

Abstract
Mutations in the inositol 5-phosphatase OCRL are responsible for Lowe syndrome, whose manifestations include mental retardation and renal Fanconi syndrome. OCRL has been implicated in membrane traffic, but disease mechanisms remain unclear. We show that OCRL visits late stage endocytic clathrin coated pits and binds the Rab5 effector APPL1 on peripheral early endosomes. The interaction with APPL1, which is mediated by the ASH-RhoGAP-like domains of OCRL and is abolished by disease mutations, provides a link to protein networks implicated in the reabsorptive function of kidney and in traffic and signaling of growth factor receptors in brain. Crystallographic studies reveal a role of the ASH-RhoGAP-like domains in positioning the phosphatase domain at the membrane interface and a clathrin box protruding from the RhoGAP-like domain. Our results support a role of OCRL in the early endocytic pathway consistent with the predominant localization of its preferred substrates, PI(4,5)P2 and PI(3,4,5)P3, at the cell surface.
Introduction
Regulated and reversible phosphorylation of the inositol ring of phosphatidylinositol generates seven “phosphoinositides,” which play major regulatory roles in cell physiology (Di Paolo and De Camilli, 2006). The human genome encodes ten inositol 5-phosphatases, i.e. enzymes that selectively dephosphorylate the 5 position of the inositol ring (Astle et al., 2006). Mutations in one of them, OCRL, cause OculoCerebroRenal Syndrome of Lowe, an X-linked disorder characterized by congenital cataracts, mental retardation, and renal Fanconi syndrome (Attree et al., 1992). OCRL mutations were also identified in a subset of Dent disease patients, a condition which, like Lowe syndrome, is associated with loss of low molecular weight proteins and electrolytes in the urine (Hoopes et al., 2005).
OCRL has a multidomain structure, with a central 5-phosphatase domain, whose preferred substrates are PI(4,5)P2 and PI(3,4,5)P3(Schmid et al., 2004; Zhang et al., 1995) followed by a sequence recently defined as an ASH domain (ASPM, SPD2, Hydin) (Ponting, 2006) and by a COOH-terminal catalytically inactive RhoGAP domain. OCRL is an effector for several Rab proteins and also binds clathrin and clathrin adaptors (Choudhury et al., 2005; Hyvola et al., 2006; Ungewickell et al., 2004).
OCRL was initially localized to the Golgi complex (Dressman et al., 2000; Olivos-Glander et al., 1995), and is recruited to membrane ruffles in response to growth factor stimulation and Rac activation (Faucherre et al., 2005). More recently it was also detected on endosomes and on clathrin-positive intracellular structures, in agreement with its binding to Rab5 and to clathrin, and it was implicated in membrane traffic from endosomes to the Golgi (Choudhury et al., 2005; Hyvola et al., 2006; Ungewickell et al., 2004). So far, however, OCRL was not detected at endocytic clathrin coated pits, in spite of its binding in vitro to the clathrin adaptor AP-2 (Ungewickell et al., 2004).
OCRL is similar to another inositol 5-phosphatase, INPP5B (also referred to as type II 5-phosphatase). The two enzymes have primarily been studied in different contexts but appear to have at least partially overlapping functions and similar enzymatic properties (Astle et al., 2006; Jefferson and Majerus, 1995). Like OCRL, INPP5B interacts with Rab5 and is recruited to plasma membrane ruffles upon growth factor stimulation (Shin et al., 2005). In mice, disruption of both genes is lethal, while disruption of a single gene produces no apparent phenotype (OCRL) or only a minor phenotype (INPP5B) (Hellsten et al., 2001; Janne et al., 1998).
How the properties and localization of OCRL relate to the pathological manifestations of Lowe syndrome remains unclear. The kidney defects observed in Lowe Syndrome and Dent disease suggest an impairment in the traffic of proteins, including receptors, implicated in reabsorption in the proximal tubule (Lowe, 2005). For example, a chloride channel mutation that produces Dent disease in mouse reduces the surface localization of megalin, a scavenger receptor (Piwon et al., 2000). Interestingly, decreased levels of the extracellular domain of megalin were observed in the urine of Lowe syndrome and Dent disease patients with OCRL mutations (Norden et al., 2002). Defects in endocytosis and recycling of receptors in the nervous system could also produce cognitive impairment, another characteristic of Lowe syndrome patients (Lowe, 2005). However, a molecular link between OCRL and cell surface receptors has not been identified so far.
Here we show that OCRL is present throughout the early endocytic pathway, including endocytic clathrin coated pits, and demonstrate a connection between OCRL and adaptor molecules implicated in the endocytic traffic of receptors in brain and kidney. In addition, we report the crystallographic structure of the COOH-terminal region of OCRL, which provides insight into protein and membrane interactions of this protein.
Results
Localization of OCRL on early endosomes and at clathrin coated endocytic pits
Analysis of endogenous OCRL immunoreactivity or of transfected (EGFP- or Myc-tagged) OCRL expressed in Cos-7 and HeLa cells revealed, as reported (Choudhury et al., 2005; Ungewickell et al., 2004), a punctate distribution throughout the cytoplasm with an accumulation of these puncta in the Golgi complex region (Figure 1A), and some accumulation at peripheral ruffles (Faucherre et al., 2005). Most peripheral puncta colocalized with Rab5, an early endosomal protein that interacts with OCRL (Hyvola et al., 2006), but not with Rab9, a late endosomal protein (Figure 1B). Partial colocalization was observed with components (SNX1 and SNX2) of the retromer (Figure 1C and not shown), a protein complex implicated in transport from early endosomes to the Golgi complex (Bonifacino and Rojas, 2006), in agreement with the reported role of OCRL in this transport reaction (Choudhury et al., 2005; Ungewickell et al., 2004).
