

Summary
Nitric oxide (NO) is generated from L-arginine by NO synthases, of which three forms have been identified: endothelial, inducible and neuronal (eNOS, iNOS and nNOS, respectively). The arginine metabolite asymmetric dimethylarginine (ADMA) is a potent, noncompetitive inhibitor of nNOS, while its congener NG-monomethyl-L-arginine (L-NMMA) is a less potent, competitive inhibitor. In rat neurons large amounts of ADMA are found, suggesting its importance in modulatin neuronal activity.
Humans generate approximately 300 μmol (∼60 mg) ADMA per day. It is released from myelin basic proteins that are highly expressed in neuronal tissue. ADMA is mainly degraded by the action of the enzyme dimethylarginine dimethylaminohydrolase (DDAH), which exists in two isoforms. DDAH1 is highly expressed in brain, suggesting specific function in this area. The presence of nNOS and DDAH1 in brain suggests that ADMA may have specific CNS activity and be more than an unregulated metabolite.
Increased NO production - either prior to or concurrently with opioid administration - results in an enhanced rate and extent of development of tolerance to morphine in mice. NO produces an alteration in the μ-opioid receptor that increases constitutive receptor activity. It thereby reduces the ability of a selective μ-opioid agonist to activate the μ-opioid receptor; these in vitro molecular effects occur in a time course consistent with the in vivo development of antinociceptive tolerance in mice. Amongst many other synthetic NOS inhibitors of varying specificity, 7-nitroindazole (7-NI) has been shown to have a high affinity (IC50 0.71 μM) to nNOS. Selective blockade of nNOS by 7-NI attenuated morphine withdrawal in opiate dependent rats, suggesting nNOS as a viable target for development of pharmacotherapies.
We hypothesize that, by inhibiting nNOS and reducing NO levels, ADMA may decrease μ-opiate receptor constitutive activity, resulting in alteration of the analgesic dose-response curve of morphine.
Keywords: addiction, opiates, heroine, nociception, NOS, Asymmetrical dimethylarginine, NG-monomethyl-L-arginine
Endogenous inhibitors of NOS in nervous tissue
Guanidino-methylated arginines (MA) including ADMA and its congener NG-monomethyl-L-arginine (L-NMMA) are potent endogenous inhibitors of NOS that were first identified in human urine in 1970 by Kakimoto et al. [1].
Although it is an analog of L-arginine, no direct route of synthesizing ADMA from the free amino acid has been identified. Instead, a rather complex process leads to the generation of ADMA. Protein-arginine methyltransferases (PRMTs) catalyze the formation of methylarginine residues from proteins that have been post-translationally methylated and subsequently hydrolysed. These proteins are largely found in the nucleolus and appear to be involved in RNA processing and transcriptional control. Because of its high content of methylated proteins, asymmetric (ADMA) and symmetric (SDMA) were first isolated from bovine brain [2].
In addition to different substrate specificities [3], two subclasses of PRMT appear to have different catalytic activities: the myelin basic protein-specific activity (type 2) catalyses the formation of L-NMMA and SDMA (symmetric dimethylarginine with no direct effect on NOS), while the non-myelin basic protein-specific activity (type 1) catalyses the formation of L-NMMA and ADMA [4-6].
It is known that myelin basic protein, which is highly expressed in neuronal tissue, is a principal target of the PRMT 2 isoform. In addition to the substrate specificity of PRMT 2, it also has specificity in its catalytic activity, with this myelin basic protein specific enzyme preferentially catalyzing the formation of NMMA [6]. Cardounel [7] and Zweier determined the concentrations of L-arginine and MA in rat cerebellar granule neurons and homogenized whole brain. Considerable MA and L-arginine concentrations were measured in neurons, with values of 11.1 ± 1.1 μmol/L for NMMA, 3.9 ± 0.6 μmol/L for ADMA, and 88.6±6.5 μmol/L for L-arginine. In freshly isolated and homogenized whole rat brain, the levels of NMMA, ADMA, and L-arginine were similar to those measured in the cerebellar neurons with values (mean ± S.E.) of 10.7 ± 1.3 μmol/L, 5.1 ± 0.6 μmol/L, and 94.0 ± 7.8 μmol/L, respectively [8].
