

Abstract
Interferons (IFNs) regulate the expression of genes that mediate their antiviral, antitumor, and immuno-modulatory actions. We have previously shown that IFN-β suppresses growth of human ovarian carcinoma xenografts in vivo and induces apoptosis of ovarian carcinoma cells in vitro. To investigate mechanisms of IFN-β-induced apoptosis we employed an antisense technical knockout approach to identify gene products that mediate cell death and have isolated several regulators of interferon-induced death (RIDs). In this investigation, we have characterized one of the RIDs, RID-2. Sequence analysis revealed that RID-2 was identical to human inositol hexakisphosphate kinase 2 (IP6K2). IP6K2 is post-transcriptionally induced by IFN-β in ovarian carcinoma cells. A mutant IP6K2 with substitutions in the putative inositol phosphate binding domain abrogates IFN-β-induced apoptosis. These studies identify a novel function for IP6K2 in cell growth regulation and apoptosis.
The interferon (IFN)1 family of cytokines stimulate antiviral, antitumor, antiproliferative, and immunoregulatory activities (1–33). Upon binding to receptors, IFNs activate a signaling cascade wherein Janus tyrosine kinases induce tyrosine phosphorylation of signal-transducing activators of transcription proteins (4, 5). These transcription factors induce expression of hundreds of genes that possess a wide range of activities (6). Although a great deal is known about antiviral actions of IFNs, mechanisms responsible for their antitumor actions are unclear. In vivo, they up-regulate expression of tumor-specific antigens, natural killer, and T cell function (7–9). IFNs also activate growth suppressive proteins such as pRb (10, 11), down-regulate c-Myc (12), and suppress activity of transcription factor E2F (13). Protein kinase R and ribonuclease L, which inhibit viral growth in IFN-treated cells (14, 15), also play a role in growth suppression (16, 17). The family of transcription factors known as IFN gene regulatory factors (IRFs) also mediates the effects of IFNs (18). Two members of this family, IRF-1 and interferon consensus sequence-binding protein (IRF-8) are up-regulated by IFN-γ. Deletion of these genes results in myelodysplasias or chronic myelogenous leukemialike disease (19, 20). Because IRFs are transcription factors, and their biological activity depends on genes they induce; characterization of downstream gene products should allow identification of critical regulators of growth suppression.
We have previously shown that IFN-β suppresses the growth of ovarian tumor xenografts in nude mice (21) and that IFN-β induces apoptosis in these cells. To identify death genes we used an antisense technical knockout approach (22). In this approach, death regulatory genes are identified by their ability, when expressed in antisense orientation, to confer resistance to death inducers. Using this technique we have identified several genes, regulators of interferon-induced death (RIDs), that enhance IFN-β-activated death. In this study we have characterized one of these genes, RID-2, and identified it as human inositol hexakisphosphate kinase 2 (IP6K2) (23). IP6K2 catalyzes the synthesis of diphosphoinositol pentakisphosphate (PP-IP5) using inositol hexaphosphate (IP6) as a substrate in the presence of ATP. Hence, PP-IP5 may cause growth suppression.
We show that cellular IP6K2 levels are post-transcriptionally enhanced by IFN-β. Overexpression of IP6K2 enhances both growth suppressive and apoptotic activities of IFN-β. A dominant negative inositol phosphate binding domain (IPBD) mutant is highly resistant to both the antiproliferative and apoptotic functions of IFN-β. Thus, our studies ascribe a novel function for IP6K2 in cell growth control via apoptosis.
EXPERIMENTAL PROCEDURES
Reagents
Human IFN-β (Serono), specific activity 2.7 ✕ 108 units/mg; IFN-α2b (Schering Plough), specific activity 3 ✕ 108 units/mg; IFN-γ (Roche Molecular Biochemicals), specific activity 2 ✕ 107 units/mg; [³H]inositol and [³H]IP6 (PerkinElmer Life Sciences); anti-PARP antibody (Biomol); anti-caspase 3 antibody (Pharmingen); anti-myc antibody (Oncogene Research Products); and horseradish peroxidase goat antirabbit IgG (Pierce) were used in these studies.
Cell Growth Assays
Cells were treated with IFNs during growth in RPMI 1640 and 5% fetal bovine serum. Growth was monitored using a calorimetric assay (24). Each treatment group contained eight replicates. Cells were fixed and stained with sulforhodamine B after 7 days. Bound dye was eluted from cells, and absorbance (Aexp) was measured at 570 nm. One plate was fixed 8 h after plating to determine the absorbance representing starting cell number (Aini). Absorbance with this plate and that obtained with untreated cells at the end of the growth period (Afin) were taken as 0 and 100% growth, respectively. Thus, percent control growth = 100% ✕ (Aexp − Aini)/(Afin − Aini) expressed as a percent of untreated controls; a decrease in cell number (death) falls on the negative scale. To determine cell cycle distribution, cells were stained with propidium iodide and analyzed by flow cytometry (Beckton Dickinson) using MultiPass software.
Construction of Antisense Expression Library
Total RNA from NIH-OVCAR-3 cells, treated with IFN-β (500 units/ml) for 0, 1, 2, 4, 8, 16, 24, 48, and 72 h, was prepared using RNAZol B (Tel-Test). RNAs were pooled, and total poly(A)+ RNA was isolated (polyAttract; Promega). cDNA libraries were constructed with a commercially available kit (Stratagene). 10 µg of mRNA was used for preparing the cDNA library, using an oligo(dT) primer and a dNTP mixture containing 5-methyl dCTP. After second-strand synthesis, Pfu thermal DNA polymerase was used to create blunt-ended cDNA. cDNAs were ligated to a bifunctional linker, 5′-GCTTGGATCCAAGC-3′. Ligated to the 3′ and 5′ ends of the cDNA, this linker generates HindIII and BamHI sites, respectively (25). The library was digested with HindIII and BamHI, purified on a Sepharose 6B column, and ligated into an episomal vector pTKO1 (from Adi Kimchi, Weizmann Institute, Rehovot, Israel), which carried markers for selection in eukaryotic and bacterial cells (22). When cloned into pTKO1 the cDNA is expressed in antisense orientation. The library was transformed into Escherichia coli DH10B, and plasmid DNA was extracted and purified on CsCl gradients. Electroporation of the library (40 µg) into NIH-OVCAR-3 cells (107) was followed by selection with hygromycin B (200 µg/ml) and IFN-β (2000 units/ml) for 4 weeks. All pTKO1-transfected cells (selected similarly) died after 14 days. After 4 weeks selection-surviving colonies were pooled and expanded, and Hirt DNA extracts were prepared. DNA was digested with DpnI and electroporated into E. coli DH10B. Resultant colonies were screened by PCR using pTKO1-specific primers to detect inserts. Episomes (20 µg) were tested for protection against IFN-β-induced death by electroporation into NIH-OVCAR-3 cells. Selection with IFN-β and hygromycin was initiated after 24 h.
