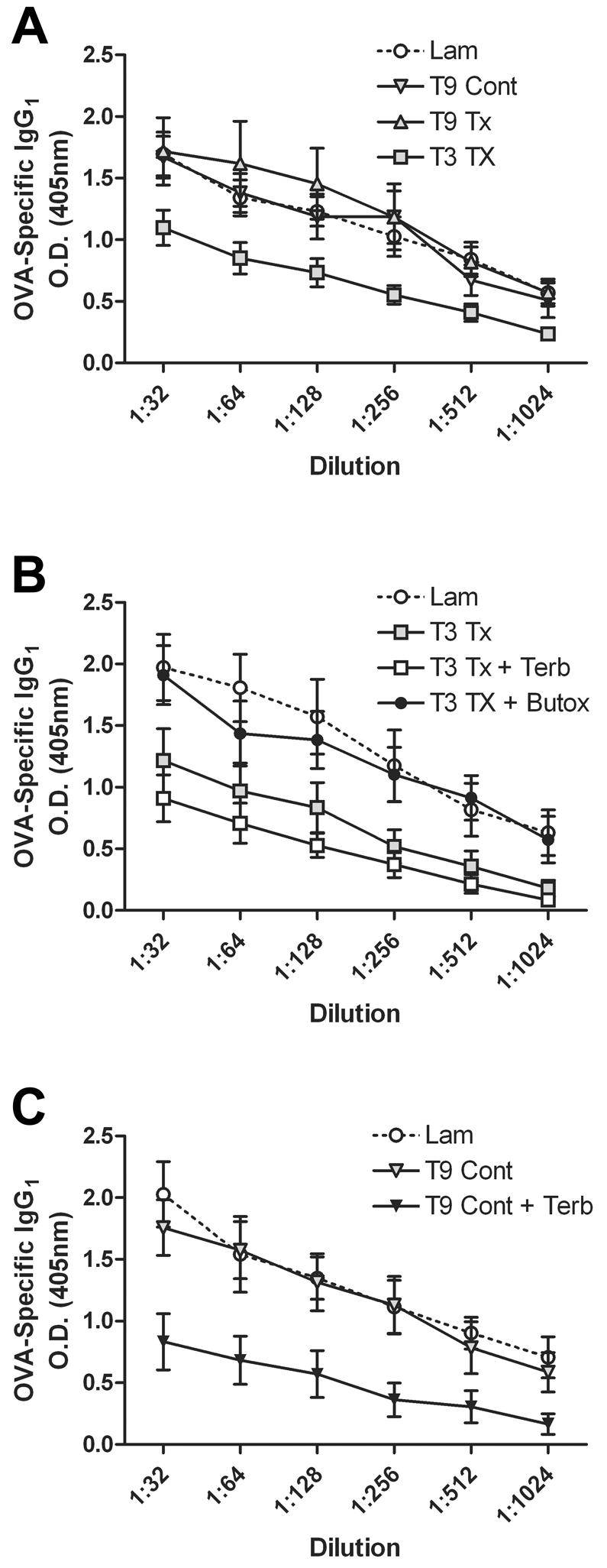
SCI-mediated reduction in OVA-specific antibody is level-dependent and is caused by NE. Data are expressed as dilution curves, with replicate samples plotted over multiple dilutions. OVA-specific IgG1 synthesis is similar after T9 spinal transection, T9 contusion or laminectomy (A). After T3 SCI, OVA-specific IgG1 is reduced (A, B; p<0.001 vs. Lam; 2-way ANOVA). When T3 SCI mice are injected with terbutaline (Terb; a β2AR agonist), no additional suppression of antibody synthesis is detected (B). However, when T3 SCI mice are injected with butoxamine (Butox; a β2AR antagonist), OVA-specific IgG1 levels are restored to control levels (B). OVA-specific IgG1 could be reduced in T9 SCI mice by providing terbutaline during immunization (C; p<0.001 vs. Lam; 2-way ANOVA). All immunizations were done at 3 dpi and antibodies analyzed at 14 dpi. n=4-8/group.
Results
SCI activates the HPA axis but disrupts circadian CORT synthesis
Previously, we demonstrated SCI-induced elevations of serum CORT in a rat model of SCI (Popovich et al., 2001). Here, we extend those findings to mice using a similar model of spinal contusion injury. Specifically, serum CORT was elevated in T9 SCI mice by 1 day post-injury (dpi), with levels returning toward baseline by 3 dpi (Fig. 1A). To determine whether SCI affected the circadian regulation of circulating CORT, sera were obtained from T9 spinal contusion injured mice at regular intervals spanning a 24 hour day/night cycle. Data in Fig. 1B confirm the 8am data shown in Fig. 1A. However, it is also evident that SCI acutely disrupts circadian CORT release (as indicated by consistently lower CORT levels in the morning with levels rising through the afternoon and peaking in the evening). Specifically, CORT release is dysregulated at 1dpi with markedly elevated CORT at 6am and 11am (**p<0.01, *p<0.05 respectively, vs. pre-injury; 2-way ANOVA). Evening CORT levels remain elevated at 3dpi with a spike at 8pm (**p<0.01). Regulation of CORT returns toward normal (both levels and rhythmicity) by 14dpi.
