

Abstract
Alzheimer's disease is a debilitating neurodegenerative disorder associated with the abnormal self-assembly of amyloid-β (Aβ) peptides into fibrillar species. N-methylated peptides homologous to the central hydrophobic core of the Aβ peptide are potent inhibitors of this aggregation process. In this work, we use fully atomistic molecular dynamics simulations to study the interactions of the N-methylated peptide inhibitor Aβ16–20m (Ac-Lys16-(Me)Leu17-Val18-(Me)Phe19-Phe20-NH2) with a model protofilament consisting of Alzheimer Aβ16–22 peptides. Our simulations indicate that the inhibitor peptide can bind to the protofilament at four different sites: 1), at the edge of the protofilament; 2), on the exposed face of a protofilament layer; 3), between the protofilament layers; and 4), between the protofilament strands. The different binding scenarios suggest several mechanisms of fibrillogenesis inhibition: 1), fibril inhibition of longitudinal growth (in the direction of monomer deposition); 2), fibril inhibition of lateral growth (in the direction of protofilament assembly); and 3), fibril disassembly by strand removal and perturbation of the periodicity of the protofilament (disruption of fibril morphology). Our simulations suggest that the Aβ16-20m inhibitor can act on both prefibrillar species and mature fibers and that the specific mechanism of inhibition may depend on the structural nature of the Aβ aggregate. Disassembly of the fibril can be explained by a mechanism through which the inhibitor peptides bind to disaggregated or otherwise free Aβ16–22 peptides in solution, leading to a shift in the equilibrium from a fibrillar state to one dominated by inhibitor-bound Aβ16–22 peptides.
INTRODUCTION
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by the presence of amyloid plaques in the brain. These extracellular deposits are fibrous masses, composed primarily of aggregates of amyloid-β (Aβ) peptides. Aβ peptides are proteolytic byproducts of the Aβ protein precursor and are most commonly composed of 40 (Aβ40) or 42 (Aβ42) amino acids. These peptides appear to be mainly unstructured in their monomeric state but aggregate to form fibrils with an ordered cross-β sheet pattern (1,2). Both small aggregates (including soluble oligomers and protofibrils) as well as mature fibrils display toxicity, although recent studies suggest that the former may be more cytotoxic (3–5).
Significant effort has been invested in finding means to combat this debilitating disease. In particular, a major research thrust has involved the development of compounds capable of inhibiting and reversing the aggregation process (6–8). Based on the observation that the hydrophobic core of the Aβ peptide (residues 17–20: Leu17-Val18-Phe19-Phe20) is critical for aggregation (9–12), Soto and co-workers designed inhibitor peptides that bind specifically to this region and prevent β-sheet formation (13). In the same spirit, a number of research groups designed several other inhibitor peptides adapted from this core sequence. These include peptides containing the 17–20 segment without alterations (12). Other strategies involved using peptides homologous to the hydrophobic core of Aβ in which elements disruptive to aggregation were inserted (14–16). In this vein, peptide N-methylation has emerged as a powerful strategy for inhibition (17–21). Meredith and co-workers have recently demonstrated that the membrane-permeable N-methylated pentapeptide Aβ16–20m (Ac-Lys16-(Me)Leu17-Val18-(Me)Phe19-Phe20-NH2) (22) is an effective fibrillogenesis inhibitor, capable of both preventing fibril growth and disassembling existing fibrils. The Aβ16–20m peptide was designed such that the N-methyl groups are placed at alternating positions along the chain. As a result of this selective replacement of the amide proton and the steric effects caused by the methyl group (21), the peptide shows one face capable of hydrogen bonding with the fibril and one in which hydrogen bonding is prevented. Because it has been shown that Aβ fibrils can grow by monomer deposition (23), it has been proposed that the N-methyl inhibitor peptide will cap the growing fibril and prevent further addition of monomer units. However, this theory does not explain how and why the fibril should disassemble.
In this article, we present a molecular dynamics (MD) investigation of the interaction of the Aβ16–20m peptide with a model Aβ16–22 protofilament. The Aβ16–22 peptide (Ac-Lys16-Leu17-Val18-Phe19-Phe20-Ala21-Glu22-NH2) is one of the shortest fragments of the Aβ peptide capable of aggregating into fibrils (24). Furthermore, these fibrils have been extensively characterized by solid-state NMR (24). Our aim is to elucidate the mechanism by which this peptide binds to the fibril, prevents its further growth, and triggers its disassembly. Understanding this process will not only enable the rational design of new N-methyl inhibitors but will also lead to insights into the factors stabilizing fibrils. A striking result from our simulations is that the inhibitor peptide can bind in multiple ways to the fibril, leading to several different mechanisms of inhibition.
MATERIALS AND METHODS
In all our simulations the GROMOS96 force field (25) was used to describe the solute and the simple point charge (spc) water model to describe the solvent (26). The temperature was maintained close to 300 K by weak coupling to an external temperature bath (27) with a coupling constant of 0.1 ps. The LINCS (28) algorithm was used to constrain bond lengths within the solute. The SETTLE (29) algorithm was used to constrain the bond lengths and the bond angle in water. The integration time step was 2 fs. A twin-range cutoff of 0.9/1.4 nm was used to evaluate the nonbonded interactions. Interactions within the short-range cutoff were evaluated every step, whereas interactions within the long cutoff were updated every five steps, together with the pair list. A reaction field (RF) correction (30) with an ɛRF = 78 was used to correct for the neglect of electrostatic interactions beyond the 1.4 nm cutoff. Charge states were set in accordance with a pH of 7 and no counterions were added (31). All simulations were performed using the GROMACS3.x software package (32,33).
Monomer Aβ16–20, Aβ16–20m, and Aβ16–22 simulations
The structure of the N-methylated Aβ16–20m was parameterized following the Gromos 96 force field manual (34) and then validated by comparing with a density functional theory-optimized structure. To sample thermodynamic-relevant states accessible to the peptide, fully atomistic MD simulations and replica-exchange MD simulations (REMD) (35) in explicit solvent were performed. Each structure was placed in a dodecahedral periodic box of volume 45 nm3. The REMD simulations were carried out at 34 temperatures ranging from 300 K to 550 K for a total of 50 ns per replica, giving a total combined time of 1.7 μs for each peptide, with swap attempts every 10 ps. Acceptance ratios for exchange moves ranged from 13% to 31%. The initial structure for each replica was taken, after equilibration, at the closest desired temperature from an inverse simulated annealing trajectory (slow heating) at constant volume. The inverse simulated annealing scheme started at 300 K. The temperature was linearly increased by 10 K during 40 ps, followed by 40 ps of simulation coupled to the new temperature and repeated until a final temperature of 550 K was reached.