Another evidence for the role of MA in neuronal activity is its elimination (Fig. 1). Since ADMA was found to be elevated in dialysis patients, renal excretion of ADMA was considered to be the main route of elimination [9]. However, an early study from McDermott in a rabbit model revealed that a catabolic pathway had to be present [10]. This major metabolic pathway is degradation by DDAH, first isolated from the rat kidney, which hydrolyses ADMA to dimethylamine and L-citrulline [11]. Vascular endothelium may be the chief tissue responsible for the catabolism of ADMA via DDAH [12].
Fig. 1.
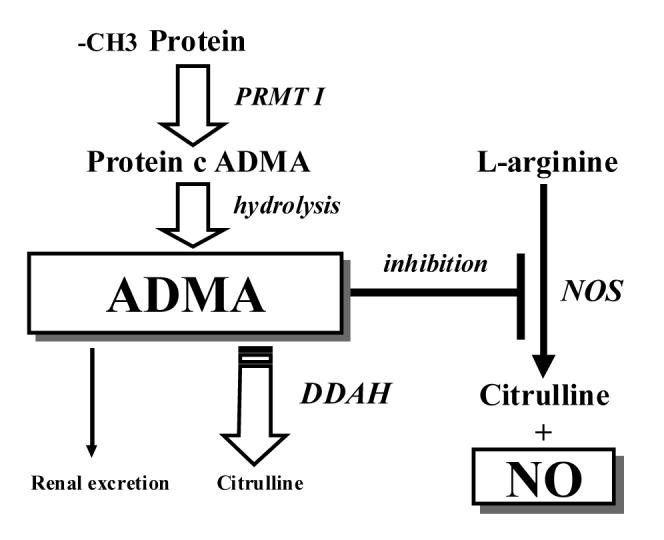
Formation, action, metabolism and elimination of ADMA. ADMA= asymmetrical dimethylarginine, DDAH= dimethylaminohydrolase, NO= nitric oxide, protein c ADMA= protein cum ADMA
Tran et al. [13] identified two different subtypes of DDAH. DDAH1 expression predominates in tissues that also express the neuronal isoform of NOS while DDAH2 expression predominates in more highly vascularized tissues. Analysis of 14 brain regions revealed that while DDAH1 expression predominated in the forebrain, significant DDAH2 expression (with lower DDAH1 expression) was apparent in the medulla and spinal cord. The functional significance of this finding is not yet known, but, given the primitive origins of the brainstem, this would be consistent with an earlier evolutionary origin of DDAH2 compared with DDAH1.
Specificity of ADMA and NMMA for neuronal NOS
NNOS is expressed both in central and peripheral neurons [14]. Both L-NMMA and ADMA exhibit prominent dose-dependent inhibition of nNOS in the presence of physiologic cellular L-arginine levels, and this inhibition was sustained for over 30 min [7]. L-NMMA inhibits NO production by 26% at 10 μmol/L, with almost complete inhibition at 100 μmol/L. These experiments were carried out in the presence of 100 μmol/L of L-arginine. With ADMA the results were comparable, with 23% inhibition at 10 μmol/L and almost complete inhibition at 100 μmol/L. While ADMA is a potent (IC50 1.5 μmol/L), noncompetitive inhibitor (Ki, 0.4 μmol/L; Kii, 1.6 μmol/L) of nNOS, L-NMMA is a competitive inhibitor (Ki, 0.65 μmol/L). In addition, ADMA is known to be preferential for nNOS over eNOS [15] (Fig. 2). In contrast to the similarities between ADMA and L-NMMA in nervous tissues, there are dissimilarities with regard to their content in plasma. It has been reported that plasma concentrations of ADMA are in the range of 0.5-5 μmol/L [6]. Teerlink et al. [16] reported that plasma from healthy volunteers (n = 53) contained 0.42 ± 0.06 μmol/L ADMA using HPLC with limits of quantification of 0.01 μmol/L for ADMA. Plasma NMMA concentrations were much lower (0.04-0.11 μmol/L), in agreement with previously reported values [17]. In general, L-NMMA levels seem to range around 10% of ADMA levels and are not very often reported in clinical studies.
Fig. 2.