Fig. 1.

SCI elevates serum CORT and disrupts diurnal CORT rhythmicity. Serum CORT is elevated within 24 hrs after T9 spinal contusion injury (A) *(p<0.05 vs. pre-injury and 3d post- laminectomy control mice; n=4; ANOVA). Analysis of mean serum CORT, as a function of time post-injury and time of day, reveals SCI-mediated dysregulation of diurnal CORT (B). Circadian CORT data (for each day post-injury) is compiled from at least two groups of mice bled at different intervals in the day. Thus, a regular two-way ANOVA (not repeated measures) was used to determine significance at specified times in the day compared to pre-injury. (**p<0.001, *p<0.05 vs pre-injury; 2-way ANOVA; n=20-28/group).
Lesion level effects serum CORT and splenic NE
The above data indicate that T9 spinal contusion injury causes acute disruption of HPA function with a return toward baseline by 3 dpi. However, as shown below (see Fig. 3), T9 SCI is not associated with acute immune suppression. Prass et al. also were unable to reverse stroke-mediated immunosuppression using glucocorticoid receptor antagonists (Prass et al., 2003). Together, these data suggest that acute activation of the HPA axis is a consequence of CNS injury but it alone is not responsible for post-traumatic immune suppression.
Because SPNs in the spinal cord are influenced by descending input from brain and given the inter-relationship between the SNS and the HPA axis, we predicted a level-dependent effect on SNS competence and HPA activation. To test this hypothesis, we compared circulating CORT, splenic NE and OVA-specific antibody responses in mice subjected to high (T3) vs. mid-thoracic (T9) SCI.
To minimize animal use, we restricted our comparative analyses of CORT and NE to 3 dpi - a time when CORT levels return toward baseline after T9 contusion injury (see Fig. 1A). In contrast to contusion and Tx injuries at T9, we found higher levels of serum CORT in mice subjected to T3 Tx injury (Fig. 2A). Splenic NE was measured as an index of SNS activity and to determine its potential availability to lymphocytes in the spleen. Only in mice with a T3 Tx was splenic NE content significantly elevated (Fig. 2B).
Fig. 2.

High (T3) but not low (T9) level SCI elevates serum CORT and splenic norepinephrine (NE). Serum CORT from T3, but not T9 SCI mice, was elevated at 3 dpi (A) (*p<0.05 vs. T3 Lam; n=4/group; ANOVA). All sera for CORT analysis was harvested at 8am. Splenic NE was increased after T3, but not T9 SCI (B) (*p<0.05 vs. T3 Lam; n=4/group; ANOVA).
A comparison of HPA/SNS function and antibody synthesis after spinal contusion injury at T9 and T3 would have been ideal. Unfortunately, our contusion model requires that the vertebrae rostral and caudal to the laminectomy site be stabilized by clamps. Pilot studies that attempted to clamp and stabilize mice via the T3 process caused mice to experience respiratory distress and excessive bleeding. This response confounded our ability to reliably compare hormonal changes in this model between the differing injury levels. However, because serum CORT, splenic NE, and antibody synthesis were not different between T9 contusion and T9 Tx injury (Fig. 2A&B; Fig. 3A), we conclude that the differences we observed between T3 and T9 TX resulted from injury level and not injury severity.
High level SCI suppresses antibody responses to non-self antigen
After SCI, the incidence of bacterial infection (e.g., pneumonia) is increased. Because of the importance of antibody synthesis in bacterial clearance/host defense, we tested whether the SCI-mediated neurohormonal changes described above were associated with changes in antigen-specific immunity. To approximate a more clinically-relevant model of vaccination, all mice were immunized at 3 dpi. Antigen-specific antibodies were evaluated 14 dpi to allow sufficient time for maximal T and B cell interactions and subsequent antibody synthesis. Sham-injured laminectomy (LAM) controls were included for each group. The data show that production of OVA-specific IgG1 was unaffected by T9 SCI (Fig. 3A). In contrast, OVA-specific IgG1 was dramatically reduced in T3 SCI mice (p<0.001; Fig. 3A&B).