Dimer and trimer simulations of Aβ16–22 and Aβ16–20m
Homodimers of Aβ16–22 and Aβ16–20m as well as mixed dimers of Aβ16–22 and Aβ16–20m and heterotrimers of two Aβ16–20m and one Aβ16–22 peptides were simulated using REMD. Each configuration was randomly generated and placed in a dodecahedral periodic box of volume 87 nm3. The REMD simulations were carried out at 34 temperatures ranging from 300 K to 550 K for a total of 50 ns per replica, with swap attempts every 100 ps. Acceptance ratios for exchange moves ranged from 6% to 33%. Two independent 50 ns REMD trajectories (each with different random starting configurations) were run, giving a total combined time of 3.4 μs for each system.
Protofilament and protofilament with inhibitor simulations
The initial structure of the model protofilament was generated by translations and rotations of a single β-strand of Aβ16–22 (Ac-Lys16-Leu17-Val18-Phe19-Phe20-Ala21-Glu22-NH2) to form a bilayer, with each sheet consisting of nine peptides. Adjacent strands in a layer were oriented antiparallel (24) to each other with an initial Cα average separation distance of 0.45 nm. The layers were oriented parallel to each other, with an initial separation distance of 0.82 nm and such that the charged side chains were pointing to the solvent (36). The volume of the dodecahedral box was 246 nm3. The density of the system was adjusted by weak coupling to an external pressure bath (27) (P0 = 1 bar, coupling time τp = 1.0 ps). To initiate the MD simulations, each system was first energy minimized using a steepest descent algorithm. The solvent was then relaxed by simulating the system with the solute positionally restrained (100 ps). Initial velocities were generated from a Maxwellian distribution at the target temperature. For the protofilament alone, two independent 100 ns trajectories at room temperature were generated and a representative structure taken as the structure with the lowest root mean-square deviation (RMSD) of main chain atom distances with respect to the average structure of the MD-generated ensemble. A protofilament is a building block of a fibril and consists itself of one or more β-sheets.
The protofilament used here has dimensions (diameter <3.5 nm) corresponding to the conventional definition of a “protofilament”. Four different configurations of the Aβ16−22 protofilament with Aβ16–20m inhibitors, named ConfA, ConfB, ConfC, and ConfD, were generated (see Fig. 5 b). The initial configuration of each conformation was built using the representative structure of the protofilament and a representative structure of the Aβ16−20m inhibitor. In conformations ConfA, ConfB, and ConfD, two inhibitors, one per protofilament layer, were placed at different distances from a single edge of the protofilament until a critical distance was found such that the inhibitor will approach the protofilament. This critical distance depends on the parameters of the force field used, the treatment of electrostatics, and the timescale of the simulations. Different orientations of the inhibitor with respect to the chain at the edge of the protofilament were tested (parallel and antiparallel) as well as different faces (N-methyl groups toward or away from the chain at the edge of the fibril). In ConfC, two inhibitors per protofilament layer were placed at both edges of the protofilament for a total of four inhibitors. Each trajectory of the protofilament with inhibitors was monitored for 50 ns.
FIGURE 5.

(a) Initial structure of the model protofilament generated by translations and rotations of a single β-strand of Aβ16–22 (Ace-Lys16 Leu17 Val18 Phe19 Phe20 Ala21 Glu22 -NH2). (b) Snapshots at 50 ns indicate four possible scenarios for Aβ16−20m peptide binding to the Aβ16−22 protofilament. In ConfA and ConfB the inhibitor binds at the edge of the protofilament via hydrogen-bond formation in either parallel or antiparallel relative orientation to the edge strand; the other inhibitor molecule shown in ConfB drifts from the edge to the lateral side of the protofilament and intercalates in between the layers; in ConfC one inhibitor binds to one of the solvent-exposed protofilament faces and across several strands of the protofilament, with N-methyl groups facing the protofilament; in ConfD, two inhibitors interact with an edge strand breaking the symmetry between protofilament layers. Animations illustrating the different binding sites are shown in the Supplementary Material.
Cluster analysis
For each monomer simulation, a series of nonoverlapping clusters of structures were obtained as described in Daura et al. (37) by calculating the backbone RMSD between all pairs of structures (sampled every 0.04 ns) after a best fit rotation. Then, the structure with the largest number of neighbors that satisfy the condition RSMD < 0.08 nm (considered the central structure of the cluster) was taken together with the neighbors to form the (first) cluster and eliminated from the pool of structures. This process was repeated until the pool of structures was empty. For the dimer and trimer simulations, basins representing the lowest energy structures were defined based on the reaction coordinates used to make the potential of mean force (PMF) plots in Figs. 3 and 4. Structures taken from replicas at all temperatures which fell into these basins were clustered based on Cα positions of nonterminal residues (residues 17–19 for Aβ16–20m and 17–21 for Aβ16–22) using a cutoff of 0.25 nm.
FIGURE 3.

Two-dimensional PMF plots at T = 300 K for the two homodimers studied: (a) the Aβ16–22 peptide and (d) the Aβ16–20m inhibitor. In each plot, the free energy is plotted in units of kJ/mol versus number of interpeptide Cα contacts (x axis) and the cosine of θ, the angle between the two peptides, defined as the angle between the vectors connecting Cα atoms of residues 17 and 19 in the Aβ16–20m inhibitor and residues 18–20 in the Aβ16–22 peptide. The boxes denote the region of phase space representing the lowest energy basins in which conformational clustering was performed. (b–c, e–f) The resulting representative structures from the most populated clusters within these basins are shown on the right. Methyl groups are shown as gray spheres, backbone interpeptide hydrogen bonds are shown as dotted lines, and the N-terminus of each strand is highlighted in orange for the Aβ16–20m inhibitor and in violet for the Aβ16–22 peptide.
FIGURE 4.

(a) Two-dimensional PMF plot at T = 300 K for the Aβ16–20m inhibitor-Aβ16–22 peptide dimer. The free energy is plotted in units of kJ/mol versus number of interpeptide Cα contacts (x axis) and the cosine of θ, the angle between the two peptides, defined as the angle between the vectors connecting Cα atoms of residues 17 and 19 in the Aβ16–20m inhibitor and residues 18–20 in the Aβ16–22 peptide. The boxes denote the region of phase space representing the lowest energy basins in which conformational clustering was performed. (b and c) The resulting representative structures from the most populated clusters within these basins are shown on the right. Methyl groups are shown as gray spheres, backbone interpeptide hydrogen bonds are shown as dotted lines, and the N-terminus of each strand is highlighted in orange for the Aβ16–20m inhibitor and in violet for the Aβ16–22 peptide. (d) Two-dimensional PMF plot for the trimer consisting of two Aβ16–20m inhibitors and one Aβ16–22 peptide. The free energy is plotted in units of kJ/mol versus number of Aβ16–20m inhibitor-Aβ16–20m inhibitor Cα contacts (x axis) and the total number of Aβ16–22 peptide-Aβ16–20m inhibitor contacts. The box denotes the region of phase space representing the lowest energy basin in which conformational clustering was performed. (e–g) The resulting representative structures from the three most populated clusters within this basin are shown on the right.
