

Abstract
We investigate Aβ17-42 protofibril structures in solution using molecular dynamics simulations. Recently, NMR and computations modeled the Aβ protofibril as a longitudinal stack of U-shaped molecules, creating an in-parallel β-sheet and loop spine. Here we study the molecular architecture of the fibril formed by spine-spine association. We model in-register intermolecular β-sheet–β-sheet associations and study the consequences of Alzheimer's mutations (E22G, E22Q, E22K, and M35A) on the organization. We assess the structural stability and association force of Aβ oligomers with different sheet-sheet interfaces. Double-layered oligomers associating through the C-terminal–C-terminal interface are energetically more favorable than those with the N-terminal–N-terminal interface, although both interfaces exhibit high structural stability. The C-terminal–C-terminal interface is essentially stabilized by hydrophobic and van der Waals (shape complementarity via M35-M35 contacts) intermolecular interactions, whereas the N-terminal–N-terminal interface is stabilized by hydrophobic and electrostatic interactions. Hence, shape complementarity, or the “steric zipper” motif plays an important role in amyloid formation. On the other hand, the intramolecular Aβ β-strand-loop-β-strand U-shaped motif creates a hydrophobic cavity with a diameter of 6–7 Å, allowing water molecules and ions to conduct through. The hydrated hydrophobic cavities may allow optimization of the sheet association and constitute a typical feature of fibrils, in addition to the tight sheet-sheet association. Thus, we propose that Aβ fiber architecture consists of alternating layers of tight packing and hydrated cavities running along the fibrillar axis, which might be possibly detected by high-resolution imaging.
INTRODUCTION
Alzheimer's disease (AD) is the most common human neurodegenerative disorder characterized by distinct neuropathological lesions, including intracellular neurofibrillary tangles and extracellular senile amyloid plaques (1,2). The major component isolated from amyloid plaques of AD is a small polypeptide, 40–42 amino acids in length (i.e., Aβ40 or Aβ42 peptide), which is derived from endoproteolytic cleavage of the transmembrane amyloid precursor protein (APP)(3,4). Before cleavage, the hydrophilic-rich N-terminus of the Aβ (residues 1–28) is exposed to the aqueous extracellular environment whereas its hydrophobic-rich C-terminus (residues 29–42) is deeply embedded in the membrane (5). Upon proteolytic cleavage, the monomeric Aβ peptides are released into the extracellular milieu, aggregating into soluble oligomers, protofibrils, and eventually into insoluble amyloid fibrils with high β-sheet content. Although it is still unclear which amyloid species, mature fibrils or intermediate oligomers, is mainly involved in neurotoxicity that is responsible for cell death, increasing in vitro evidence (6–8) has shown that soluble oligomeric intermediates are cytotoxic whereas either monomers or mature fibrils are apparently harmless. Determination of high-resolution molecular structures of soluble oligomeric intermediates is a challenging task due to their small size and dynamic, short-lived nature (9,10) unlike mature amyloid fibrils. In addition, recent experiments have shown that Aβ and calcitonin aggregates commonly exhibit multiple distinct morphologies with different molecular structures when exposed to different environmental conditions (i.e., pH, ionic strength, temperature, agitation, and solvent) that may be correlated with different neuronal toxicities (11,12). Thus, to shed light on the relationship between the Aβ oligomers' intrinsic conformational properties and fibril morphology and neurotoxicity it is important to characterize their molecular architectures.
Aβ peptides have been studied extensively using a variety of experimental and theoretical methods at the functional and structural levels. Lührs et al. (13) recently presented the three-dimensional structure of Aβ42 fibrils based on hydrogen/deuterium-exchange NMR data (Protein Data Bank code, 2BEG). This emerging structure with a U-turn bent β-sheet validates both the computational model of Ma and Nussinov (14) and the experimental model of Tycko and his colleagues (15), including the side-chain orientation where the side chain of I32 points in toward the β-turn whereas the side chain of M35 points outward. All the computational and experimental models remarkably agree that the buried salt bridge between residues D23 and K28 stabilizes the turn, although each model has distinct turn structure (turn location, S26–I31 for the Lührs model, V24–N27 for the Ma model, and D23–G29 for the Tycko model). Experiment further revealed an important role of M35 in the oxidative stress and neurotoxic properties of the Aβ peptide (16,17). The oxidation of methionine affects the aggregation properties of the peptide, leading to an altered oligomer size distribution and inhibition of fibril formation (18,19). Ciccotosto et al. (17) reported that Aβ peptide with a methionine at position 35 to valine substitution (AβM35V) was more neurotoxic than wild-type Aβ42 in cortical cell cultures, although there were no quantifiable differences in the sizes of the oligomers for AβM35V and Aβ42 in phosphate buffer saline solution. Sato et al. (20) recently found a tightly formed steric zipper via the intermolecular M35–M35 association between antiparallel double-layered Aβ-sheets. This steric zipper in the Aβ amyloid fibril provides a key element for the rational design of inhibitors to prevent fibril formation. By attacking M35–M35 contacts, peptide inhibitors designed by Sato et al. (20) not only disrupted the sheet-to-sheet packing and prevented the Aβ fibril formation, but also significantly reduced the toxicity on neuronal cell cultures.
The achievement of atomic-resolution structures has opened a window to investigate computationally the mechanism of amyloid formation using molecular modeling methods and to directly compare computational modeling with experimental results. Most computer simulations of amyloid-forming peptides fall into two categories. All-atom molecular dynamics (MD) simulations have been applied to study amyloid oligomer stability alone by testing different candidate β-sheet arrangements of preformed oligomers mimicking possible nucleus seeds at the very early stage of fibril formation (14,21,22). This approach can determine the most stable conformation for minimal nucleus seeds, but cannot predict the aggregation scenario of amyloid intermediates/fibril growth since aggregation is an extremely slow process on the timescale of minutes to days, which is typically beyond the timescale of nanoseconds for conventional MD simulations. To overcome computational limitations, alternative computer simulations using low-resolution models (e.g., coarse-grained protein models and implicit solvent models) have been used to directly study the formation of oligomers (small species) and even fibrils (large species) (9,23). These simulations can roughly map out the kinetic pathways of protein aggregation, but cannot adequately capture different interactions, such as hydrophobic interactions, electrostatic interactions, and hydrogen bonding.
