

Abstract
The interactions of three therapeutic agents, viz. the antipsychotics HPD and CPZ, and the antineoplastic anthracycline DOX, with oxidatively modified phospholipids were studied by monitoring the quenching of fluorescence of an incorporated pyrene-labeled lipid derivative. All three drugs bound avidly to the two oxidized PCs bearing either an aldehyde or carboxylic function at the end of the sn-2 nonanoyl chain, with the highest affinity measured between CPZ and the latter oxidized lipid. Subsequent dissociation of the above drugs from the oxidized lipids by DNA, acidic phospholipids, and NaCl revealed the binding of these drugs with the aldehyde lipid to be driven by hydrophobicity similarly to their binding to lysophosphatidylcholine, whereas a significant contribution of electrostatics was evident for the lipid with the carboxylic moiety. These results connect to previous experimental data, demonstrating the induction by these drugs of oxidative stress and binding to membrane phospholipids. These issues are elaborated with reference to their clinical use and side effects.
INTRODUCTION
The mechanism(s) of drug action are conventionally rationalized in terms of their binding as agonists or antagonists to specific proteins. However, for compounds acting on the membrane level other modes of action are also possible, owing to the principles of biomembrane operations as many body systems composed of lipids and associated proteins (1). In brief, there is a wealth of evidence for the control of the function of both integral and peripheral membrane proteins by direct interactions with specific phospholipids. Accordingly, membrane-partitioning compounds may directly interfere with lipid-protein interactions (2). Membranes also exhibit a rich scale of cooperative phenomena, responsible for the dynamic microheterogeneity connected to biological functions (3). Together with physical factors such as temperature, the organization of these complex entities is amenable to perturbation by membrane-associating molecules (4). Along these lines, we have shown that lipid-binding drugs alter the lateral organization of membranes and may thus influence their functions, without direct interactions with membrane proteins (5,6).
The number of different lipid species in eukaryotic membranes has been estimated at ∼1200 (4). However, even this figure is an underestimate, as it excludes lipids generated by peroxidation, forming upon unsaturated acyl chains reacting with the so-called ROS (7). The latter, e.g., superoxide radical anion (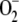 ), hydroxyl radical (·OH), and hydrogen peroxide (H2O2), are produced in oxidative processes of cellular metabolism and accumulate in tissues upon aging as well as under pathophysiological conditions such as inflammation, leading to a state known as oxidative stress (8). These compounds are highly reactive toward cellular macromolecules, unsaturated lipids in particular, and their overproduction has been linked with several major diseases such as cancer, diabetes, schizophrenia, and neurodegenerative disorders, most notably both Parkinson's and Alzheimer's disease (9–12). The changes in the chemical composition of the lipid bilayer caused by phospholipid peroxidation include shortening of the acyl chains and a decrease in the degree of their unsaturation, together with the modification of the acyl chains by polar carbonyl, carboxyl, hydroxide, and peroxide moieties. The above can be readily expected to result in alterations in several key physical properties of the bilayer, including lipid-lipid interactions, the polarity profile, chain order, lateral pressure profile, as well as thermal phase behavior, lateral organization, and membrane asymmetry. These modifications of the membrane lipid matrix can be anticipated to modulate the functions and lipid interactions of the various membrane-associated proteins in addition to possible ROS-induced direct chemical modification of the proteins themselves (4,13).
), hydroxyl radical (·OH), and hydrogen peroxide (H2O2), are produced in oxidative processes of cellular metabolism and accumulate in tissues upon aging as well as under pathophysiological conditions such as inflammation, leading to a state known as oxidative stress (8). These compounds are highly reactive toward cellular macromolecules, unsaturated lipids in particular, and their overproduction has been linked with several major diseases such as cancer, diabetes, schizophrenia, and neurodegenerative disorders, most notably both Parkinson's and Alzheimer's disease (9–12). The changes in the chemical composition of the lipid bilayer caused by phospholipid peroxidation include shortening of the acyl chains and a decrease in the degree of their unsaturation, together with the modification of the acyl chains by polar carbonyl, carboxyl, hydroxide, and peroxide moieties. The above can be readily expected to result in alterations in several key physical properties of the bilayer, including lipid-lipid interactions, the polarity profile, chain order, lateral pressure profile, as well as thermal phase behavior, lateral organization, and membrane asymmetry. These modifications of the membrane lipid matrix can be anticipated to modulate the functions and lipid interactions of the various membrane-associated proteins in addition to possible ROS-induced direct chemical modification of the proteins themselves (4,13).
We report here on the interactions of three currently used drugs: the antipsychotics HPD and CPZ, and the antineoplastic compound DOX with two biologically pertinent oxidatively modified phospholipids. More specifically, we studied the binding of the above compounds to PazePC and PoxnoPC (Fig. 1), monitored by a semiquantitative fluorescence quenching assay utilizing a pyrene-labeled phospholipid derivative. Of the above lipids, PazePC is present in LDL (14), whereas PoxnoPC has been recognized as the main product of ozone-mediated oxidation of lung surfactant extract (15). Binding of these drugs to lysoPC was employed as a control. Notably, for the above drugs the mechanisms of action responsible for their clinical and side effect profiles have long been subjects of considerable controversy, yet membrane interactions and oxidative stress-linked lipid peroxidation have been suggested to be involved (16,17). Here we show these three drugs to bind avidly to oxPLs, with both hydrophobicity and electrostatics (for PazePC) accounting for the interaction. To our knowledge, this constitutes the first demonstration of a direct interaction of clinically used drugs with oxPLs.
