

Abstract
We have developed a new method, using a nanopipette, for controlled voltage-driven delivery of individual fluorescently labeled probe molecules to the plasma membrane which we used for single-molecule fluorescence tracking (SMT). The advantages of the method are 1), application of the probe to predefined regions on the membrane; 2), release of only one or a few molecules onto the cell surface; 3), when combined with total internal reflection fluorescence microscopy, very low background due to unbound molecules; and 4), the ability to first optimize the experiment and then repeat it on the same cell. We validated the method by performing an SMT study of the diffusion of individual membrane glycoproteins labeled with Atto 647-wheat germ agglutin in different surface domains of boar spermatozoa. We found little deviation from Brownian diffusion with a mean diffusion coefficient of 0.79 ± 0.04 μm2/s in the acrosomal region and 0.10 ± 0.02 μm2/s in the postacrosomal region; this difference probably reflects different membrane structures. We also showed that we can analyze diffusional properties of different subregions of the cell membrane and probe for the presence of diffusion barriers. It should be straightforward to extend this new method to other probes and cells, and it can be used as a new tool to investigate the cell membrane.
INTRODUCTION
Currently, a major problem in membrane biology is to understand how the heterogeneous distribution of component lipids and proteins is generated and maintained against the randomizing forces of diffusion, especially in highly polarized cells with specialized functions (1). Progress in addressing this problem has depended to a large extent on the application of novel biophysical methods, particularly methods that have sufficient resolution to interrogate the dynamics of submicron structures like lipid rafts or transient confinement zones. This is achievable only by tracking and analyzing the diffusion of single molecules in real time, where the centroid of the diffraction-limited optical image from the molecule can be determined with a precision greater than 50 nm. Gold-conjugated molecules have been widely employed, although the size of this tag (10–40 nm) may perturb their motion (2). More recently, fluorophores (e.g., green fluorescent protein proteins, dye-conjugated lipids) have been used as labels as they are less perturbative but have the disadvantage of reduced observation times as eventually they photobleach (3–6). Single-molecule tracking experiments observe motion from random positions on the cell surface. Presently, there is no capability for delivering the molecule to a predefined position to systematically investigate diffusional properties in different areas of the cell surface. This is particularly important when a fluorophore-labeled probe is used since the length of the trajectory is limited by photobleaching. This means that without control of the region explored by the probe it is not possible to systematically explore the diffusion in different regions since the probe is unlikely to reach the region of interest before photobleaching. In addition, it would be advantageous to control the number of labeled molecules diffusing over the cell surface to avoid problems caused by tracking intersecting trajectories. The topography of the cell surface is not determined or known in many experiments, making it difficult to correlate the measured trajectory to the presence of a particular cell structure.
It is clear all current biophysical methods have limitations and so there is a pressing need to develop new investigative tools. To overcome this problem, we are developing a method to probe the structure and function of the membrane of live cells by measuring the diffusion of fluorophore-labeled molecules, such as lipids and glycoproteins, at the single-molecule level beginning at a defined region. Our aim is to exploit advances in scanning ion-conductance microscopy (SICM) (7,8) to controllably deliver individual fluorophore-labeled probes to a defined position on the cell surface using a nanopipette operating under physiological buffer. The diffusion of the individual fluorophore-tagged molecules will then be followed in real time using single-molecule fluorescence detection and a sensitive charge-coupled device (CCD) camera. In this way we can directly interrogate the diffusional properties of different regions of the cell membrane and the presence of barriers to diffusion.
A second issue is to perform the biophysical studies on well-characterized and reproducible cell surfaces. We are currently using the sperm plasma membrane as a paradigm for mammalian cell surface membranes in general. Spermatozoa are a tractable system for investigating many membrane phenomena and have several advantages over other cell types. First, they are available as single cells and hence are readily accessible to scanning nanopipettes. Second, they remain viable in a suitable diluent for 3–4 days at room temperature, allowing prolonged study. Third, they have a highly polarized and reproducible structure and topography, allowing us to compare data from different cells. In particular, the plasma membrane overlying the boar sperm head, which is ∼9 μm in length and ∼4 μm at its widest point, has three well-defined surface macrodomains that are antigenically different. These are known as the anterior acrosome, equatorial segment, and postacrosome (see Fig. 2 C) (9). We postulate that these differences are maintained by diffusion barriers or organizational disparities in membrane structure. Fourth, spermatozoa are transcriptionally inactive, and hence protein synthesis and intracellular trafficking are not confounding issues. Fifth, their surface is relatively flat as measured by atomic force microscopy (AFM) (10) and SICM (8), making them more amenable to single-molecule tracking studies than somatic cells which contain microvilli and surface projections of various dimensions. Sixth, the sperm plasma membrane undergoes a series of developmental changes (collectively known as capacitation) in preparation for fusion with the egg that can be controlled experimentally by sequestering membrane cholesterol with cyclodextrins (11). Thus differences in diffusional properties of the macrodomains can potentially be related to physiological function. As a paradigm for a polarized cell, therefore, spermatozoa have considerable merits.
FIGURE 2.

(A and B) Two live boar spermatozoa labeled with Alexa 488-WGA from solution. Note the uniform fluorescence over the entire plasma membrane. Scale bar = 10 microns. (C) Outline of the head from a boar spermatozoa showing the different regions or macrodomains.
In previous work we used a nanopipette as a nanopen to deliver biomolecules in ionic solution to functionalized surfaces (7,12–17). An ion current that flows between electrodes in the pipette and the bath is used to control the distance between nanopipette tip and surface. The number of delivered molecules is controlled by the sign and magnitude of the applied voltage (12,13). The biomolecules are always in buffered solution and maintain their biological activity. We have recently shown that we can deliver controlled numbers of functional biomolecules, such as DNA and antibodies, to a surface, that the antibody-antigen interactions are specific, and that we can address a specific feature on the surface with a second biomolecule (14).
In this investigation we describe the use of a nanopipette to deliver individual molecules of a fluorescent probe to preselected sites on the plasma membrane of live boar spermatozoa to track the diffusion of membrane glycoproteins in different macrodomains.