In this work, we performed all-atom MD simulations to study the structural stability and conformational dynamics of Aβ17-42 oligomeric models for both wild-type and the Alzheimer-related mutants, based on recent NMR data (13). To gain an insight into how two β-sheets associate along the direction perpendicular to the fibril axis, we developed double-layered models (Fig. 1), which consist of five-parallel strands within the sheets while maintaining antiparallel organization between the sheets through sheet-to-sheet interfaces (i.e., C-terminal–C-terminal and N-terminal–N-terminal interfaces). To generate the most likely stable double-layered models, we attempted to achieve either maximal overlap of the hydrophobic region (I31–I41) for the C-terminal–C-terminal interface, or a combination of both the hydrophobic region (V18–F20) and the salt bridge (K16–E22) for the N-terminal–N-terminal interface between two neighboring β-sheets. The double-layered models (Fig. 1) were used to examine: 1), the association forces between β-sheets, by considering various sheet-to-sheet interfaces; and 2), the effect of specific single point mutations on the association forces and structural stability. These simulations allowed us to obtain detailed information about the oligomeric structures and driving forces critical in the formation of amyloid fibrils. We further studied intramolecular morphological features of the amyloid organization, providing a more complete atom-level picture of the fibrillar architecture. Thus, rather than the β-sheets being equally spaced longitudinally, the detailed fibril architecture predicted by the simulations consisted of tight intermolecular sheet-sheet packing and intramolecular hydrated cavities along the fibrillar axis. The hydrated cavity allowed optimal packing at the sheet-sheet interface. Lastly, we outlined some common structural motifs observed in amyloid-forming polypeptides. It should be noted that the large conformational changes (i.e., peptide folding and self-assembling process) leading to the amyloid aggregation is beyond the scope of this work.
FIGURE 1.

Double-layered structural models of Aβ oligomers. Two β-sheets were associated together via a CC (upper) or NN (lower) interface along the direction perpendicular to fibril axis. Color identifications are: backbone (green); negatively charged residues (red); positively charged residues (blue); hydrophobic residues (orange).
MATERIALS AND METHODS
Single- and double-layered models of Aβ17-42 and Aβ15-42
Initial coordinates of Aβ17-42 hexamer were extracted from 10 NMR structures (Protein Data Bank code, 2BEG), derived from quenched hydrogen/deuterium-exchange NMR (13), whereas the initial conformations of Aβ15-42 hexamer were constructed by adding two residues (Gln-15 and Lys-16) from the Ma et al. model (14) to the N-terminus of each corresponding Aβ17-42 monomer. The two added residues maintained the same side-chain orientation and backbone conformation as those in the Ma model. For dimeric structures (10-mers) with or without mutations, double-layered oligomers were built by placing two five-strand β-sheets of Aβ17-42 and Aβ15-42 together with different termini facing each other. Specifically, we examined two possible distinct interfaces (i.e., C-terminal–C-terminal and N-terminal–N-terminal interfaces) when stacking two β-sheets onto each other in an antiparallel fashion (Fig. 1). To generate the most likely stable double-layered models, we attempted to achieve maximum overlap either of the hydrophobic region (I31–I41) for the CC interface or combination of both the hydrophobic region (V18–F20) and the salt bridge (K16–E22) for the NN interface in two neighboring β-sheets. All starting structures of the mutants were built from the wild-type Aβ17-42 and Aβ15-42 by replacing the side chains of the targeted residues, but without changing the backbone conformations and side-chain orientations. The structure of the designed mutant was first minimized for 500 steps using the steepest decent algorithm with the backbone of the protein restrained before being subjected to the following system setup and production runs. The N- and C-termini were blocked by acetyl and amine groups, respectively. As shown in Fig. 1, the Aβ42 model proposed by Lührs et al. (13) consists of two β-strands, β1 (residues V17–S26) and β2 (residues I31–A42), connected by a U-bent turn spanning four residues N27–A30.
MD simulation protocol
All simulations were performed in the NPT ensemble using the NAMD simulation package (24) with the all-atom CHARMM27 force field and the TIP3P water model (25). The pressure (1 atm) was maintained by a Langevin piston with a decay period of 100 fs and a damping time of 50 fs, whereas the temperature (330 K) was controlled by a Langevin thermostat with a damping coefficient of 5 ps−1. The short-range van der Waals (VDW) interactions were calculated by the switch function with a twin range cutoff of 10.0 and 12.0 Å and the long-range electrostatic interactions were calculated by the force-shift function with a cutoff of 12.0 Å. The integration time step was 2 fs. Periodic boundary conditions with the minimum image convention were applied to the systems in all directions.
All initial Aβ structures were first energy minimized and then solvated in a TIP3P water box with a minimum distance of at least 8 Å from any edge of the box to any Aβ atom. Any water molecule within 2.4 Å of the Aβ was removed. Counterions (Na+ and Cl−) were added at random locations to neutralize the Aβ charge, with ion concentration of ∼150 mM, close to the physiological value. The resulting solvated systems were energy minimized for 5000 conjugate gradient steps, where the protein was fixed and water molecules and counterions were allowed to move, followed by additional 3000 conjugate gradient steps, where all atoms were allowed to move. After minimization, the systems were heated from 30 to 330 K for 100 ps and equilibrated at 330 K for 500 ps. All simulations ran for 20 ns and structures were saved every 2 ps for analysis.
Sheet-to-sheet binding energy
For double-layered Aβ-sheets, Aβ trajectories were first extracted from explicit MD trajectories by excluding water molecules. The solvation energies of double-layered Aβ-sheets and each single-layered Aβ-sheet were calculated using the generalized Born method with molecular volume (GBMV) (26). In the GBMV calculation, the dielectric constant of water is set to 80 and no distance cutoff is used. The binding energy between two β-sheets was calculated by
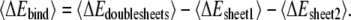 |
Analysis details
The relative structural stability of the oligomers is measured by root mean-squared deviation (RMSD) of the backbone atoms with respect to initial minimized crystal structure throughout the simulations.
The root mean-squared atomic fluctuation (RMSF) was calculated for each individual residue by aligning all trajectory structures with the averaged structure from the trajectory.
The twist angle is measured by averaging over the angles between two vectors connecting the first Cα atom to the last Cα atom in the same β-strand portion between two neighboring β-strands within a β-sheet.
The shape complementarity (Sc) is used to measure the geometric surface complementarity of protein-protein interfaces between two adjacent layers, where 1.0 represents a perfect match between the interfaces, whereas 0.0 represents two unrelated interfaces. Sc is calculated by using the program SC of the CCP4 with default parameters (27).