FIGURE 1.

The chemical structures of the two oxidatively modified phospholipids used in this study, viz. PazePC and PoxnoPC.
MATERIALS AND METHODS
Materials
PazePC, PoxnoPC, POPC, POPS, and lysoPC were obtained from Avanti Polar Lipids (Alabaster, AL) and PPDPC from K&V Bioware (Espoo, Finland). HPD, CPZ, DOX, NaCl, Hepes, EDTA, calf thymus DNA, and polyphosphate were from Sigma (St. Louis, MO) and CaCl2 and DMSO from Merck (Darmstadt, Germany). The purity of the lipids was checked by thin layer chromatography on silicic acid-coated plates (Merck), using chloroform/methanol/water/ammonia (65:20:2:2, v/v) as the eluent. Examination of the plates after iodine staining or, when appropriate, for fluorescence upon UV illumination revealed no impurities. Concentrations of the nonfluorescent phospholipids were determined gravimetrically with a high-precision electrobalance (Cahn Instruments, Cerritos, CA), and the concentration of pyrene containing PPDPC was obtained spectrophotometrically using the molar extinction coefficient ɛ = 42,000 at 344 nm.
Lipid dispersions
Lipid stock solutions were prepared in chloroform and mixed in this solvent to obtain the desired compositions. Unless otherwise indicated PPDPC (XPPDPC = 0.01) was included as the fluorescent lipid probe. The solvent was removed under a stream of nitrogen and the lipid residue subsequently maintained under reduced pressure for at least 2 h. The dry lipid films were then hydrated at 60°C for 30 min in 20 mM Hepes, 0.1 mM EDTA, pH 7.4 to yield a final concentration of 200 μM. To assure efficient dispersion of the lipids the solutions were further placed in a bath-type sonicator for 20 min. During the above procedure the samples were vortexed several times, and minimal exposure to light was ensured. Subsequently, the dispersions of oxPLs or lysoPC were divided into proper aliquots and diluted with the buffer to a final lipid concentration of 25 μM for the subsequent use in fluorescence experiments. This concentration was used as our Langmuir-balance experiments (see below) revealed PoxnoPC and PazePC to have CMC of 21.6 and 18.7 μM, respectively, at pH 7.4 (Table 1). For lysoPC a CMC of 13.8 μM was measured. Large unilamellar POPC/POPS vesicles were prepared essentially as described above, except omitting sonication and instead subjecting the hydrated lipid solution to 19 passes through a polycarbonate filter (100 nm pore size, Nucleopore, Pleasanton, CA) using a LiposoFast small volume homogenizer (Avestin, Ottawa, Canada).
TABLE 1.
Surface chemical characteristics of the two oxidatively modified phospholipids
| CMC, μM | Ksw × 10−3, M−1 | AS, Å2 (apparent) | |
|---|---|---|---|
| PazePC | 18.7 | 202 | 16.0 |
| PoxnoPC | 21.6 | 164 | 14.0 |
Steady-state fluorescence measurements
The binding of HPD, CPZ, and DOX to oxPLs was assessed by monitoring the efficiency of quenching of the fluorescence emission of pyrene containing phospholipid analog PPDPC by these molecules (2,18,19). The measurements were conducted with a Perkin-Elmer (Foster City, CA) LS50B spectrofluorometer using 5.0 nm band-passes for both the excitation and emission beams of 344 and 394 nm, respectively. Two milliliters of 25 μM lipid dispersion were placed into a magnetically stirred four-window quartz cuvette in a holder thermostated at 25°C with a circulating water bath. Subsequently, 2.5 μl aliquots of 800 μM solution (CPZ and DOX in water and HPD in DMSO) of one of the drugs were added and the quenching of pyrene fluorescence was observed. Thereafter, proper aliquots of 5 M NaCl, 2 mM DNA, or 1 mM POPC/POPS (XPOPS = 0.20) LUVs were added to yield the indicated final concentrations. Changes in the pyrene fluorescence between additions were allowed to stabilize for ∼5–20 min, whereafter the intensity value was recorded and subsequently corrected for sample dilution. The merits as well as limitations of the use of pyrene-labeled lipids in energy transfer measurements have been discussed elsewhere (20).
Dynamic light scattering measurements
Zetasizer Nano ZS (Malvern Instruments, Malvern, UK) was used to measure DLS from lipid dispersions in 10 × 10 mm disposable cuvettes. Data were analyzed with dedicated software provided by the instrument manufacturer. Temperature of the sample compartment was controlled by a Peltier element and set to 25°C.