Figure 1. OCRL and the early endocytic pathway.

A: Immunofluorescence of the intracellular distribution of Myc-tagged OCRL in Cos-7 cells and comparison with the distribution of co-transfected Xpress-tagged INPP5B. B: RFP-OCRL (and RFP-INPP5B) colocalizes with EGFP-rab5 positive endosomes but not with EGFP-Rab9-positive organelles. C: EGFP-OCRL positive spots are directly adjacent to endogenous retromer (SNX1) immunoractivity suggesting budding of retromer positive membrane from OCRL positive endosomes. D: Modular structure of OCRL and INPP5B. E: TIRF microscopy of Cos-7 cells transfected with EGFP-clathrin light chain together with RFP-OCRL (left column) or RFP-INPP5B (right column). OCRL, but not INPP5B, colocalizes with a subset of clathrin spots in these single short exposure (200 msec) frames. F: Percentage of clathrin coated pits positive for INPP5B or for OCRL during their lifetime. G and H: Sequential TIRF microscopy images (8 sec intervals)(G) and time course of fluorescence intensity (H) for two clathrin-coated pits from cells co-transfected with RFP-clathrin and EGFP-OCRL. Note: In H, fluorescence is measured at a specific position so that both loss of fluorescence and lateral movement of the fluorescent spot away from such position result in a loss of signal intensity. I: Interaction of clathrin heavy chain with OCRL but not with INPP5B as revealed by western blotting of material affinity-purified by GST-OCRL or GST-INPP5B from a rat brain lysate (Bars = A, 10 μm; B, 2.5 μm; C, 5 μm; E, 5 μm; F, 1 μm).
Observation of Cos-7 cells coexpressing EGFP-OCRL and RFP-clathrin light chain by total internal reflection fluorescence (TIRF) microscopy (Zoncu et al., 2007) revealed that OCRL was also detected transiently at a fraction of endocytic clathrin coated pits (38%) (Figures 1E and F). Its residence at the pits generally occurred during the declining phase of the clathrin fluorescence thought to reflect internalization of the pit. Often some OCRL fluorescence persisted and moved away from the location of the original clathrin spot suggesting a continued association with the uncoated vesicle (Figures 1G and H, Movie 1).
An overall similar localization was observed for RFP-INPP5B (Figures 1A and B). However, INPP5B does not contain the clathrin box and the AP-2 binding motif, and was not detected at endocytic clathrin pits (Figures 1D-F). The interaction of clathrin heavy chain with OCRL but not INPP5B was confirmed using GST-fusions of OCRL or INPP5B (Figure 1I), or of their COOH-terminal regions (ASH-RhoGAP-like domains) (not shown) as baits in pulldowns from rat brain lysate.
OCRL and INPP5B bind APPL1
The pulldown assays (see above) used to analyze the clathrin binding of OCRL and INPP5B showed that the COOH-terminal regions of both proteins, but not GST, bound a 90 kDa band (Figure 2A). The band was identified by mass spectrometry, and then western blotting, as APPL1 (DIP13alpha)(Figure 2B). Interestingly, APPL1, like OCRL and INPP5B, is a Rab5 effector and is localized on a subpopulation of peripheral early endosomes (Miaczynska et al., 2004). Thus, this interaction appeared to be physiologically important and was further investigated. Binding of APPL1 to OCRL/INPP5B is direct because recombinant APPL1 bound GST-fusions of OCRL and INPP5B (Figure 2C). APPL1 has a close homologue, APPL2. However, GST-pulldowns from extracts of Cos-7 cells expressing Myc-tagged APPL proteins demonstrated that both OCRL and INPP5B interact selectively with APPL1 (Figure 2D)
Figure 2. The Rab5 effector protein APPL1 interacts, and colocalizes with, OCRL and INPP5B.

A: A 90 kd protein binds OCRL and INPP5B in pulldown experiments. GST and GST fusion proteins of the COOH-terminal regions of OCRL and INPP5B were incubated with rat brain lysate and bound proteins were analyzed by SDS-PAGE followed by Coomassie blue staining. B: Anti-APPL1 western blotting of the samples shown in A as well as bound proteins from pulldowns in which GST-fusions of full length INPP5B and OCRL were used as baits. C: The interaction between OCRL/INPP5B and APPL1 is direct. His-tagged recombinant APPL1 was incubated with the indicated GST-fusion proteins and bound proteins were analyzed by western-blotting. D: OCRL and INPP5B interact with APPL1 but not with APPL2 as shown by GST-pulldowns from transfected cells followed by western blotting. E: Coprecipitation of OCRL with APPL1 from extracts of transfected Cos-7 cells. Western blotting of the starting lysate and of the immunoprecipitates are shown. F: Epifluorescence images demonstrating that APPL1 colocalizes with OCRL and INPP5B on endosomes in transfected Cos-7 cells. G: H and I: TIRF microscopy sequential images (12 sec intervals) of spots positive for RFP-clathrin, EGF-OCRL and EGFP- or RFP-APPL1 at the cell surface (Bars = F, 2.5 μm; G, H and I = 1μm).
The binding of OCRL/INPP5B to APPL1 was validated by coprecipitation of HA-tagged OCRL or INPP5B with Myc-tagged APPL1 from extracts of Cos-7 cells (Figure 2E and not shown) and supported by colocalization studies. EGFP-APPL1 and endogenous APPL1 immunoreactivity colocalized with a subset of peripheral endosomes positive for RFP-INPP5B or RFP-OCRL (Figure 2F, movie 2 and 3, see Figure 6C and not shown). In addition, the recruitment of OCRL and INPP5B to the large endosomes induced by constitutively active Rab5 [Figure S2 (a and b) (f and g)] was more robust in cells also overexpressing APPL1, more so in the case of INPP5B, which was less efficiently colocalized with Rab5 alone [Figure S2 (c–e) (h–j)]. .