Inhibition of eNOS and nNOS activity in isolated preparations by ADMA. The IC50 values are estimated to be about 1.5 μmol/L for nNOS and 12 μmol/L for eNOS. eNOS and nNOS activity was measured as nitrite production rate.
Morphine tolerance and NO
There is evidence that NO is involved in the behavioral effects induced by morphine and cocaine, e.g., behavioral sensitization [18-20], kindling [21] and conditioned place preference (CPP) [22, 23]. Furthermore, hippocampal injection of NG-nitro-L-arginine methyl ester (L-NAME), a synthetic NOS inhibitor, blocks the enhancing effect of L-arginine on the expression of the morphine-induced conditioned place preference [23]. Increases in NO content in rats occur in a close temporal relationship with the loss of morphine’s antinociceptive effect [24]. Thus, NO appears to be a key modulator of morphine tolerance, and may be involved in the receptor-based adaptations observed with chronic μ-opioid receptor agonist administration.
Heinzen and Pollack [24] investigated the effects of NO on μ-opioid receptor agonist and antagonist binding and Giα activation in the presence and absence of morphine in rat brain tissue. Systemic infusion of the NO precursor L-arginine, in the absence of in vivo morphine administration (NO control) and infusion of morphine alone (tolerant group) resulted in a dose-dependent increase in efficacy and potency of the selective μ-opioid receptor agonist [D-Ala2, N-MePhe4, Gly-ol5]enkephalin (DAMGO) to produce Giα activation. The enhanced efficacy of DAMGO is not due to NO-induced increases in μ-opioid receptor expression. Animals pretreated with L-arginine, followed by a prolonged morphine infusion (supertolerant group) evidenced compromised efficacy of DAMGO to stimulate Giα binding when compared to control, NO control, and tolerant groups. These molecular effects occurred with a time course consistent with the development of antinociceptive tolerance and are observed as a consequence of the enhanced μ-opioid receptor constitutive activity that is apparent in the supertolerant state and which is measured as Giα binding in the absence of agonist [25]. In the presence of enhanced basal stimulation, opioid peptides cause reduced agonist-stimulated G protein activation [26], thereby minimizing antinociceptive effects. This could increase the risk of dependence by requiring use of higher doses. Recently, in rats, morphine was shown capable of stimulating the release of NO from limbic tissues (hippocampus and amygdala) in a naloxone- and L-NAME-sensitive manner in rats [27]. This feedback mechanism could account for the phenomenon of tolerance.
Inhibition of nNOS and nociceptive tolerance
The molecular signal of opiate tolerance is μ-opioid receptor down-regulation. Inhibition or lack of the inducible form of NOS has been shown to reduce μ-opioid receptor up-regulation in induced intestinal inflammation in mice [28]. We postulate that inhibiting nNOS could produce down-regulation in μ-opiate receptor function. In contrast, opiate dependence and withdrawal lead to an up-regulation of nNOS in normal mice [29] and even more so in μ-opioid receptor deficient mice [30]. Therefore, an opiate-tolerant system with down-regulated μ-opioid receptors and increased nNOS activity should be even more sensitive to nNOS inhibition. Elevated MA levels should protect against the development of tolerance to (and possibly dependence on) morphine. Blockade of nNOS by a selective synthetic inhibitor, i.e. 7-NI, attenuated morphine withdrawal in rats [31], suggesting nNOS as a viable target for development of treatment options [32]. In addition to severe irritation of the mucous membranes by 7-NI, aromatic nitro compounds have been found to be both mutagenic and carcinogenic [33] and are not tested in humans.
Recently it was demonstrated that in vivo inhibition of nNOS through repeated 7-NI administration attenuates the decreased μ-opioid receptor signaling (i.e., tolerance) observed in the locus coeruleus (LC) after morphine treatment in mice [34]. The inhibitory effect of the opioid agonist Met5-enkephalin (ME) on the cell firing rate was evaluated by single-unit extracellular recordings of noradrenergic neurons in the locus coeruleus from brain slices, and the antinociceptive effect of morphine was measured by tail-flick techniques: 7-NI administration in sham-treated rats failed to change the effect induced by morphine in the tail-flick test as compared to vehicle groups. Another study showed changes of catecholaminergic metabolism in the locus coeruleus during morphine withdrawal using microdialysis in freely moving rats [35].