Construction of RID-2 Mutants
RID-2 cDNA was digested from pTKO1 and cloned into the pCXN2myc mammalian expression vector (26) in which the chicken actin promoter regulates expression of the Myc-His-tagged insert. A substitution mutant (SUB) of the putative IPBD was created using PCR-based site-directed mutagenesis (Stratagene) with full-length RID-2 as template, replacing the highly conserved (bold) core consensus IPBD (PCVLDLKMG) with point mutations at 7 of 9 residues (to yield amino acid residues ACTANLAAA) using primers 5′-pAACCTCGCGGCAGCAACACGACAACATGGTGAT-3′ and 5′-pAGCGGTACAAGCCACCTCATAGCGGGAAGT-3′ (p indicates phosphorylated 5′ nucleotide). PCR products were digested with DpnI, ligated, and then transformed into JM109 E. coli, and plasmids were isolated. An antisense mutant (ANTI) was created by ligating RID-2 open reading frame into pCXN2myc in antisense orientation. Mutations were confirmed by sequencing. Constructs were electroporated into NIH-OVCAR-3 cells, and stable transfectants were selected with G418. After 3 weeks of selection, surviving clones were pooled for further studies. Expression of mutants was monitored by Western blotting.
Northern Blot Analysis
Total RNA (20 µg) was separated on 1% formaldehyde-agarose gels, transferred to nylon membrane, and probed with the 32P-labeled PCR product of RID-2 cDNA.
Western Blot Analysis
Total cell protein (20 µg) was separated on 10% SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane. Membranes were incubated with rabbit polyclonal antibody, made in our laboratory, raised against full-length bacterially expressed RID-2. After washing, membranes were incubated with anti-rabbit IgG antibody conjugated to horseradish peroxidase and developed using ECL reagents (Amersham Pharmacia Biotech).
Assays for Apoptosis
Apoptotic cells were detected using a commercially available kit (Pharmingen) and staining with annexin V-FITC and propidium iodide (PI). Cells were analyzed by flow cytometry. DNA fragmentation was detected using the APO-BRDU™ kit (Pharmingen). Cells were labeled with bromo-dUTP using terminal deoxynucleotidyl-transferase, stained with FITC-conjugated anti-bromodeoxyuridine monoclonal antibody followed by RNase-PI. The percentage of FITC-positive cells was determined by flow cytometry.
Inositol Hexakisphosphate Kinase Enzymatic Assay
IP6K activity (27) was determined in whole cell extracts of 107 NIH-OVCAR-3 cells by Dounce homogenization on ice in 100 mm KCl, 20 mm NaCl, 1 mm EGTA, 20 mm HEPES, pH 7.4, and adjusted to 1.5 mg of protein/ml. Using 0.15-ml extracts, kinase assays were run in 0.5 ml of 100 mm KCl, 25 mm HEPES, pH 7.2, 5 mm Na2ATP, 6 mm MgSO4, 10 mm phosphocreatine, 20 U creatine phosphokinase, 0.1 mg of saponin using [³H]IP6 (PerkinElmer Life Sciences) as substrate. Reactions were incubated at 37 °C for 20 min. Reactions were terminated on ice and then quenched with 0.25 ml of 6% v/v perchloric acid and 0.5 mg/ml IP6 (Calbiochem) followed by extraction with 1:1 freon/octylamine and concentration by Speedvac. Products were separated using polyethyleneimine cellulose thin-layer chromatography (PEI-TLC; Merck). The reaction was spotted onto PEI-TLC plates and developed in 1.1 m KH2PO4, 0.8 m K2HPO4, and 2.3 m HCl. Lanes were divided into 1-cm fractions, and the PEI cellulose matrix was scraped from the TLC plates, shaken with 0.5 ml of 16 m HCl, mixed with 0.5 ml of H2O, 3 ml of scintillant, and counted. ~80% of applied ³H was recovered by this method. IP6 and PP-IP5 migrated with an Rf migration ratio of ~0.75 and ~0.45, respectively, and comigrated with standard preparations. [³H]PP-IP5 standard was prepared by incubation of 20 ng of recombinant IP6K2 and [³H]IP6 in a kinase reaction as above (60-min incubation), followed by high pressure liquid chromatography purification on a 4.6 ✕ 125-mm Partisphere strong anion exchange column (Whatman). Gradient elution utilized Buffer A (1 mm Na2EDTA) and Buffer B (Buffer A and 1.3 m (NH4)2HPO4). On PEI-TLC plates, area under the curve (AUC) was used to determine total radioactivity of each species.
RESULTS
IFN-β Is Cytotoxic to Ovarian Carcinoma Cells
We have previously shown that human IFN-β induces regression of NIH-OVCAR-3 human ovarian carcinoma in athymic nude mice (21). To directly demonstrate that IFN-β was cytotoxic in vitro, we treated tumor cell lines with various IFNs. After 1 week, cell growth was measured using a colorimetric assay based on binding of the chromophore sulforhodamine B (24). Increasing doses of IFN-β caused significant growth inhibition in NIH-OVCAR-3 cells in vitro, with complete cytostasis occurring at ~300 units/ml (Fig. 1A) and death occurring at 500–1000 units/ml. IFN-β was significantly more potent than IFN-α at growth inhibition. IFN-α resulted in only 39% inhibition at the highest dose, whereas IFN-β was cytocidal. IFN-γ had negligible effect on cell growth even at 1000 units/ml (Fig. 1A). Hence IFN-β was most effective of the type I IFNs at induction of death.
FIG. 1. IFN-β but not IFN-α or IFN-γ induces ovarian carcinoma cell death.
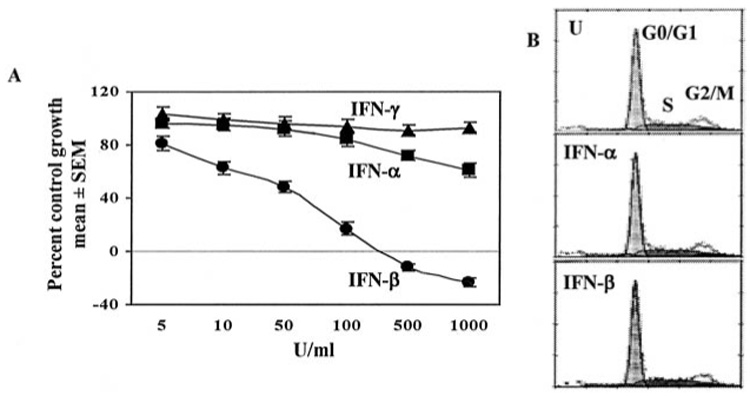
A, NIH-OVCAR-3 cells were grown in the presence of 5–1000 units/ml IFN-β, IFN-α, or IFN-γ. After 7 days cells were fixed and stained with sulforhodamine B. Absorbance (570 nm) of bound dye was measured and expressed as the percent of untreated controls. Each data point represents mean ± S.E. of eight replicates. Values on the negative scale indicate death of initially plated cells. B, cells were left untreated (U) or treated with IFN-β or IFN-α (200 units/ml) for 2 days. Cells were stained with propidium iodide and subjected to flow cytometry and cell cycle analysis (MultiPass).