MATERIALS AND METHODS
Sperm preparation
Ejaculated spermatozoa were obtained fresh from boars maintained at the Babraham Institute or supplied by JSR Genetics (Southburn, East Yorkshire, UK). All procedures involving animals were carried out within UK Home Office approved guidelines. Spermatozoa were diluted in HEPES-buffered Tyrode's medium with added lactate and pyruvate (TALP) medium (18) minus Ca2+ and 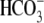 and washed once by centrifugation through a Percoll step gradient (35%:75% Percoll in TALP). This procedure removes dead cells and debris (19). The live fraction obtained is >90% viable on the basis of their motility and exclusion of DNA-binding dyes such as propidium iodide (20). The live sperm fraction at the bottom of the gradient was resuspended in 1 ml TALP, centrifuged, and the pellet resuspended to ∼107 sperm/ml with TALP. Glass coverslips (16 mm diameter) were glued onto 30 mm petri dishes to overlie 10 mm central holes, coated with poly-L-lysine (0.1%, Sigma, Poole, Dorset, UK) and washed with MilliQ water (Millipore, Bedford, MA). Live sperm suspensions were applied to the coverslips in the presence of sodium azide (0.003% or 0.549 mM final concentration). This low concentration of azide uncouples motility from metabolism (21) with the result that sperm are immobilized but continue to produce sufficient ATP to remain alive. The azide effect is reversible and was confirmed for these experiments (data not shown). After 15 min, unbound spermatozoa were washed away and the dishes filled with 3 ml TALP buffer. All experiments were performed at room temperature (22°C–23°C).
and washed once by centrifugation through a Percoll step gradient (35%:75% Percoll in TALP). This procedure removes dead cells and debris (19). The live fraction obtained is >90% viable on the basis of their motility and exclusion of DNA-binding dyes such as propidium iodide (20). The live sperm fraction at the bottom of the gradient was resuspended in 1 ml TALP, centrifuged, and the pellet resuspended to ∼107 sperm/ml with TALP. Glass coverslips (16 mm diameter) were glued onto 30 mm petri dishes to overlie 10 mm central holes, coated with poly-L-lysine (0.1%, Sigma, Poole, Dorset, UK) and washed with MilliQ water (Millipore, Bedford, MA). Live sperm suspensions were applied to the coverslips in the presence of sodium azide (0.003% or 0.549 mM final concentration). This low concentration of azide uncouples motility from metabolism (21) with the result that sperm are immobilized but continue to produce sufficient ATP to remain alive. The azide effect is reversible and was confirmed for these experiments (data not shown). After 15 min, unbound spermatozoa were washed away and the dishes filled with 3 ml TALP buffer. All experiments were performed at room temperature (22°C–23°C).
Lectin labeling of spermatozoa
To a vial containing ∼0.3 mg Atto 647-NHS ester (ATTO-TEC, Siegen-Weidenau, Germany) was added 0.5 mL WGA solution (2 mg/mL, Cambridge Bioscience, Cambridge, UK) and 50 μL sodium bicarbonate buffer (1 M, pH 8.3) to adjust the solution pH to ∼8.3. The resulting solution was thoroughly mixed and then gently stirred at room temperature for 2 h, after which the labeling reaction was terminated by addition of 100 μL hydroxylamine solution (1.5 M, pH 8.5). The mixture was then loaded on a gel filtration column and washed with phosphate buffer saline buffer (pH 7.2). The labeled WGA was collected as the first eluted blue band. The degree of labeling was determined by measuring the ultraviolet-visible absorbance at 280 and 644 nm, based on the molecular extinction coefficients of 78,000 cm−1 M−1 for WGA at 280 nm and 150,000 cm−1 M−1 for Atto-647 at 644 nm. The average labeling was 1.5 Atto647 fluorophores per WGA molecule. For details of suspension labeling and Western blotting, see Supplementary Material.
Pipettes, SICM construction, and probe delivery
Nanopipettes were routinely fabricated from glass capillaries (borosilicate glass, inner diameter 0.58 mm, outer diameter 1.0 mm, Intracell, Royston, UK) using a laser-based pipette puller (P-2000, Sutter Instruments, Novato, CA). The tip opening has a 90–130 nm inner diameter and 240–280 nm outer diameter, as measured by scanning electron microscopy (15). Pipettes were backfilled with ∼8 μL solution of 100 nM Atto 647-WGA in TALP buffer with 10% ethanol using a microfiller (MF34G, World Precision Instruments, Sarasota, FL). An AgCl-coated Ag wire was inserted into the pipette to form an Ag/AgCl electrode. The pipettes were immersed into a bath of HEPES/TALP buffer solution containing an Ag/AgCl reference electrode. A voltage of 0.5 V was applied across the electrodes in the bath and in the pipette (throughout this article it is always the potential of the bath electrode which is given). The resulting current of ∼5 nA was amplified by a home-built amplifier with 108 gain. We used mechanical translation stages for coarse positioning and a three-axis piezoelectric stage (Tritor38, Piezosystem Jena, Jena, Germany) for fine positioning of the pipettes. Distance control between the pipette tip and the cell membrane was achieved using an M44 digital signal processing (DSP) card (Innovative Integration, Simi Valley, CA) and software developed in house using distance modulation (22). The M44 card was also used to apply negative voltage pulses of −10 V and 0.1–0.5 s for delivery of probe molecules to the cell membrane. A short script can be executed which approaches the surface under distance control and on application of a trigger signal switches the control off, applies a voltage pulse, and withdraws the pipette. To test the WGA delivery, microscope coverglasses were treated with poly-L-lysine and then with 5 mg/ml hyaluronic acid (HA) sodium salt (Fluka, Milwaukee, WI; No. 53747) to form a surface with N-acetyl-D-glucosamine groups (23).
Microscopy
The apparatus used is shown schematically in Fig. 1. The pipette holder is mounted on an inverted microscope (Eclipse TE 200; Nikon, Tokyo, Japan) with an epifluorescence attachment. Fluorescence was excited using 5 mW power of a He-Ne laser at 632.8 nm (LHP 925, Melles Griot, Carlsbad, CA). The laser passed a spatial filter before it was focused onto the back aperture of the oil-immersion objective (Plan APO TIRF 60×, numerical aperture 1.45, Nikon). A laser line filter (Z488-633DBX; Chroma Technology, Rockingham, VT) was used to reject unwanted plasma emission, and a dichroic (Z488-633RPC, Chroma) was used to separate laser excitation and fluorescence. The width of the Gaussian beam profile was adjusted to 28 μm, which gave a peak power density measured under a normal incidence of 260 W/cm2. The incident angle can be adjusted for objective-type total internal reflection fluorescence (TIRF). On the relatively flat surface of boar sperm heads (8,10), it was possible to do TIRF at the level of the plasma membrane (see Supplementary Material and Sako et al. (24)). The fluorescence light was collected through the objective and filtered with a band-pass filter HQ700/60m (Chroma) mounted in a dual-view image splitter (Optical Insights, Tucson, AZ).