Potential of mean force
One- and two-dimensional potentials of mean force along a number of reaction coordinates at different temperatures were built using the weighted histogram analysis method (38,39), which provides an optimal estimate of the density of states. The reaction coordinates used include the number of Cα contacts and the angle between the strands. A Cα contact is defined if any two Cα atoms have a separation less than 0.65 nm. For the angle between the strands, a vector was formed for each strand by connecting the Cα atoms of two residues (18 and 20 in Aβ16–22 or 17 and 19 in Aβ16–20m), and the angle, θ, between the vectors was calculated such that θ = 0° for parallel strands and θ = 180° for antiparallel strands.
3J-coupling constants
3J-coupling constants were calculated from the trajectories using the Karplus relation:
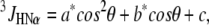 |
where a, b, and c were chosen equal to 6.4 Hz, −1.4 Hz, and 1.9 Hz, respectively, and θ = φ-60°, with φ the backbone dihedral angle (40).
RESULTS AND DISCUSSION
N-methylation rigidifies the Aβ16–20m peptide
Experimentally, the Aβ16−20m inhibitor peptide has been characterized using circular dichroism (CD) spectroscopy and two-dimensional NMR (22). These studies indicate that the N-methylated peptide adopts an extended or β-strand conformation, whereas its unmethylated counterpart adopts a random coil structure. The Aβ16–20m peptide does not form fibrils and its conformation is remarkably stable in different solvent conditions and resists chemical and thermal denaturation.
We characterized the conformational space sampled by the Aβ16–20m inhibitor peptide using REMD simulations in explicit solvent, with a cumulative simulation time of 1.7 μs. To further establish the effect of N-methylation on the structure of the peptide, a reference REMD simulation of the unmethylated Aβ16–20 was performed.
Conformations obtained from the REMD simulations were clustered according to mutual RMSD. The end-to-end distance distribution (a measure of the size of the peptide), along with representative structures of the Aβ16–20 and Aβ16–20m peptides obtained from the clustering analysis at room temperature, are shown in Fig. 1. The conformational space accessible to the Aβ16–20m peptide is more restricted than for the unmethylated peptide, with the Aβ16–20m peptide exhibiting a narrow peak at distances corresponding to extended conformations (∼1.24 nm). The unmethylated peptide, on the other hand, shows a broader distribution of end-to-end distances, corresponding to a greater variety of possible conformations for the peptide. JNHα coupling constants calculated from the REMD trajectory at room temperature for Aβ16–20m fall in the >7 Hz range characteristic of β-strand conformation (22).
FIGURE 1.

Plot of the distribution of the end-to-end distance and of the central structures of the most populated conformational clusters of the Aβ16–20 and Aβ16–20m peptides. The N-methyl groups are highlighted in magenta. There is a clear peak in the end-to-end distance distribution of the Aβ16–20m peptide indicating that this peptide resides preferentially in an extended conformation. This dominant conformation exhibits a relative population of ∼50% for the peptide ensemble sampled.
Two-dimensional potentials of mean force, plotted at room temperature as a function of the backbone dihedral φ and ψ angles for Aβ16–20 and Aβ16–20m, illustrate the conformational restrictions arising from the N-methylation (Fig. 2). In particular, the difference in dihedral space sampled by Lys16 and Val18 is striking. In the methylated peptide these two residues are clearly confined to the β-strand region, whereas in the unmethylated peptide these two residues are free to sample a broader region in dihedral space. The backbone dihedral angle φ sampled by the methylated residues, mLeu17 and mPhe19, populates a significant region in the range φ > 0 (although no turn structure was observed), another manifestation of steric effects introduced by the methyl group. The coexistence of the diverse ensemble of conformers for Aβ16–20 likely accounts for the random-coil CD seen experimentally, whereas the restricted conformations observed here for Aβ16–20m are consistent with the shifted β-strand CD profile (22).
FIGURE 2.

Ramachandran plots for the (top row: a, c, e, and g) Aβ16–20 peptides and (bottom row: b, d, f, and h) Aβ16–20m peptides. a and b correspond to Lys16, c and d to Leu17, e and f to Val18, and g and h to Phe19. The conformational effect induced by the N-methylation results in the restriction of the dihedral space of residues (b) Lys16 and (f) Val18.
Binding of the inhibitor peptide Aβ16–20m to free Aβ16–22 peptides
The ability of the inhibitor peptide to bind to free Aβ16–22 peptides in solution was investigated using replica exchange MD simulations. An initial “benchmark” simulation was performed to determine the preferred dimer conformations for the Aβ16–22 peptide. The ability of Aβ16–22 to dimerize and form larger aggregates has been previously investigated by Gnanakaran et al. (41) and Nguyen et al. (42). The PMF for the Aβ16–22 dimer at 300 K is shown in Fig. 3 a. The PMF as a function of the number of Cα contacts and the angle between the two peptides shows two basins, a deeper one corresponding to an antiparallel orientation (Fig. 3 c) and a shallower one corresponding to a parallel orientation of the two strands (Fig. 3 b). The antiparallel configuration is the lowest in energy. Aβ16–22 homodimers display a range of registries with a clear preference for the lysine residue to be bound, whereas the glutamic acid residue favors a more solvated conformation. Other higher energy dimer configurations correspond to less-ordered, “bent” assemblies. Temperature-dependent studies of the PMF projected onto the number of interpeptide Cα contacts (see Supplementary Material) indicate that the Aβ16–22 dimer is the preferred conformation over the entire range of temperatures studied (300–550), with the Aβ16–22 monomer basin residing much higher in energy.
A second benchmark simulation was performed to investigate the inhibitor peptide's ability to self-dimerize. Fig. 3 d shows the PMF of the Aβ16–20m dimer at 300 K as a function of the number of Cα contacts and the angle between the two peptides. Representative dimer configurations are shown in Fig. 3, e and f. As in the case of Aβ16–22, the dimers can adopt a parallel or antiparallel orientation, with the antiparallel configuration lower in energy. Inhibitor homodimers accommodate mainly in-registry with a lower population of out-of-registry dimers. The less-ordered, bent configurations seen for the Aβ16–22 dimers are not observed here. This is a direct result of the rigidification imparted by the N-methylation. In all dimer configurations, the N-methyl groups point to the solvent. Hence, these dimers correspond to end-products, to which no further monomer can add on. This is consistent with the experimental observation that the N-methylated peptides cannot form fibrils (22).
Unlike the Aβ16–22 peptide that preferentially adopts a dimer conformation, the Aβ16–20m peptide coexists between monomeric and dimeric forms. In fact, the dimeric states of Aβ16–20m are stable only at temperatures below 310 K. At higher temperatures, diverse dimer conformations which are neither parallel nor antiparallel are short lived, and the monomer state becomes dominant (see Supplementary Material). This finding is consistent with the inability of the Aβ16–20m inhibitor to form fibrils. The Aβ16–22 dimers, on the other hand, display two well-defined temperature-dependent basins, with a basin representing monomeric states that develops at slightly higher temperature than 300 K and remaining at high temperatures but always higher in free energy than the dimeric basin.