Microscopic examination of NIH-OVCAR-3 cells treated with IFN-β revealed a dose-dependent cytotoxicity, with condensed nuclei detected by intense DAPI staining (Fig. 2A, arrows). Untreated cells had a diffuse pattern of DAPI staining. To identify apoptotic cells annexin V binding assays were performed. IFN-β was much more efficient than IFN-α at causing phosphatidyl serine translocation in the plasma membrane, but IFN-γ did not cause increased annexin V binding (Table I). IFN-β at low concentrations (200 units/ml) caused 61% apoptosis after 3 days of exposure, whereas high concentrations of IFN-α (1000 units/ml) induced only 16% apoptosis. Maximum apoptosis induced by IFN-γ (2000 units/ml) was 8%, no different from untreated cells. TUNEL assays confirmed that chromosomal fragmentation occurred as early as 48 h after treatment with 200 units/ml IFN-β (Fig. 2B). The degree of chromosomal fragmentation detected by TUNEL assay mirrored the extent of annexin V staining (Table I). IFN-β induced highest levels of TUNEL staining, IFN-α generated intermediate levels, and IFN-γ did not cause an increase over baseline.
FIG. 2. IFN-β induces apoptosis in NIH-OVCAR-3 cells.
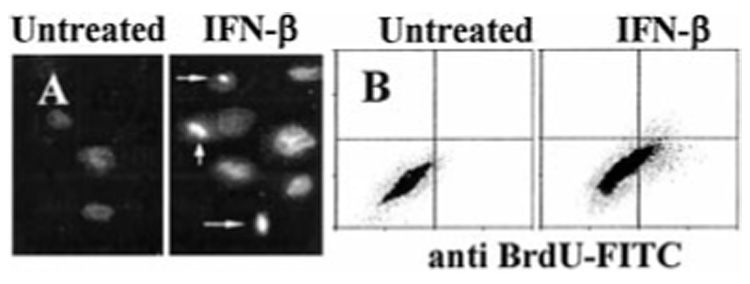
A, cells were treated with IFN-β (200 units/ml) for 2 days. Condensed, DAPI-stained apoptotic nuclei are visible (arrows). B, TUNEL assay. Cells were treated with IFN-β as above, labeled with bromo-dUTP using terminal deoxynucleotidyltransferase, and stained with FITC-conjugated anti-bromodeoxyuridine monoclonal antibody (x axis) and PI (y axis). The percentage of FITC-positive cells (in upper right and lower right quadrants) was determined by flow cytometry.
TABLE I. Effect of IFNs on induction of apoptosis in NIH-OVCAR-3 cells.
Numbers indicate % of positive cells determined by flow cytometry, mean of three separate experiments.
| Treatment | Units/ml | Hours | Annexin V | TUNEL |
|---|---|---|---|---|
| None | 6.8 | 5.4 | ||
| IFN-β | 200 | 48 | 44.6 | 31.6 |
| 200 | 72 | 61.5 | 58.8 | |
| IFN-α | 500 | 48 | 7.1 | 6.5 |
| 1000 | 72 | 16.3 | 12.7 | |
| IFN-γ | 1000 | 48 | 7.3 | 6.1 |
| 2000 | 72 | 8.0 | 7.4 |
To determine whether cell cycle arrest preceded death, cells were treated with IFN-β for 1–3 days, stained with PI, and analyzed by flow cytometry. Interestingly, neither IFN-β nor IFN-α caused cell cycle arrest (Fig. 1B). The proportion of cells in G0/G1, S, and G2/M phases was unchanged after IFN-β or IFN-α treatment (200 units/ml). Thus, death occurred independently of cell cycle arrest. Unlike IFN-α or IFN-γ, only IFN-β caused death in NIH-OVCAR-3 cells at low doses (200 units/ml). Similarly, in WM9 melanoma and KU2 renal carcinoma IFN-β strongly induced apoptosis compared with IFN-α (not shown). IFN-γ (2000 units/ml) did not inhibit growth or induce apoptosis in any cell line tested (NIH-OVCAR-3, MCF-7, WM-9, KU2, MDA-MB-231, HT-29, and A375). To confirm that caspases were activated by IFN-β, Western blotting for caspase 3 and PARP was performed. After 8 h of IFN-β treatment, cleavage of caspase 3 was evident, followed by PARP cleavage at 16 h (Fig. 3, arrows). Hence, at least one effector caspase and its downstream substrate necessary for cell viability were proteolytically cleaved following IFN-β treatment.
FIG. 3. IFN-β activates caspase 3 during apoptosis.
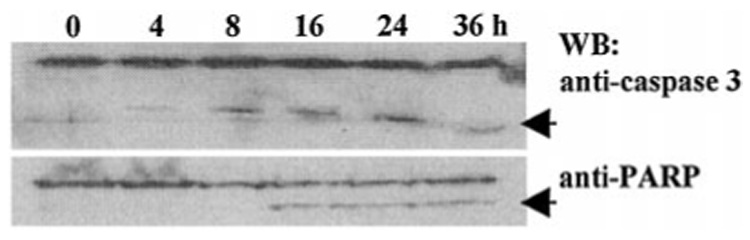
Western blots (WB) of caspase 3 (upper) and PARP (lower). Lysates (40 µg of protein) of NIH-OVCAR-3 cells that were treated with 0–36 h of IFN-β (200 units/ml) are shown. Caspase 3 cleavage products (top arrow) appear earlier than PARP cleavage products (bottom arrow).
Genetic Approach to Isolate Cell Death Genes
Because NIH-OVCAR-3 cells contain non-functional p53 (28, 29), apoptosis appears to occur via p53-independent mechanisms. We hypothesized that hitherto unidentified molecules may transmit the death signal to the core apoptotic machinery resulting in activation of caspases. Therefore we sought to identify gene products using an antisense knockout approach (22). In this technique, a death-regulatory gene can be isolated by antisense inactivation. Cells are transfected with an antisense cDNA library derived from tumor cells. Antisense RNA inhibits expression of the endogenous death gene. Consequently, only those cell clones that express the death-related antisense mRNA will survive in the presence of death inducer. Rescued episomes are retransfected individually into ovarian carcinoma cells to express the antisense mRNA and eliminate false positives isolated in the first round of transfection.