CMC determination
CMCs, AS, and apparent air-water partitioning coefficients for PazePC, PoxnoPC, and lysoPC were determined using serial dilutions of the above lipids in 20 mM Hepes, 0.1 mM EDTA, pH 7.4 up to 48, 45, or 40 μM lipid (lysoPC, PazePC, and PoxnoPC, respectively) made into 96-well cuvettes. The surface tensions γ of these solutions were measured by the du Noüy maximum pull technique using a computer-controlled 8-channel analyzer (Delta-8, Kibron, Helsinki, Finland). Data were retrieved and analyzed using dedicated software (Delta-8 Manager, Kibron). In brief, Gibbs adsorption isotherm defined by Eq. 1 was used to relate the surface excess Γ to the chemical potential μ of the molecule under study.
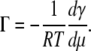 |
(1) |
In dilute solutions dμ ≈ d ln c, and thus
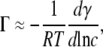 |
(2) |
where c indicates concentration, T temperature, and R = 8.314 J K−1 mol−1. Molecular interfacial cross sectional area AS can be subsequently obtained from
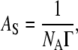 |
(3) |
where NA is Avogadro's number. The value for CMC is determined by the intersection of lines fitted to the slope of the decreasing part of surface tension versus bulk concentration graph and the following plateau (Fig. 2), whereas the reciprocal of the 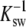 is given by extrapolating the slope to a surface tension of 72.8 mN/m recorded for the aqueous buffer.
is given by extrapolating the slope to a surface tension of 72.8 mN/m recorded for the aqueous buffer.
FIGURE 2.

Surface tension γ versus concentration of (A) PoxnoPC and (B) PazePC in 20 mM Hepes, 0.1 mM EDTA, pH 7.4. Values in Table 1 were obtained by fitting the Gibbs adsorption isotherm to the measured data. Experiments were carried out at ambient temperature of ∼23°C. The data points represent the mean of three or four measurements with the respective standard deviations.
Determination of drug-membrane molar partition coefficients
The values for Kp were determined from the fluorescence-quenching data by the method described by Parry et al. (21). In brief, increasing [drug] was titrated into a dispersion of PazePC, PoxnoPC or lysoPC including the fluorescent phospholipid analog PPDPC whereafter fluorescence intensity values corresponding to each drug/oxPL ratio were recorded (series 1). Subsequently, aliquots of unlabeled oxPL or lysoPC dispersion were added and again the intensity values were collected (series 2). Using the cubic interpolation function of Matlab (The MathWorks, Natick, MA) values for [drug] that would produce identical values of intensity in series 2 to those measured in series 1 were obtained, assuming each measured intensity value (i.e., quenching efficiency) to correspond to a single mole fraction X of the drug in the lipid phase, defined by
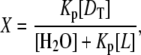 |
(4) |
where [DT] and [L] stand for the total drug and lipid concentrations, respectively. Next, the numerical value of Kp was retrieved by minimizing the difference between the values of X produced by series 1 and 2 by varying Kp.
RESULTS
Quenching due to HPD, CPZ, and DOX of the fluorescence from the pyrene-labeled phospholipid derivative PPDPC incorporated in PazePC and PoxnoPC dispersions was observed, revealing the binding of these drugs to both oxPL species (Figs. 3 A and 4 A). Upon deprotonation of the carboxylic function of PazePC this lipid bears a net negative charge, thus allowing for electrostatic interactions with cationic drugs. In keeping with the above, all three drugs were found to bind PazePC with a higher apparent affinity compared to the zwitterionic PoxnoPC. An increase in ionic strength led to their partial dissociation from PazePC, as judged from the RFQ due to added NaCl (Fig. 3 B). However, the fact that considerable amounts of all three drugs remain associated with PazePC at [NaCl] far exceeding the physiological 150 mM suggests the involvement of hydrophobicity. This is substantiated by the data on these drugs and PoxnoPC, from which essentially no dissociation of the three compounds was observed even at ∼0.8 M NaCl (Fig. 4 B). Furthermore, lysoPC, which due to the absent sn-2 acyl chain bears a structural resemblance to the two oxPLs, displayed behavior essentially identical to that of PoxnoPC with respect to both drug binding and lack of their subsequent dissociation with increasing [NaCl] (Figs. 5, A and B). The formation of submicellar aggregates of bile salts and fluorescently labeled lysophosphatidylethanolamines have been shown to be driven by hydrophobic interactions (22). Considering that these bile salts (cholate and taurodeoxycholate) share the amphipathic nature of the drugs used in this study, similar molecular interactions can be expected to account for the binding of the drugs to lysoPC.
FIGURE 3.

(A) Binding of DOX (▪), CPZ (•), and HPD (▴) to PazePC as revealed by the decrease in the RFI of PPDPC as a function of [drug]. Subsequent detachment of the bound drugs by added (B) NaCl, (C) POPC/POPS = 8:2 LUVs, or (D) DNA, evident as RFQ compared to the initial value before addition of the indicated solute. Total lipid concentration in 20 mM Hepes, 0.1 mM EDTA, pH 7.4 was 25 μM. Temperature was maintained at 25°C.
FIGURE 4.

(A) Binding of DOX (▪), CPZ (•), and HPD (▴) to PoxnoPC as revealed by the decrease in the RFI of PPDPC as a function of [drug]. Subsequent detachment of the bound drugs by added (B) NaCl, (C) POPC/POPS = 8:2 LUVs, or (D) DNA, evident as RFQ compared to the initial value before addition of the indicated solute. The experimental conditions were as described in the legend for Fig. 3.