Aberrant β2AR stimulation suppresses antibody synthesis to non-self antigen after T3 SCI
Based on the above findings and data showing NE-mediated immunosuppression in a model of stroke (Prass et al., 2003), we predicted a causal role for NE in suppressing antibody synthesis after T3 SCI. To test this hypothesis, we inhibited NE signaling in T3 SCI mice at 3 dpi, i.e., the time of OVA immunization and increased NE in spleen (see Fig. 2B). Butoxamine, a selective β2AR antagonist, restored circulating levels of OVA-specific IgG1 to those found in immunized mice without SCI (Fig. 3B). Additional β2AR stimulation with terbutaline, a selective β2AR agonist, failed to exacerbate the SCI-mediated suppression of IgG1 synthesis.
To further evaluate the immunosuppressive potential of NE after SCI, T9 SCI mice were given terbutaline. This treatment was meant to mimic increased levels of NE found in T3 SCI spleens (see Fig. 2B). When T9 SCI mice were injected with terbutaline, OVA-specific IgG1 antibodies were markedly reduced compared to T9 SCI alone (p<0.001; Fig. 3C). Interestingly, the magnitude of immunosuppression induced by terbutaline was similar to that found after T3 SCI (compare Figs. 3A&C).
T3 SCI induces splenocyte cell death and B cell apoptosis
When the spleens of T3 SCI mice were initially observed post-mortem, they were noticeably smaller than T9 SCI or laminectomy controls. Indeed, spleen weights of non-bled T3 SCI mice were reduced by ∼50% and were associated with fewer total splenocytes when compared to sham-operated mice (p<0.05; 3 dpi; Fig. 4A,B). Since elevated concentrations of catecholamines can trigger lymphocyte cell death (Harris et al., 1995; Gu et al., 2000; Stevenson et al., 2001; Wahle et al., 2002; del Rey et al., 2003), we tested whether the reduction in spleen size and splenocyte number after T3 SCI was a result of increased immune cell apoptosis. Western blot analyses of homogenized spleen showed elevated levels of activated caspase-3 (a primary effecter of apoptosis) only after T3 SCI; thereby confirming our initial hypothesis (p<0.05; Fig. 4C,D).
Fig. 4.

T3 spinal transection induces splenocyte apoptosis. The post-injury decrease in spleen weight (A) and splenocyte count (B) corresponds with an increase in splenocyte apoptosis at 3 dpi (C, D). Splenocyte counts (B) were quantified using compiled data from two independent experiments and are expressed as percent of laminectomy control (using the appropriate laminectomy level and experiment). A Wilcoxon signed-rank test was used to compare experimental groups versus a control value of 100% (*p<0.05). Densitometric analysis (C) of Western blots for activated caspase-3 (D) reveal enhanced splenocyte apoptosis only after T3 SCI (*p<0.05 vs. laminectomy control mice; ANOVA; blot is representative of 2 independent experiments). n=4-8/group.
To extend this observation, we next determined the phenotype of dying cells. Using flow cytometry, we enumerated numbers of dendritic cells, T cells and B cells (Fig. 5A). After T3 SCI, all immune cells were decreased relative to laminectomy control values. Further analysis of Annexin V labeling (for apoptosis) confirmed the activated caspase-3 western blots (Fig. 4) and showed the largest degree of apoptosis occurring in splenic B cells (Fig 5B&C). Although T cells and dendritic cells were decreased in the spleen after T3 SCI (Fig. 5A), these cells did not exhibit enhanced Annexin V labeling at 3 dpi.
Fig. 5.

T3 spinal transection reduces splenic lymphocytes and induces B cell apoptosis. Although all splenic immune cells were decreased in number after T3 SCI, B cells were affected to the greatest extent (A). The large decline in total B cell number was accompanied by a detectable increase in Annexin V+ B cells, indicating apoptosis (B, C). Cell counts in spleen (A) were obtained by multiplying percent of cells observed by total splenocyte count. Since similar CD4 and CD8+ T cell reductions were observed, their values were summated and expressed as “T Cells” for ease of presentation (*p<0.05, **p<0.01; independent t tests). The percentage of Annexin V+ (apoptotic) B cells was quantified from gates of all CD19+ cells (B; *p<0.05; t test). Representative plots (C) used for quantitative analysis are shown. n=5/group.
Discussion
Here we show that immune suppression after SCI is level-dependent and involves NE acting at β2ARs. Indeed, only in mice with high level (T3) SCI was the concentration of splenic NE increased and antibody synthesis decreased relative to sham-injured or T9 SCI mice. Although the precise mechanism of immune suppression remains unclear, our results implicate aberrant β2AR-mediated signaling in lymphocytes. Indeed, only in T3 SCI mice was increased splenocyte apoptosis noted and immune suppression reversed by β2AR antagonists.