To isolate death-regulatory genes we prepared antisense cDNA libraries cloned in the episomal vector, pTKO1. An IFN-stimulated gene promoter drives expression of antisense RNAs in this vector. This library was electroporated into NIH-OVCAR-3 cells (~50% transfection efficiency) and selected for resistance to hygromycin B and human IFN-β. Cell clones surviving 4 weeks of double selection were pooled, and Hirt extracts were prepared. DNA was digested with DpnI (to inactivate unreplicated input DNA) and electroporated into E. coli DH10B. 18 episomes were rescued in the first round. Each episome was individually transfected into NIH-OVCAR-3 cells and examined for cell protection against IFN-β-induced death. After two rounds of screening, seven episomes consistently conferred resistance to IFN-β-induced death. We named them RIDs. We chose RID-2 for further characterization. Transfection of the RID-2 antisense episome clearly conferred protection against IFN-β-induced death (Fig. 4). No surviving colonies were seen in cells transfected with pTKO1 vector alone.
FIG. 4. Protection of NIH-OVCAR-3 cells by antisense IP6K2 episome.
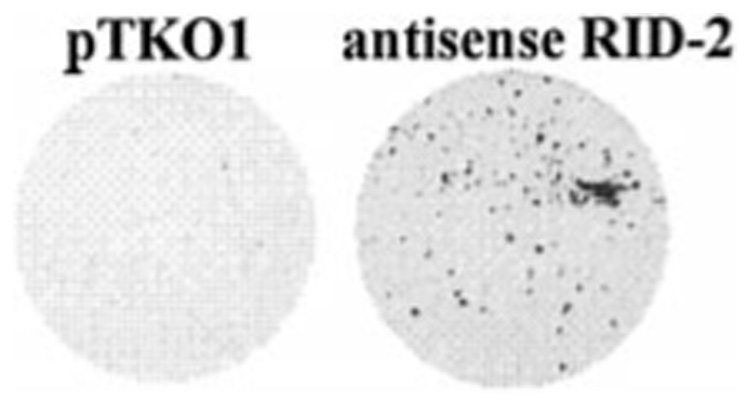
Cells were electroporated with 20 µg of pTKO1 (left) or antisense IP6K2 episomes (right) and selected for 4 weeks with IFN-β and hygromycin B. Surviving cells were fixed and stained with sulfor-hodamine B.
Identification of RID-2 as IP6K2
The RID-2 insert was completely sequenced on both strands. The predicted open reading frame codes for a 49.2-kDa protein. Sequence analysis (Fig. 5) revealed that this cDNA was identical to human IP6K2, GenBank™ accession number AAF15057 (23). A putative (Fig. 5, underlined) is located between amino acids 201 and 232 of this protein. The IPBD motif consensus sequence is (LV)(LA)(DE)X(3,8)PX(VAI)(ML)DXK(ML)G, where X can be any amino acid. The most highly conserved residues are shown in bold. This sequence is required for the catalytic activity of inositol 1,4,5 triphosphate kinase (30). Among the notable features are the presence of a large number of methionine residues and the high concentration of acidic residues in the C terminus. Henceforth, RID-2 will be referred to as IP6K2.
FIG. 5. Identification of RID-2 as human IP6K2.

The amino acid sequence of IP6K2, GenBank™ accession number AAF15057, is shown. Putative IPBD is underlined with most highly conserved residues shown in bold.
Effect of IFN-β on Expression of IP6K2
To study the effect of IFN-β on IP6K2, we first examined whether its mRNA levels were inducible. IFN-β did not induce IP6K2 mRNA in NIH-OVCAR-3 cells even after prolonged exposure (Fig. 6A). Reprobing blots with glyceraldehyde 3-phosphate dehydrogenase cDNA demonstrated the presence of comparable amounts of RNA in each lane. Similarly, IP6K2 mRNA was not induced in CaOv-3 or Hey ovarian carcinoma cells (not shown). To examine whether IFN-β enhanced expression of IP6K2 protein, Western blot analysis was performed on whole cell lysates. IFN-β induced IP6K2 protein in a time-dependent manner. There was no detectable induction until 4 h post-treatment. Between 4 and 24 h, IP6K2 protein levels remained elevated (Fig. 6B). Densitometric quantitation of this blot revealed that maximal elevation of IP6K2 protein levels occurred at 16 h of IFN-β treatment (~5-fold induction) and declined thereafter (Fig. 6C).
FIG. 6. Effects of IFN-β on IP6K2 expression.
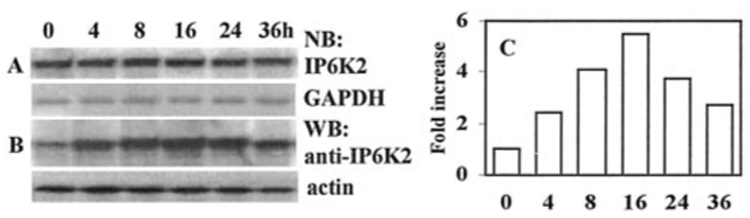
(A), Northern blot (NB) analysis. Total RNA (40 µg) derived from NIH-OVCAR-3 cells after treatment with IFN-β (200 units/ml for 0–36 h) are shown; blots were hybridized with IP6K2 cDNA probe and glyceraldehyde-3-phosphate dehydrogenase as control. B, Western blot (WB) analysis of whole cell lysates (70 µg) prepared after IFN-β treatment as above. Blots were probed with anti-IP6K2 antibody and anti-actin as control. C, densitometric quantitation of IP6K2 protein levels from B. The x axis indicates time of IFN-β exposure (0–36 h), and the y axis indicates band intensity normalized to actin.
Effect of Antisense IP6K2 mRNA on Protein Expression
Because the rescued episome clearly conferred protection from death (Fig. 4), it was important to confirm that this effect was because of expression of antisense IP6K2 mRNA and subsequent down-regulation of IP6K2 protein. Northern blot analysis (Fig. 7) of cells transfected either with vector alone (V) or antisense IP6K2 (ANTI) was performed. In vector cells only one band, representing endogenous (E) IP6K2 mRNA was seen. ANTI cells also express endogenous mRNA and, in addition, express another mRNA species that migrated more slowly (arrow). This second band, absent in vector cells, corresponds to antisense IP6K2 mRNA transcribed from the trans gene. We then determined whether anti-mRNA expression resulted in down-regulation of protein. Western blot analysis showed that ANTI cells contained lower levels of IP6K2 protein compared with vector cells (Fig. 7).
FIG. 7. Expression of antisense IP6K2 mRNA reduces IP6K2 protein levels.