Figure 6. OCRL point mutations of Lowe syndrome patients abolish the interaction with APPL1.
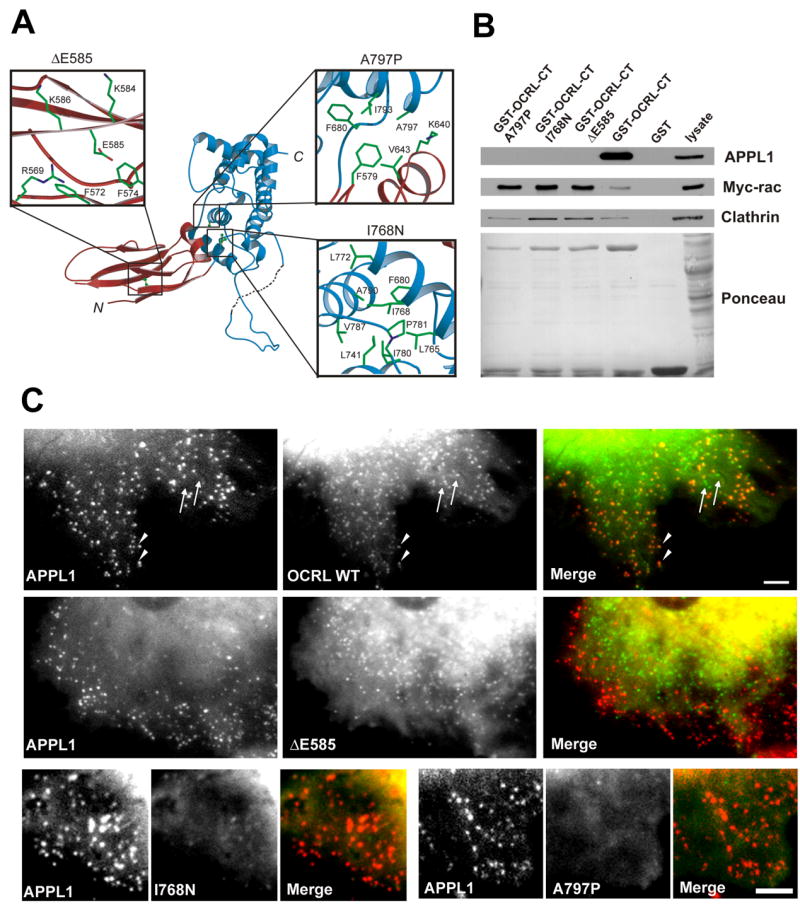
A: Close view of the local structural environment of three single amino acid patient mutations in the COOH-terminal regio n of OCRL. B: GST-pulldowns from extracts of rat brain (APPL1, clathrin) or transfected Cos-7 cells (Myc-RacV12) on wildtype and mutant fusion proteins, demonstrating that each of the three mutations abolish binding to APPL1 but not to clathrin or Rac (Myc-RacV12). Bound proteins were detected by western blotting. C: Two colour fluorescence of cells expressing RFP-APPL1 and EGFP-OCRL (wildtype or mutant). Wildtype OCRL is localized on spots throughout the cell. The more peripheral spots (arrowheads), but not all spots (arrows), colocalize with APPL1. Each of three patient mutations abolishes the colocalization with APPL1, although APPL1 positive spots at the periphery persist. Note that at least for the Δ585 mutation, OCRL still has a punctate subcellular distribution.
In contrast to OCRL, APPL1 did not appear to be a resident protein of coated pits, although a relationship of EGFP-APPL1-positive puncta with such pits was observed. In some cases, pre-existing APPL1 puncta transiently approached clathrin coated pits and then moved away (Figure 2G). Of note, endosomes were recently shown to transiently approach sites of endocytic vesicle formation in yeast (Toshima et al., 2006). In many other cases, APPL1-positive puncta appeared at the location where a clathrin coated pit had just disappeared (Figure 2H) and, as often seen for OCRL (see Figure 1G), moved away from these sites. These spots are likely to represent association of APPL1 with OCRL on newly formed endocytic vesicles, because APPL1 fluorescence was often preceded by the appearance of OCRL fluorescence (Figure 2I, movie 3). The dynamic pattern of EGFP-Rab5 fluorescence, as revealed by TIRF, was very similar to that of APPL1 (not shown).
A protein network implicated in receptor endocytosis and recycling
APPL1 is an adaptor/signaling protein that binds the membrane (via a BAR and a PH domain), several transmembrane receptors including the nerve growth factor receptor TrkA (via a PTB domain), and the oligomeric endocytic adaptor protein GIPC (via a COOH-terminal PDZ binding motif)(Figure 3A) (Lin et al., 2006; Liu et al., 2002; Mao et al., 2006; Varsano et al., 2006). GIPC also binds TrkA (Figure 3A) and, together with APPL1, participates in the regulation of endocytic trafficking of this receptor (Lin et al., 2006; Varsano et al., 2006). We found that GIPC was present together with APPL1 in the material affinity-purified from rat brain lysate on a GST fusion of the COOH-terminal region of OCRL (Figure 3B, top), indicating that a complex of these proteins may occur in vivo. It is therefore possible that the neurological and cognitive manifestations of Lowe syndrome may be mediated, at least in part, by an effect of impaired OCRL function on TrkA signaling.
Figure 3. OCRL and APPL1 are components of a protein network implicated in receptor trafficking and signalling.