Infection is a leading cause of death after SCI (DeVivo et al., 1989). Most assume that the increased incidence of infection after SCI is a consequence of poor respiratory function, mechanical ventilation or chronic catheterization. However, other factors must contribute since ventilator-associated pneumonia occurs in ∼20% of patients without CNS injury (Chastre and Fagon, 2002) but rises to ∼60% in cases of CNS trauma (Woratyla et al., 1995; Ewig et al., 1999). The mechanisms responsible for heightened post-traumatic morbidity and mortality are unknown but defects in cellular immunity are suspected. Indeed, suppression of function in natural killer cells, neutrophils, macrophages and lymphocytes has been documented after SCI (Cruse et al., 1992; Campagnolo et al., 1994; Cruse et al., 1996; Campagnolo et al., 1997). In contrast, little is known about how SCI influences humoral (antibody) immunity.
Recently, our group showed that T9 SCI causes B cell activation and increased synthesis of autoantibodies (Ankeny et al., 2006). Also, we previously showed that T9 SCI activates autoreactive T cells (Jones et al., 2002). Collectively, these data indicate that SCI does not universally suppress immune function. However, after a high-thoracic (T3) SCI, when the majority of supraspinal control of sympathetic preganglionic neurons is disrupted, immune responses to exogenous antigens are impaired. This is important clinically since a robust antibody response is critical for neutralizing and lysing pathogens (Luster et al., 1993; Robbins et al., 1995). Also, prophylactic or therapeutic vaccines (e.g., pneumococcal vaccines or anti-myelin vaccines, respectively) require intact humoral immunity (T and B cell interactions). To our knowledge, only one other study has evaluated antibody responses to exogenous antigen after SCI (Vega et al., 2003). Using a high thoracic spinal contusion injury model in rats, Vega et al. showed transient reduction of antibody synthesis to ovalbumin infused into the CNS. Antibody synthesis could be restored by pre-treating rats with nadolol (a non-specific βAR blocker). We now show that SNS-mediated suppression of humoral immunity is level-dependent and extends to antigens administered systemically. Also, this suppression is due in part to aberrant β2AR stimulation and the induction of lymphocyte apoptosis.
Massive and immediate activation of the sympathetic nervous system, with subsequent release of systemic (from adrenal glands) and tissue NE (from nerve fibers), is common to high-level SCI, stroke, traumatic brain injury and shock (Tibbs et al., 1979; Woiciechowsky et al., 1998; Molina, 2001; Prass et al., 2003). In each case, NE is believed to be responsible for causing or exacerbating lymphocyte apoptosis and splenic atrophy over a period of up to 10 days (Oberbeck et al., 2002; Prass et al., 2003; Offner et al., 2006). However, because extracellular NE is metabolized rapidly (Mitchell et al., 1994) and given that high-level SCI is associated with SNS hypoactivity (Krum et al., 1990; Schmid et al., 2000), it is difficult to reconcile the high splenic NE levels that we observed 3 days after T3 SCI (see Fig. 2).
It is possible that high resting levels of splenic NE and impaired immune function are an indirect consequence of splenic atrophy. Splenic contraction and atrophy are common after severe trauma or stress and represent a physiological adaptation by the organism to regulate blood volume distribution (Hurford et al., 1996). Moreover, a decrease in sympathetic tone to peripheral vasculature after high- but not low-level SCI would cause hypotension and reduced filling of the spleen. Accumulation of NE may also occur if mechanisms of NE reuptake into nerve terminals is impaired after T3 SCI. Regardless of the mechanism, if we predict that the number of noradrenergic fibers innervating the spleen is unchanged after SCI but spleen size decreases, lymphocytes would be exposed to higher NE concentrations on a per cell basis.
In parallel with a decrease in spleen size after T3 SCI, we noted a marked decrease in the number of splenic T and B lymphocytes and dendritic cells (see Fig. 5). In fact, the nearly 75% decrease in B cell number that occurs after T3 spinal transection, represents a loss of ∼50×106 B cells per spleen. This alone could explain the impaired antibody synthesis documented in Fig. 3. A similar magnitude of B cell apoptosis was recently described in a model of stroke (Offner et al., 2006). Experiments are in progress to determine the mechanisms that predispose B cells to catecholamine-induced apoptosis; however, additional studies are needed to determine the duration of splenic atrophy, whether other immune cells are impaired by high-level SCI and the extent to which these cells repopulate peripheral lymphoid organs. Also, it will be important to determine if splenic noradrenergic innervation is affected in chronically injured animals. Indeed, our data illustrate the acute immunosuppressive effects of high-level SCI. If similar mechanisms persist in animals or humans, aberrant sympathetic reflexes and repeated exposure of peripheral immune cells to catecholamines could place individuals at increased risk for opportunistic infection. Clinical data indicate this occurs, but the mechanisms remain poorly defined (Tvede et al., 1994). Because β2ARs are expressed on most immune cells (Sanders et al., 1997), recurrent catecholamine “storms” due to periodic bouts of autonomic dysreflexia would be expected to impair multiple axes of host defense after SCI. In our study, even though SCI mice are unlikely to experience autonomic dysreflexia while resting in their cages, exaggerated sympathetic reflexes and acute spikes in splenic NE may be frequently initiated during daily bladder care.