FIGURE 5.

(A) Binding of DOX (▪), CPZ (•), and HPD (▴) to lysoPC as revealed by the decrease in the RFI of PPDPC as a function of [drug]. (B) Subsequent detachment of the bound drugs by added NaCl evident as RFQ compared to the initial value before addition of the salt. The experimental conditions were as described in the legend for Fig. 3.
All three drugs have been previously demonstrated to bind with high affinity to acidic phospholipids (e.g., 6,18,19). It was therefore of interest to see if they can be dissociated from the oxPLs by PC/PS mixed vesicles. First, drugs were titrated into oxPL/PPDPC dispersions. Subsequently, unlabeled phospholipid vesicles were added and the reversal of the quenching of PPDPC residing in the oxPL assemblies was recorded. Addition of POPC/POPS (XPOPS = 0.20) LUVs into 25 μM PazePC solution led to a slight dissociation of all three drugs from the latter lipid as evidenced by an increase in fluorescence (Fig. 3 C), most likely reflecting a competition between the carboxylic function of this oxPL and the anionic PS headgroup as binding sites for these drugs. Unexpectedly, for PoxnoPC and HPD and CPZ, the addition of POPC/POPS LUVs caused a further decrease in PPDPC fluorescence (Fig. 4 C). This could result from Coulombic attraction between the negatively charged PC/PS LUVs and PoxnoPC aggregates, the latter bearing a positive net charge because of the bound cationic drugs. This in turn may induce lipid mixing between the aggregates and liposomes, including transfer of the fluorescent pyrene-labeled lipid analog from PoxnoPC aggregates to LUVs, and because of higher affinities of the drugs to PC/PS LUVs, leading to augmented quenching of PPDPC in these vesicles.
The higher affinities of the drugs to PC/PS vesicles and hence the more efficient quenching are likely to result from the lack of electrostatic interaction between the drugs and PoxnoPC. Changes in particle size upon putative lipid mixing were assessed by DLS, whereupon a decrease was seen in the average aggregate diameter from 89.2 nm (corresponding to 100 μM solution of neat POPC/POPS LUVs) to 78.1 nm upon the addition of PoxnoPC preincubated with HPD (with final concentrations of 25 and 7 μM, respectively). A similar decrease was evident for CPZ (data not shown). Importantly, no additional peaks in the size distribution appeared upon adding PoxnoPC-drug complexes, thus suggesting that the diminished (apparent) aggregate size results from the complexes interacting with the LUVs. The reason for a decrease instead of an increase in the aggregate size could be due to morphological changes induced in the liposomes upon fusion with the PoxnoPC-drug aggregates, thus resulting in a nonspherical shape (see Discussion).
Quantitative analysis of the affinities of the drugs to the two oxPLs and lysoPC was carried out by determining their respective molar lipid/water partition coefficients (Table 2). Interestingly, the coefficient for partitioning of CPZ to PazePC was too high to be reliably determined with this technique, and accordingly, only a rough estimate of >1 × 108 could be obtained, indicating at least one order of magnitude higher affinity than measured between this drug and POPS, 2.2 × 107 (21). However, a stringent comparison of these two Kp values has to be made with caution since they were determined for two structurally different membrane systems. Yet, these data together with the estimated three orders of magnitude higher Kp of CPZ to PazePC compared to lysoPC do reveal that the affinity of CPZ to this lipid is indeed very high. For PoxnoPC the Kp for HPD could not be determined since the addition of unlabeled PoxnoPC aggregates into a solution of PPDPC-labeled lipid with bound HPD increased the fluorescence intensity above the level recorded in the absence of the drug. The reason behind this behavior may lie in the lyotropic effect induced by an increase in PoxnoPC concentration together with the presence of HPD, whereupon the environment of PPDPC pyrenes residing in the aggregates is altered to increase their quantum yield.
TABLE 2.
The Kp between the aqueous phase and the lipid phase composed by PazePC, PoxnoPC, or lysoPC for the three drugs used in this study
| Drug | PazePC | PoxnoPC | lysoPC |
|---|---|---|---|
| CPZ | >1 × 108 | 1.70 × 106 | 7.45 × 105 |
| DOX | 1.18 × 106 | 1.92 × 106 | 5.95 × 105 |
| HPD | 4.29 × 105 | – | 3.50 × 105 |
The values were obtained after the drug-induced quenching of pyrene-labeled phospholipid analog (see Materials and Methods).
For lysoPC the obtained partition coefficients for CPZ and DOX were 2–3 times lower compared to PoxnoPC, and only a minor difference in the Kp of HPD between lysoPC and PazePC was evident. To explore if PPDPC had an influence on the formation of aggregates of oxPLs or their ability to bind these drug molecules, the association of CPZ with POPC/POPS/PPDPC (XPOPS = 0.20, XPPDPC = 0.01) LUVs in the presence of neat PazePC was studied. The association of CPZ to the LUVs diminished progressively with increasing [PazePC] (Fig. 6), thus revealing that the binding of this drug to PazePC is not due to the presence of PPDPC in the oxPL aggregates.