After the benchmark homodimer simulations, we performed a series of simulations to probe how the inhibitor peptide binds free Aβ16–22 peptides in solution. We consider first the interaction of one inhibitor Aβ16–20m peptide with one Aβ16–22 peptide, followed by the interaction of two inhibitor Aβ16–20m peptides with one Aβ16–22 peptide. The latter simulation mimics experimental conditions in which the inhibitor/peptide ratio is at least 2:1 (typical ratios used in the experiment ranged between 2:1 and 10:1) (22).
For the system of one Aβ16–20m peptide with one Aβ16–22 peptide, we find that at room temperature, the preferred state is one in which the inhibitor peptide is bound to Aβ16–22. Parallel and antiparallel relative orientations with different backbone registries generate a rich pool of mixed dimer conformations. Fig. 4 a shows a two-dimensional PMF as a function of the number of peptide-inhibitor Cα contacts and the angle between the two peptides, as well as representative dimer conformations in Fig. 4, b and c. Interestingly, the degree of conformational restriction of the mixed dimers, as measured by the chosen reaction coordinates, lies in between that of the inhibitor homodimer and the Aβ16–22 homodimer, which is consistent with the relative degree of flexibility of the two peptides.
Finally, we studied the interaction of two Aβ16–20m peptides with one Aβ16–22 peptide. At room temperature, a dominant free energy basin is present in the PMF plotted as a function of the number of peptide-inhibitor Cα contacts and the number of Cα contacts between the inhibitors (Fig. 4 d). A projection of the free energy in Fig. 4 d onto the number of Cα inhibitor-peptide contacts when the number of inhibitor-inhibitor contacts is zero is shown in Fig. 2 of the Supplementary Material. This figure shows that the minimum free energy occurs at a number of Cα inhibitor-peptide contacts of 30. The lowest energy basin corresponds to a structure in which the Aβ16–22 peptide is sandwiched between two inhibitor peptides (snapshots of the centroids of the three most populated clusters, C1, C2, and C3 are shown in Fig. 4, e–g).
In all three representative structures, one inhibitor molecule is bound to the Aβ16–22 peptide via five (C1 and C2) or six (C3) hydrogen bonds in an antiparallel alignment with both N-methyl groups pointed toward the solvent. Likewise, in all three structures, the second inhibitor molecule is bound to the Aβ16–22 peptide on the side opposite the first. In the two most populated clusters (C1 and C2, Fig. 4, e and f) the second inhibitor is bound across to the peptide with its methyl groups either solvent exposed (C1) or pointing toward the peptide (C2) and held in place by side-chain interactions, in particular Phe-Phe interactions (C1) and Lys-Phe, Val-Phe, and Leu-Leu interactions (C2). In the third most populated cluster (Fig. 4 g), the second inhibitor is bound to the Aβ16–22 peptide via five hydrogen bonds in a parallel orientation with its methyl groups solvent exposed. C4 (not shown) is the all-antiparallel counterpart of C3 and comprises 6% of the structures in this basin. The higher-energy basins present in Fig. 4 d correspond to structures present mainly in the higher-temperature replicas in which the peptide binds to one inhibitor or to neither inhibitor.
A plausible mechanism of action of the N-methylated peptides would be to bind to the free Aβ16–22 monomers in solution, thus effectively sequestering these peptides from the pool of available monomers for fibril growth. Although a direct competitive experiment involving a pool of free monomers and a protofilament is not computationally tractable using fully atomistic, solvated simulations, this mechanism is supported by the nature of the structures belonging to the lowest free-energy basin. Not only would this process prevent fibril growth, it could also lead to fibril disassembly. Fibrils are dynamic entities, with monomers at the edges attaching and detaching. The inhibitor peptides could bind to the departing monomers and remove them from active circulation, shifting the equilibrium from aggregate to inhibitor-bound monomer.
Binding of the inhibitor Aβ16–20m to ordered Aβ16–22 aggregates
Structure of the Aβ16–22 protofilament
The nature of the arrangements of β-strands in fibrils of Aβ16–22 has been investigated by solid-state NMR by Tycko and co-workers (24). Their results reveal that the peptides adopt an antiparallel arrangement, consistent with other experimental (43) and theoretical investigations (41,44–46). Based on the available NMR data, we constructed a model protofilament consisting of two parallel sheets, each composed of nine antiparallel Aβ16–22 β-strands. Charged side chains (Lys16 and Glu22) point toward the solvent, whereas the Leu17, Phe19, and Ala21 residues form the hydrophobic interior of the protofilament. A snapshot of an equilibrated protofilament structure is shown in Fig. 5 a. We did not observe a significant number of water molecules at the interior of the protofilament. The equilibrated protofilament displays a twist of average angle ∼16° between adjacent strands and a helical pitch of ∼16 nm. The separation between the two layers of the protofilament ranged from 0.89 nm to 0.95 nm during the course of two different 100-ns-long simulations. The separation between each individual peptide chain in a layer ranged from 0.43 nm to 0.53 nm, with an average value of 0.47 nm. Both layer and strand separation distances are consistent with the solid-state NMR values (0.99 nm and 0.47 nm, respectively (24)). The distribution of φ and ψ backbone dihedral angles of Leu17, Val18, Phe19, Phe20, and Ala21 (data not shown) remains in the characteristic range for antiparallel β-sheet structure during the course of the simulations (47).
The overall in-register network of hydrogen bonds is stable throughout both trajectories. There are about four backbone hydrogen bonds between adjacent strands within each layer in locations that repeat every two strands. The distance between the Cα atoms of the first and last residue of a given strand is on average ∼1.91 nm, slightly more extended than monomer Aβ16–22 in solution (basin of the extended conformation centered at ∼1.54 nm, see Supplementary Material). Root mean-squared fluctuations of the atomic positions indicate that the flexibility of the protofilament is most pronounced for the strands located at the extremities where the packing of the side chains is less dense than in the center of the protofilament (data not shown). The twisting and bending seen in our simulations contribute to the overall stability of the structure, as has been observed for other model protofilaments (48). Finally, we note that a number of other strand and sheet arrangements have been proposed for fibrils of Aβ16–22 (such as, for instance, an antiparallel strand arrangement in which the side chains of Lys16, Val18, Phe20, and Glu22 point to the interior of the fibril) (49,50). However, the model used here has proved to be the lowest energy and the most stable one within our simulation protocol.
Binding of the inhibitor to the protofilament
The interaction of the inhibitor peptide with the protofilament was monitored through 13 50-ns simulations (10 trajectories for the system protofilament—2 inhibitors and 3 trajectories for the system protofilament—4 inhibitors) and 4 20-ns simulations (for the system protofilament—2 inhibitors). The different simulations were initiated with either two or four inhibitor peptides placed at different distances and orientations from one edge of the protofilament. These stoichiometric ratios were chosen for their computational tractability and are much lower (by ninefold in the case of two inhibitor molecules and by 4.5-fold in the case of four inhibitor molecules) than even the lowest inhibitor/Aβ16–22 peptide ratios tested experimentally (22).