If ADMA blunts development of opiate tolerance in a similar way the therapeutic efficacy of μ-opiates could be improved (Fig 3). To the extent that tolerance and dependence are related, inhibition of nNOS could decrease the risk of developing opiate dependence. One potential mechanism to achieve an increase in ADMA levels for attenuation of withdrawal symptoms or to prevent development of opioid dependence could be the use of a novel DDAH inhibitor, L-291, which has been originally developed and tested in rats with endotoxemia [36]. There is an abundance of literature about increases of ADMA levels in cardiovascular deseases and metabolic syndromes (see [37] for an overview), although the majority of subjects tested in these studies have not been characterized for their alcohol and drug use. Studies on ADMA infusions in healthy volunteers have shown that an increase of ADMA is associated with the risk of decreased cerebral blood flow. Future research should concentrate on changes in ADMA and NO levels during development of opiate tolerance and dependence and during and after withdrawal in humans to clarify the role of ADMA in the etiology of opiate dependence and tolerance, while animal studies will elucidate the possible beneficial effects of ADMA increases as a therapeutic intervention.
Fig. 3.

NO pathway and the effects on antinociceptive properties of morphine. - and + mean inhibition and activation, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Kakimoto Y, Akazawa S. Isolation and identification of N-G, N-G- and N-G, N’-G-dimethyl-arginine, N-epsillon-mono-, di-, and trimethyllysine, and glucosylgalactosyl- and galactosyl-delta-hydroxylysine from human urine. J. Biol. Chem. 1970;245:5751–5758. [PubMed] [Google Scholar]
- [2].Nakajima T, Matsuoka Y, Kakimoto Y. Isolation and identification of N-G-monomethyl, N-G, N-G-dimethyl- and N-G, N’G-dimethylarginine from the hydrolysate of proteins of bovine brain. Biochim. Biophys. Acta. 1971;230:212–222. doi: 10.1016/0304-4165(71)90206-6. [DOI] [PubMed] [Google Scholar]
- [3].Najbauer J, Johnson B, Young A, Aswad D. Peptides with sequences similar to glycine, arginine-rich motifs in proteins interacting with RNA are efficiently recognized by methyltransferase(s) modifying arginine in numerous proteins. J. Biol. Chem. 1993;268:10501–10509. [PubMed] [Google Scholar]
- [4].Ghosh S, Paik W, Kim S. Purification and molecular identification of two protein methylases I from calf brain myelin basic protein-and histone-specific enzyme. J. Biol. Chem. 1988;263:19024–19033. [PubMed] [Google Scholar]
- [5].Rajpurohit R, Paik W, Kim S. Enzymatic methylation of heterogenous nuclear ribonucleoprotein in isolated liver nuclei. Biochim. Biophys. Acta. 1992;1122:183–188. doi: 10.1016/0167-4838(92)90322-5. [DOI] [PubMed] [Google Scholar]
- [6].Leiper J, Vallance P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc. Res. 1999;43:542–548. doi: 10.1016/s0008-6363(99)00162-5. [DOI] [PubMed] [Google Scholar]
- [7].Cardounel AJ, Zweier JL. Endogenous methylarginines regulate neuronal nitric-oxide synthase and prevent excitotoxic injury. J. Biol.Chem. 2002;277:33995–34002. doi: 10.1074/jbc.M108983200. [DOI] [PubMed] [Google Scholar]
- [8].Cardounel AJ, Xia Y, Zweier JL. Endogenous methylarginines modulate superoxide as well as nitric oxide generation from neuronal nitric-oxide synthase: differences in the effects of monomethyl- and dimethylarginines in the presence and absence of tetradihydrobiopterin. J. Biol. Chem. 2005;280:7540–7549. doi: 10.1074/jbc.M410241200. [DOI] [PubMed] [Google Scholar]