FIGURE 1.

Schematic of single-molecule tracking experiment with nanopipette delivery. Only the HeNe laser and the red channel of the image splitter were used in these experiments.
Only the red channel of the image splitter was used. The image was magnified 1.6× using a magnifying lens (DL-tube; Infinity Photo-optical, Göttingen, Germany) to achieve 170 nm pixel size on a back-thinned electron multiplying CCD camera (Cascade II 512B, Photometrics, Tucson, AZ). Frame rates of 40 frames/s were used to record image sequences of a 256 × 256 pixel region. The camera control software sent out a trigger signal to the M44 DSP card to apply a voltage pulse to start the experiment. The average signal/noise ratio in these experiments was found to be 10. This should allow a position accuracy of 40 nm based on nonlinear least square fitting of Gaussians (see Supplementary Material and Bobroff (25) and Thompson et al. (26)). The template-matching algorithm implemented in the MetaMorph imaging software (Universal Imaging Corporation, Downington, PA) was used to track fluorescent molecules, and the position accuracy was determined experimentally to be below 40 nm. For the analysis of diffusion boundaries, an automated tracking algorithm based on Crocker and Grier (27) was programmed in MATLAB (The MathWorks, Natick, MA).
Data analysis
For individual trajectories of N points (xi, yi) measured at time intervals Δt, the mean-square displacement (MSD) for a time lag nΔt was calculated using a running average along the trajectory (28).
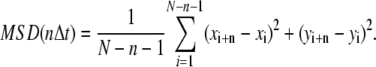 |
(1) |
For plots of averaged MSD versus time lag, the mean MSD and the standard error of the mean of all trajectories were calculated for a time lag nΔt. In the case of Brownian diffusion the MSD increases linearly with time lag. The diffusion coefficient can be obtained from the slope; in two dimensions the relation is
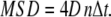 |
(2) |
To plot histograms of the diffusion coefficient in a given data set, the slope was determined from the first three data points of the MSD curves of the individual trajectories. The restriction to just three points was used because the error in MSD increases with time lag as the number of independent measurements decreases (29). The histograms were compared to a probability distribution p(De) of apparent diffusion coefficients De for a given true diffusion coefficient D0 and a given number of independent data points N = Ntotal/(nΔt) calculated according to Vrljic et al. (4):
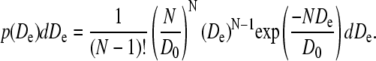 |
(3) |
To orientate the cells to the same position and rotation, images of the cells under white light illumination were recorded together with the fluorescence image sequences. The position and rotation of each cell relative to the position of an idealized outline of a boar spermatozoa was then determined. The outline including the boundaries of the different compartments were taken from the AFM image in Fig. 3 of James et al. (9). Using this information the individual trajectories were then rotated and translated so that they match the outline. Home-written software was used to identify trajectories or subsections of trajectories in the different regions, which were then analyzed separately.
FIGURE 3.

Deposition of Atto 647-WGA onto a coated glass slide. (A) Fluorescence image of molecules delivered by one pulse (−10 V, 300 ms) from the pipette. The scale bar is 5 μm. (B) Line scan through the fluorescent spot with Gaussian fit, showing a feature size of 0.9 μm full width at half-maximum. (C) Intensity versus time traces from the low-intensity spots surrounding the bright feature in A, showing single- and double-step photobleaching. (D) Intensity versus time trace obtained by confocal illumination and detection at the pipette tip under application of voltage pulses (−10 V, 300 ms). The beginning of the pulses are marked by arrows. The inset shows a region with high time resolution so that bursts caused by individual molecules are visible.
RESULTS
Wheat germ agglutin
Wheat germ agglutinin (WGA) conjugated with fluorescent dyes has affinity for N-acetyl-β-D-glucosaminyl and sialic acid sugars of membrane glycoproteins. Western blot analysis of horseradish peroxidase-conjugated WGA showed at least six clearly resolvable bands (Fig. S1 in Supplementary Material), indicating that WGA binds to several different glycoproteins of different molecular weights. A bulk labeling experiment with Alexa 488-conjugated WGA showed that the lectin binds uniformly over the entire surface of live boar spermatozoa as shown in Fig. 2. Atto 647-labeled WGA was used throughout the single-molecule tracking experiments.
Pipette delivery
We found in previous studies that proteins loaded into the pipette are delivered on application of a negative voltage to the electrode in the bath (13,14). The same is true for the Atto 647-WGA used in this experiment. Atto 647 is slightly hydrophobic, which can lead to aggregation of the labeled protein in the pipette tip. To prevent this, 10% ethanol was included in the solution in the pipette. Fig. 3 A shows the deposition of Atto 647-WGA onto a surface coated with HA, which contains N-acetyl-D-glucosamine groups. One voltage pulse of −10 V and 300 ms duration was used to deliver the molecules onto the surface, and the pipette to surface distance was ∼100 nm. A bright feature caused by the Atto 647-WGA is visible in the image surrounded by a few low-intensity spots. The full width at half-maximum of the central spot is just 0.9 μm, which shows the accuracy with which the molecules can be delivered to a specific location. From the size, intensity, and the single-step photobleaching of the low-intensity spots (Fig. 3 B), we conclude that these are single WGA molecules labeled with one Atto 647 or sometimes two Atto 647 fluorophores. The fluorescence in the central spot is on average 90 times brighter than a single molecule (from n = 9 images), which suggests that 90 molecules attach to the HA-coated surface.
In a further experiment the pipette was filled with a lower concentration of Atto 647-WGA (10 nM) and illuminated with a focused laser beam. Fluorescence was detected in a confocal mode and avalanche photodiodes for higher time resolution as described in Ying et al. (12). In this way fluorescence bursts of individual Atto 647-WGA molecules exiting the tip during applied voltage pulses (−10 V, 300 ms) could be detected (Fig. 3 D, inset). By dividing the overall fluorescence intensity from molecules excited during one burst by the average intensity from a single molecule, an estimate of the number of delivered molecules was possible. For 10 nM Atto 647-WGA in the pipette, this is 200 molecules. For 100 nM Atto 647-WGA, the concentration used for the deposition experiment above and the cell experiments, the fluorescence increases ∼9 times (data not shown). There are ∼1800 molecules that exit the pipette under these conditions. This shows a capture efficiency for the HA surface of ∼5%.