Binding of the inhibitor to the protofilament was seen in all but one simulation. Competition simulations in which both inhibitor peptide and free Aβ16–22 peptide were placed in solution with the fibril only showed binding of the inhibitor to the fibril during the duration of the simulations (see Fig. 4 of the Supplementary Material). Three distinct binding scenarios emerge.
Fibril termination in the direction of monomer deposition
The most common scenario seen in our simulations involves the binding of the inhibitor peptide to the edge of the protofilament via hydrogen bonding, with the N-methyl groups exposed to the solvent (Fig. 5 b, Conf A). This binding event constitutes a means of terminating fibril growth in the longitudinal direction: additional Aβ16–22 monomers cannot form hydrogen bonds with the exposed N-methylated side of the bound Aβ16–20m peptide, and hence no further deposition of Aβ16–22 will take place.
Interestingly, the bound inhibitor shows backbone dihedral angles consistent with β-strand secondary structure, with all three nonmethylated residues involved in hydrogen bonds with the protofilament. The registry of backbone hydrogen bonds, however, slightly differs in each binding event observed, which is consistent with the varied pool of conformations observed in our mixed di- and trimerization simulations. The mLeu17, Val18, mPhe19, and Phe20 side chains of the inhibitor form a stable hydrophobic cluster with either the Leu17, Val18, Phe19, Phe20, or to a lesser extent with the Lys16 and Ala21 residues of the Aβ16–22 β-strand of the protofilament. The side chain of residue Glu22 does not form a significant number of side-chain contacts with the inhibitor. In the process of binding, there is no clear distinction in terms of inhibitor-protofilament interaction energy for parallel or antiparallel orientation. The antiparallel arrangement has been proposed as the most probable orientation in fibrils formed of small peptides with low amphiphilicity (51), but, interestingly, our REMD simulations discussed here as well as recent REMD simulations by Garcia show that the Aβ16–22 peptide can form both parallel and antiparallel dimers (41). Our simulations suggest that the binding of the Aβ16−20m inhibitor to the fibril can be accommodated in both orientations, i.e., parallel and antiparallel, thus maximizing the possibilities of interaction of the peptide with the fibril (Fig. 5 b, ConfA and ConfB). Such degeneracy in our model may arise from the high affinity of the short hydrophobic sequence, whose side chains easily pack to each other without favoring any particular chain orientation. It is also possible that the parallel orientation of the inhibitor with respect to the protofilament peptide chain is metastable and en route in the path toward antiparallel binding, as supported by our dimer REMD simulations in which the parallel basin is shallow whereas the antiparallel is deep.
We postulate that the rigidity of Aβ16–20m favors binding since the peptide end-to-end distance is already adjusted to fit the homologous sequence in the protofilament. Indeed, the end-to-end distance of the Aβ16–20m in free monomeric form is ∼1.24 nm, comparable to its end-to-end distance in dimers and trimers (∼1.3 nm) and when bound to the protofilament (∼1.24–1.28 nm). Furthermore, this distance is consistent with the average distance of ∼1.27 nm between the Cα atom of residues Lys16 and Phe20 for each strand of the protofilament. The Aβ16–20m peptide hence appears to be prestructured in its monomeric form in a state commensurate for binding to the protofilament. This property could enable the Aβ16–20m to compete effectively for binding with free Aβ16–22 peptides in solution. The rigid N-methylated peptide would display a smaller loss of conformational entropy and hence more favorable free energy upon binding to the fibril than a flexible Aβ16–22 peptide. The Aβ16–22 peptide would presumably have to either first extend before binding to the strand at the edge of the protofilament or bind and subsequently rearrange its structure.
Fibril inhibition in the direction of lateral growth
Binding at the edge of the protofilament is not the only way in which the inhibitor peptide can interact with the protofilament. Our simulations reveal two other binding modes that can lead to inhibition in the direction of lateral, rather than longitudinal, growth.
The first scenario involves the drifting of the inhibitor peptide from the edge of the protofilament to the side until it is positioned between the layers (Fig. 5 b, ConfB), with the Lys16 residue pointing to the solvent or, in other words, with the hydrophobic residues inserted in between the protofilament layers. The interstrand distance for the two chains closer to the site where the inhibitor has intercalated (in the case of Fig. 5 b, ConfB strands number 6 and 7) considerably increases from ∼0.50 nm to ∼0.61 nm in one layer but decreases in the other from ∼0.45 nm to ∼0.43 nm (Fig. 6, ConfB), altering the geometry of the protofilament. These observations indicate that the inhibitor has the ability to interact via its side chains in the dimension of lateral fibril growth, perpendicular to the elongation dimension. This is an interesting possibility for inhibition because this binding site could potentially stop further addition of layers, thus preventing the formation and stabilization of mature fibrils. Two new simulations were started from the same initial configuration that Fig. 5 b ConfB was started from, but with different initial velocities. In both new trajectories we observed that, after 20ns, one of the inhibitors had hydrogen bonded at the edge strand of the protofilament and that the other inhibitor had positioned itself between the layers. This further confirms the plausibility of this binding scenario.
FIGURE 6.

Interstrand distance for the model Aβ16−22 protofilament in the absence and presence of the inhibitor for each of the configurations (ConfA, B, C, and D) shown in Fig. 5 b. For the protofilament alone, the average value of the interstrand distance in our trajectories converges to the value reported by Tycko and co-workers (24). In the cases where the inhibitor simply binds to the edge of the protofilament (Conf A) or binds on the exposed face of the protofilament (Conf C), the interstrand distances remain mostly unaffected. In binding scenarios B and D, where the inhibitor intercalates between the layers and strands, respectively, the distance between protofilament strands is perturbed.
A second scenario of lateral growth inhibition involves the binding of the Aβ16–20m peptide on a solvent-exposed face of the protofilament, rather than between the layers of the protofilament (Fig. 5 b, ConfC). This configuration differs from ConfA, ConfB, and ConfD in that there are four inhibitors. Initially, each inhibitor is positioned at the bottom edge of each protofilament layer with the N-methyl side pointing toward the solvent. During the simulation, one of the Aβ16–20m peptides drifts toward the center of the protofilament solvent-exposed face and rotates such that it binds across several (three to four) protofilament strands with the N-methylated side of the inhibitor facing the protofilament. Due to fluctuations in the position of the inhibitor throughout the simulation, some hydrogen bonds are made between the inhibitor and the fibril; however, the binding of this inhibitor is mainly stabilized by contacts between the Aβ16–20m side chains and the protofilament. Although all inhibitor residues form a relatively high number of contacts with the side chain of residue Phe20 of the protofilament strands, the highest number of side-chain contacts is between mPhe19 of the inhibitor and Phe20 of the protofilament. These simulations suggest that the binding of the inhibitor via side-chain interactions on the face of the protofilament can block fibril growth in the lateral direction. Two additional 20 ns simulations started from the same initial coordinates and different initial velocities confirmed this binding event. The experiments of Meredith and co-workers (22) were performed with a higher ratio of inhibitor/Aβ16–22 peptide than used in our simulations, and it is likely that under such conditions more than one inhibitor could bind to the face of the aggregate. This mechanism could be highly effective at inhibiting the growth of not only prefibrillar species but of much larger fibrils as well.