FIGURE 6.

The effect of PazePC on the binding of CPZ to LUVs composed of 79:20:1 POPC/POPS/PPDPC (total lipid concentration 25 μM in 20 mM Hepes, 0.1 mM EDTA, pH 7.4). Quenching of RFI of PPDPC is illustrated in the absence (▪) and in the presence of 25 (•), 50 (▴), and 100 (▾) μM PazePC. Temperature was maintained at 25°C.
Several of the putative mechanisms of action ascribed to the antitumor effects of DOX involve interactions with DNA (17). It was therefore of interest to observe, in accordance with previous studies revealing a high affinity of DOX to polynucleotides (19,23), that while the binding of DOX to both oxPLs persisted in physiological saline, this drug was readily scavenged from oxPLs by DNA, in contrast to HPD and CPZ (Figs. 3 D and 4 D). Notably, if DNA was replaced by polyphosphate (average chain length of 62 residues) essentially no dissociation of DOX from PazePC was observed even when the sample phosphate concentration was 10-fold higher than the highest concentration present in similar experiments using DNA (calculated from its backbone phosphates; data not shown).
DISCUSSION
Recent studies by our laboratory demonstrated that the two oxPLs (PazePC and PoxnoPC, Fig. 1) used in this investigation profoundly modify the organization and structural dynamics of a host phospholipid matrix (13). Based on monolayer compression isotherms and surface potential data, it was concluded that the polar functional groups at the ends of the oxidized sn-2 acyl chains of these lipids reside in the interfacial region. This location is a consequence of the oxidized lipids adapting the so-called “extended” lipid conformation (24,25), in which the sn-2 acyl chains reverse their orientation to loop back from the membrane hydrocarbon region, accommodating in the interface and contacting the aqueous phase. At high lateral packing densities oxPLs became solubilized from the monolayers, most likely dissolving into the subphase as micellar aggregates (13). Accordingly, it was of interest to investigate if the above features of oxPLs would facilitate membrane association of ligands, such as drugs, by virtue of direct interaction with the oxidatively modified phospholipids. At this stage we chose for this purpose three currently widely used compounds, HPD, CPZ, and DOX. Although the characterization of the molecular aggregates formed by the two oxPLs above their respective CMCs in aqueous solutions is currently underway in our laboratory, at present their exact structural characteristics remain unresolved.
Based on fixed angle DLS measurements comicelles of lysoPC and oxPLs retain a size comparable to neat lysoPC micelles, with the bulk of the sample volume consisting of aggregates with a diameter of ∼6 nm up to XoxPL of 0.5 (Fig. 7). However, further increase in the content of the two oxidized PCs causes the diameter measured by DLS to drop below 1 nm, a value too small to be realistic for a spherical (micellar) aggregates. Keeping the above in mind, it is plausible that the oxPLs in aqueous solutions form nonspherical aggregates and may induce a similar morphology in other membrane systems if present above a certain critical mole fraction. Techniques such as small-angle x-ray scattering are needed to elucidate this issue. Further, surface tension versus concentration data for PazePC and PoxnoPC, while readily yielding their respective CMCs, could not be properly fitted with the Gibbs adsorption isotherm, yielding unrealistically low AS values for these molecules bearing a phosphocholine headgroup (Fig. 2, Table 1). Importantly, however, these complications do not undermine the conclusions of this work, since effects arising from aggregate size and shape (e.g., variation in curvature) should influence the drug binding only in a quantitative manner, not affecting the observed interactions between the drugs and the two oxPLs.
FIGURE 7.

Size distributions by volume of aggregates of lysoPC and indicated binary mixtures with oxPLs measured by DLS. LysoPC (▪), lysoPC/PazePC = 8:2 (•), lysoPC/PoxnoPC = 8:2 (▴), lysoPC/PazePC = 1:1 (▾), and lysoPC/PoxnoPC = 1:1 (♦). The total lipid concentration in 20 mM Hepes, 0.1 mM EDTA, pH 7.4 was 200 μM in all measurements. Temperature was maintained at 25°C.
Two of the drugs shown here to interact with oxidatively modified PCs are used in the treatment of schizophrenia. The pathogenesis of this disease has remained elusive, yet changes in membrane lipids (26) and elevated activity of phospholipase A2 (27) have been implicated. At this stage it would certainly be premature to assign even a putative role to the interaction between the oxPLs and the two antipsychotic drugs in their mechanism(s) of action. Yet, these interactions could be relevant and could also be involved in their known side effects. More specifically, increased oxidative stress has been associated with the pathogenesis of tardive dyskinesia and other extrapyramidal side effects of the antipsychotic treatment of schizophrenia (28,29). Intriguingly, chronic exposure to HPD and CPZ has been shown to decrease the expression and activity of key antioxidant enzymes and to elevate the level of lipid peroxides in rat brain (30), the changes induced by HPD being more pronounced. In light of the results here, it could be speculated that this reflects the lower affinity of HPD to oxPLs (at physiological saline). Accordingly, avid binding of CPZ to phospholipid oxidation products may hamper the propagation of free radical chain reactions and the spreading of oxidative damage due to ROS, thus possibly explaining at least in part the differences in the antioxidant status and level of lipid peroxides after the administration of HPD or CPZ.