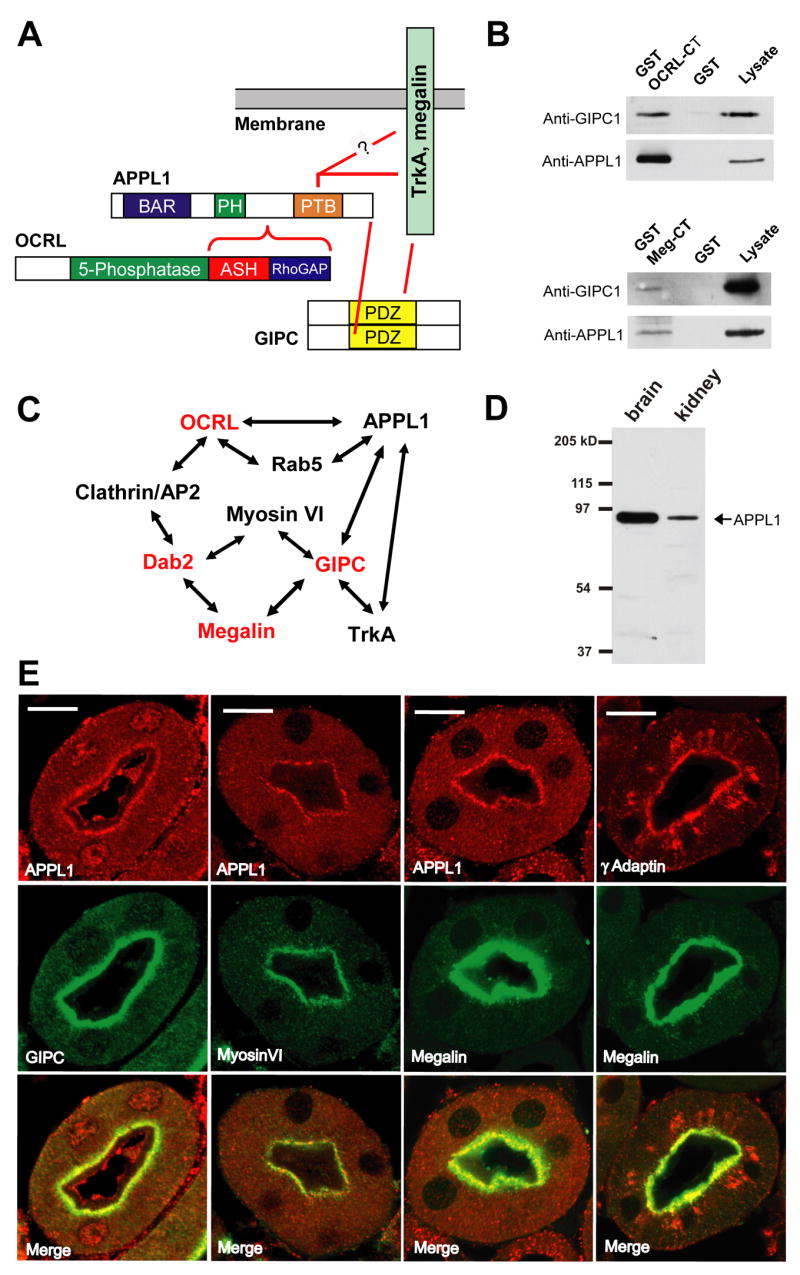
A: Schematic drawing indicating direct and indirect interactions of OCRL described in this study or reported in the literature. The PDZ domain of GIPC binds the COOH-terminus of both the TrkA receptor and megalin. The PTB domain of APPL1 also binds the cytoplasmic domain of TrkA. A direct interaction of this PTB domain with megalin remains hypothetical. B: GST pulldowns of a rat brain lysate on GST or GST fusions of the COOH-terminal region of OCRL (top) or of the cytoplasmic domain of megalin (bottom). The western blots shown demonstrate presence of both APPL1 and GIPC in the materials bound both baits. C: Protein network involving OCRL and APPL1 and potentially relevant to the kidney and brain defects observed in Lowe syndrome. Loss of function mutations of proteins indicated in red lead to renal proximal tubule reabsorption defects either in human (OCRL) or in mouse (Megalin, GIPC and Dab2)(see text). D: Presence of APPL1 in both rat brain and kidney lysates (western blot). E: Immunofluorescence of cross-sectioned (cryostat sections) mouse kidney proximal tubules showing enrichment of APPL1 at the apical region of the epithelium, where it colocalizes with GIPC, myosin VI, and megalin, and also, partially with γ-adaptin. (Bar = 10 μm)
The interaction of APPL1 with GIPC is also of special significance in the context of the kidney defects observed in patients with OCRL mutations because GIPC directly binds megalin (Lou et al., 2002) (Figure 3A). Interestingly, not only GIPC, but also APPL1, was recovered in a pull-down from a rat brain extract with a GST fusion of the cytoplasmic domain of megalin (Figure 3B, bottom). These results point to a molecular network that in kidney links OCRL to the reabsorption machinery of the proximal tubule (Figure 3C). Indeed, GIPC knock out mice, as well as mice defective in megalin or in another megalin adaptor, Dab2, exhibit low molecular weight proteinuria (Leheste et al., 1999; Naccache et al., 2006; Nagai et al., 2005). To provide evidence for a role of APPL1 in this network, we investigated expression and localization of APPL1 in kidney.