Protofilament strand removal by the inhibitor
The third scenario observed in our simulations involves two inhibitors simultaneously attacking the terminal strand residing at the edge of the protofilament (Fig. 5 b, ConfD). The first inhibitor binds to the edge strand of the fibril, whereas the second inhibitor intercalates between the edge strand and the previous one. This configuration would lead to a scenario strikingly similar to the stable trimer shown in Fig. 4 g in which an Aβ16–22 peptide is isolated by two Aβ16–20m inhibitor molecules. This process could potentially lead to disassembly of the fibril by strand removal, one by one. This is a particularly interesting scenario, as the protofilament is believed to grow in the longitudinal direction by monomer deposition, and our inhibitor would be effectively reversing this process. This mechanism of targeted strand removal is quite plausible, given the high ratios of inhibitor required in the experiments of Meredith and co-workers (22).
Although a complete unbinding event was not observed on the timescale of our simulation, we could nonetheless see evidence of destabilization of the terminal strand. For the case of the binding event shown in Fig. 5 b, ConfD, the interstrand distance between the terminal strand (number 9) and the preceding strand (number 8), considerably increases from ∼0.47 nm to ∼0.68 nm. Interestingly, this disruption also affected the interstrand distance between strands 6 and 7 (Fig. 6, ConfD). The charged group Lys16 of the inhibitor inserts deeply between the strands (Fig. 5 b, ConfD). Insertion is driven both by side-chain interaction with neighboring strands and by the rigid nature of the inhibitor that eases intercalation between the protofilament strands. It is possible that this mode of binding is more likely to occur in prefibrillar species than in mature fibrils. Indeed the loose-packing present in early oligomeric forms would lend itself more easily to insertion of inhibitor molecules between strands.
The breaking of symmetry between the layers resulting from strand removal in one layer may induce a destabilization of the entire suprastructure of the fibril. Simulations were performed in which one or more strands were manually removed from one layer of the protofilament. During the time course of our simulations (20 ns) the morphology of the protofilament became highly distorted, suggesting its eventual disruption (see Fig. 3 in Supplementary Material).
CONCLUSIONS
Our simulations reveal a much more complex mechanism of inhibition for N-methylated peptides than was previously thought. Our simulations suggest that the inhibitor can act in two manners: 1), by binding to free Aβ16–22 monomers in solution, depleting the pool of available fibril “building blocks”, and shifting the equilibrium from fibrillar to monomer; and 2), by binding to the protofilament. We see different binding sites along the protofilament structure, consistent with the presence of multiple binding sites identified in spectroscopic studies of ligands binding to Aβ1–40 fibrils (52,53). Binding at different sites can lead to different mechanisms by which fibril growth is stopped, its structure destabilized, and disassembly induced.
The dominant binding site of the N-methyl inhibitor Aβ16−20m with the Aβ16–22 protofilament is at the edge of the protofilament via hydrogen bonds to prevent further elongation. The binding sites which occurred less frequently in our simulations correspond to insertion of the inhibitor in between protofilament layers and on the exposed face of a protofilament, both of which can stop lateral growth of the fibril. Finally, two inhibitors can act in concert to remove peptide strands one by one from the edge of the protofilament. In all instances, binding of the inhibitor perturbed the symmetry between layers and had an overall destabilizing effect on the morphology of the fibril. We believe that the prestructuring of the N-methyl inhibitor favors binding since the peptide is already accommodated to fit the homologous sequence in the Aβ16–22 protofilament. Our trimer simulations of two inhibitors with one Aβ16–22 peptide show that the inhibitor would rather bind to Aβ16–22 than to itself, or remain in monomeric form. Furthermore, the dimer and trimer structures which are formed (Fig. 4, e–g) in the REMD simulations mimic the binding modes observed in the MD simulation of the fibril and inhibitors. Specifically, parallel and antiparallel backbone hydrogen bonding of the inhibitor molecules to the Aβ16–22 peptide as well as binding via interaction of hydrophobic side chains were observed in both the REMD and MD simulations. In addition, competition simulations in which we placed both inhibitor and Aβ16–22 peptides in solution with the fibril only showed binding of the inhibitor to the fibril in the timeframe of our simulation (50 ns or less) (see Fig. 4 of the Supplementary Material).
Disassembly of the fibril can be triggered by a destabilization of the fibril upon binding of the inhibitor or by the sequential removal of the end strands (Fig. 5 b, Conf D). Complete disassembly of the fibril cannot be seen in our simulations, as this process occurs on timescales that are not computationally accessible. The inhibitors can prevent reassembly of the fibril by binding to the disaggregated (or otherwise free) Aβ16−22 peptides in solution (Fig. 4). Fibril disassembly can hence be rationalized in terms of a population shift from aggregate species to monomeric species. The high concentration of inhibitors required in the experiment is consistent with this idea, with the inhibitors binding to Aβ16−22 monomers or even Aβ16−22 prefibrillar species in solution. The end result is a shift from a fibril-dominated ensemble to one populated by Aβ16–22 monomers bound to inhibitor peptides.
The fibrillogenesis mechanisms we observed suggest that the N-methylated inhibitor peptide can act on both prefibrillar forms and on mature fibrils of Aβ16–22. These oligomeric species coexist in solution, and the precise mode of action of the inhibitor may depend on the nature of the aggregate morphology. The structures in which binding of the inhibitor at the edge of the protofilament takes place could be the dominant aggregate population. Less populated protofilament morphologies may facilitate lateral or interstrand inhibitor binding. That N-methyl inhibitors act via different mechanisms, not only by stopping strand deposition, has also been suggested by Doig and co-workers to explain the effect of N-methyl inhibitors on Aβ25–35 fibrillization (17). Ongoing research in our group involves the investigation of the binding of Aβ16–20m to model protofilaments of the longer Aβ9–40 peptide (54).
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Stephen Meredith and Gal Bitan for very helpful discussions, and Alejandro Ramirez for performing the quantum mechanical calculations of Aβ16–20m.
Simulations were performed using the computational resources of the California NanoSystems Institute (National Science Foundation grant CHE-0321368) and Texas Advanced Computing Center Cray-Dell Linux Cluster (National Science Foundation Teragrid MCA05S027). Support from the National Science Foundation (No. MCB 0642086), the David and Lucile Packard Foundation, and the A. P. Sloan Foundation are gratefully acknowledged. P.S. is grateful for support from an American Association of University Women postdoctoral fellowship.