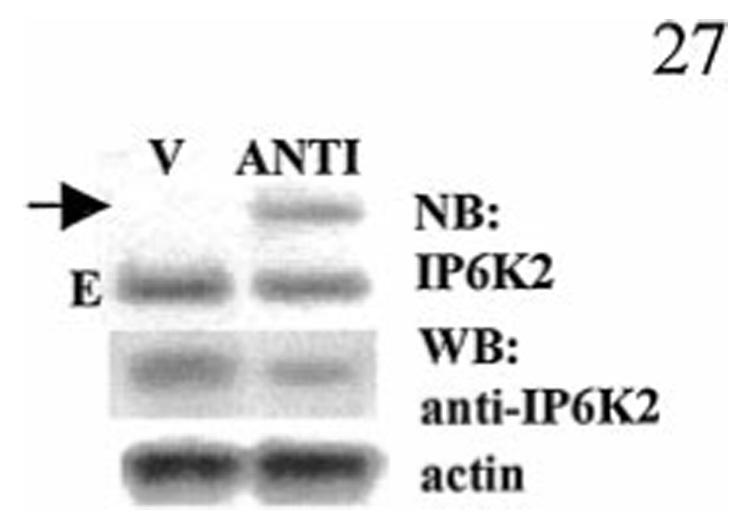
Upper panel, Northern blot (NB) analysis of pools (~100 clones each) of NIH-OVCAR-3 cells stably transfected with vector (V) or antisense IP6K2 (ANTI). The arrow indicates the position of mRNA in transgene-expressing cells. E indicates endogenous mRNA. Lower panel, IP6K2 Western blot (WB) of same cells and actin as control.
IFN-β Induces IP6K Enzymatic Activity
We next determined whether the rise in IP6K2 protein levels following IFN-β correlated with an increase in IP6K enzymatic activity. In vitro kinase assays utilizing [³H]IP6 as substrate were performed to measure IP6K activity in whole cell homogenates of untreated and IFN-β-treated cells, using equal amounts of total cell protein. In the reaction, PP-IP5 was synthesized by IP6K, using ATP and [³H]IP6 as substrates. Indeed, enzymatic activity of IP6K was increased after 4 h of IFN-β exposure, reaching a peak at 8 h and beginning to decrease from maximum by 24 h (Fig. 8). Enzymatic activity was consistent with IP6K2 protein concentration as determined by Western blotting (Fig. 6C). When normalized for immunoreactive IP6K2, enzyme-specific activity was unchanged by IFN-β treatment. Untreated cells displayed an activity of 8 ± 2 pmol/mg protein/min. IFN-α2b or IFN-γ did not enhance IP6K activity at concentrations up to 1000 units/ml (not shown). ANTI cells expressing antisense IP6K2 mRNA assayed at the 8-h time point displayed reduced enzymatic activity compared with untransfected cells, indicated by reduced (~2-fold) levels of the PP-IP5 product.
FIG. 8. Time course of IP6K activity in IFN-β-stimulated NIH-OVCAR-3 cells.
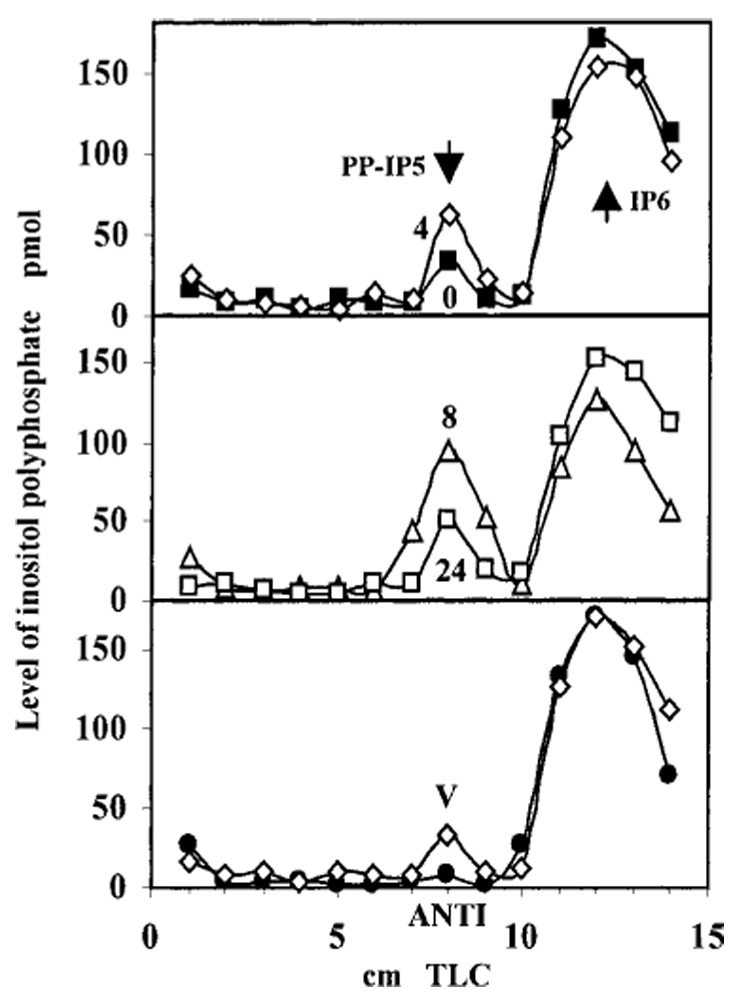
Cells were treated with IFN-β (200 units/ml). Numbers next to curves in top and middle panels (0, 4, 8, and 24) indicate treatment duration (h). IP6K enzymatic activity in whole cell homogenates was determined using [³H]IP6 as substrate in kinase reactions in vitro and detected by PEI-TLC. Total radioactivity contained in the [³H]PP-IP5 product, represented as AUC, for IFN-β-treated cells at various time points was 70 (0 h), 120 (4 h), 209 (8 h), and 101 (24 h). Lower panel, empty vector (V) and antisense IP6K2 (ANTI)-expressing cells. AUC for V cells was 72, and for ANTI cells it was 32. Arrows indicate Rf for PP-IP5 and IP6.
A Mutant IP6K2 Inhibits IFN-β-induced Cell Death
As described above (Fig. 5) IP6K2 possesses a putative IPBD. Therefore, we examined the functional relevance of this domain by generating an SUB. In this mutant, 7 of 9 conserved residues in the IPBD were mutated at positions 216, 218–220, and 222–224. Expression of this mutant open reading frame in the pCXN2myc vector allowed detection of the SUB protein with anti-Myc antibody. This vector was transfected into NIH-OVCAR-3 cells, and stable cell lines were established. Full-length (FL) IP6K2 was cloned into the same vector. During generation of FL stable transfectants, 70% fewer clones were isolated, and they exhibited growth rates of 50% compared with empty vector transfectants (not shown). Expression of the SUB mutant was confirmed by Western blot (Fig. 9). Comparable amounts of each protein were present in both cell lines. No Myc-tagged proteins were detected in cells expressing empty vector.