Both OCRL (Janne et al., 1998) and APPL1 (Figure 3D) are expressed in kidney. APPL1 immunoreactivity in kidney proximal tubules appeared as an apical band of tightly apposed puncta just below the brush border (Figure 3E). This is the region where endocytic and recycling vesicles implicated in reabsorption from the primary urine are localized. Accordingly, APPL1 immunoreactivity strikingly overlapped with megalin and with other endocytic proteins concentrated in this region, including clathrin (Rodman et al., 1984), GIPC (Lou et al., 2002), Dab2 (Nagai et al., 2005), Rab5, and myosin VI, a GIPC and Dab2 interactor and a marker of clathrin coated and newly uncoated vesicles (Naccache et al., 2006; Nagai et al., 2005) (Figure 3E and not shown). Available antibodies did not allow for the reliable detection of OCRL in kidney proximal tubules by immunocytochemistry. However, the localization of the major binding partners of OCRL at the apical pole of these cells (APPL1, Rab5, clathrin) strongly suggests localization of OCRL as well in this region. It is of interest that even the AP-1 clathrin adaptor complex (as visualized by immunofluorescence of γ-adaptin), which was shown to colocalize with OCRL in the Golgi complex area (Choudhury et al., 2005), extends into the apical region containing APPL1 positive endosomes in kidney proximal tubule cells (Figure 3E). These findings are consistent with the hypothesis that kidney defects produced by OCRL mutations reflect abnormal endocytic traffic and/or signaling at the apical pole of proximal tubule cells. Given the potential importance of the binding of OCRL to APPL1 in disease, a molecular analysis of this interaction was performed.
A short peptide in APPL1 mediates binding to OCRL and INPP5B
Interaction surfaces between APPL1 and OCRL/INPP5B were analyzed using protein fragments as baits in GST pulldown assays from brain lysates. APPL1 deletion constructs defined the minimal binding site as an 11-mer peptide within the region comprised between the PH and the PTB domain (Figures 4A and B). Mutation to alanine of the phenylalanine at the +2 position of this peptide (F404A) completely abolished binding (Figure 4B). Interestingly, this short sequence is highly conserved across species but is not present in APPL2 (Figure 4A), thus explaining the selective binding of OCRL and INPP5B to APPL1 (Figure 2D). The affinity (Kd) of the interaction between the COOH-terminal region of ORCL and the 11-mer APPL1 peptide, as measured by isothermal titration calorimetry (ITC), was 20 μM, while the F404A peptide did not show any measurable binding (Figure 4C).
Figure 4. Mapping of the binding interfaces between APPL1 and OCRL.

A: Modular structure and interactions of APPL1. The figure also shows an alignment of the 11-mer peptide of human APPL1 with the corresponding sequence of APPL from other species, demonstrating its evolutionary conservation. The sequence is not conserved in human APPL2. Potential phosphorylation sites within the 11-mer peptide are indicated by asterisks. B: GST pulldowns from extracts of Cos-7 expressing either Myc-OCRL or Xpress-INPP5B define an 11 amino acid (residues 403–413) stretch of human APPL1 as the minimal region necessary and sufficient for binding. Bound proteins were identified by western blotting. Numbers indicate the amino acid boundaries of the APPL1 fragment fused to GST. The F404A mutation abolished binding. C: ITC analysis of the binding of the wildtype APPL1 11-mer peptide and of the F404A-mutant peptide. Raw data are shown in the upper panels, and plots of the total heat released as a function of the molar ratio of each ligand are shown in the bottom panels. D: GST-fusions of the wildtype or mutant (S403D and S410D) minimal binding region (11-mer peptide) were incubated with the catalytic subunit of PKA or with type II Ca2+-calmodulin dependent kinase (CamKII) in the presence of γ-32P-ATP. Proteins were separated by SDS-PAGE and the corresponding autoradiography is shown. E: Co-immunoprecipitation of full length wildtype and mutant (S410D) APPL1 with OCRL. F: PKA overexpression impairs binding of APPL1 to OCRL in vivo. For E and F Cos-7 cells were transfected with the indicated expression constructs. Protein complexes were immunoprecipitated using anti-GFP antibody and the starting lysates and the immunoprecipitates were analyzed by western blotting.
The 11-mer peptide of APPL1 and adjacent regions contain a number of potential serine/threonine phosphorylation sites (Figure 4A). A phosphorylation-dependent regulation of the interaction between APPL1 and OCRL/INPP5B would support its physiological significance. In vitro phosphorylation of the GST- 11-mer peptide with several protein kinases revealed strong phosphorylation by the catalytic subunit of protein kinase A (PKA) but not other kinases (Figure 4D and not shown). Interestingly, the reabsorption rate of kidney proximal tubules is regulated by manipulations that enhance PKA activity, such as forskolin or parathyroid hormone (PTH) (Gekle et al., 1997). Replacing the two serines within the 11-mer peptide (S403 and S410) with aspartate mapped the phosphorylation site to S410 (Figure 4D). The phosphomimetic S410D mutation strongly inhibited the binding of the peptide to OCRL both in GST pulldowns (not shown) and in coprecipitation experiments (Figure 4E). Likewise, overexpression of PKA together with HA-OCRL and EGFP-APPL1 in Cos-7 cells reduced the coprecipitation of the two proteins (Figure 4F), supporting a physiological role of this phosphorylation reaction.
Efforts to narrow down the APPL1-interacting region within the COOH-terminal region of OCRL and INPP5B revealed that both COOH-terminal and NH2-terminal truncations of this protein fragment abolished binding (Figure S2). Hence, this fragment appears to function as a folded unit that recognizes APPL1 in a module-peptide type of interaction. To gain structural insight into the COOH-terminal region of OCRL, X-Ray crystallography studies were performed.