When a positive voltage of 0.5 V is applied, it is possible to control the pipette 100 nm above the membrane surface without molecules being delivered (Fig. 4). When a pulse of −10 V is applied for 0.1–0.5 s, molecules are ejected from the pipette tip. Although most delivered molecules will diffuse rapidly into the buffer solution, we found that a small number (<10) will attach to the cell membrane, giving a binding efficiency of ∼0.5% or less. These are visible by TIRF fluorescence microscopy under laser excitation as diffraction-limited spots (Fig. 5 and see videos in Supplementary Material). The pipette loaded with the fluorescent molecules is visible as a bright spot in the fluorescence images until it is withdrawn rapidly by the piezo stage. Diffusion of individual Atto 647-WGA molecules on the cell membrane can then be monitored until they photobleach in a single or two steps, allowing the trajectory of the molecule over the cell surface to be determined. This experiment can then be repeated on the same cell. During the first experiment, the TIRF incidence angle and the focus of the microscope objective could be optimized for each individual cell, and the repeats (usually 10 times) are used to collect data. In a control experiment 10 μM N-acetyl-D-glucosamine was added to the Atto 647-WGA with 10% ethanol in the pipette. The WGA, which binds the sugar under these conditions, no longer labeled the cell membrane.
FIGURE 4.

Schematic of the experiment showing how the probe molecules are delivered to a defined position on the sperm cell surface using the nanopipette. (A) The pipette is controlled at 100 nm distance from the cell membrane at positive potential (electrode in the bath) so that the probe molecules do not exit. (B) A negative voltage pulse is applied to deliver molecules to the cell membrane, distance control is switched off for the duration of the pulse, and fluorescence imaging starts. (C) The pipette is withdrawn rapidly at positive potential to prevent further molecules exiting the tip; fluorescence imaging continues.
FIGURE 5.

(A) Image of the head of the sperm cell attached to the coverslip under white light illumination. (B) Fluorescence image showing a single Atto 647-labeled WGA molecule (arrow) on the cell membrane. Shown is one frame of a video (available as Supplementary Material). (C) Overlay of the white light and fluorescence images. Scale bars are 5 μm. (D) Typical trajectory on the anterior acrosomal region obtained by tracking a single WGA molecule, the square and triangle show the start and end of the trajectory, respectively. The idealized outline of a typical boar sperm cell is also shown. (E) Typical trajectory on the postacrosomal region.
This shows the specificity of the probe under the delivery conditions. Furthermore, it is possible to select specific regions on the cell to which the probe molecules are delivered, allowing us to study the diffusional properties of different domains. The number of molecules delivered from the pipette can be controlled by the applied voltage and the duration of the pulse to find optimum conditions for single-fluorophore tracking. Molecules which do not bind to the membrane are diluted rapidly in the buffer solution or attach to the glass coverslip. Due to this dilution, there is no need to wash away surplus probe molecules, and the fluorescence background due to molecules in solution was found to be very low. Furthermore, it is possible to photobleach background fluorescence from the glass and autofluorescence from the cell effectively before the start of the experiment. Control experiments, in which spermatozoa were stained with the lipid reporter 5-(N-octadecanoyl)aminofluoresein (ODAF) after photobleaching showed that ∼90% had live pattern staining (30), indicating that they were unaffected by this laser exposure (data not shown).
When a potential drop of 200–300 mV is created across a cell membrane, there is a possibility of electroporating the membrane (31). In an experiment similar to Ryttsen et al. (32) we carefully checked pore formation under our delivery conditions. The fluorescence of the cell was monitored before and after application of 20 pulses (−10 V, 300 ms) with 1 μM fluorescein present in the bath. Pulses were applied without laser illumination to prevent photobleaching, and the medium was replaced after the pulses. The experiment was repeated several times, but no increase in fluorescence was detected under these conditions (data not shown). In contrast, when using a pipette with a fractured tip, which can pierce the cell membrane, the cells showed bright fluorescence due to fluorescein entering the cell. We conclude that under our delivery conditions there is no significant pore formation. Sperm are well known to be unusually tolerant of up to 5% ethanol (21) and to recover their motility when the ethanol is removed by washing (data not shown). Thus, in these experiments the brief local exposure to ethanol (concentration estimated to be 4%–5% over 1 μm2 of membrane for 0.5 s) is unlikely to damage the sperm.
Analysis of single-molecule diffusion
Fig. 5, D and E, shows typical trajectories of WGA molecules delivered to the anterior acrosomal and postacrosomal regions, respectively, of an ejaculated spermatozoon. During these experiments, 65 trajectories were recorded on the acrosome of 15 different cells. On the postacrosome, 37 trajectories were recorded on 13 different cells. In both cases the average length of a trajectory was 84 steps, which amounts to 2.1 s recording time.
The averaged (MSD) (28) for increasing time lags was calculated from these data (Fig. 6 A). For short time lags below 0.25 s, the MSD increases linearly, which is interpreted as free Brownian diffusion of the WGA molecules. However diffusion coefficients were different for molecules in the two regions. In the acrosomal region (which in this case includes the equatorial segment), the linear fit gives a diffusion coefficient of 0.79 ± 0.04 μm2/s; and in the postacrosomal region it is 0.10 ± 0.02 μm2/s. For longer times, the MSD is smaller than the linear relation would predict; however, the linear curve is still inside the error bars. The intercept of the linear fit A0 is related to the position accuracy with standard deviation σ by the relation A0 = 2σ 2 (see Martin et al. (33)). With a position accuracy σ of 40 nm (see Materials and Methods) the value for A0 should be 0.003 μm2/s. For the postacrosome data the measured value is 0.04 μm2/s. This high value might be caused by a faster diffusion for shorter time lags, which is not resolved in our experiment. For longer time lags the particle then follows Brownian motion but is much slower. This might be a hint of some form of corralling of the molecules on the postacrosome.
FIGURE 6.

Analysis of trajectories. (A) MSD versus time lag for the WGA molecules diffusing on the acrosome and postacrosome. For a given time lag the average of the MSD of 65 individual trajectories for the acrosome and 37 trajectories for the postacrosome region is shown. The data points and error bars give the mean ± SE. The diffusion coefficients were determined from the slope of linear fits through data points 2–10 (shown in the graph). (B and C) Histogram of diffusion coefficients of individual trajectories in the (B) acrosomal region, and (C) postacrosomal region of the sperm cell. A Gaussian fit to the histogram (continues gray curve) was added as a guide to the eye. A theoretical probability distribution (black dash dot curve) for trajectories with 84 data points is shown for comparison. This has been multiplied with the number of events and the bin width and divided by 3 for presentational purposes.