The cytotoxicity of DOX is likely to involve several mechanisms in addition to those involving its intercalation into DNA. Generation of ROS and subsequent lipid peroxidation are known to be involved in the triggering of apoptosis (31). DOX has been demonstrated to induce apoptosis (32). Intriguingly, DOX bound to polymer beads and only contacting the outer surface of the plasma membrane of cultured cells has also been shown to be cytotoxic (33). Increased levels of membrane lipid unsaturation due to dietary supplementation of polyunsaturated fatty acids have been shown to attenuate the growth of human breast cancer cells ((34) and references therein) and to increase their susceptibility to the cytotoxic effects of DOX (35), including DNA intercalation, inhibition of topoisomerase II, and augmented ROS production (17). To this end, DOX may act as an electron acceptor in reactions catalyzed by intracellular or cell surface-bound oxoreductive enzymes (36), thus resulting in the formation of a semiquinone free radical, which can subsequently autooxidize back to DOX with a concomitant superoxide radical generation. Similar redox cycling should be possible for lipid-bound DOX, thus allowing complexation with oxPLs to build up high drug concentrations after initial membrane oxidative damage and a local oxidative attack, especially in the heart with abundance of mitochondria and low antioxidant levels (37).
The anticancer activity of DOX may thus be directly related to its side effects. Development of acute congestive heart failure with cumulative DOX doses limits its clinical utility (38). DOX-induced cardiomyopathy has been suggested to be caused by the high affinity of this drug to cardiolipin, free radical generation, and increased oxidative stress (36,39). To this end, it is of interest that CPZ has also been demonstrated to be cytotoxic to cancer cells (40). It is plausible that increased ROS production and membrane oxidative damage by DOX act in parallel to its effects inside the nucleus, perhaps also by providing means for crossing the lipid bilayer barriers. This is in keeping with studies revealing the tendency of oxPLs to dissolve from phospholipid monolayers at slightly elevated lipid packing densities, with subsequent aggregation (13). Accordingly, complexation with oxPLs could therefore aid the diffusion of DOX into the nucleus through cellular membranes.
Although adding to the complexity of the molecular pharmacology of processes involving the modification of lipids by ROS, these results certainly warrant further investigation of the interactions of drugs with oxidized lipids. For comparison, lysoPC also retained the three compounds studied driven by hydrophobicity. However, except for the intestinal lumen, the concentrations of this lipid in cells and tissues are generally very low. In contrast, although quantitative studies are unfortunately still lacking, one may readily expect during oxidative stress the accumulation of significant amounts of PoxnoPC and PazePC, as these are the oxidation products formed from the most abundant membrane lipid constituents, PCs bearing unsaturated acyl chains (viz. linoleyl, linolenyl) in the sn-2 position of the glycerol backbone. As the oxPLs have very different physicochemical properties from those of their nonoxidized parent molecules, including their partitioning into membranes (13), these lipids and their complexes with drugs can be readily expected to have a significant impact on the distribution of drugs in the various cellular and tissue compartments, for instance. Along these lines, drugs may further either enhance or retard the solubilization of oxPLs from biomembranes. These issues are now being explored in our laboratory.
Acknowledgments
The authors wish to thank Dr. Juha-Matti Alakoskela for rewarding discussions and his help with determination of the drug partition coefficients. The expert technical assistance of Kaija Niva and Kristiina Söderholm is appreciated.
The Helsinki Biophysics and Biomembrane Group is supported by the Finnish Academy and Sigrid Jusélius Foundation.
HPD, haloperidol; AS, apparent molecular cross sectional area; CMC, critical micelle concentration; DLS, dynamic light scattering; DMSO, dimethyl sulfoxide; DOX, doxorubicin; CPZ, chlorpromazine; Kp, molar partition coefficient; Ksw, apparent surface-water partition coefficient; LDL, low density lipoprotein; LUV, large unilamellar vesicle; lysoPC, 1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine; oxPL, oxidized phospholipid; PazePC, 1-palmitoyl-2-azelaoyl-sn-glycero-3-phosphocholine; PC, phosphatidylcholine; POPC, 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine; POPS, 1-palmitoyl-2-oleyl-sn-glycero-3-phospho-L-serine; PoxnoPC, 1-palmitoyl-2-(9′-oxo-nonanoyl)-sn-glycero-3-phosphocholine; PPDPC, 1-palmitoyl-2-[10-(pyren-1-yl)decanoyl]-sn-glycero-3-phosphocholine; PS, phosphatidylserine; RFI, relative fluorescence intensity; RFQ, reversal of fluorescence quenching; ROS, reactive oxygen species; UV, ultraviolet; XY, mole fraction of compound Y
Editor: Petra Schwille.
References
- 1.Mouritsen, O. G. 2005. Life as a Matter of Fat: The Emerging Science of Lipidomics. Springer-Verlag, Berlin, Heidelberg.