Editor: Ruth Nussinov.
References
- 1.Eanes, E. D., and G. G. Glenner. 1968. X-ray diffraction studies on amyloid filaments. J. Histochem. Cytochem. 16:673–677. [DOI] [PubMed] [Google Scholar]
- 2.Kirschner, D., C. Abraham, and D. Selkoe. 1986. X-ray diffraction from intraneuronal paired helical filaments and extra-neuronal amyloid fibres in Alzheimer's disease indicates cross β conformation. Proc. Natl. Acad. Sci. USA. 83:503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirkitadze, M. D., G. Bitan, and D. B. Teplow. 2002. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J. Neurosci. Res. 69:567–577. [DOI] [PubMed] [Google Scholar]
- 4.Klein, W. L. 2002. ADDLs & protofibrils—the missing links? Neurobiol. Aging. 23:231–233. [DOI] [PubMed] [Google Scholar]
- 5.Caughey, B., and P. T. Lansbury Jr. 2003. Protofibrils, pores, fibrils and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 26:267–298. [DOI] [PubMed] [Google Scholar]
- 6.Soto, C. 2003. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4:49–60. [DOI] [PubMed] [Google Scholar]
- 7.Mason, J. M., N. Kokkoni, K. Stott, and A. J. Doig. 2003. Design strategies for anti-amyloid agents. Curr. Opin. Struct. Biol. 13:526–532. [DOI] [PubMed] [Google Scholar]
- 8.Sciarretta, K. L., D. J. Gordon, and S. C. Meredith. 2006. Peptide-based inhibitors of amyloid assembly. Methods Enzymol. 413:273–312. [DOI] [PubMed] [Google Scholar]
- 9.Hilbich, C., B. Kisters-Woike, C. L. Masters, and K. Beyruether. 1992. Substitutions of hydrophobic amino-acids reduce the amylodogenicity of Alzheimer's-disease β-A4 peptides. J. Mol. Biol. 219:460–473. [DOI] [PubMed] [Google Scholar]
- 10.Wood, S. J., R. Wetzel, J. D. Martin, and M. R. Hurle. 1995. Prolines and amyloidogenicity in fragments of the Alzheimer's peptide β/A4. Biochemistry. 34:724–730. [DOI] [PubMed] [Google Scholar]
- 11.Esler, W. P., E. R. Stimson, J. R. Ghilardi, Y. A. Lu, A. M. Felix, H. V. Vinters, P. W. Mantyh, J. P. Lee, and J. E. Maggio. 1996. Point substitution in the central hydrophobic cluster of a human-amyloid congener disrupts peptide folding and abolishes plaque competence. Biochemistry. 35:13914–13921. [DOI] [PubMed] [Google Scholar]
- 12.Tjernberg, L. O., J. Naslund, F. Lindquist, J. Johansson, A. R. Karlstrom, J. Thybrg, L. Terenius, and C. Nordstedt. 1996. Arrest of b-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem. 271:8545–8549. [DOI] [PubMed] [Google Scholar]
- 13.Soto, C., E. M. Sigurdsson, L. Morelli, R. A. Kumar, E. M. Castano, and B. Frangione. 1998. β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy. Nat. Med. 4:822–826. [DOI] [PubMed] [Google Scholar]
- 14.Soto, C., M. S. Kindy, M. Baumann, and B. Frangione. 1996. Inhibition of Alzheimer's amyloidosis by peptides that prevent b-sheet conformation. Biochem. Biophys. Res. Commun. 226:672–680. [DOI] [PubMed] [Google Scholar]
- 15.Lowe, T. L., A. Strzelec, L. L. Kiessling, and R. M. Murphy. 2001. Structure-function relationships for inhibitors of β-amyloid toxicity containing the recognition sequence KLVFF. Biochemistry. 40:7882–7889. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe, K., K. Nakamura, S. Akikusa, T. Okada, M. Kodaka, T. Konakahara, and H. Okuno. 2002. Inhibitors of fibril formation and cytotoxicity of b-amyloid peptide composed of KLVFF recognition element and flexible hydrophilic disrupting element. Biochem. Biophys. Res. Commun. 290:121–124. [DOI] [PubMed] [Google Scholar]
- 17.Hughes, E., R. M. Burke, and A. J. Doig, A. J. 2000. Inhibition of toxicity in the b-amyloid peptide fragment b-(25–35) using n-methylated derivatives. J. Biol. Chem. 275:25109–25115. [DOI] [PubMed] [Google Scholar]
- 18.Kapurniotu, A., A. Schmauder, and K. Tenidis. 2002. Structure-based design and study of non-amyloidogenic, double N-methylated IAPP amyloid core sequences as inhibitors of IAPP amyloid formation and cytotoxicity. J. Mol. Biol. 315:339–350. [DOI] [PubMed] [Google Scholar]
- 19.Yan, L.-M., M. Tatarek-Nossol, A. Velkova, A. Kazantzis, and A. Kapurniotu. 2006. Design of a mimic of nonamyloidogenic and bioactive human islet amyloid polypeptide (IAPP) as nanomolar affinity inhibitor of IAPP cytotoxic fibrillogenesis. Proc. Natl. Acad. Sci. USA. 103:2046–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bodles, A. M., O. M. A. El-Agnaf, B. Greer, D. J. S. Guthrie, and G. B. Irvine. 2004. Inhibition of fibril formation and toxicity of a fragment of alpha-synuclein by an N-methylated peptide analogue. Neurosci. Lett. 359:89–93. [DOI] [PubMed] [Google Scholar]
- 21.Gordon, D. J., K. L. Sciarreta, and S. C. Meredith. 2001. Inhibition of β-amyloid(40) fibrillogenesis and disassembly of β-amyloid(40) fibrils by short β-amyloid congeners containing N-methyl amino acids at alternate residues. Biochemistry. 40:8237–8245. [DOI] [PubMed] [Google Scholar]
- 22.Gordon, D. J., R. Tappe, and S. C. Meredith. 2003. Design and characterization of a membrane permeable N-methyl amino acid-containing peptide that inhibits A β (1–40) fibrillogenesis. J. Pept. Res. 60:37–55. [DOI] [PubMed] [Google Scholar]
- 23.Tseng, B. P., W. P. Esler, C. B. Clish, E. R. Stimson, J. R. Ghilardi, H. V. Vinters, P. W. Mantyh, J. P. Lee, and J. E. Maggio. 1999. Deposition of monomeric, not oligomeric, Ab mediates growth of Alzheimer's disease amyloid plaques in human brain preparations. Biochemistry. 38:10424–10431. [DOI] [PubMed] [Google Scholar]
- 24.Balbach, J. J., Y. Ishii, O. N. Antzutkin, R. D. Leapman, N. W. Rizzo, F. Dyda, J. Reed, and R. Tycko. 2000. Amyloid fibril formation by Ab16–22, a seven-residue fragment of the Alzheimer's b-amyloid peptide, and structural characterization by solid state NMR. Biochemistry. 39:13748–13759. [DOI] [PubMed] [Google Scholar]
- 25.Scott, W. R. P., P. H. Hunenberger, I. G. Tironi, A. E. Mark, S. R. Billeter, J. Fennen, A. E. Torda, T. Huber, P. Kruger, and W. F. van Gunsteren. 1999. The GROMOS biomolecular simulation program package. J. Phys. Chem. A. 103:3596–3607. [Google Scholar]
- 26.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, and J. Hermans. 1981. Interaction models for water in relation to protein hydration. In Intermolecular Forces. B. Pullman, editor. Reidel, Dordrecht, The Netherlands. 331–342.