RESULTS AND DISCUSSION
Single-layered Aβ-sheets
A quantitative measure of conformational dynamics and local flexibility of Aβ peptides is provided by the backbone RMSD and Cα RMSF from the initial structure, respectively. In Fig. 2, for both five- and 10-stranded parallel β-sheets, the RMSDs for all residues rose quickly to a plateau of ∼5 Å within 2 ns and then oscillated around this value for the remainder of the simulations, whereas the RMSDs for loop residues (S26–I31) were maintained at ∼2 Å throughout the entire simulations. This suggests that the large structural deviation was not from loop residues. The RMSF profile showed that the most flexible regions of the Aβ peptide were the edge residues of the N- and C-termini and the loop residues of S26 and I31 connecting the β-strand and the β-turn (Fig. 3). The β-strand at the C-terminal (residues I31–A42) is two residues longer than that at N-terminal (residues V17–S26). This fact leads to the conclusion that residues of I41 and A42 at the C-terminal do not have direct contacts with hydrophobic residues of I17 and V18 at the N-terminal, thus displaying high flexibility in the single-layered model. Visual inspection of the trajectories together with RMSD profiles suggested that the overall Aβ structures were stable, with the β-strands tightly packed on top of each other without dissociation and the secondary structures of the strand-loop-strand motif were well preserved. The large structural deviation originates from the edge residues in the β-strand region, leading to the twisted β-sheets. The loop is stabilized by the salt bridge between residues K28 and D23 and hydrophobic interaction between residues V24 and K28, whereas the edge residues in the β-strands were largely exposed to the solvent, thus experiencing large fluctuations. As expected, the RMSD and RMSF values in the five-β-stranded simulation are slightly higher than the corresponding values in the 10-β-stranded simulation, indicating that higher-order aggregates of amyloid fibrils generally exhibit more stable molecular structures, i.e., the stability increases with size. It should be noted that both five- and 10-stranded Aβ models have an acceptable interior diameter of ∼8–10 Å, which allows small molecules such as water molecules and ions to penetrate. Similar water channels formed by Aβ9-40 peptides were also observed by Buchete and co-workers (28).
FIGURE 2.

Backbone RMSDs of single-layered Aβ17-42 models with 5 and 10 β-strands, respectively. The RMSD curves are shown for all residues (black line) and loop residues (red line).
FIGURE 3.

Average RMSF values for 5 β-strands (black line) and 10 β-strands (red line) in single-layered Aβ17-42 models.
Double-layered Aβ-sheets
For double-layered Aβ-sheets, the C-terminal–C-terminal (CC) interface consists of highly hydrophobic patches of I31, I41, and M35, whereas the N-terminal–N-terminal (NN) interface consists of both hydrophobic patches of V18 and F20 and K16–E22 salt bridges. Backbone RMSDs of all wild-type and mutants with the CC interface generally experienced smaller conformational deviations than those of corresponding oligomers with the NN interface, as shown in Fig. 4. As expected, in all cases the terminal residues have higher RMSF values (data not shown) as compared to the central residues. The average intermolecular distances between two β-sheets (see Fig. 6) are ∼8.75 ± 0.25 Å for all CC interface models and 12.0 ± 1.0 Å for all NN interface models other than the E22K mutant. The interaction energies of two β-sheets for various interfaces and mutations are shown in Fig. 5.
FIGURE 4.

Backbone RMSDs of double-layered Aβ models with (a) CC interfaces and (b) NN interfaces for both wild-type and mutants (E22G, E22Q, E22K, and M35A). The RMSD curves are shown for all residues (black line), β-sheet 1 residues (red line), β-sheet 2 residues (green line), loop1 residues (blue line), and loop2 residues (light-blue line).
FIGURE 6.

Averaged mass-center distances between two facing β-sheets associated through (a) the CC interface and (b) the NN interface. The distance is measured between backbone residues of 31–42 for CC interface and 15–25 for NN interface in two facing β-sheets.
FIGURE 5.

Interaction energies for the wild-type and mutated sequences between two β-sheets associated through (a) the CC interface and (b) the NN interface. The interaction energy is calculated by the GBMV implicit solvation model in the CHARMM program.
In the double-layered Aβ-sheets with the CC interface, the M35 hydrophobic side chain on one β-sheet packs against the corresponding M35 of the mating sheet, forming an intersheet steric zipper. The sheet-to-sheet packing is stabilized by this nonpolar zipper through VDW and hydrophobic interactions. The presence of M35 has large effects on the CC interface association between two Aβ-sheets. Substitution of M35 by a small, short Ala not only disrupts this shape-complementary steric zipper, but also weakens hydrophobic interactions between residues M35–G37 (Fig. 7 e), leading to an increase in packing energy (∼100 kcal/mol) between the two β-sheets relative to the wild-type with the same CC interface (Fig. 5). This result is consistent with the experimental observation that oxidation of M35 in Aβ42 prevents fibril formation (29). Since G22 is not located at the CC interface, removal of G22 by mutation to Lys, Gln, or Gly has little effect on structural stability and binding affinity, as indicated by RMSDs, interaction energy, and visual inspection (Fig. 7, b–d).
FIGURE 7.

Snapshots from simulations of double-layered models of (a) CC; (b) CC-E22G; (c) CC-E22K; (d) CC-E22Q; (e) CC-M35A; (f) NN; (g) NN-E22G; (h) NN-E22K; (i) NN-E22Q; and (j) NN-M35A at 20 ns. Color identifications are: backbone (green); negatively charged residues (red); positively charged residues (blue); hydrophobic residues (orange).
For the NN interface models, the disruption of the intersheet salt bridge of K16–E22 by replacing E22 led to unfavorable interaction energy and large structural deviations (Figs. 4 and 5). Especially in the case of the E22K mutant, the unfavorable positively charged Lys residues were introduced and packed against K16 residues at the NN interface, resulting in the loss of the stabilizing salt bridges between E22 and Lys-16. It is unlikely for β-sheets with E22K mutations to associate via the NN interface. Interestingly, although two β-sheets in the E22K mutant were separated from each other, the secondary structures of both β-sheets were still preserved. The E22G mutant experienced slightly larger structural deviations than E22Q due to the flexible backbone of Gly, but both E22G and E22Q mutants showed similar interaction energy profiles. The M35A mutation occurring in the opposite side of the NN interface was not sensitive to the structure and dynamics of oligomers.
Sheet-to-sheet interactions
Protofibrils or fibrils not only grow in the fibril axis direction, but also stack in the lateral direction normal to the fibril axis, in which fibril growth corresponds to intrasheet interactions, while fibril stacking corresponds to intersheet interactions. Fibril growth and stacking are competitive with each other in these two directions. Our simulation results showed that the Dutch (E22Q), Italian (E22K), and Arctic (E22G) mutations occurring at the NN interface were less stable than the wild-type. This fact may imply that the weaker lateral association could lead to more efficient growth along the fibril axis by reducing the kinetic barrier for the early-stage oligomerization, consistent with experimental observations (30,31) that the Dutch, Italian, and Arctic mutants at position 22 show higher aggregation rates and stronger neurotoxicity than the wild-type Aβ42 (31), but do not significantly affect the morphology and structure of the resulting amyloid fibrils. It should be noted that the rapid conformational transition from random coil to β-strand-loop–β-strand structure could be another crucial step in the kinetics of amyloid formation.