- 2.Jutila, A., M. Rytömaa, and P. K. J. Kinnunen. 1998. Detachment of cytochrome c by cationic drugs from membranes containing acidic phospholipids: comparison of lidocaine, propranolol, and gentamycin. Mol. Pharmacol. 54:722–732. [PubMed] [Google Scholar]
- 3.Mouritsen, O. G., and P. K. J. Kinnunen. 1996. Role of lipid organization and dynamics for membrane functionality. In Biological Membranes: Role of Lipid Organization and Dynamics for Membrane Functionality. K. M. Merz and B. Roux, editors. Birkhäuser, Boston. 463–502.
- 4.Kinnunen, P. K. J. 1991. On the principles of functional ordering in biological membranes. Chem. Phys. Lipids. 57:375–399. [DOI] [PubMed] [Google Scholar]
- 5.Söderlund, T., J. Y. A. Lehtonen, and P. K. J. Kinnunen. 1999. Interaction of cyclosporin A with phospholipid membranes. Mol. Pharmacol. 55:32–38. [DOI] [PubMed] [Google Scholar]
- 6.Jutila, A., T. Söderlund, A. L. Pakkanen, M. Huttunen, and P. K. J. Kinnunen. 2001. Comparison of the effects of clozapine, chlorpromazine, and haloperidol on membrane lateral heterogenity. Chem. Phys. Lipids. 112:151–163. [DOI] [PubMed] [Google Scholar]
- 7.Marathe, G. K., K. A. Harrison, R. C. Murphy, S. M. Prescott, G. A. Zimmerman, and T. M. McIntyre. 2000. Bioactive phospholipid oxidation products. Free Radic. Biol. Med. 28:1762–1770. [DOI] [PubMed] [Google Scholar]
- 8.Coyle, J. T., and P. Puttfarcken. 1993. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 262:689–695. [DOI] [PubMed] [Google Scholar]
- 9.Lenaz, G. 1998. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta. 1366:53–67. [DOI] [PubMed] [Google Scholar]
- 10.Butterfield, D. A., and C. M. Lauderback. 2002. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 32:1050–1060. [DOI] [PubMed] [Google Scholar]
- 11.Mahadik, S. P., and S. Mukherjee. 1996. Free radical pathology and antioxidant defense in schizophrenia: a review. Schizophr. Res. 19:1–17. [DOI] [PubMed] [Google Scholar]
- 12.Halliwell, B., and J. M. C. Gutteridge. 1990. Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol. 186:1–85. [DOI] [PubMed] [Google Scholar]
- 13.Sabatini, K., J. P. Mattila, F. M. Megli, and P. K. J. Kinnunen. 2006. Characterization of two oxidatively modified phospholipids in mixed monolayers with DPPC. Biophys. J. 90:4488–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davies, S. S., A. V. Pontsler, G. K. Marathe, K. A. Harrison, R. C. Murphy, J. C. Hinshaw, G. D. Prestwich, A. St. Hilaire, S. M. Prescott, G. A. Zimmerman, and T. M. McIntyre. 2001. Oxidized alkyl phospholipids are specific, high affinity peroxisome proliferator-activated receptor γ ligands and agonists. J. Biol. Chem. 276:16015–16023. [DOI] [PubMed] [Google Scholar]
- 15.Uhlson, C., K. Harrison, C. B. Allen, S. Ahmad, C. W. White, and R. C. Murphy. 2002. Oxidized phospholipids derived from ozone-treated lung surfactant extract reduce macrophage and epithelial cell viability. Chem. Res. Toxicol. 15:896–906. [DOI] [PubMed] [Google Scholar]
- 16.Parikh, V., M. M. Khan, and S. P. Mahadik. 2003. Differential effects of antipsychotics on expression of antioxidant enzymes and membrane lipid peroxidation in rat brain. J. Psychiatr. Res. 37:43–51. [DOI] [PubMed] [Google Scholar]
- 17.Gewirtz, D. A. 1999. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 57:727–741. [DOI] [PubMed] [Google Scholar]
- 18.Mustonen, P., and P. K. J. Kinnunen. 1991. Activation of phospholipase A2 in vitro. Role of drug-lipid interactions. J. Biol. Chem. 266:6302–6307. [PubMed] [Google Scholar]
- 19.Mustonen, P., and P. K. J. Kinnunen. 1993. On the reversal by deoxyribonucleic acid of the binding of adriamycin to cardiolipin-containing liposomes. J. Biol. Chem. 268:1074–1080. [PubMed] [Google Scholar]
- 20.Kinnunen, P. K. J., A. Kõiv, and P. Mustonen. 1993. Pyrene-labelled lipids as fluorescent probes in studies on biomembranes and membrane models. In Fluorescence Spectroscopy: New Methods and Applications. O. S. Wolbeis, editor. Springer-Verlag, Berlin, Heidelberg. 159–171.