- [9].Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of and endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572–575. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- [10].McDermott J. Studies on the catabolism of Ng-methylarginine, N-G, N-G-dimethylarginine and N-G, N-G-dimethylarginine in the rabbit. Biochem. J. 1976;154:179–184. doi: 10.1042/bj1540179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ogawa T, Kimoto M, Sasaoka K. Purification and properties of a new enzyme, N-G, N-G-dimethylarginine dimethylaminohydrolase, from rat kidney. J. Biol. Chem. 1989;264:10205–10209. [PubMed] [Google Scholar]
- [12].McCarty MF. Vascular endothelium is the organ chiefly responsible for the catabolism of plasma asymmetric dimethylarginine--an explanation for the elevation of plasma ADMA in disorders characterized by endothelial dysfunction. Med. Hypotheses. 2004;63:699–708. doi: 10.1016/j.mehy.2002.11.008. [DOI] [PubMed] [Google Scholar]
- [13].Tran CT, Fox MF, Vallance P, Leiper JM. Chromosomal localization, gene structure, and expression pattern of DDAH1: comparison with DDAH2 and implications for evolutionary origins. Genomics. 2000;68:101–105. doi: 10.1006/geno.2000.6262. [DOI] [PubMed] [Google Scholar]
- [14].Schuman EM, Madison DV. Nitric oxide and synaptic function. Annu. Rev. Neurosci. 1994;17:153–183. doi: 10.1146/annurev.ne.17.030194.001101. [DOI] [PubMed] [Google Scholar]
- [15].Tsikas D, Boger RH, Sandmann J, Bode-Boger SM, Frolich JC. Endogenous nitric oxide synthase inhibitors are responsible for the L-arginine paradox. FEBS Lett. 2000;478:1–3. doi: 10.1016/s0014-5793(00)01686-0. [DOI] [PubMed] [Google Scholar]
- [16].Teerlink T, Nijveldt R, deJong S, VanLeeuwen P. Determination of Arginine, Asymmetric Dimethylarginine, and Symmetric Dimethylarginine in Human Plasma and Other Biological Samples by High-Performance Liquid Chromatography. Anal. Biochem. 2002;303:137–137. doi: 10.1006/abio.2001.5575. [DOI] [PubMed] [Google Scholar]
- [17].Anderstam B, Katzarski K, Bergstrom J. Serum levels of NG, NG-dimethyl-L-arginine, a potential endogenous nitric oxide inhibitor in dialysis patients. J. Am. Soc. Nephrol. 1997;8:1437–1442. doi: 10.1681/ASN.V891437. [DOI] [PubMed] [Google Scholar]
- [18].Itzhak Y. Modulation of cocaine- and methamphetamine-induced behavioral sensitization by inhibition of brain nitric oxide synthase. J. Pharmacol. Exp. Ther. 1997;282:521–527. [PubMed] [Google Scholar]
- [19].Itzhak Y, Ali S, Martin J, Black M, Huang P. Resistance of neuronal nitric oxide synthase-deficient mice to cocaine-induced locomotor sensitization. Psychopharmacology. 1998;140:378–386. doi: 10.1007/s002130050779. [DOI] [PubMed] [Google Scholar]
- [20].Zarrindast M, Gholami A, Sahraei H, Haeri-Rohani A. Role of nitric oxide in the acquisition and expression of apomorphine- or morphine-induced locomotor sensitization. Eur. J. Pharmacol. 2003;482:205–213. doi: 10.1016/j.ejphar.2003.10.006. [DOI] [PubMed] [Google Scholar]
- [21].Itzhak Y. Attenuation of cocaine kindling by 7-nitroindazole, an inhibitor of brain nitric oxide synthase. Neuropharmacology. 1996;35:1065–1073. doi: 10.1016/s0028-3908(96)00037-8. [DOI] [PubMed] [Google Scholar]
- [22].Itzhak Y, Martin J, Black M, Huang P. The role of neuronal nitric oxide synthase in cocaine-induced conditioned place preference. Neuroreport. 1998;9:2485–2488. doi: 10.1097/00001756-199808030-00011. [DOI] [PubMed] [Google Scholar]
- [23].Karami M, Zarrindast M, Sepehri H, Sahraei H. Role of nitric oxide in the rat hippocampal CA1 area on morphine-induced conditioned place preference. Eur. J. Pharmacol. 2002;449:113–119. doi: 10.1016/s0014-2999(02)01991-x. [DOI] [PubMed] [Google Scholar]