For the preformed β-strand-loop–β-strand motifs, we observed that the double-layered Aβ oligomeric structures are stabilized not only by nonspecific hydrophobic interactions and specific electrostatic interactions (salt bridges), but also by favorable shape-complementary side-chain packing. Previous studies (32,33) showed that Aβ amyloid fibril formation is predominantly driven by hydrophobic interactions. As shown in Fig. 8, for both CC and NN interfaces, the two sublayers interact primarily through their β-strand portions, creating an antiparallel β-sheet organization. The CC interface contains three clusters of hydrogen bonds formed between I31–I41, G37–M35, and G33–V39. These hydrogen bonds are almost evenly distributed along the interface. This interface is also stabilized by hydrophobic contacts between I310–I41 and M35–M35. As compared to the CC interface, hydrophobic interactions are reduced and only occur between F20 residues, whereas electrostatic interactions contributed by salt bridges between E22 and K16 are largely enhanced. The intrasheet salt bridges between residues D23 and K28 enhance the turn stability, whereas intersheet salt bridges between residues E22 and K16 improve the NN termini association between two stacked β-sheets. The formation of salt bridges also implies that the Aβ-sheets tend to stack in an antiparallel fashion, maximizing the number of hydrophobic and electrostatic interactions. Moreover, the alignment of aromatic residues F19 and F20 between adjacent strands provided additional π-π interactions stabilizing the Aβ structures at both interfaces.
FIGURE 8.

Structures and interactions of double-layered Aβ models for the (a) CC interface and the (b) NN interface. A hydrophobic contact is identified when the mass center distance between a pair of hydrophobic side chains is <4.0 Å. In these models, the hydrogen bonds are red; hydrophobic contacts are blue; and salt bridges are black.
Mutations at the β-sheet–β-sheet interface greatly alter interactions such as hydrophobic, electrostatic, and hydrogen bonding and thus the interaction energies. For example, when the salt bridge pair of E22–K16 at the NN interface was disrupted by the E22K, E22Q, and E22G mutations (Fig. 7, g–i), the two β-sheets lost the attractive electrostatic interactions, leading to large interaction energy and sheet-to-sheet distance. This phenomenon was even more pronounced in the case of the E22K mutant where repulsive forces between K16 and K22 pushed the two β-sheets away from each other (Fig. 7 h). As described in the previous section, the M35A mutation led to less favorable interaction energy at the CC interface. The specific hydrophobic and van der Waals interactions involving M35 residues were reduced. Similarly, the hydrogen bonding between the side chains of M35 and G37 was also affected when substituted by alanines.
Ion and water conductible channel
Table 1 summarizes the average interior pore sizes for all Aβ models. The pore size is characterized approximately by a pocket of side chains consisting of Ala-21, Asp-23, Lys-28, Ala-30, Ile-32, and Leu-34. As seen in Table 1, the average interior pore sizes of the CC and NN models are ∼7.1 and 6.0 Å, respectively, which are large enough for small molecules like water and ions to conduct through. Fig. 9 shows the occupancy profile of water molecules as a function of time for all single and double-layered Aβ models. It should be noted that all starting configurations did not have water molecules in the interior of Aβ structures. As shown in Fig. 9, the water profiles inside the Aβ hydrophobic pore have a similar trend for all models, regardless of their interfaces (CC or NN interfaces) and layers (single or double layers), because all interior channel structures include the U-shaped loop and the side-chain orientations are the same. Initially, during the first 6 ns the number of water molecules rises, ultimately leveling off to a more constant plateau, indicating that water molecules were able to gradually penetrate into the hydrophobic pore from both ends of the channel. It took an average of ∼6–7 ns for water molecules to fill the pore. Due to the limited interior space and restricted side-chain movement, the entering water molecules were aligned into an almost contiguous chain along the fibril axis, but they cannot accommodate clusters as in the bulk. The water molecules that are inside the pore couple with the pore-facing side chains via hydrogen bonding interactions (Fig. 10). Similarly, we also observed that Cl− ions could enter the hydrophobic pore, but without hydration shell around them. Penetrating water molecules act as a hydration shell of charged D23–K28 residues and interact with side chains via hydrogen bonding, assisting in maintaining and stabilizing the oligomeric pore structures, consistent with the observations of Buchete and co-workers (28,34) that the interior cavities of Aβ9-40 were hydrated by gradually penetrating water molecules. The desolvation of the interior of the Aβ oligomers would impose a large energy and entropic penalty on the oligomers. It has been shown that the hydration free energy of nonpolar groups stabilize native structures (35). It should be mentioned that we recently performed additional MD simulations to study the structural dynamics of K3 oligomers from β2-microglobulin fragments (residues 22–41). Initial coordinates of K3 peptide derived from solid-state NMR were kindly provided by the Goto (36) lab. Similar to Aβ40 and Aβ42 peptides, K3 peptide has a U-shaped strand-loop-strand motif consisting of an internal hydrophobic pore of ∼6 Å, in which water molecules can penetrate into and conduct through this pore, as observed in simulations.
TABLE 1.
Aβ structural parameters for all models
| Twist angle (°)
|
Pore size (Å)
|
||||
|---|---|---|---|---|---|
| Simulations | Sheet 1 | Sheet 2 | Sheet 1 | Sheet 2 | Sc |
| Single-layered | |||||
| Five strands | 21.2 ± 2.2 | – | 8.8 ± 0.7 | – | – |
| Ten strands | 17.5 ± 1.8 | – | 7.2 ± 1.0 | – | – |
| Double-layered | |||||
| CC-WT | 8.8 ± 0.7 | 10.5 ± 0.6 | 7.2 ± 0.8 | 7.3 ± 0.4 | 0.67 ± 0.06 |
| CC-E22G | 11.9 ± 1.0 | 7.8 ± 0.8 | 7.0 ± 1.3 | 6.7 ± 0.9 | 0.66 ± 0.02 |
| CC-E22K | 9.9 ± 0.8 | 10.4 ± 0.9 | 8.0 ± 0.7 | 6.9 ± 1.3 | 0.64 ± 0.04 |
| CC-E22Q | 9.6 ± 1.0 | 9.7 ± 0.6 | 6.8 ± 0.8 | 6.9 ± 0.7 | 0.67 ± 0.04 |
| CC-M35A | 12.7 ± 0.7 | 14.7 ± 0.7 | 6.9 ± 0.4 | 6.8 ± 0.3 | 0.55 ± 0.07 |
| NN-WT | 8.1 ± 2.7 | 13.0 ± 1.7 | 6.2 ± 0.7 | 5.8 ± 1.2 | 0.58 ± 0.05 |
| NN-E22G | 11.8 ± 3.5 | 16.5 ± 3.7 | 6.7 ± 1.2 | 6.5 ± 1.0 | 0.56 ± 0.03 |
| NN-E22K | 3.6 ± 0.9 | 20.0 ± 1.8 | 5.5 ± 1.2 | 5.9 ± 1.1 | – |
| NN-E22Q | 10.5 ± 2.8 | 16.7 ± 2.3 | 5.7 ± 1.1 | 6.0 ± 1.1 | 0.53 ± 0.05 |
| NN-M35A | 11.2 ± 3.7 | 13.0 ± 3.2 | 6.2 ± 1.5 | 5.8 ± 1.5 | 0.54 ± 0.04 |
FIGURE 9.