FIG. 9. Expression of mutant IP6K2 in NIH-OVCAR-3 cells.
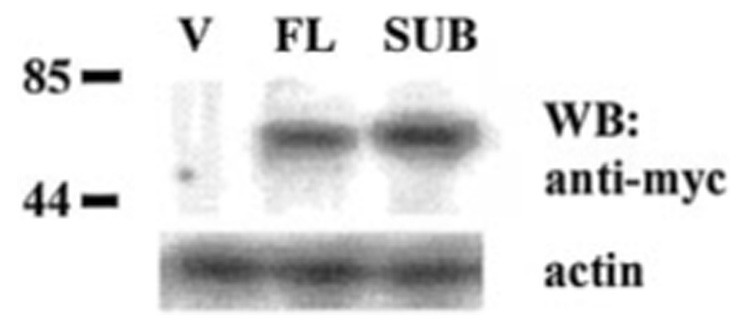
Lysates (80 µg) from cell lines were separated on SDS-polyacrylamide gel electrophoresis and probed with antibody directed against Myc tag (Invitrogen). Labels indicate plasmids transfected into cells. V, pCXN2myc vector; FL, full-length IP6K2. Predicted molecular mass (kDa) of tagged IP6K2 proteins is 51.7 for FL and 51.5 for SUB. Western blots (WB) were stripped and reprobed with actin as control. Tagged proteins were detected in FL and SUB but not in vector (V) cells.
Growth assays were performed in NIH-OVCAR-3 cells that expressed various constructs. In each case, pools of clones (~100 clones) were used, so changes in growth characteristics could not be attributed to clonal effects. As expected, the FL IP6K2-expressing cell line displayed enhanced suppression of growth in response to IFN-β (Fig. 10). The SUB mutant was resistant to antiproliferative effects of IFN-β. SUB cells were inhibited only 45% by 100 units/ml IFN-β compared with empty vector cells, which were inhibited 78%. At doses of IFN-β that killed vector cells (500–1000 units/ml) SUB cells still grew, albeit slowly. As expected, cells that expressed IP6K2 mRNA in antisense orientation (ANTI) were most resistant to IFN-β, displaying only 34% growth inhibition at 1000 units/ml, the highest dose tested (Fig. 10). Despite the near total suppression of IP6K enzymatic activity in ANTI cells (Fig. 8), their growth was still partially suppressed by IFN-β (Fig. 10), suggesting that additional factors may mediate the antiproliferative effects of IFN-β in these cells.
FIG. 10. Effect of IFN-β on growth of NIH-OVCAR-3 cells expressing SUB mutant.
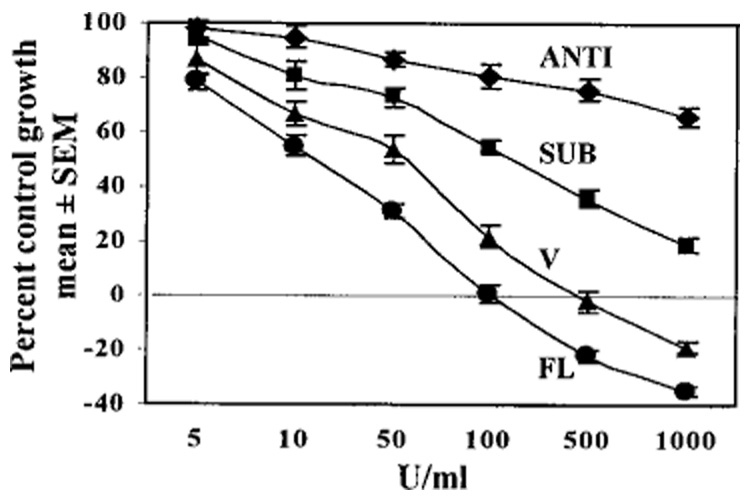
Growth assays were performed as described in legend for Fig. 1. Each point represents mean ± S.E. of eight replicates. Notations are similar to those in Fig. 9. Cells expressing ANTI IP6K2 mRNA were most resistant to IFN-β; cells overexpressing full-length IP6K2 were most sensitive.
Induction of apoptosis by IFN-β was measured using annexin V assays (Fig. 11A). Cells overexpressing FL IP6K2 demonstrated the highest percentage of apoptosis following treatment with IFN-β (200 units/ml). Annexin V staining was ~2-fold higher in FL cells (86.4%) compared with untransfected cells (41.3%) or vector-transfected cells (40.9%). Expression of FL IP6K2 did not induce apoptosis in the absence of IFN-β but rather sensitized cells to death induction by IFN-β. Importantly, SUB cells displayed decreased IFN-β-induced apoptosis (18%) when compared with vector cells, a reduction of 55%. Therefore, an intact IPBD is critical for induction of apoptosis.
FIG. 11. Apoptosis and IP6K enzymatic activity in transfected cell lines.
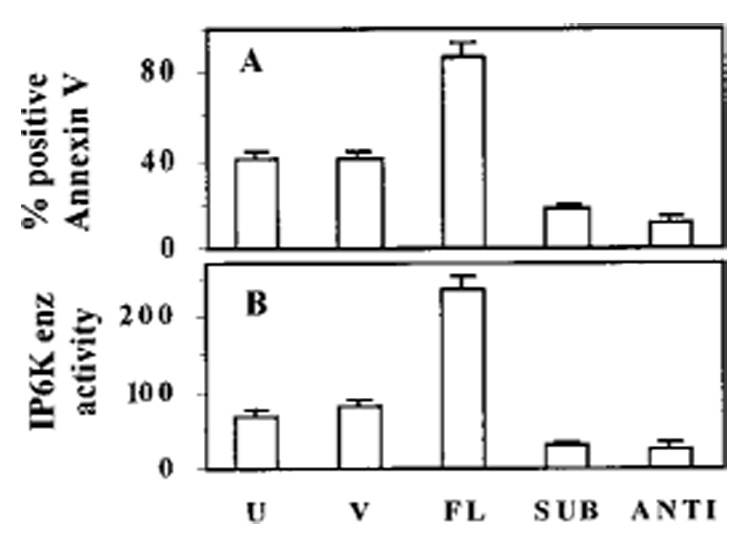
A, U indicates untransfected NIH-OVCAR-3 cells. Other notations are similar to those in Fig. 9. Cell lines were treated with IFN-β (200 units/ml) and subjected to annexin staining. The percentage of apoptotic cells was determined by flow cytometry. B, in parallel, IP6K enzymatic activity was determined in whole cell lysates. IP6K enzyme activity is represented as total area under PP-IP-5 curve. Annexin V and enzymatic data are shown as mean ± S.E. of three separate experiments.