Although post-SCI elevations of serum CORT did not appear to predict immunological impairment, acute surges in circulating CORT can increase β2AR expression, ligand binding affinity, and simultaneously prevent β2AR down-regulation (Davies and Lefkowitz, 1981; Mak et al., 1995). Therefore, while abnormally high levels of NE, even for brief periods of time, can predispose lymphocytes to apoptosis (Gu et al., 2000; Wahle et al., 2002; del Rey et al., 2003), this effect could be exacerbated by post-traumatic elevations of circulating CORT. If this occurs after SCI, acute rises in CORT may “sensitize” lymphocytes to subsequent NE exposure and cell death. Work in progress is testing whether low-dose glucocorticoid receptor antagonists can reverse splenic atrophy and lymphocyte apoptosis.
Immune suppression and increased frequency of infection are common in individuals living with SCI. While a dysfunctional autonomic nervous system has been suspected as a mechanism of immune impairment, this has never been proven. Here, we show for the first time that the maintenance of immune function after SCI is critically influenced by the level at which the spinal injury occurs. Moreover, SCI-mediated suppression of antibody synthesis is caused in part by NE acting at β2ARs. Based on these data, selective β2AR blockers may be useful in reversing SCI-induced immune suppression -- particularly in individuals who endure periodic catecholamine “storms” during bouts of autonomic dysreflexia. Also, selective antagonism of β2ARs may prove useful as an adjunct therapy for prophylactic vaccination (e.g., pneumonia).
Acknowledgments
The authors thank Ming Wang, Zhen Guan, Pat Walters, Violeta McGaughy, and Susan Moseley for their technical assistance. The authors also thank Daniel Ankeny, Kristina Kigerl and Dana McTigue for their critical review. Funding was provided by NIH T32 AI55411 (KML), NIH AI37326 (VMS), NIH NS047175 (PGP) and P30-NSO45758.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ankeny DP, Lucin KM, Sanders VM, McGaughy VM, Popovich PG. Spinal cord injury triggers systemic autoimmunity: evidence for chronic B lymphocyte activation and lupus-like autoantibody synthesis. Journal of Neurochemistry. 2006;99:1073–1087. doi: 10.1111/j.1471-4159.2006.04147.x. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin. Sci. (Lond) 1998;94:557–572. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- Bennett B, Check IJ, Olsen MR, Hunter RL. A comparison of commercially available adjuvants for use in research. J. Immunol. Methods. 1992;153:31–40. doi: 10.1016/0022-1759(92)90302-a. [DOI] [PubMed] [Google Scholar]
- Bloom SR, Edwards AV, Jones CT. The adrenal contribution to the neuroendocrine responses to splanchnic nerve stimulation in conscious calves. J. Physiol. 1988;397:513–526. doi: 10.1113/jphysiol.1988.sp017016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campagnolo DI, Bartlett JA, Chatterton R, Keller SE. Adrenal and pituitary hormone patterns after spinal cord injury. Amer. J. Phys. Med. Rehab. 1999;11:361–366. doi: 10.1097/00002060-199907000-00013. [DOI] [PubMed] [Google Scholar]
- Campagnolo DI, Bartlett JA, Keller SE, Sanchez W, Oza R. Impaired phagocytosis of Staphylococcus aureus in complete tetraplegics. Am. J. Phys. Med. Rehabil. 1997;76:276–280. doi: 10.1097/00002060-199707000-00005. [DOI] [PubMed] [Google Scholar]
- Campagnolo DI, Keller SE, DeLisa JA, Glick TJ, Sipski ML, Schleifer SJ. Alteration of immune system function in tetraplegics. A pilot study. Am. J. Phys. Med. Rehabil. 1994;73:387–393. doi: 10.1097/00002060-199411000-00003. [DOI] [PubMed] [Google Scholar]
- Cano G, Sved AF, Rinaman L, Rabin BS, Card JP. Characterization of the central nervous system innervation of the rat spleen using viral transneuronal tracing. J. Comp Neurol. 2001;439:1–18. doi: 10.1002/cne.1331. [DOI] [PubMed] [Google Scholar]
- Chastre J, Fagon JY. Ventilator-associated pneumonia. Am. J. Respir. Crit Care Med. 2002;165:867–903. doi: 10.1164/ajrccm.165.7.2105078. [DOI] [PubMed] [Google Scholar]