- [24].Heinzen E, Pollack G. Pharmacodynamics of morphine-induced neuronal nitric oxide production and antinociceptive tolerance development. Brain Research. 2004;1023:175–184. doi: 10.1016/j.brainres.2004.07.015. [DOI] [PubMed] [Google Scholar]
- [25].Heinzen E, Brooth R, Pollack G. Neuronal nitric oxide modulates morphine antinociceptive tolerance by enhancing constitutive activity of the μ-opioid receptor. Biochem. Pharmacol. 2005;69:479–488. doi: 10.1016/j.bcp.2004.11.004. [DOI] [PubMed] [Google Scholar]
- [26].Liu J, Prather P. Chronic exposure to μ-opioid agonists produces constitutive activation of mu-opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Mol. Pharmacol. 2001;60:53–62. doi: 10.1124/mol.60.1.53. [DOI] [PubMed] [Google Scholar]
- [27].Zhu W, Ma Y, Bell A. Presence of morphine in rat amygdala: evidence for the 3 opiate receptor subtype via nitric oxide release in limbic structures. Med. Sci. Monit. 2004;10:BR433–BR439. [PubMed] [Google Scholar]
- [28].Pol O, Sasaki M, Jimenez N, Dawson VL, Dawson TM, Puig MM. The involvement of nitric oxide in the enhanced expression of μ-opioid receptors during intestinal inflammation in mice. Br. J. Pharmacol. 2005;145:758–766. doi: 10.1038/sj.bjp.0706227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cuellar B, Fernandez AP, Lizasoain I, Moro MA, Lorenzo P, Bentura ML, Rodrigo J, Leza JC. Up-regulation of neuronal NO synthase immunoreactivity in opiate dependence and withdrawal. Psychopharmacology (Berl) 2000;148:66–73. doi: 10.1007/s002130050026. [DOI] [PubMed] [Google Scholar]
- [30].Yoo JH, Cho JH, Lee SY, Lee S, Loh HH, Ho IK, Jang CG. Differential effects of morphine- and cocaine-induced nNOS immunoreactivity in the dentate gyrus of hippocampus of mice lacking mu-opioid receptors. Neurosci. Lett. 2006;395:98–102. doi: 10.1016/j.neulet.2005.10.089. [DOI] [PubMed] [Google Scholar]
- [31].Vaupel DB, Kimes AS, London ED. Comparison of 7-nitroindazole with other nitric oxide synthase inhibitors as attenuators of opioid withdrawal. Psychopharmacology (Berl) 1995;118:361–368. doi: 10.1007/BF02245935. [DOI] [PubMed] [Google Scholar]
- [32].Vaupel DB, Kimes AS, London ED. Nitric oxide synthase inhibitors. Preclinical studies of potential use for treatment of opioid withdrawal. Neuropsychopharmacology. 1995;13:315–322. doi: 10.1016/0893-133X(95)00138-4. [DOI] [PubMed] [Google Scholar]
- [33].Rosenkranz HS, Mermelstein R. Mutagenicity and genotoxicity of nitroarenes. All nitro-containing chemicals were not created equal. Mutation Research. 1983;114:217–267. doi: 10.1016/0165-1110(83)90034-9. [DOI] [PubMed] [Google Scholar]
- [34].Santamarta MT, Ulibarri I, Pineda J. Inhibition of neuronal nitric oxide synthase attenuates the development of morphine tolerance in rats. Synapse. 2005;57:38–46. doi: 10.1002/syn.20151. [DOI] [PubMed] [Google Scholar]
- [35].Javelle N, Bérod A, Renaud B, Lambás-Señas L. NO synthase inhibitors attenuate locus coeruleus catecholamine metabolism and behavior induced by morphine withdrawal. Neuroreport. 2002;13:725–728. doi: 10.1097/00001756-200204160-00037. [DOI] [PubMed] [Google Scholar]
- [36].Leiper J, Nandi M, Torrondel B, Murray-Rust J, Malaki M, O’Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill A, McDonald NQ, Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nature medicine. 2007;13:198–2003. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- [37].Kielstein JT, Zoccali C. Asymmetric dimethylarginine: a cardiovascular risk factor and a uremic toxin coming of age? Am J Kidney Dis. 2005;46:186–202. doi: 10.1053/j.ajkd.2005.05.009. [DOI] [PubMed] [Google Scholar]