Atomic structure of the COOH-terminal region of OCRL
The crystal structure of the selenomethionine-substituted COOH-terminal region (residues 564–901) of human OCRL (GI: 57209431) was determined by the single anomalous dispersion (SAD) method (Supplemental Table 1. This region of OCRL is composed of an NH2 terminal module (ASH domain, see below) (residue 564 to 678), represented primarily by β strands and a COOH-terminal RhoGAP-like domain (residue 679 to 901) (Figures 5A and 5B). The two domains are closely apposed to each other in a rigid orientation determined by a number of polar and apolar interactions that bury a total solvent accessible surface of 1056.2 Å2. These interactions may help stabilize the structure of the ASH domain, which was not stable in solution when expressed alone.
Figure 5. Structure of the COOH-terminal region of OCRL.

A: Ribbon diagram with the ASH domain (red) and the RhoGAP-like domain (blue). Residues of the clathrin box motif are in sticks. B: Stereo view of the Cα backbone traces. Trace color ramped from the NH2 terminus (red) to the COOH-terminus (blue). Every 20 residues are labeled and indicated with small filled circles. C: (Left) RhoGAP like domain of OCRL (blue) superimposed on the RhoGAP domain of P50RhoGAP (PDB ID: 1AM4). Bound Cdc42 is shown in gold. (Right) Biochemical interaction of the RhoGAP-like domain of OCRL and INPP5B with Rac and Cdc42. GST pulldowns from lysates of Cos-7 cells expressing tagged versions of Rac, Rho or Cdc42 with GST-fusions of the RhoGAP-like domain of OCRL or INPP5B. Bound proteins were analyzed by western-blotting. D: Alignment of the ASH domain of OCRL and INPP5B with other members of the MSP/VAP domain protein family. The E585 deletion mutation found in a patient is marked by a black triangle. Secondary structure elements are drawn above the alignment. Entrez database accession numbers are as follows: OCRL_Hs, GI: 57209431; Inpp5_Hs, GI: 59803021; OCRL_Dr, GI: 68374521; Inpp5_Dm, GI: 54642833; Inpp5_Ce, GI: 17505597; Inpp5_Dd, GI: 66828629; MSP_Ce, GI: 21730216; Vap-A_Rn, GI: 73535850. (Hs, Homo sapiens; Dr, Danio rerio; DM, Drosophila melanogaster; CE, Caenorhabditis elegans; Dd, Dictyostelium discoideum; Rn, Rattus norvegicus).
RhoGAP-like domain
The RhoGAP-like domain is very similar to previously characterized RhoGAPs (Peck et al., 2002) (Figure 5C). When compared to p50RhoGAP, the RMSD value was 1.42 Å over a total of 146 residues. However, the catalytic arginine is replaced by a glutamine (Figure S3 and S4) and the F’ helix (Figure 5C) is missing. These differences explain why the RhoGAP-like domain of OCRL has no detectable GAP activity against a number of G-proteins tested, including its known interactors Rac and Cdc42 [(Lichter-Konecki et al., 2006) and our unpublished results]. A striking characteristic of the RhoGAP-like domain of OCRL is the presence of an extended (approximately 40 a.a.) loop between αA0 and αA (Figures 5A and 5B). The electron density of most of this loop is visible due to the crystal packing, which stabilizes an interaction of the loop with adjacent molecules “in trans”. The loop contains the type I clathrin binding motif (clathrin box) 702LIDLE706 (Figures 5A and E, and Figure S5), which was previously thought to lay outside of the RhoGAP-like domain. Such loop is subject to alternative splicing (Lowe, 2005) –exclusion or inclusion of 8 residues downstream of the clathrin box motif that contain a predicted casein kinase II phosphorylation site - and is shorter in INPP5B, which lacks the clathrin box.
The insertion of the long loop after the αA0 helix has previously blurred the definition of the boundaries of the RhoGAP-like domain based on primary sequence. This may explain why, in contrast to what has been described for OCRL, no evidence for an interaction of INPP5B with Rac or CDC42 was reported. Indeed, when tested in pulldown experiments from extracts of Cos-7 cells expressing constitutively active Rac1 (Rac1V12), CDC42 (CDC42V12), or Rho (RhoV12), the RhoGAP-like domains of both OCRL and INPP5B (GI: 30231213) bound Rac and CDC42 but not Rho (Figure 5C). Similar to OCRL, INPP5B also interacted with dominant negative RacN17, indicating nucleotide independent binding (Figure 5C).
ASH domain
The NH2-terminal module is comprised by two layers of β-sheets (parallel strand β1, β6, antiparallel strand β5, and anti-parallel strands β2, β4, β3) (Figure 5A and 5B). Hydrophobic side chains from the two layers occupy the interior of the sandwich. There was no interpretable electron density for the tip of this module distal to the RhoGAP-like domain, most likely due to its flexible nature and to the lack of crystal packing contacts around this region. The overall folding of this module is closely related to that of the MSP and VAP domains, members of the family of immunoglobulin-like β sandwiches with s-type topology (Bork et al., 1994; Kaiser et al., 2005). This structure is in agreement with a recent bioinformatics study that, based on a remote primary sequence similarity, included these regions of OCRL and INPP5B into the newly defined ASH (ASPM, SPD2 and Hydin) domain family (Figure 5D) (Ponting, 2006).
The VAP domain, which is found in several endoplasmic reticulum proteins, interacts with the conserved FFAT motif, present in a family of oxysterol binding proteins (Loewen et al., 2003). A 10 residues peptide comprising the FFAT motif, binds to a groove perpendicular to the main axis of the VAP domain (Kaiser et al., 2005). It is tempting to speculate that the 11-mer peptide of APPL1 binds in a similar fashion to the ASH domain of INPP5B and OCRL. However, as shown above, binding of the 11-mer peptide requires both the intact ASH and the RhoGAP-like domains. This may be due to a role of the RhoGAP-like domain in the stabilization of the ASH domain (see above) or by a binding interface contributed by both domains. Attempts to co-crystallize the 11-mer peptide with the ASH-RhoGAP-like domains were unsuccessful.