To investigate the variation in diffusion coefficients, we analyzed the trajectories separately. The diffusion coefficient for an individual molecule was determined from the slope of the first three data points of the MSD curve of its trajectory. Fig. 6 B shows histograms of the diffusion coefficients for both regions. On the acrosome the diffusion coefficients show a broad distribution. A Gaussian fit is centered at 0.60 μm2/s and has a width of 0.6 μm2/s. The fit to the histogram for the postacrosome data are centered at 0.11 and the width is 0.17 μm2/s. Because the trajectories are of finite length and Brownian diffusion can produce a large variety of trajectories with a broader distribution of apparent diffusion coefficients (29,34), it is interesting to compare the measured values with the expected probability distribution for trajectories of 84 points centered at the same peak value (4). This distribution was calculated using Eq. 3 and a time lag of three steps (28 independent measurements). The width of this distribution is about half the measured value: 0.27 μm2/s for the acrosome and 0.06 μm2/s for the postacrosome. In the case of the acrosome, the probability distribution is practically zero for diffusion coefficients above 1 μm2/s or below 0.2 μm2/s, but there is a significant number of measured trajectories which show diffusion coefficients outside this region. For the postacrosome region we can also find trajectories with diffusion coefficients higher than predicted by the probability distribution. This is a good indication for real variation in diffusion coefficients of the glycoproteins identified by WGA. This variation may be due to the probe binding to different glycoproteins, as indicated by the Western blot experiments (Fig. S1).
We overlaid all the trajectories analyzed on the sperm to check that the molecules diffused over all parts of the macrodomain in question and that there was no bias in the sampling of the cell membrane surface (Fig. 7). In both cases the distribution of trajectories is highly uniform with slightly higher densities in the small region to which the Atto 647-WGA was initially deposited by the nanopipette.
FIGURE 7.

Overlay map generated by adding up all trajectories and color coding the resulting hits per pixel. Arrows show the starting position of the pipette for delivery of Atto 647-WGA to (A) acrosome and (B) postacrosome.
Subregions
Further analysis was carried out to discriminate between the diffusion coefficients in the equatorial segment and the anterior acrosomal cap from the collective acrosome data set. Because every cell has a different orientation on the coverslip, it was necessary to match images taken from the different cells with an idealized outline of a sperm cell. Once the relative orientation of the individual cells was determined, it was straightforward to rotate and translate the individual trajectories to find their position relative to the cell outline. Using this it was possible to analyze the trajectories in the different regions separately and to divide trajectories crossing from one region to another to analyze the subsections individually (Fig. 8 B). Averaged MSD curves for trajectories in the equatorial segment and the anterior acrosomal cap show very similar behavior. The diffusion coefficients determined from a linear fit to the first five data points was found to be 0.74 ± 0.05 μm2/s for the equatorial segment and gives 0.68 ± 0.05 μm2/s for the anterior acrosomal cap. Both values are indistinguishable within the error limits and very close to the value determined for the full acrosomal data set but clearly different from the postacrosomal trajectories.
FIGURE 8.

(A) Trajectories crossing the boundary between anterior acrosomal cap and equatorial segment were divided into different tracks. The tracks in the anterior acrosomal cap and equatorial segment were analyzed separately. (B and C) Plots of the averaged MSD curves. The diffusion coefficients were calculated from the slope of a fit to the first five data points.
Diffusion boundaries
Much insight is expected from the analysis of diffusional boundaries on spermatozoa. Because of the high spatial and temporal resolution, single-molecule trajectories provide an excellent tool for this analysis. For the measurement of diffusion coefficients as discussed above, only those trajectories were analyzed that did not cross the boundary between acrosome and postacrosome. To probe this boundary, other trajectories were analyzed from the same image sequences using an automated tracking program (see Materials and Methods). The crossing between the two regions was analyzed by introducing a border region to count all attempted crossing events. Successful crossing events were then counted for all trajectories and divided by the number of attempted crossing events to get a crossing probability (Fig. 9). This probability depends on the width of the border region. For a thin border region the crossing probability is high and every trajectory diffusing along the boundary can introduce a high number of wrongly counted crossings. For a thick boundary, the crossing probability is low, because the particle is less likely to diffuse the longer distance. Therefore the probability was compared with an artificially introduced border dividing the cell in two halves (Fig. 9 C) and with the boundary between anterior acrosomal cap and equatorial segment (Fig. 9 B). For molecules delivered to the postacrosome region, the probability to cross into the equatorial segment is ∼27%, based on 14 successful out of 51 attempted crossings. This is not much different from the probability of 24% to cross from left to right (or right to left). The same holds for molecules delivered to the anterior acrosomal cap: the probability to cross into the equatorial segment is 39% (21 crossings, 54 attempts) compared to 40% to cross from left to right. The crossing probability measured on the anterior acrosome is higher because of the different diffusion coefficients.
FIGURE 9.

Schematic showing how diffusional boundaries were probed using the measured trajectories. A six pixel wide boundary region 3 (black line) was inserted between region 1 (white) and region 2 (gray). When a molecule moves into the boundary region, this was counted as an attempted crossing event. When the molecule moves from region 1 to region 2 (or vice versa) this was counted as a successful crossing event. The crossing probability was calculated by dividing successful crossing events by attempted crossings.
Another data set was analyzed where we delivered molecules onto the equatorial segment. The probability of crossing onto the anterior acrosome was found to be 40%, based on 93 successful out of 237 attempted crossings. This is not much different from the probability of 50% to cross from left to right (or right to left). The probability to cross into the postacrosome was found to be 50%, based on 167 successful out of 347 attempted crossings, which is the same as the 50% to cross from left to right (or right to left). These measurements show that glycoproteins recognized by Atto647-WGA can diffuse without obvious hindrance between the equatorial segment and postacrosomal regions.
Sperm maturation
Spermatozoa leaving the testis are immotile and infertile and only acquire these properties during passage through the epididymal duct, a process known as maturation. Once in the cauda epididymidis spermatozoa can survive in a fertile state for several weeks before they are ejaculated. During maturation the sperm plasma membrane undergoes substantial remodeling, both in terms of its lipid and protein composition and redistribution of specific antigens. Therefore, we compared the mobility of Atto 647-WGA-labeled glycoproteins on the acrosomal regions of testicular and cauda spermatozoa. Fig. 10 shows that on testicular spermatozoa glycoprotein diffusion falls into two groups: those (∼70%) that are almost stationary (mean = 0.012 μm2/s) and those with very slow diffusion coefficients (mean = 0.093 μm2/s. Averaged MSD curves analyzed separately for both groups indicated Brownian diffusion. In contrast, diffusion coefficients on cauda spermatozoa showed a wide distribution with an overall mean of 0.29 μm2/s. A small proportion (∼10%) had diffusion coefficients similar to ejaculated spermatozoa (i.e., ∼0.8 μm2/s). Again MSD plots indicated random diffusion with hints of confinement at longer times.