Water occupancy profiles inside a hydrophobic cavity of (a) single-layered models, (b) double-layered models with CC interface, and (c) double-layered model with NN interface. The hydrophobic cavity is defined by the inwarding side chains of Ala-21, Asp-23, Lys-28, Ala-30, Ile-32, and Leu-34. Any water molecule within 3.0 Å cutoff of this cavity is counted as hydration number.
FIGURE 10.

Interior hydration of Aβ models. Top panel is side view perpendicular to fibril axis, whereas bottom panel is top view along fibril axis. Broken red lines indicate H-bonds between water molecules and side chains of inward-pointing residues, whereas broken blue lines indicate H-bonds between penetrated water molecules.
The water self-diffusion coefficient (D), measured by the Einstein equation via the mean-squared displacement, provides insight into the water dynamics and mobility in the hydrophobic cavity. A slow self-diffusion coefficient indicates that water molecules are tightly bound. As listed in Table 2, averaged water self-diffusion coefficients (∼0.183 × 10−5 cm2/s) in the hydrophobic cavities were at least one order of magnitude smaller than that of bulk TIP3P water (5.06 × 10−5 cm2/s) (37). Results showed that hydrophobic cavity with limited space could slow down the mobility of water molecules through breaking/reforming hydrogen bonding interactions. The minor difference of D among different models was mainly due to local movement and conformation of inward-pointing side chains in the hydrophobic cavities.
TABLE 2.
Water self-diffusion coefficient (D) in the hydrophobic Aβ cavity
| Simulations | D (10−5 cm2/s) |
|---|---|
| Single-layered | |
| Five strands | 0.403 |
| Ten strands | 0.211 |
| Double-layered | |
| CC-WT | 0.137 |
| CC-E22G | 0.103 |
| CC-E22K | 0.277 |
| CC-E22Q | 0.141 |
| CC-M35A | 0.291 |
| NN-WT | 0.063 |
| NN-E22G | 0.203 |
| NN-E22K | 0.103 |
| NN-E22Q | 0.191 |
| NN-M35A | 0.074 |
Universal structural motifs in amyloid fibrils
Aβ amyloids are not unique (38). Although sequence-specific effects are always involved, features illustrated by the Aβ amyloids are observed in other amyloid systems (36,39,40). This leads us to enumerate these general features, and to examine the Aβ fibril organization and morphology in this light.
Shape complementarity (or “steric zipper”)
To characterize how two interfaces geometrically fit to each other, we analyzed the shape complementarity (Sc) of the β-sheet–β-sheet interface (excluding water), using the program SC of the CCP4 with default parameters (27). Sc of 1.0 represents a perfect match between the interfaces, whereas a value of 0.0 represents two unrelated interfaces. As shown in Table 1, CC interfaces including wild-type and mutants have relatively higher Sc values (from 0.55 to 0.67) than the corresponding NN interfaces (from 0.53 to 0.58). For CC interfaces, residues M35 at the two facing β-sheets were packed against each other, forming a shape complimentary steric zipper. When M35 residues were replaced by the small alanines, Sc was greatly reduced from 0.66 to 0.55. There is no difference in Sc values for other mutants (i.e., E22G, E22Q, and E22K). All NN interfaces displayed weak shape complementary where Sc values were 0.58 for wild-type, 0.56 for E22G, 0.53 for E22Q, and 0.54 for M35A. Sc of the E22K mutant was not presented here since the initial NN interface was not available after 10 ns. Structural investigation of various interfaces and mutations suggests that Aβ aggregates with high shape complementarity display strong binding affinity and high structural stability. Amino acid substitutions that either disrupt the steric zipper or eliminate favorable electrostatic interactions have large impact on shape complementarity, interaction energy, and structural stability.
Naturally occurring protein surfaces exhibit either concave or convex curvatures. To form highly ordered amyloid fibrils with common cross-β spine, shape complementarity of correctly matched side chains (41,42), tightly interdigitating into a steric zipper is essential for ordered protein aggregates. Shape complementary steric zipper motifs have been often observed in different amyloid-forming sequences. Eisenberg and co-workers (43) recently determined the crystal structure of the cross-β spine of the yeast prion Sup-35 fragment (GNNQQNY) using x-ray microcrystallography. This structure consists of a pair of β-sheets, with each sheet formed by parallel β-strands in-register. Between the two facing β-sheets, the polar side chains (Asn-2, Gln-4, and Asn-6) are tightly interdigitated at a dry interface, forming a self-complementary steric zipper, whereas within the sheet each strand is linked to others by backbone and side-chain hydrogen bonds. The shape complementarity of these two facing β-sheets was very good (Sc = ∼0.85) (44) and the disruption of this steric zipper at the β-sheet–β-sheet interfaces led to unstable oligomers (21). Ferguson et al. (39) also found that the human CA150 amyloidogenic peptide contains a steric zipper formed by the interdigitations of side-chain contacts between T13-T18, V5-R24, V5-L26, and T3-S28 using magic-angel-spinning NMR. It should be noted that the steric zipper in human CA150 is formed within a single sheet, whereas GNNQQNY and Aβ42 peptides form zippers between β-sheets. It thus appears that the steric zipper may be a generic structural motif of amyloid protofilaments (39,40), because the zipper feature not only provides favorable van der Waals interactions, but also constrains side-chain movement due to geometrical fit, which explains why amyloid fibrils are as stable and persistent as they are. Recently, Eisenberg and co-workers (45) investigated 30 short segments (less than seven residues) from fibril-forming proteins and they found that 13 of these microcrystal structures reveal varied steric zippers. However, if the amyloid-forming peptides are longer, while the shape complementarity is still expected to persist, it may not be uniformly as tight as in observed in crystals of short segments. Some variability might be expected.