- Cruse JM, Keith JC, Bryant ML, Jr., Lewis RE., Jr. Immune system-neuroendocrine dysregulation in spinal cord injury. Immunology Research. 1996;15:306–314. doi: 10.1007/BF02935314. [DOI] [PubMed] [Google Scholar]
- Cruse JM, Lewis RE, Bishop GR, Kliesch WF, Gaitan E. Neuroendocrine-immune interactions associated with loss and restoration of immune system function in spinal cord injury and stroke patients. Immunology Research. 1992;11:104–116. doi: 10.1007/BF02918615. [DOI] [PubMed] [Google Scholar]
- Cruse JM, Lewis RE, Jr., Bishop GR, Kliesch WF, Gaitan E, Britt R. Decreased immune reactivity and neuroendocrine alterations related to chronic stress in spinal cord injury and stroke patients. Pathobiology. 1993;61:183–192. doi: 10.1159/000163790. [DOI] [PubMed] [Google Scholar]
- Davies AO, Lefkowitz RJ. Agonist-promoted high affinity state of the beta-adrenergic receptor in human neutrophils: modulation by corticosteroids. J. Clin. Endocrinol. Metab. 1981;53:703–708. doi: 10.1210/jcem-53-4-703. [DOI] [PubMed] [Google Scholar]
- del Rey A, Kabiersch A, Petzoldt S, Besedovsky HO. Sympathetic abnormalities during autoimmune processes: potential relevance of noradrenaline-induced apoptosis. Ann. N. Y. Acad. Sci. 2003;992:158–167. doi: 10.1111/j.1749-6632.2003.tb03146.x. [DOI] [PubMed] [Google Scholar]
- DeVivo MJ, Kartus PL, Stover SL, Rutt RD, Fine PR. Cause of death for patients with spinal cord injuries. Arch. Intern. Med. 1989;149:1761–1766. [PubMed] [Google Scholar]
- Engeland WC. Functional innervation of the adrenal cortex by the splanchnic nerve. Horm. Metab Res. 1998;30:311–314. doi: 10.1055/s-2007-978890. [DOI] [PubMed] [Google Scholar]
- Ewig S, Torres A, El Ebiary M, Fabregas N, Hernandez C, Gonzalez J, Nicolas JM, Soto L. Bacterial colonization patterns in mechanically ventilated patients with traumatic and medical head injury. Incidence, risk factors, and association with ventilator-associated pneumonia. Am. J. Respir. Crit Care Med. 1999;159:188–198. doi: 10.1164/ajrccm.159.1.9803097. [DOI] [PubMed] [Google Scholar]
- Felten DL, Ackerman KD, Wiegand SJ, Felten SY. Noradrenergic sympathetic innervation of the spleen: I. Nerve fibers associate with lymphocytes and macrophages in specific compartments of the splenic white pulp. J. Neurosci. Res. 1987;18:28–21. doi: 10.1002/jnr.490180107. [DOI] [PubMed] [Google Scholar]
- Felten SY, Olschowka J. Noradrenergic sympathetic innervation of the spleen: II. Tyrosine hydroxylase (TH)-positive nerve terminals form synapticlike contacts on lymphocytes in the splenic white pulp. J. Neurosci. Res. 1987;18:37–48. doi: 10.1002/jnr.490180108. [DOI] [PubMed] [Google Scholar]
- Gu C, Ma YC, Benjamin J, Littman D, Chao MV, Huang XY. Apoptotic signaling through the beta -adrenergic receptor. A new Gs effector pathway. Journal of Biological Chemistry. 2000;275:20726–20733. doi: 10.1074/jbc.M000152200. [DOI] [PubMed] [Google Scholar]
- Harris TJ, Waltman TJ, Carter SM, Maisel AS. Effect of prolonged catecholamine infusion on immunoregulatory function: implications in congestive heart failure. J. Am. Coll. Cardiol. 1995;26:102–109. doi: 10.1016/0735-1097(95)00123-h. [DOI] [PubMed] [Google Scholar]
- Hauben E, Ibarra A, Mizrahi T, Barouch R, Agranov E, Schwartz M. Vaccination with a Nogo-A-derived peptide after incomplete spinal-cord injury promotes recovery via a T-cell-mediated neuroprotective response: comparison with other myelin antigens. Proc. Natl. Acad. Sci. U. S. A. 2001;98:15173–15178. doi: 10.1073/pnas.011585298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes K, Yardley CP, Weaver LC. Evidence for descending tonic inhibition specifically affecting sympathetic pathways to the kidney in rats. J. Physiol. 1991;434:295–306. doi: 10.1113/jphysiol.1991.sp018470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, McKerracher L, Braun PE, David S. A therapeutic vaccine approach to stimulate axon regeneration in the adult mammalian spinal cord. Neuron. 1999;24:639–647. doi: 10.1016/s0896-6273(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Hurford WE, Hochachka PW, Schneider RC, Guyton GP, Stanek KS, Zapol DG, Liggins GC, Zapol WM. Splenic contraction, catecholamine release, and blood volume redistribution during diving in the Weddell seal. J. Appl. Physiol. 1996;80:298–306. doi: 10.1152/jappl.1996.80.1.298. [DOI] [PubMed] [Google Scholar]