FIGURE 10.

Histogram of diffusion coefficients of individual trajectories on the acrosomal region of (A) testicular and (B) cauda epididymidal spermatozoa. Note the different scale for the diffusion coefficients. Gaussian fits to the data and mean values of the diffusion coefficients are shown. The insets show typical trajectories, the upper inset in A represents an almost immobile molecule, and the lower trajectory a slowly moving molecule. These measurements were done by labeling the cells from solution.
DISCUSSION
The methodology described in this article has enabled us to examine the role of putative diffusion barriers in the sperm membrane. When viewed by transmission electron microscopy the sperm plasma membrane has a classic bilayer structure with no obvious distinguishing features between surface domains. However, localized binding of antibody and lectin probes (35) as well as freeze-fracture (36,37), AFM (10,38,39), and SICM analyses (8) have demonstrated substantial topographical differences between the anterior acrosome, equatorial segment, and postacrosome. The boundaries between these domains are sharp and abrupt, suggesting major compositional and/or organizational differences and have long been thought to represent diffusion barriers preventing free exchange of lipids and proteins. Recent work, however, does not support this simplistic view of the sperm membrane. Polarized migration of a GPI-anchored hyaluronidase (known variously as PH20, 2B1, or SPAM1) (40,41) from the tail to the acrosome of rat spermatozoa during capacitation (a developmental process stimulated by cholesterol efflux from the plasma membrane) indicates that some molecules can be spatially repositioned from one domain to another, frequently against concentration gradients.
Conversely, short chain saturated lipid probes (e.g., DiIC14) have been shown to exchange rapidly between all domains on the sperm head and tail (9). The nature of the lipid, however, seems to be important. The longer acyl chain probe DiIC16 forms aggregates or particles ∼300 nm diameter in the membrane and are confined to the acrosomal and equatorial segment domains, leading to the hypothesis that molecular “filters”, rather than immutable barriers, are present in sperm membranes and that transfer of a lipid or protein from one domain to another depends on whether it diffuses as single molecules (as with DiIC12) or as part of a larger assembly (as with DiIC16). The results here do not disagree with this hypothesis. The expected finding that WGA lectin binds to a variety of glycoproteins could account for the variation in diffusion coefficients on the anterior acrosome, but it is not known if these glycoproteins behave as single entities or if they are sequestered into larger complexes. The uniform fluorescence observed over the sperm surface (Fig. 2, A and B) could be indicative of single glycoprotein molecules but small lipid rafts, 10–20 nm diameter, could also be present that would behave differently and would not be resolvable by light microscopy.
A detailed MSD analysis of individual trajectories was consistent with random diffusion, and there was no evidence for anomalous diffusion (42) or for a diffusion barrier between the anterior acrosome and the equatorial segment, despite the known differences in membrane topography. By contrast, diffusion coefficients on the postacrosome were 5–7 times slower with less variation or scatter. This could be due to a more restricted range of glycoproteins binding Atto 647-WGA, or it could be caused by the postacrosomal plasma membrane existing in a more liquid-ordered phase, promoted by the influence of the membrane skeleton. In support of the latter possibility is our earlier observation that DiIC16, a saturated lipid probe which partitions preferentially into liquid-ordered phases, stains the postacrosome in a uniformly dense manner (9). Additionally, actin and its regulatory proteins, destrin and thymosin β10, localize to the postacrosomal and equatorial segment domains (43) where they are correctly positioned to influence transmembrane proteins and formation of molecular assemblies. The slowly diffusing or immobile WGA-binding glycoproteins on testicular spermatozoa is consistent with the physiological immaturity of these cells (e.g., cholesterol efflux does not trigger the downstream tyrosine phosphorylation processes typical of cauda or ejaculated spermatozoa (11)) and reflects the complex remodeling that takes place during epididymal maturation to produce a fully competent spermatozoon.
The procedures developed in this work have the following advantages over random labeling from solution: they allow us to deliver probe molecules to specific regions of the cell surface; the number of molecules delivered depends on the applied voltage and duration of the pulse so that we can controllably deliver a single or a few fluorescently labeled molecules; the background due to unbound probe molecules is low because of rapid dilution in solution; the experiment can be repeated many times on the same cell, allowing one to first optimize the experimental setup and then record many sets of data under optimal conditions; and since the application of the voltage pulse is computer controlled, this allows the laser illumination and CCD imaging to be turned on at the same time, allowing efficient data collection and single-molecule trajectories of maximum length. Collectively, these developments greatly increase the precision and reproducibility of single-molecule fluorescence tracking.
FRAP data on spermatozoa show that there is less than 7% variation in diffusion coefficients measured on different cells (data not shown). Hence we can obtain a large data set where the probe molecules are diffusing over a cell membrane of the same topography by combining data taken on different spermatozoa. In cases where the cell topography is not known, it would be possible to use SICM for noncontact imaging of the cell topography before performing single-molecule tracking experiments since SICM has been demonstrated to allow reliable imaging of cell topography under physiological conditions (44) and can be used to image a wide range of different cell types (22,45,46). This will allow us to extend our studies in future to more complex cells. We have previously shown that pipette delivery is applicable to a wide range of biomolecules, including antibodies (14), so this method should be very general. Multibarrel pipettes have also been shown to allow the local delivery of different fluorophore-labeled biomolecules from each barrel (17), so our method can therefore be extended to deliver two probes simultaneously to the same region of the cell surface and then follow their subsequent diffusion. This could be used to compare the diffusion of different membrane classes, e.g., a lipid raft component versus a transmembrane protein versus a phospholipid, in the same region of the same sperm cell and follow their relative and absolute motion. We could also use our controlled delivery to test more rigorously the hypothesis of a molecular filter and its size limitations and whether or not it is uni- or bidirectional.
CONCLUSIONS
We have developed a new biophysical tool to controllably deliver fluorophore-labeled probe molecules to defined regions of the cell surface using a nanopipette and then performing single-molecule tracking. This allows us to perform repeat experiments on the same cell, to probe different macrodomains and subregions within the plasma membrane, and to investigate the existence of putative diffusion barriers. This is a new and general method that can be used to elucidate the structure of the cell membrane and how lateral heterogeneity is maintained in differentiated cells.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Andrew I. Shevchuk for programming the DSP card.