Parallel versus antiparallel β-sheets
Recent experimental studies have shown that many amyloid fibrils consist of parallel β-sheet structures at least for longer protein chains or peptides, including Alzheimer's amyloid-β42 (13), Sup-35 prion domain (43), human CA-150 (39), β2-microglobulin (36), Ure2p prion domain (46), IAPP amylin (47), and α-synuclein (48). In a parallel β-sheet structure, identical residues stack on top of each other in an in-register way. These identical paired residues could lead to Asn or Gln ladders, aromatic stacking, and continuous hydrogen bonding, suggesting that amyloid fibrils of longer polypeptide chains may have general structural features of an in-register, parallel-stranded sheet organization (40,49). Shuffling the sequence is not likely to disrupt those residue pairs, thus has a little impact on parallel β-sheet structures (50). For Aβ with parallel β-sheet topology, the stacked aromatic residues of Phe-19 and Phe-20 in a parallel orientation in adjacent peptides provide additional stabilization force to maintain oligomeric structures. Nevertheless, antiparallel or mixed parallel-antiparallel β-sheets have also been observed in a variety of amyloid sequences under different experimental conditions (e.g., pH concentration), such as human calcitonin (51), Syrian hamster prion protein109-122 (52), human tropoelastin (53), and Aβ14-23 (54). In an antiparallel β-sheets topology, although hydrophobic interactions and hydrogen bonds are still two major dominant driving forces in stabilizing the protein structure, amyloid β-sheet formation could be more sensitive to mutations in the amyloid-forming sequences.
β-strand-loop–β-strand
The β-strand-loop–β-strand motif is formed by two β-strands with nonnative register linked by a flexible loop. As first predicted by the simulations of the Aβ (14) and then confirmed by experiments (55), the β-strand-loop–β-strand motif was recently shown in the amyloid protofilaments of human CA150 (39) and the β2-microglobulin (36). This motif consists of two sheets whose side chains zip against each other in an antiparallel fashion, where each sheet consists of a parallel arrangement of the β-strands. The loop is stabilized by a salt bridge in the Aβ and covalent bonds in CA150 and β2-microglobulin. Saiki et al. (56) reported that the capability to form amyloids was significantly reduced when a double-stranded sequence consisting of a turn region was altered to a single-stranded sequence. Wu and co-workers (57) suggested that a strand-loop-strand structure could be an important binding intermediate for Aβ fibril growth. The loop-induced double-β-strand motif has an intrastrand interaction via hydrophobic interactions, hydrogen bonds, or steric zippers, which is absent in a single-strand peptide. Thus, a loop-induced motif is advantageous, as it may lead to a zipper-motif resembling the tightly packed β-sheets of shorter peptides.
Twisted cross-β-sheets
Amyloid fibrils exhibit twisted β-sheets, as observed by electron microscopy and solid state NMR. As shown in Table 1, Aβ-sheets twist by ∼15°. The twisted sheets are not unique to Aβ; rather, similar twisted β-sheets were observed in other amyloid peptides such as GNNQQNY from the yeast prion protein (21), the human islet amyloid polypeptide IAPP22-27 (NFGAIL), KFFE, KVVE, KLLE, and KAAE (58), the human calcitonin hormone (residues 15–19, DFNKF), and NHVTLSQ from human β2-microglobulin. Since twisted β-sheets optimize the hydrogen bonds, side-chain stacking, and electrostatic interactions, it is commonly accepted that twisted sheets are more stable than flat ones. While twisting, the β-sheets pairs are still compatible with the steric zipper.
CONCLUSIONS
We have performed MD simulations to investigate the conformational and thermodynamic properties of Aβ oligomers. Current NMR data provide structural models of protofibrils consisting of single Aβ molecules stacked on top of each other to create parallel, in-register β-sheets and loops. However, the detailed atomistic models of the lateral organization of the fibrils are not available. Here, we have modeled the organization by comparing two different types of oligomers: protofibrils packed CC and NN, both oriented in an antiparallel fashion. We further tested the effects of some Alzheimer mutations on these oligomers. Conformational analysis and interaction energy estimation indicate that both the CC and NN interfaces exhibit stable structures, but the CC interface appears more energetically favorable than the NN interface due to large hydrophobic contacts and shape-complementary steric zipper. This suggests that elongated Aβ protofibrils could be mainly associated with one another via the CC interface along a direction perpendicular to the fibril axis. The intermolecular steric zipper arrangement via the M35-M35 contacts at the CC interface has a strong tendency to stabilize and to form amyloid fibrils. Disruption of this steric zipper leads to a large energy penalty and structural destabilization. All mutations except E22K at the NN interface have comparable structural stability as the wild-type regardless of the CC or NN interface type, indicating that they all have the potential to form stable amyloid fibrils. Further, our Aβ simulations also show that all oligomers with the U-bent strand-loop-strand structural motif have an inner hydrophobic cavity of 6–7 Å in diameter that allows small molecules such as ions and water molecules to conduct through. The hydrated hydrophobic cavity is expected to facilitate an optimal intermolecular sheet-sheet packing. Since the U-bent structure has recently been shown to be a recurring motif in amyloids (36,39,40), this leads us to propose that in solution this could be a general feature of amyloids. This prediction is in agreement with the alternating dry and wet interfaces in microcrystals of a Sup-35 fragment (43). We thus hypothesize that the molecular architecture of the amyloid fibril may consist of U-shaped molecules, with in-parallel twisted β-sheets. The sheets of laterally stacked molecules are tightly packed. Intramolecular hydrated cavities run in the fibril axis direction. It will be interesting to see if high-resolution AFM or using x-ray diffraction (59) images will be able to distinguish between the alternating, tighter interfaces and the ion and water conducting pores. We add a cautious remark that no protein kinetic factors were considered in this study.
Acknowledgments
This study used the high-performance computational capabilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov). We thank the Frederick Advanced Biomedical Computing Center for computational facilities used in this work. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract No. NO1-CO-12400. This research was supported (in part) by the Intramural Research Program of National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Jie Zheng's present address is Dept. of Chemical and Biomolecular Engineering, University of Akron, Akron, Ohio 44325.
Editor: John E. Straub.