APPL1 binding and human mutations
Deletions of the COOH-terminal region of OCRL as well as three distinct single amino acid changes in this region have been associated with Lowe syndrome (Monnier et al., 2000). The three single amino acid mutations (ΔE585, I768N, A797P) were mapped on the crystal structure (Figure 6A). E585 is located in the β2 strand of the ASH domain, and its deletion may be disruptive of the correct structure of this domain. I768 is located in the middle of αA1 of the Rho-GAP-like domain with its side chain pointing to the hydrophobic core formed by αA, αA1, and αB. Mutation of this nonpolar residue to the polar residue asparagine is also likely to affect folding. A797 is located at the end of αB, at the interface between the RhoGAP-like and ASH domains, thus in a position that may affect the interaction between these two domains. ASH-RhoGAP-like domains harboring these mutations were tested for APPL1 binding in GST pulldown assays. None of these mutant proteins bound to APPL1, while binding to Rac and clathrin was preserved (Figure 6B). Accordingly, immunofluorescence revealed no colocalization of EGFP-OCRL harboring these three mutations with RFP-APPL1 (Fig 6C) although APPL1 still had a punctate peripheral distribution. These findings support the hypothesis that an impairment of the interaction between OCRL and APPL1 may play a role in disease.
Discussion
The localization and interactions of OCRL reported in this study suggest a role of OCRL at early steps of the endocytic pathway and its action, via its binding to APPL1, in a protein network closely linked to some of the phenotypic manifestations of Lowe syndrome and Dent disease. Our structural studies demonstrate that the entire COOH-terminal region of OCRL functions as a folded unit, thus explaining why truncations of this region at either end abolish binding to APPL1. The structure of this region reveals how its multiple interactions may help position the catalytic site of the inositol 5-phosphatase module at the membrane interface. It also shows the first structure of an ASH domain.
OCRL and the early endocytic pathway
Recent studies had demonstrated localizations of OCRL not limited to the Golgi complex area, as originally reported, but also on endosomes, and functional studies suggested its role in transport between endosomes and the trans-Golgi complex (Choudhury et al., 2005; Ungewickell et al., 2004). We now show that OCRL is also present at a subpopulation of endocytic clathrin coated pits and that one of its important binding partners is a protein, APPL1, implicated in signaling and sorting of cell surface receptors which is localized, together with OCRL, on a subset of peripheral early endosomes (Miaczynska et al., 2004). Recent studies have implicated INPP5B as well in endosomal function (Shin et al., 2005). Together, these findings solve the apparent paradox raised by a selective concentration of OCRL in the Golgi complex: the discrepancy between this localization and the predominant concentration of its preferred substrates, PI(4,5)P2 and PI(3,4,5)P3 (Schmid et al., 2004) (see also Figure S6), at the plasma membrane (Di Paolo and De Camilli, 2006; Stauffer et al., 1998) They strongly suggest that OCRL functions in the coupling of endocytic traffic to PI(4,5)P2 and PI(3,4,5)P3 dephosphorylation, as reported for INPP5B (Shin et al., 2005) and synaptojanin (Cremona et al., 1999). This scenario does not exclude roles of the two phosphatases at their other intracellular locations where they may function to prevent ectopic or abnormal accumulation of these phosphoinositides.
A structural model of OCRL
Our crystallographic results demonstrate the striking structural similarity of the COOH-terminal portion of OCRL to RhoGAP domains, in spite of the lack of a catalytic arginine, reveal the first structure of an ASH domain, and support the assignment of this domain to the MSP/VAP domain superfamily (Ponting, 2006). Since both OCRL and INPP5B metabolize inositol phospholipids, it is of interest to note that mutations of the VAP domain protein Scs2 in S. cerevisiae confer inositol auxotrophy (Kagiwada et al., 1998), and more generally, that VAP domains bind lipid metabolizing enzymes (Loewen et al., 2003).
The structure of the ASH-RhoGAP-like domains of OCRL, together with the predicted structure of the inositol 5-phosphatase domain (based on the 5-phosphatase domain of S. Pombe synaptojanin (Tsujishita et al., 2001), allows modeling of OCRL and INPP5B at the membrane interface (Figure 7). The COOH-terminus of INPP5B is farnesylated (Jefferson and Majerus, 1995) and is thus expected to be adjacent to the membrane. Although the COOH-terminal region of OCRL lacks a motif predictive of farnesylation, its strong overall similarity to INPP5B suggests a similar localization/orientation relative to the membrane. Such orientation is compatible with an interaction of the RhoGAP-like domains of both phosphatases with membrane bound small G proteins, which may thus provide an indirect anchor at the membrane for OCRL and a second anchor for INPP5B. The rigid angle between the ASH and Rho-GAP-like domains may guide the ASH domain away from the bilayer and make it dock to the inositol 5-phosphatase module in a way that allows for the juxtaposition of the catalytic site to the membrane. Three residues whose mutations abolish Rab5 binding (D555E, S564P, G664D (Hyvola et al., 2006)) form a cluster (solid line oval) that is separated from the bilayer by a distance that can accommodate membrane bound Rab5. The model also shows how the clathrin box motif projects away from other domains, thus being accessible to the foot of clathrin triskelia, i.e. the clathrin heavy chain module that binds clathrin boxes (ter Haar et al., 2000).
Figure 7. Structural model of the interaction of OCRL and INPP5B with the membrane.