This work was funded by the Biotechnology and Biological Sciences Research Council, UK.
Editor: Thomas Schmidt.
References
- 1.Kusumi, A., C. Nakada, K. Ritchie, K. Murase, K. Suzuki, H. Murakoshi, R. S. Kasai, J. Kondo, and T. Fujiwara. 2005. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 34:351–378. [DOI] [PubMed] [Google Scholar]
- 2.Daumas, F., N. Destainville, C. Millot, A. Lopez, D. Dean, and L. Salome. 2003. Confined diffusion without fences of a G-protein-coupled receptor as revealed by single particle tracking. Biophys. J. 84:356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schütz, G. J., G. Kada, V. P. Pastushenko, and H. Schindler. 2000. Properties of lipid microdomains in a muscle cell membrane visualized by single molecule microscopy. EMBO J. 19:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vrljic, M., S. Y. Nishimura, S. Brasselet, W. E. Moerner, and H. M. McConnell. 2002. Translational diffusion of individual class II MHC membrane proteins in cells. Biophys. J. 83:2681–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harms, G. S., L. Cognet, P. H. M. Lommerse, G. A. Blab, H. Kahr, R. Gamsjager, H. P. Spaink, N. M. Soldatov, C. Romanin, and T. Schmidt. 2001. Single-molecule imaging of L-type Ca2+ channels in live cells. Biophys. J. 81:2639–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lommerse, P. H. M., G. A. Blab, L. Cognet, G. S. Harms, B. E. Snaar-Jagalska, H. P. Spaink, and T. Schmidt. 2004. Single-molecule imaging of the H-Ras membrane-anchor reveals domains in the cytoplasmic leaflet of the cell membrane. Biophys. J. 86:609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ying, L. M., A. Bruckbauer, D. J. Zhou, J. Gorelik, A. Shevehuk, M. Lab, Y. Korchev, and D. Klenerman. 2005. The scanned nanopipette: a new tool for high resolution bioimaging and controlled deposition of biomolecules. Phys. Chem. Chem. Phys. 7:2859–2866. [DOI] [PubMed] [Google Scholar]
- 8.Shevchuik, A. I., G. I. Frolenkov, D. Sanchez, P. S. James, N. Freedman, M. J. Lab, R. Jones, D. Klenerman, and Y. E. Korchev. 2006. Imaging proteins in membranes of living cells by high-resolution scanning ion conductance microscopy. Angew. Chem. Int. Ed. Engl. 45:2212–2216. [DOI] [PubMed] [Google Scholar]
- 9.James, P. S., C. Hennessy, T. Berge, and R. Jones. 2004. Compartmentalisation of the sperm plasma membrane: a FRAP, FLIP and SPFI analysis of putative diffusion barriers on the sperm head. J. Cell Sci. 117:6485–6495. [DOI] [PubMed] [Google Scholar]
- 10.Ellis, D. J., S. Shadan, P. S. James, R. M. Henderson, J. M. Edwardson, A. Hutchings, and R. Jones. 2002. Post-testicular development of a novel membrane substructure within the equatorial segment of ram, bull, boar, and goat spermatozoa as viewed by atomic force microscopy. J. Struct. Biol. 138:187–198. [DOI] [PubMed] [Google Scholar]
- 11.Shadan, S., P. S. James, E. A. Howes, and R. Jones. 2004. Cholesterol efflux alters lipid raft stability and distribution during capacitation of boar spermatozoa. Biol. Reprod. 71:253–265. [DOI] [PubMed] [Google Scholar]
- 12.Ying, L. M., A. Bruckbauer, A. M. Rothery, Y. E. Korchev, and D. Klenerman. 2002. Programmable delivery of DNA through a nanopipet. Anal. Chem. 74:1380–1385. [DOI] [PubMed] [Google Scholar]
- 13.Bruckbauer, A., L. M. Ying, A. M. Rothery, D. J. Zhou, A. I. Shevchuk, C. Abell, Y. E. Korchev, and D. Klenerman. 2002. Writing with DNA and protein using a nanopipet for controlled delivery. J. Am. Chem. Soc. 124:8810–8811. [DOI] [PubMed] [Google Scholar]
- 14.Bruckbauer, A., D. J. Zhou, L. M. Ying, Y. E. Korchev, C. Abell, and D. Klenerman. 2003. Multicomponent submicron features of biomolecules created by voltage controlled deposition from a nanopipet. J. Am. Chem. Soc. 125:9834–9839. [DOI] [PubMed] [Google Scholar]
- 15.Ying, L. M., S. S. White, A. Bruckbauer, L. Meadows, Y. E. Korchev, and D. Klenerman. 2004. Frequency and voltage dependence of the dielectrophoretic trapping of short lengths of DNA and dCTP in a nanopipette. Biophys. J. 86:1018–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruckbauer, A., D. J. Zhou, D. J. Kang, Y. E. Korchev, C. Abell, and D. Klenerman. 2004. An addressable antibody nanoarray produced on a nanostructured surface. J. Am. Chem. Soc. 126:6508–6509. [DOI] [PubMed] [Google Scholar]
- 17.Rodolfa, K. T., A. Bruckbauer, D. J. Zhou, A. I. Schevchuk, Y. E. Korchev, and D. Klenerman. 2006. Nanoscale pipetting for controlled chemistry in small arrayed water droplets using a double-barrel pipet. Nano Lett. 6:252–257. [DOI] [PubMed] [Google Scholar]
- 18.Parrish, J. J., J. Suskoparrish, M. A. Winer, and N. L. First. 1988. Capacitation of bovine sperm by heparin. Biol. Reprod. 38:1171–1180. [DOI] [PubMed] [Google Scholar]
- 19.Harrison, R. A. P., P. J. C. Ashworth, and N. G. A. Miller. 1996. Bicarbonate/CO2, an effector of capacitation, induces a rapid and reversible change in the lipid architecture of boar sperm plasma membranes. Mol. Reprod. Dev. 45:378–391. [DOI] [PubMed] [Google Scholar]
- 20.Harrison, R. A. P., and S. E. Vickers. 1990. Use of fluorescent-probes to assess membrane integrity in mammalian spermatozoa. J. Reprod. Fertil. 88:343–352. [DOI] [PubMed] [Google Scholar]
- 21.Mann, T. 1964. The Biochemistry of Semen and of the Male Reproductive Tract. Methuen, London.