Finally, we examined whether ablation of IFN-β-induced apoptosis by the IPBD mutant was because of a decrease in enzymatic activity of IP6K. In vitro IP6 kinase assays were performed to detect these differences (Fig. 11B). Total radioactivity incorporated into the PP-IP5 product was expressed as AUC. Cells that expressed FL IP6K2 had the highest enzymatic activity (235). Compared with FL cells, SUB cells had an ~8-fold lower level of activity (30). Thus, the SUB construct functioned as a dominant negative mutant. Antisense ANTI cells had the lowest level of activity (27), which was 3-fold less than activity of vector cells (83). Thus, ablation of IP6K enzymatic activity correlated well with loss of growth suppression and blunted induction of apoptosis by IFN-β. These data confirm the role of IP6K2 as a mediator of growth inhibition and apoptosis in response to IFN-β treatment.
DISCUSSION
IFN-β was the most potent inducer of cell death of the type I IFNs. IFN-γ did not cause death at comparable doses. These observations suggest that type I and type II IFNs employ distinct pathways to exert anti-tumor actions. The fact that IFN-β induced death independently of cell cycle arrest suggests that different gene products regulate growth arrest and apoptosis. Because NIH-OVCAR-3 cells lack functional p53 (28,29), it appears that IFN-β-induced apoptosis occurs via p53-independent mechanisms. Furthermore, because caspases are activated by a variety of death stimuli (fas, tumor necrosis factor, perforins, and many others) it is unclear what molecules modulate the core apoptosis machinery in response to IFN-β.
To this end, we employed an antisense technical knockout strategy (22) and identified the RIDs. The library used was generated using mRNA isolated from untreated cells, as well as those treated with IFN-β. Thus, genes expressed in all stages of apoptosis were included in the library. Inactivation of Janus tyrosine kinases or signal-transducing activators of transcription cannot account for the observed protection, because expression of antisense inserts is driven by an IFN-stimulated promoter (22). Indeed, cDNAs corresponding to these signaling components have not been rescued in our studies (not shown). The RID-2 cDNA characterized in this study is identical to IP6K2. Because IFN-β enhances protein levels but not mRNA levels, it appears that a post-transcriptional regulatory mechanism controls IP6K2 expression. The protective effect of anti-sense IP6K2 was because of a down-regulation of IP6K2 protein and enzymatic activity. Similarly, expression of SUB also conferred resistance to IFN-β-induced apoptosis.
Inositol PP include PP-IP5 (7 phosphates/inositol), bis-PP-IP4 (8 phosphates/inositol) (31), PP-IP4 (6 phosphates/inositol), and bis-PP-IP3 (7 phosphates/inositol) (32). The specific functional roles of PP-IP5, bis-PP-IP4, and their precursor, IP6, are not clearly understood. IP6 was previously believed to be an inactive metabolite. Several laboratories have demonstrated that IP6 functions as a substrate in mammalian cells, yeast, and protozoa (23, 31, 33) to generate a family of high energy pyrophosphates. IP6 is phosphorylated to yield PP-IP5, which in mammalian cells is subsequently phosphorylated by a second kinase to yield bis-PP-IP4 (34). The fact that basal IP6 concentration in cells is relatively high and that IP6 functions as a substrate in synthesis of small pools of high energy pyrophosphates of short half-life (35) suggests that these pyrophosphates play an important regulatory role. IP6 is not the only substrate for IP6K1 and IP6K2. Both IP6Ks can also phosphorylate IP5 to yield PP-IP4 (32). Because PEI-TLC could not clearly resolve PP-IP5 and PP-IP4, it is possible the latter compound may also contribute to growth inhibition and apoptosis.
The inositol pyrophosphates do not appear to play a role in the metabolism of membrane-associated inositol phospholipids (phosphoinositides), responsible for Ca2+ release and protein kinase C activation. Links between phospholipid metabolism and cell growth regulation have recently been identified. The type 3 inositol 1,4,5-trisphosphate receptor actively participates in apoptosis during differentiation (36). In these studies, reduction of type 3 inositol 1,4,5-trisphosphate receptor expression by antisense oligonucleotides selectively blocked apoptosis. Inositol polyphosphate 4-phosphatase mediates hydrolysis of phosphatidylinositol 3,4-bisphosphate. Overexpression of this enzyme markedly reduces growth of NIH3T3 fibroblasts (37). Our studies implicate inositol pyrophosphates as regulators of IFN-β-induced apoptosis.
IP6K1 and IP6K2 have recently been cloned (23). Although biochemically and genetically distinct from IP6K2, IP6K1 may also play a role in growth regulation. Expression of the IP6K2 dominant negative SUB mutant abrogated only ~50% of growth inhibition and apoptosis induced by IFN-β. ANTI cells and SUB cells both demonstrated an ~70% inhibition of IP6K enzymatic activity. Yet ANTI cells were more resistant than SUB cells to IFN-β-mediated growth inhibition, suggesting that additional, IP6K2-independent pathways may be inhibited in the ANTI cells. It remains to be determined whether blockade of both IP6K1 and IP6K2 function further increases the IFN-β resistance of these cells.
Footnotes
These studies were supported in part by a grant from the Cleveland Clinic Foundation bridge grant program (to D. J. L.) and by NCI, National Institutes of Health Grants CA71401 and CA78282 (to D. V. K.).
The abbreviations used are: IFN(s), interferon(s); RID(s), regulator(s) of interferon-induced death; IPBD, inositol phosphate binding domain; IP6K, inositol hexakisphosphate kinase; PP-IP5, diphosphoinositol pentakisphosphate; PARP, poly(ADP-ribose) polymerase, a caspase 3 substrate; DAPI, 4,6-diamidino-2-phenylindole; TUNEL, terminal dUTP nick-end labeling; IRF(s), IFN gene regulatory factor(s); IP6, inositol hexaphosphate; PCR, polymerase chain reaction; SUB, substitution mutant; ANTI, antisense mutant; FITC, fluorescein isothiocyanate; PI, propidium iodide; PEI, polyethyleneimine; TLC, cellulose thin-layer chromatography; AUC, area under the curve; FL, full-length; PP, pyrophosphate.