- Jakeman LB, Guan Z, Wei P, Ponnappan R, Dzwonczyk R, Popovich PG, Stokes BT. Traumatic spinal cord injury produced by controlled contusion in mouse. J Neurotrauma. 2000;17:299–319. doi: 10.1089/neu.2000.17.299. [DOI] [PubMed] [Google Scholar]
- Jones TB, Basso DM, Sodhi A, Pan JZ, Hart RP, MacCallum RC, Lee S, Whitacre CC, Popovich PG. Pathological CNS autoimmune disease triggered by traumatic spinal cord injury: implications for autoimmune vaccine therapy. J. Neurosci. 2002;22:2690–2700. doi: 10.1523/JNEUROSCI.22-07-02690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller SE, Weiss JM, Schleifer SJ, Miller NE, Stein M. Stress-induced suppression of immunity in adrenalectomized rats. Science. 1983;221:1301–1304. doi: 10.1126/science.6612346. [DOI] [PubMed] [Google Scholar]
- Kohm AP, Sanders VM. Suppression of antigen-specific Th2 cell-dependent IgM and IgG1 production following norepinephrine depletion in vivo. J. Immunol. 1999;162:5299–5308. [PubMed] [Google Scholar]
- Krum H, Brown DJ, Rowe PR, Louis WJ, Howes LG. Steady state plasma [3H]-noradrenaline kinetics in quadriplegic chronic spinal cord injury patients. J. Auton. Pharmacol. 1990;10:221–226. doi: 10.1111/j.1474-8673.1990.tb00021.x. [DOI] [PubMed] [Google Scholar]
- Luster MI, Portier C, Pait DG, Rosenthal GJ, Germolec DR, Corsini E, Blaylock BL, Pollock P, Kouchi Y, Craig W. Risk assessment in immunotoxicology. II. Relationships between immune and host resistance tests. Fundam. Appl. Toxicol. 1993;21:71–82. doi: 10.1006/faat.1993.1074. [DOI] [PubMed] [Google Scholar]
- Maisel AS. Beneficial effects of metoprolol treatment in congestive heart failure. Reversal of sympathetic-induced alterations of immunologic function. Circulation. 1994;90:1774–1780. doi: 10.1161/01.cir.90.4.1774. [DOI] [PubMed] [Google Scholar]
- Mak JC, Nishikawa M, Shirasaki H, Miyayasu K, Barnes PJ. Protective effects of a glucocorticoid on downregulation of pulmonary beta 2-adrenergic receptors in vivo. J. Clin. Invest. 1995;96:99–106. doi: 10.1172/JCI118084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melmon KL, Bourne HR, Weinstein Y, Shearer GM, Kram J, Bauminger S. Hemolytic plaque formation by leukocytes in vitro. Control by vasoactive hormones. J. Clin. Invest. 1974;53:13–21. doi: 10.1172/JCI107530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell K, Oke AF, Adams RN. In vivo dynamics of norepinephrine release-reuptake in multiple terminal field regions of rat brain. J. Neurochemistry. 1994;63:917–926. doi: 10.1046/j.1471-4159.1994.63030917.x. [DOI] [PubMed] [Google Scholar]
- Molina PE. Noradrenergic inhibition of TNF upregulation in hemorrhagic shock. Neuroimmunomodulation. 2001;9:125–133. doi: 10.1159/000049016. [DOI] [PubMed] [Google Scholar]
- Moraska A, Deak T, Spencer RL, Roth D, Fleshner M. Treadmill running produces both positive and negative physiological adaptations in Sprague-Dawley rats. Am. J. Physiol Regul. Integr. Comp Physiol. 2000;279:R1321–R1329. doi: 10.1152/ajpregu.2000.279.4.R1321. [DOI] [PubMed] [Google Scholar]
- Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr. Rev. 1984;5:25–44. doi: 10.1210/edrv-5-1-25. [DOI] [PubMed] [Google Scholar]
- Nash MS. Known and plausible modulators of depressed immune functions following spinal cord injuries. J Spinal Cord. Med. 2000;23:111–120. doi: 10.1080/10790268.2000.11753518. [DOI] [PubMed] [Google Scholar]
- Oberbeck R, van Griensven M, Nickel E, Tschernig T, Wittwer T, Pape HC. Influence of beta-adrenoceptor antagonists on hemorrhage-induced cellular immune suppression. Shock. 2002;18:331–335. doi: 10.1097/00024382-200210000-00007. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J. Immunol. 2006;176:6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- Podojil JR, Sanders VM. Selective regulation of mature IgG1 transcription by CD86 and beta 2-adrenergic receptor stimulation. J. Immunol. 2003;170:5143–5151. doi: 10.4049/jimmunol.170.10.5143. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Stuckman S, Gienapp IE, Whitacre CC. Alterations in immune cell phenotype and function after experimental spinal cord injury. J. Neurotrauma. 2001;18:957–966. doi: 10.1089/089771501750451866. [DOI] [PubMed] [Google Scholar]