- 22.Shevchuk, A. I., J. Gorelik, S. E. Harding, M. J. Lab, D. Klenerman, and Y. E. Korchev. 2001. Simultaneous measurement of Ca2+ and cellular dynamics: combined scanning ion conductance and optical microscopy to study contracting cardiac myocytes. Biophys. J. 81:1759–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khademhosseini, A., K. Y. Suh, J. M. Yang, G. Eng, J. Yeh, S. Levenberg, and R. Langer. 2004. Layer-by-layer deposition of hyaluronic acid and poly-L-lysine for patterned cell co-cultures. Biomaterials. 25:3583–3592. [DOI] [PubMed] [Google Scholar]
- 24.Sako, Y., S. Minoghchi, and T. Yanagida. 2000. Single-molecule imaging of EGFR signalling on the surface of living cells. Nat. Cell Biol. 2:168–172. [DOI] [PubMed] [Google Scholar]
- 25.Bobroff, N. 1986. Position measurement with a resolution and noise-limited instrument. Rev. Sci. Instrum. 57:1152–1157. [Google Scholar]
- 26.Thompson, R. E., D. R. Larson, and W. W. Webb. 2002. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 82:2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crocker, J. C., and D. G. Grier. 1996. Methods of digital video microscopy for colloidal studies. J. Colloid Interface Sci. 179:298–310. [Google Scholar]
- 28.Ritchie, K., X. Y. Shan, J. Kondo, K. Iwasawa, T. Fujiwara, and A. Kusumi. 2005. Detection of non-Brownian diffusion in the cell membrane in single molecule tracking. Biophys. J. 88:2266–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong, Q. A., M. P. Sheetz, and E. L. Elson. 1991. Single-particle tracking: analysis of diffusion and flow in 2-dimensional systems. Biophys. J. 60:910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfe, C. A., P. S. James, A. R. Mackie, S. Ladha, and R. Jones. 1998. Regionalized lipid diffusion in the plasma membrane of mammalian spermatozoa. Biol. Reprod. 59:1506–1514. [DOI] [PubMed] [Google Scholar]
- 31.Nolkrantz, K., C. Farre, A. Brederlau, R. I. D. Karlsson, C. Brennan, P. S. Eriksson, S. G. Weber, M. Sandberg, and O. Orwar. 2001. Electroporation of single cells and tissues with an electrolyte-filled capillary. Anal. Chem. 73:4469–4477. [DOI] [PubMed] [Google Scholar]
- 32.Ryttsen, F., C. Farre, C. Brennan, S. G. Weber, K. Nolkrantz, K. Jardemark, D. T. Chiu, and O. Orwar. 2000. Characterization of single-cell electroporation by using patch-clamp and fluorescence microscopy. Biophys. J. 79:1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin, D. S., M. B. Forstner, and J. A. Kas. 2002. Apparent subdiffusion inherent to single particle tracking. Biophys. J. 83:2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saxton, M. J. 1997. Single-particle tracking: the distribution of diffusion coefficients. Biophys. J. 72:1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Primakoff, P., and D. G. Myles. 1983. A map of the guinea-pig sperm surface constructed with monoclonal-antibodies. Dev. Biol. 98:417–428. [DOI] [PubMed] [Google Scholar]
- 36.Friend, D. S., and D. W. Fawcett. 1974. Membrane differentiations in freeze-fractured mammalian sperm. J. Cell Biol. 63:641–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suzuki, F. 1990. Morphological aspects of sperm maturation: modification of the sperm plasma membrane during epididymal transit. In Fertilization in Mammals. B. D. Banister, J. Cummins, and E. R. S. Roldan, editors. Serono Symposium, Norwell, MA. 65–75.
- 38.Allen, M. J., E. M. Bradbury, and R. Balhorn. 1995. The natural subcellular surface structure of the bovine sperm cell. J. Struct. Biol. 114:197–208. (Erratum in J. Struct. Biol. 1995. 115:338–341.) [DOI] [PubMed] [Google Scholar]
- 39.Takano, H., and K. Abe. 2000. Changes in the surface structure of the hamster sperm head associated with maturation, in vitro capacitation and acrosome reaction: an atomic force microscopic study. J. Electron. Microsc. (Tokyo). 49:437–443. [DOI] [PubMed] [Google Scholar]
- 40.Cowan, A. E., D. G. Myles, and D. E. Koppel. 1987. Lateral diffusion of the Ph-20 protein on guinea-pig sperm: evidence that barriers to diffusion maintain plasma-membrane domains in mammalian sperm. J. Cell Biol. 104:917–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones, R., R. Shalgi, J. Hoyland, and D. M. Phillips. 1990. Topographical rearrangement of a plasma-membrane antigen during capacitation of rat spermatozoa in vitro. Dev. Biol. 139:349–362. [DOI] [PubMed] [Google Scholar]
- 42.Cherry, R. J., P. R. Smith, I. E. G. Morrison, and N. Fernandez. 1998. Mobility of cell surface receptors: a re-evaluation. FEBS Lett. 430:88–91. [DOI] [PubMed] [Google Scholar]
- 43.Howes, E. A., S. M. Hurst, and R. Jones. 2001. Actin and actin-binding proteins in bovine spermatozoa: potential role in membrane remodeling and intracellular signaling during epididymal maturation and the acrosome reaction. J. Androl. 22:62–72. [PubMed] [Google Scholar]
- 44.Gorelik, J., A. I. Shevchuk, G. I. Frolenkov, I. A. Diakonov, M. J. Lab, C. J. Kros, G. P. Richardson, I. Vodyanoy, C. R. W. Edwards, D. Klenerman, and Y. E. Korchev. 2003. Dynamic assembly of surface structures in living cells. Proc. Natl. Acad. Sci. USA. 100:5819–5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Klenerman, D., and Y. Korchev. 2006. Potential biomedical applications of the scanned nanopipette. Nanomed. 1:107–114. [DOI] [PubMed] [Google Scholar]
- 46.Gorelik, J., Y. C. Gu, H. A. Spohr, A. I. Shevchuk, M. J. Lab, S. E. Harding, C. R. W. Edwards, M. Whitaker, G. W. J. Moss, D. C. H. Benton, D. Sanchez, A. Darszon, I. Vodyanoy, D. Klenerman, and Y. E. Korchev. 2002. Ion channels in small cells and subcellular structures can be studied with a smart patch-clamp system. Biophys. J. 83:3296–3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.