REFERENCES
- 1.Gresser I, Bourali C, Levy JP, Fontaine-Brouty-Boye D, Thomas MT. Proc. Natl. Acad. Sci. U. S. A. 1969;63:51–57. doi: 10.1073/pnas.63.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evinger M, Rubinstein M, Pestka S. Arch. Biochem. Biophys. 1981;210:319–329. doi: 10.1016/0003-9861(81)90195-8. [DOI] [PubMed] [Google Scholar]
- 3.Kalvakolanu DV. Histol. Histopathol. 2000;15:523–537. doi: 10.14670/HH-15.523. [DOI] [PubMed] [Google Scholar]
- 4.Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. Ann. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 5.Darnell JEJ. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 6.Der SD, Zhou A, Williams BR, Silverman RH. Proc. Natl. Acad. Sci. U. S. A. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ortaldo JR, Mason A, Rehberg E, Moschera J, Kelder B, Pestka S, Herberman RB. J. Biol. Chem. 1983;258:15011–15015. [PubMed] [Google Scholar]
- 8.Greiner JW, Hand PH, Noguchi P, Fisher PB, Pestka S, Schlom J. Cancer Res. 1984;44:3208–3214. [PubMed] [Google Scholar]
- 9.Greiner JW, Guadagni F, Noguchi P, Pestka S, Colcher D, Fisher PB, Schlom J. Science. 1987;235:895–898. doi: 10.1126/science.3580039. [DOI] [PubMed] [Google Scholar]
- 10.Kumar R, Atlas I. Proc. Natl. Acad. Sci. U. S. A. 1992;89:6599–6603. doi: 10.1073/pnas.89.14.6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Resnitzky D, Tiefenbrun N, Berissi H, Kimchi A. Proc. Natl. Acad. Sci. U. S. A. 1992;89:402–406. doi: 10.1073/pnas.89.1.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raveh T, Hovanessian AG, Meurs EF, Sonenberg N, Kimchi A. J. Biol. Chem. 1996;271:25479–25484. doi: 10.1074/jbc.271.41.25479. [DOI] [PubMed] [Google Scholar]
- 13.Melamed D, Tiefenbrun N, Yarden A, Kimchi A. Mol. Cell. Biol. 1993;13:5255–5265. doi: 10.1128/mcb.13.9.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samuel CE, Kuhen KL, George CX, Ortega LG, Rende-Fournier R, Tanaka H. Int. J. Hematol. 1997;65:227–237. doi: 10.1016/s0925-5710(96)00544-0. [DOI] [PubMed] [Google Scholar]
- 15.Silverman RH. In: Ribonucleases: Structure and Functions. D’Alessio G, Riordan JF, editors. New York: Academic Press; 1997. pp. 515–551. [Google Scholar]
- 16.Barber GN, Wamback M, Thompson S, Jagus R, Katze MG. Mol. Cell. Biol. 1995;15:3138–3146. doi: 10.1128/mcb.15.6.3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hassel BA, Zhou A, Sotomayor C, Maran A, Silverman RH. EMBO J. 1993;12:3297–3304. doi: 10.1002/j.1460-2075.1993.tb05999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harada H, Kitagawa M, Tanaka N, Yamamoto H, Harada K, Ishihara M, Taniguchi T. Science. 1993;259:971–974. doi: 10.1126/science.8438157. [DOI] [PubMed] [Google Scholar]
- 19.Willman CL, Sever CE, Pallavicini MG, Harada H, Tanaka N, Slovak ML, Yamamoto H, Harada K, Meeker TC, List AF, Taniguchi T. Science. 1993;259:968–971. doi: 10.1126/science.8438156. [DOI] [PubMed] [Google Scholar]
- 20.Holtschke T, Lohler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, Lou J, Knobeloch KP, Gabriele L, Waring JF, Bachmann MF, Zinkernagel RM, Morse HC, III, Ozato K, Horak I. Cell. 1996;87:307–317. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 21.Lindner DJ, Borden EC. J. Interferon Cytokine Res. 1997;17:681–693. doi: 10.1089/jir.1997.17.681. [DOI] [PubMed] [Google Scholar]
- 22.Deiss LP, Kimchi A. Science. 1991;252:117–120. doi: 10.1126/science.1901424. [DOI] [PubMed] [Google Scholar]
- 23.Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH. Curr. Biol. 1999;9:1323–1326. doi: 10.1016/s0960-9822(00)80055-x. [DOI] [PubMed] [Google Scholar]
- 24.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. J. Natl. Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 25.Meissner PS, Sisk WP, Berman ML. Proc. Natl. Acad. Sci. U. S. A. 1987;84:4171–4175. doi: 10.1073/pnas.84.12.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinoshita S, Suzuki H, Ito K, Kume K, Shimizu T, Sugiyama Y. Pharm. Res. 1998;15:1851–1856. doi: 10.1023/a:1011953906065. [DOI] [PubMed] [Google Scholar]
- 27.Voglmaier SM, Bembenek ME, Kaplin AI, Dorman G, Olszewski JD, Prestwich GD, Snyder SH. Proc. Natl. Acad. Sci. U. S. A. 1996;93:4305–4310. doi: 10.1073/pnas.93.9.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yaginuma Y, Westphal H. Cancer Res. 1992;52:4196–4199. [PubMed] [Google Scholar]
- 29.Ramet M, Castren K, Jarvinen K, Pekkala K, Turpeenniemi-Hujanen T, Soini Y, Paakko P, Vahakangas K. Carcinogenesis. 1995;16:2117–2124. doi: 10.1093/carcin/16.9.2117. [DOI] [PubMed] [Google Scholar]
- 30.Togashi S, Takazawa K, Endo T, Erneux C, Onaya T. Biochem. J. 1997;326:221–225. doi: 10.1042/bj3260221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stephens L, Radenberg T, Thiel U, Vogel G, Khoo KH, Dell A, Jackson TR, Hawkins PT, Mayr GW. J. Biol. Chem. 1993;268:4009–4015. [PubMed] [Google Scholar]
- 32.Saiardi A, Caffrey JJ, Snyder SH, Shears SB. J. Biol. Chem. 2000;275:24686–24692. doi: 10.1074/jbc.M002750200. [DOI] [PubMed] [Google Scholar]
- 33.Menniti FS, Miller RN, Putney JW, Jr, Shears SB. J. Biol. Chem. 1993;268:3850–3856. [PubMed] [Google Scholar]
- 34.Huang CF, Voglmaier SM, Bembenek ME, Saiardi A, Snyder SH. Biochemistry. 1998;37:14998–15004. doi: 10.1021/bi981920l. [DOI] [PubMed] [Google Scholar]
- 35.Shears SB, Ali N, Craxton A, Bembenek ME. J. Biol. Chem. 1995;270:10489–10497. doi: 10.1074/jbc.270.18.10489. [DOI] [PubMed] [Google Scholar]
- 36.Blackshaw S, Sawa A, Sharp AH, Ross CA, Snyder SH, Khan AA. FASEB J. 2000;14:1375–1379. doi: 10.1096/fj.14.10.1375. [DOI] [PubMed] [Google Scholar]
- 37.Vyas P, Norris FA, Joseph R, Majerus PW, Orkin SH. Proc. Natl. Acad. Sci. U. S. A. 2000;97:13696–13701. doi: 10.1073/pnas.250476397. [DOI] [PMC free article] [PubMed] [Google Scholar]