References
- 1.Buxbaum, J. D., E. H. Koo, and P. Greengard. 1993. Protein phosphorylation inhibits production of Alzheimer amyloid beta/A4 peptide. Proc. Natl. Acad. Sci. USA. 90:9195–9198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin, H., R. Bhatia, and R. Lal. 2001. Amyloid beta protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 15:2433–2444. [DOI] [PubMed] [Google Scholar]
- 3.Fraser, P. E., L. Levesque, and D. R. McLachlan. 1993. Biochemistry of Alzheimer's disease amyloid plaques. Clin. Biochem. 26:339–349. [DOI] [PubMed] [Google Scholar]
- 4.Wertkin, A. M., R. S. Turner, S. J. Pleasure, T. E. Golde, S. G. Younkin, J. Q. Trojanowski, and V. M. Lee. 1993. Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular beta-amyloid or A4 peptides. Proc. Natl. Acad. Sci. USA. 90:9513–9517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamour, Y. 1994. Alzheimer's disease: a review of recent findings. Biomedecine & Pharmacotherapy. 48:312–318. [DOI] [PubMed] [Google Scholar]
- 6.Kayed, R., E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman, and C. G. Glabe. 2003. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 300:486–489. [DOI] [PubMed] [Google Scholar]
- 7.Bucciantini, M., E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson, and M. Stefani. 2002. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 416:507–511. [DOI] [PubMed] [Google Scholar]
- 8.Cleary, J. P., D. M. Walsh, J. J. Hofmeister, G. M. Shankar, M. A. Kuskowski, D. J. Selkoe, and K. H. Ashe. 2005. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 8:79–84. [DOI] [PubMed] [Google Scholar]
- 9.Mousseau, N., and P. Derreumaux. 2005. Exploring the early steps of amyloid peptide aggregation by computers. Acc. Chem. Res. 38:885–891. [DOI] [PubMed] [Google Scholar]
- 10.Makabe, K., D. McElheny, V. Tereshko, A. Hilyard, G. Gawlak, S. Yan, A. Koide, and S. Koide. 2006. Atomic structures of peptide self-assembly mimics. Proc. Natl. Acad. Sci. USA. 103:17753–17758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Petkova, A. T., R. D. Leapman, Z. Guo, W.-M. Yau, M. P. Mattson, and R. Tycko. 2005. Self-propagating, molecular-level polymorphism in Alzheimer's beta-amyloid fibrils. Science. 307:262–265. [DOI] [PubMed] [Google Scholar]
- 12.Naito, A., M. Kamihira, R. Inoue, and H. Saito. 2004. Structural diversity of amyloid fibril formed in human calcitonin as revealed by site-directed 13C solid-state NMR spectroscopy. Magn. Reson. Chem. 42:247–257. [DOI] [PubMed] [Google Scholar]
- 13.Luhrs, T., C. Ritter, M. Adrian, D. Riek-Loher, B. Bohrmann, H. Dobeli, D. Schubert, and R. Riek. 2005. 3D structure of Alzheimer's amyloid-beta (1–42) fibrils. Proc. Natl. Acad. Sci. USA. 102:17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma, B., and R. Nussinov. 2002. Stabilities and conformations of Alzheimer's beta-amyloid peptide oligomers (Abeta 16–22, Abeta 16–35, and Abeta 10–35): sequence effects. Proc. Natl. Acad. Sci. USA. 99:14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petkova, A. T., W. M. Yau, and R. Tycko. 2006. Experimental constraints on quaternary structure in Alzheimer's beta-amyloid fibrils. Biochemistry. 45:498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butterfield, D. A., and A. I. Bush. 2004. Alzheimer's amyloid beta-peptide (1–42): involvement of methionine residue 35 in the oxidative stress and neurotoxicity properties of this peptide. Neurobiol. Aging. 25:563–568. [DOI] [PubMed] [Google Scholar]
- 17.Ciccotosto, G. D., D. Tew, C. C. Curtain, D. Smith, D. Carrington, C. L. Masters, A. I. Bush, R. A. Cherny, R. Cappai, and K. J. Barnham. 2004. Enhanced toxicity and cellular binding of a modified amyloid {beta} peptide with a methionine to valine substitution. J. Biol. Chem. 279:42528–42534. [DOI] [PubMed] [Google Scholar]
- 18.Hou, L., I. Kang, R. E. Marchant, and M. G. Zagorski. 2002. Methionine 35 oxidation reduces fibril assembly of the amyloid A[beta]-(1–42) peptide of Alzheimer's disease. J. Biol. Chem. 277:40173–40176. [DOI] [PubMed] [Google Scholar]
- 19.Palmblad, M., A. Westlind-Danielsson, and J. Bergquist. 2002. Oxidation of methionine 35 attenuates formation of amyloid beta-peptide 1–40 oligomers. J. Biol. Chem. 277:19506–19510. [DOI] [PubMed] [Google Scholar]
- 20.Sato, T., P. Kienlen-Campard, M. Ahmed, W. Liu, H. Li, J. I. Elliott, S. Aimoto, S. N. Constantinescu, J. N. Octave, and S. O. Smith. 2006. Inhibitors of amyloid toxicity based on beta-sheet packing of Abeta40 and Abeta42. Biochemistry. 45:5503–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng, J., B. Ma, C.-J. Tsai, and R. Nussinov. 2006. Structural stability and dynamics of an amyloid-forming peptide GNNQQNY from the yeast prion sup-35. Biophys. J. 91:824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai, H.-H., M. Reches, C.-J. Tsai, K. Gunasekaran, E. Gazit, and R. Nussinov. 2005. Energy landscape of amyloidogenic peptide oligomerization by parallel-tempering molecular dynamics simulation: significant role of Asn ladder. Proc. Natl. Acad. Sci. USA. 102:8174–8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nguyen, H. D., and C. K. Hall. 2006. Spontaneous fibril formation by polyalanines: discontinuous molecular dynamics simulations. J. Am. Chem. Soc. 128:1890–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kale, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- 25.Brooks, B. R., R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- 26.Lee, M. S., M. Feig, F. R. Salsbury Jr., and C. L. Brooks III. 2003. New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J. Comput. Chem. 24:1348–1356. [DOI] [PubMed] [Google Scholar]
- 27.Lawrence, M. C., and P. M. Colman. 1993. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234:946–950. [DOI] [PubMed] [Google Scholar]
- 28.Buchete, N.-V., R. Tycko, and G. Hummer. 2005. Molecular dynamics simulations of Alzheimer's beta-amyloid protofilaments. J. Mol. Biol. 353:804–821. [DOI] [PubMed] [Google Scholar]
- 29.Bitan, G., B. Tarus, S. S. Vollers, H. A. Lashuel, M. M. Condron, J. E. Straub, and D. B. Teplow. 2003. A molecular switch in amyloid assembly: Met35-protein and amyloid beta-protein oligomerization. J. Am. Chem. Soc. 125:15359–15365. [DOI] [PubMed] [Google Scholar]
- 30.Nilsberth, C., A. Westlind-Danielsson, C. B. Eckman, M. M. Condron, K. Axelman, C. Forsell, C. Stenh, J. Luthman, D. B. Teplow, S. G. Younkin, J. Naslund, and L. Lannfelt. 2001. The ‘Arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat. Neurosci. 4:887–893. [DOI] [PubMed] [Google Scholar]