- Prass K, Meisel C, Hoflich C, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, Volk HD, Meisel A. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J. Exp. Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JB, Schneerson R, Szu SC. Perspective: hypothesis: serum IgG antibody is sufficient to confer protection against infectious diseases by inactivating the inoculum. J. Infect. Dis. 1995;171:1387–1398. doi: 10.1093/infdis/171.6.1387. [DOI] [PubMed] [Google Scholar]
- Sanders VM, Baker RA, Ramer-Quinn DS, Kasprowicz DJ, Fuchs BA, Street NE. Differential expression of the beta2-adrenergic receptor by Th1 and Th2 clones: implications for cytokine production and B cell help. J. Immunol. 1997;158:4200–4210. [PubMed] [Google Scholar]
- Schmid A, Halle M, Stutzle C, Konig D, Baumstark MW, Storch MJ, Schmidt-Trucksass A, Lehmann M, Berg A, Keul J. Lipoproteins and free plasma catecholamines in spinal cord injured men with different injury levels. Clin. Physiol. 2000;20:304–310. doi: 10.1046/j.1365-2281.2000.00263.x. [DOI] [PubMed] [Google Scholar]
- Stevenson JR, Westermann J, Liebmann PM, Hortner M, Rinner I, Felsner P, Wolfler A, Schauenstein K. Prolonged alpha-adrenergic stimulation causes changes in leukocyte distribution and lymphocyte apoptosis in the rat. J. Neuroimmunol. 2001;120:50–57. doi: 10.1016/s0165-5728(01)00417-9. [DOI] [PubMed] [Google Scholar]
- Strack AM, Sawyer WB, Platt KB, Loewy AD. CNS cell groups regulating the sympathetic outflow to adrenal gland as revealed by transneuronal cell body labeling with pseudorabies virus. Brain Res. 1989;491:274–296. doi: 10.1016/0006-8993(89)90063-2. [DOI] [PubMed] [Google Scholar]
- Taylor RB, Weaver LC. Dorsal root afferent influences on tonic firing of renal and mesenteric sympathetic nerves in rats. Am. J. Physiol. 1993;264:R1193–R1199. doi: 10.1152/ajpregu.1993.264.6.R1193. [DOI] [PubMed] [Google Scholar]
- Tibbs PA, Young B, Ziegler MG, McAllister RG., Jr. Studies of experimental cervical spinal cord transection. Part II: Plasma norepinephrine levels after acute cervical spinal cord transection. J. Neurosurg. 1979;50:629–632. doi: 10.3171/jns.1979.50.5.0629. [DOI] [PubMed] [Google Scholar]
- Tvede N, Kappel M, Klarlund K, Duhn S, Halkjaer-Kristensen J, Kjaer M, Galbo H, Pedersen BK. Evidence that the effect of bicycle exercise on blood mononuclear cell proliferative responses and subsets is mediated by epinephrine. Int. J. Sports Med. 1994;15:100–104. doi: 10.1055/s-2007-1021028. [DOI] [PubMed] [Google Scholar]
- Vega JL, Ganea D, Jonakait GM. Acute down-regulation of antibody production following spinal cord injury: role of systemic catecholamines. Journal of Neuropathology and Experimental Neurology. 2003;62:848–854. doi: 10.1093/jnen/62.8.848. [DOI] [PubMed] [Google Scholar]
- Wahle M, Pierer M, Krause A, Kolker S, Baerwald CG. Decreased catecholamine-induced cell death in B lymphocytes from patients with rheumatoid arthritis. Ann. N. Y. Acad. Sci. 2002;966:425–428. doi: 10.1111/j.1749-6632.2002.tb04243.x. [DOI] [PubMed] [Google Scholar]
- Wan W, Vriend CY, Wetmore L, Gartner JG, Greenberg AH, Nance DM. The effects of stress on splenic immune function are mediated by the splenic nerve. Brain Res. Bull. 1993;30:101–105. doi: 10.1016/0361-9230(93)90044-c. [DOI] [PubMed] [Google Scholar]
- Woiciechowsky C, Asadullah K, Nestler D, Eberhardt B, Platzer C, Schöning B, Glöckner F, Lanksch WR, Volk H-D, Döcke W-D. Sympathetic activation triggers systemic interleukin-10 release in immunodepression induced by brain injury. Nature Med. 1998;4:808–813. doi: 10.1038/nm0798-808. [DOI] [PubMed] [Google Scholar]
- Woratyla SP, Morgan AS, Mackay L, Bernstein B, Barba C. Factors associated with early onset pneumonia in the severely brain-injured patient. Conn. Med. 1995;59:643–647. [PubMed] [Google Scholar]