- 21.Parry, M. J., A. Jutila, P. K. J. Kinnunen, and J.-M. Alakoskela. 2007. A versatile method for determining the molar ligand-membrane partition coefficient. J. Fluoresc. 17:97–103. [DOI] [PubMed] [Google Scholar]
- 22.Shoemaker, D. G., and J. W. Nichols. 1990. Hydrophobic interaction of lysophospholipids and bile salts at submicellar concentrations. Biochemistry. 29:5837–5842. [DOI] [PubMed] [Google Scholar]
- 23.Frederick, C. A., L. D. Williams, G. Ughetto, G. A. van der Marel, J. A. van Boom, A. Rich, and A. H. Wang. 1990. Structural comparison of anticancer drug-DNA complexes: adriamycin and daunomycin. Biochemistry. 29:2538–2549. [PubMed] [Google Scholar]
- 24.Kinnunen, P. K. J. 1992. Fusion of lipid bilayers: a model involving mechanistic connection to HII phase forming lipids. Chem. Phys. Lipids. 63:251–258. [DOI] [PubMed] [Google Scholar]
- 25.Tuominen, E. K. J., C. J. A. Wallace, and P. K. J. Kinnunen. 2002. Phospholipid-cytochrome c interaction: evidence for the extended lipid anchorage. J. Biol. Chem. 277:8822–8826. [DOI] [PubMed] [Google Scholar]
- 26.Fenton, W. S., J. Hibbeln, and M. Knable. 2000. Essential fatty acids, lipid membrane abnormalities, and the diagnosis and treatment of schizophrenia. Biol. Psychiatry. 47:8–21. [DOI] [PubMed] [Google Scholar]
- 27.Gattaz, F., M. Köllisch, T. Thuren, J. A. Virtanen, and P. K. J. Kinnunen. 1987. Increased plasma phospholipase A2 activity in schizophrenic patients: reduction after neuroleptic therapy. Biol. Psychiatry. 22:421–426. [DOI] [PubMed] [Google Scholar]
- 28.Cadet, J. L., and J. B. Lohr. 1989. Possible involvement of free radicals in neuroleptic-induced movement disorders. Evidence from treatment of tardive dyskinesia with vitamin E. Ann. N. Y. Acad. Sci. 570:176–185. [DOI] [PubMed] [Google Scholar]
- 29.Peet, M., J. Laugharne, N. Rangarajan, and G. P. Reynolds. 1993. Tardive dyskinesia, lipid peroxidation, and sustained amelioration with vitamin E treatment. Int. Clin. Psychopharmacol. 8:151–153. [DOI] [PubMed] [Google Scholar]
- 30.Pillai, A., V. Parikh, A. V. Terry Jr., and S. P. Mahadik. 2007. Long-term antipsychotic treatments and crossover studies in rats: differential effects of typical and atypical agents on the expression of antioxidant enzymes and membrane lipid peroxidation in rat brain. J. Psychiatr. Res. 41:372–386. [DOI] [PubMed] [Google Scholar]
- 31.Slater, A. F., C. Stefan, I. Nobel, D. J. van den Dobbelsteen, and S. Orrenius. 1995. Signalling mechanisms and oxidative stress in apoptosis. Toxicol. Lett. 82–83:149–153. [DOI] [PubMed] [Google Scholar]
- 32.Skladanowski, A., and J. Konopa. 1993. Adriamycin and daunomycin induce programmed cell death (apoptosis) in tumour cells. Biochem. Pharmacol. 46:375–382. [DOI] [PubMed] [Google Scholar]
- 33.Tritton, T. R., and G. Yee. 1982. The anticancer agent adriamycin can be actively cytotoxic without entering cells. Science. 217:248–250. [DOI] [PubMed] [Google Scholar]
- 34.Das, U. N. 1999. Essential fatty acids and their metabolites and cancer. Nutrition. 15:239–240. [DOI] [PubMed] [Google Scholar]
- 35.Mahéo, K., S. Vibet, J. P. Steghens, C. Dartigeas, M. Lehman, P. Bougnoux, and J. Goré. 2005. Differential sensitization of cancer cells to doxorubicin by DHA: a role for lipoperoxidation. Free Radic. Biol. Med. 39:742–751. [DOI] [PubMed] [Google Scholar]
- 36.Keizer, H. G., H. M. Pinedo, G. J. Schuurhuis, and H. Joenje. 1990. Doxorubicin (adriamycin): a critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol. Ther. 47:219–231. [DOI] [PubMed] [Google Scholar]
- 37.Doroshow, J. H., G. Y. Locker, and C. E. Myers. 1980. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: alterations produced by doxorubicin. J. Clin. Invest. 65:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Green, M. D., J. L. Speyer, and F. M. Muggia. 1984. Cardiotoxicity of anthracyclines. Eur. J. Cancer Clin. Oncol. 20:293–296. [DOI] [PubMed] [Google Scholar]
- 39.Goormaghtigh, E., P. Huart, M. Praet, R. Brasseur, and J. M. Ruysschaert. 1990. Structure of the adriamycin-cardiolipin complex. Role in mitochondrial toxicity. Biophys. Chem. 35:247–257. [DOI] [PubMed] [Google Scholar]
- 40.Nordenberg, J., E. Fenig, M. Landau, R. Weizman, and A. Weizman. 1999. Effects of psychotropic drugs on cell proliferation and differentiation. Biochem. Pharmacol. 58:1229–1236. [DOI] [PubMed] [Google Scholar]