- 31.Murakami, K., K. Irie, A. Morimoto, H. Ohigashi, M. Shindo, M. Nagao, T. Shimizu, and T. Shirasawa. 2003. Neurotoxicity and physicochemical properties of Abeta mutant peptides from cerebral amyloid angiopathy: implication for the pathogenesis of cerebral amyloid angiopathy and Alzheimer's disease. J. Biol. Chem. 278:46179–46187. [DOI] [PubMed] [Google Scholar]
- 32.Klimov, D. K., and D. Thirumalai. 2003. Dissecting the assembly of Abeta16–22 amyloid peptides into antiparallel beta sheets. Structure. 11:295–307. [DOI] [PubMed] [Google Scholar]
- 33.Baumketner, A., S. L. Bernstein, T. Wyttenbach, N. D. Lazo, D. B. Teplow, M. T. Bowers, and J.-E. Shea. 2006. Structure of the 21–30 fragment of amyloid beta-protein. Protein Sci. 15:1239–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buchete, N.-V., and G. Hummer. 2007. Structure and dynamics of parallel beta-sheets, hydrophobic core, and loops in Alzheimer's Abeta fibrils. Biophys. J. 92:3032–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makhatadze, G., and P. Privalov. 1995. Energetics of protein structure. Adv. Protein Chem. 47:307–425. [DOI] [PubMed] [Google Scholar]
- 36.Iwata, K., T. Fujiwara, Y. Matsuki, H. Akutsu, S. Takahashi, H. Naiki, and Y. Goto. 2006. 3D structure of amyloid protofilaments of beta2-microglobulin fragment probed by solid-state NMR. Proc. Natl. Acad. Sci. USA. 103:18119–18124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michael, W. M., and L. J. William. 2001. Diffusion constant of the TIP5P model of liquid water. J. Chem. Phys. 114:363–366. [Google Scholar]
- 38.Makin, O. S., and L. C. Serpell. 2005. Structures for amyloid fibrils. FEBS J. 272:5950–5961. [DOI] [PubMed] [Google Scholar]
- 39.Ferguson, N., J. Becker, H. Tidow, S. Tremmel, T. D. Sharpe, G. Krause, J. Flinders, M. Petrovich, J. Berriman, H. Oschkinat, and A. R. Fersht. 2006. General structural motifs of amyloid protofilaments. Proc. Natl. Acad. Sci. USA. 103:16248–16253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zheng, J., B. Ma, and R. Nussinov. 2006. Consensus features in amyloid fibrils: sheet-sheet recognition via a (polar or nonpolar) zipper structure. Physical Biology. 3:P1–P4. [DOI] [PubMed] [Google Scholar]
- 41.Zanuy, D., B. Ma, and R. Nussinov. 2003. Short peptide amyloid organization: stabilities and conformations of the islet amyloid peptide NFGAIL. Biophys. J. 84:1884–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zanuy, D., and R. Nussinov. 2003. The sequence dependence of fiber organization. a comparative molecular dynamics study of the islet amyloid polypeptide segments 22–27 and 22–29. J. Mol. Biol. 329:565–584. [DOI] [PubMed] [Google Scholar]
- 43.Nelson, R., M. R. Sawaya, M. Balbirnie, A. O. Madsen, C. Riekel, R. Grothe, and D. Eisenberg. 2005. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 435:773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esposito, L., C. Pedone, and L. Vitagliano. 2006. Molecular dynamics analyses of cross-beta-spine steric zipper models: beta-sheet twisting and aggregation. Proc. Natl. Acad. Sci. USA. 103:11533–11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sawaya, M. R., S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. O. Madsen, C. Riekel, and D. Eisenberg. 2007. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 447:453–457. [DOI] [PubMed] [Google Scholar]
- 46.Chan, J. C. C., N. A. Oyler, W. M. Yau, and R. Tycko. 2005. Parallel beta-sheets and polar zippers in amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p. Biochemistry. 44:10669–10680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jayasinghe, S. A., and R. Langen. 2004. Identifying structural features of fibrillar islet amyloid polypeptide using site-directed spin labeling. J. Biol. Chem. 279:48420–48425. [DOI] [PubMed] [Google Scholar]
- 48.Jao, C. C., A. Der-Sarkissian, J. Chen, and R. Langen. 2004. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. USA. 101:8331–8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai, H.-H., K. Gunasekaran, and R. Nussinov. 2006. Sequence and structure analysis of parallel beta helices: implication for constructing amyloid structural models. Structure. 14:1059–1072. [DOI] [PubMed] [Google Scholar]
- 50.Ross, E. D., A. Minton, and R. B. Wickner. 2005. Prion domains: sequences, structures and interactions. Nat. Cell Biol. 7:1039–1044. [DOI] [PubMed] [Google Scholar]
- 51.Akira Naito, M. K. R. I. H. S. 2004. Structural diversity of amyloid fibril formed in human calcitonin as revealed by site-directed 13C solid-state NMR spectroscopy. Magn. Reson. Chem. 42:247–257. [DOI] [PubMed] [Google Scholar]
- 52.Petty, S. A., and S. M. Decatur. 2005. Intersheet rearrangement of polypeptides during nucleation of beta-sheet aggregates. Proc. Natl. Acad. Sci. USA. 102:14272–14277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tamburro, A. M., A. Pepe, B. Bochicchio, D. Quaglino, and I. P. Ronchetti. 2005. Supramolecular amyloid-like assembly of the polypeptide sequence coded by exon 30 of human tropoelastin. J. Biol. Chem. 280:2682–2690. [DOI] [PubMed] [Google Scholar]
- 54.Bu, Z., Y. Shi, D. J. E. Callaway, and R. Tycko. 2007. Molecular alignment within beta-sheets in Abeta14–23 fibrils: solid-state NMR experiments and theoretical predictions. Biophys. J. 92:594–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Petkova, A. T., Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio, and R. Tycko. 2002. A structural model for Alzheimer's beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 99:16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saiki, M., T. Konakahara, and H. Morii. 2006. Interaction-based evaluation of the propensity for amyloid formation with cross-beta structure. Biochem. Biophys. Res. Commun. 343:1262–1271. [DOI] [PubMed] [Google Scholar]
- 57.Han, W., and Y. D. Wu. 2005. A strand-loop-strand structure is a possible intermediate in fibril elongation: long time simulations of amyloid-beta peptide (10–35). J. Am. Chem. Soc. 127:15408–15416. [DOI] [PubMed] [Google Scholar]
- 58.Wei, G., N. Mousseau, and P. Derreumaux. 2004. Sampling the self-assembly pathways of KFFE hexamers. Biophys. J. 87:3648–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Makin, O. S., P. Sikorski, and L. C. Serpell. 2006. Diffraction to study protein and peptide assemblies. Curr. Opin. Chem. Biol. 10:417–422. [DOI] [PubMed] [Google Scholar]