- 27.Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. Di Nola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 28.Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comp. Chem. 18:1463–1472. [Google Scholar]
- 29.Miyamoto, S., and P. A. Kollman. 1992. SETTLE: an analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comp. Chem. 13:952–962. [Google Scholar]
- 30.Tironi, I. G., R. Sperb, P. E. Smith, and W. F. van Gunsteren. 1995. A generalized reaction field method for molecular dynamics simulations. J. Chem. Phys. 102:5451–5459. [Google Scholar]
- 31.Villa, A., A. E. Mark, G. A. A. Saracino, U. Cosentino, D. Pitea, G. Moro, and M. Salmona. 2006. Conformational polymorphism of the PrP106–126 peptide in different environments: a molecular dynamics study. J. Phys. Chem. B. 110:1423–1428. [DOI] [PubMed] [Google Scholar]
- 32.Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: a message-passing parallel molecular dynamics implementation. Comp. Phys. Comm. 91:43–56. [Google Scholar]
- 33.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Mod. 7:306–317. [Google Scholar]
- 34.van Gunsteren, W. F., S. R. Billeter, A. A. Eising, P. H. Hunenberger, P. Kruger, A. E. Mark, W. R. P. Scott, and I. G. Tironi. 1996. Biomolecular Simulation: The Gromos96 Manual and User Guide. Hochschuverlag AG an der ETH, Zurich.
- 35.Sugita, Y., and Y. Okamoto. 1999. Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314:141–151. [Google Scholar]
- 36.Lu, K., J. Jacob, P. Thiyagarajan, V. P. Conticello, and D. G. Lynn. 2003. Exploiting amyloid fibril lamination for nanotube self-assembly. J. Am. Chem. Soc. 125:6391–6393. [DOI] [PubMed] [Google Scholar]
- 37.Daura, X., K. Gademann, B. Jaun, D. Seebach, W. F. van Gunsteren, and A. E. Mark. 1999. Peptide folding: when simulation meets experiment. Angew. Chem. Int. Ed. Engl. 38:236–240. [Google Scholar]
- 38.Ferrenberg, A. M., and R. H. Swendsen. 1989. Optimized Monte Carlo data analysis. Phys. Rev. Lett. 63:1195–1198. [DOI] [PubMed] [Google Scholar]
- 39.Kumar, S., D. Bouzida, R. H. Swendsen, P. A. Kollman, and J. M. Rosenberg. 1992. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J Comp Chem. 13:1011–1021. [Google Scholar]
- 40.Pardi, A., M. Billeter, and K. Wüthrich. 1984. Calibration of the angular dependence of the amide proton-Cα proton coupling constants, 3JHNα in a globular protein. Use of 3JHNα for identification of helical secondary structure. J. Mol. Biol. 180:741–751. [DOI] [PubMed] [Google Scholar]
- 41.Gnanakaran, S., R. Nussinov, and A. E. Garcia. 2006. Atomic-level description of amyloid b-dimer formation. J. Am. Chem. Soc. 128:2158–2159. [DOI] [PubMed] [Google Scholar]
- 42.Nguyen, P. H., M. S. Li, G. Stock, J. E. Straub, and D. Thirumalai. 2007. Monomer adds to preformed structured oligomers of Abeta-peptides by a two-stage dock-lock mechanism. Proc. Natl. Acad. Sci. USA. 104:111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petty, S. A., and S. M. Decatur. 2005. Experimental evidence for the reorganization of β-strands within aggregates of the Aβ (16–22) peptide. J. Am. Chem. Soc. 127:13488–13489. [DOI] [PubMed] [Google Scholar]
- 44.Klimov, D. K., and D. Thirumalai. 2003. Dissecting the assembly of Aβ (16–22) amyloid peptides into antiparallel β sheets. Structure. 11:295–307. [DOI] [PubMed] [Google Scholar]
- 45.Santini, S., G. H. Wei, N. Mousseau, and P. Derreumaux. 2004. Pathway complexity of Alzheimer's β-amyloid Aβ (16–22) peptide assembly. Structure. 12:1245–1255. [DOI] [PubMed] [Google Scholar]
- 46.Favrin, G., A. Irback, and S. Mohanty. 2004. Oligomerization of amyloid Aβ (16–22) peptides using hydrogen bonds and hydrophobicity forces. Biophys. J. 87:3657–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Munoz, V., and L. Serrano. 1994. Intrinsic secondary structure propensities of the amino acids, using statistical φ-ψ matrices: comparison with experimental scales. Proteins. 20:301–311. [DOI] [PubMed] [Google Scholar]
- 48.Soto, P., J. Cladera, A. E. Mark, and X. Daura. 2005. Stability of SIV gp32 fusion-peptide single-layer protofibrils as monitored by molecular-dynamics simulations. Angew. Chem. Int. Ed. Engl. 44:1065–1067. [DOI] [PubMed] [Google Scholar]
- 49.Ma, B., and R. Nussinov. 2002. Stabilities and conformations of Alzheimer's beta-amyloid peptide oligomers (Aβ (16–22′) Aβ (16–35′) and Aβ (10–35)): sequence effects. Proc. Natl. Acad. Sci. USA. 99:14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rohrig, U. F., A. Laio, N. Tantalo, M. Parrinello, and R. Petronzio. 2006. Stability and structure of oligomers of the Alzheimer peptide Ab16–22: from the dimer to the 32-mer. Biophys. J. 91:3217–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gordon, D. J., J. J. Balbach, R. Tycko, and S. C. Meredith. 2004. Increasing the amphiphilicity of an amyloidogenic peptide changes the β-sheet structure in the fibrils from antiparallel to parallel. Biophys. J. 86:428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen, Z., G. Krause, and B. Reif. 2006. Structure and orientation of peptide inhibitors bound to β-amyloid fibrils. J. Mol. Biol. 354:760–776. [DOI] [PubMed] [Google Scholar]
- 53.Levine III, H. 2005. Multiple ligand binding sites on Ab(1–40) fibrils. Amyloid. 12:5–14. [DOI] [PubMed] [Google Scholar]
- 54.Buchete, N. V., R. Tycko, and G. Hummer. 2005. Molecular dynamics simulations of Alzheimer's β-amyloid protofilaments. J. Mol. Biol. 353:804–821. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}