The ASH-RhoGAP-like domains of OCRL were docked to the structure of the inositol 5-phosphastase domain from S. Pombe determined by Hurley and coworkers (Tsujishita et al., 2001). The NH2-terminal domain of OCRL, whose structure remains unknown, is not included. OCRL and INPP5B form an arc that is anchored to the membrane via the catalytic site of the inositol 5-phosphatase domain (shown here in a complex with the head group of a phosphoinositide) (PDB ID: 1I9Z) and the COOH-terminal region of the RhoGAP-like domain, which in INPP5B is farnesylated. Three mutations which abolish Rab5 binding are located within a small region (solid oval line), whose distance from the bilayer can accommodate a small GTPase such as Rab5. Rac or Cdc42 could also be accommodated next to the RhoGAP-like domain. The precise binding interface for APPL1 (dashed oval line) remains to be defined. The clathrin binding site protrudes from the structure.
OCRL, Lowe syndrome and Dent disease
The identification of APPL1 as a new interactor of OCRL and INPP5B places these two phosphatases in protein networks that are likely to be relevant for the functional defects characteristic of Lowe syndrome and Dent disease (Figure 3C). APPL1 is an adaptor/signaling protein whose PTB domain binds directly cell surface receptors, including DCC (deleted in colon cancer), adiponectin, FSH receptor, and TrkA (Lin et al., 2006; Liu et al., 2002; Mao et al., 2006; Nechamen et al., 2004). Its COOH-terminus binds GIPC (Lin et al., 2006), an oligomeric adaptor that provides an additional link to a variety of receptors, including TrkA (Lou et al., 2001) and megalin (Lou et al., 2002), a scavenger receptor in the kidney proximal tubule. GIPC also binds myosin VI, a molecular motor implicated in the motility of endocytic vesicles, including megalin containing ones in kidney (Naccache et al., 2006). Mice lacking expression of megalin, GIPC, or Dab2, another endocytic adaptor for megalin, display urinary defects similar to those observed in Lowe syndrome and Dent disease patients (Leheste et al., 1999; Naccache et al., 2006; Nagai et al., 2003). In kidney proximal tubule cells, APPL1is present together with these proteins in the apical region, where endocytic vesicles that internalize and recycle cell surface proteins implicated in reabsorption are also localized. Furthermore, an interaction of OCRL with GIPC, most likely mediated by APPL1, could be detected by pulldown experiments.
Thus, given the importance of DCC and TrkA signaling in brain and of megalin function in the kidney (megalin also has important functions in brain), themental retardation and kidney reabsorption defects observed in Lowe syndrome may reflect a role of OCRL in the regulation of receptor trafficking via the APPL1/GIPC complex. The decreased shedding of megalin in the urine of patients with OCRL mutations provides genetic evidence for this hypothesis (Norden et al., 2002). Importantly, the interaction with APPL1 is the only one shown to be selectively disrupted by all three published disease relevant mutations in the COOH-terminal region of OCRL. Changes in the phosphoinositide composition of endosomal membranes resulting from the absence of OCRL, such as an ectopic accumulation of PI(4,5)2 and PI(3,4,5)P3, may impact sorting of receptors in multiple ways, including retrograde traffic to the Golgi complex, as reported (Choudhury et al., 2005; Ungewickell et al., 2004).
In parallel to effects on trafficking, OCRL mutations may also produce defects in signaling pathways downstream of PI(4,5)P2 and/or PI(3,4,5)P3. Activation of TrkA results in the generation of PI(3,4,5)P3 and subsequent Akt activation (Kaplan and Miller, 2000). Megalin was reported to bind Akt (Caruso-Neves et al., 2006). It is therefore of interest that OCRL dephosphorylates PI(3,4,5)P3 (Schmid et al., 2004) (Figure S6) and that APPL1 interacts with AKT2 (Mitsuuchi et al., 1999). The localization of OCRL and INPP5B on endocytic membranes and binding of these proteins to APPL1 may affect the duration and localization of AKT signaling on such membranes as their receptor cargo progresses along the endocytic pathway. It is also possible that the activity of OCRL in kidney cells may be needed to prevent accumulation of PI(3,4,5)P3 at the apical pole, given the recent finding that PI(3,4,5)P3 is excluded from the apical surface (Martin-Belmonte et al., 2007). Finally, molecular defects in OCRL may affect, directly or indirectly, the nuclear-cytoplasmic shuttling of APPL1 (Miaczynska et al., 2004), and thus the nuclear actions of this protein.
Collectively, our findings provide a new context in which to interpret some of the defects observed in patients with OCRL mutations and provide new important information toward the formulation of testable hypotheses concerning the physiological function of OCRL and its role in disease.
Experimental Procedures
Antibodies, plasmids and critical reagents
Rabbit polyclonal antibodies were raised against his-tagged fusion proteins of mouse OCRL (amino acids 1–220) and INPP5B (amino acids 1–256 of) or against a synthetic peptide (CINKPDEESYCKQWLTARPSKG) of INPP5B. The rabbit polyclonal anti-megalin antibody (anti-MC-220) was described (Zou et al., 2004). Antibodies purchased from commercial sources are listed in the supplemental material. Other antibodies were from our lab. Full-length cDNAs for mouse INPP5B, human OCRL, human APPL1 and APPL2, and mouse rab5 were amplified by PCR from brain cDNA libraries (Clontech) and subcloned into the expression vectors pEGFP-C1 or pcDNA3.1 harboring either a Myc-tag or an RFP-NH2-terminal tag. Phosphoinositides were purchased from Echelon Research Laboratories. The following reagents were kind gifts: Myc-rab5Q79L in pcDNA3 from Hong Chen in our lab; EGFP-rab9 from Suzane Pfeffer (Stanford Univ.), RFP-clathrin light chain from James Keen (Thomas Jefferson Univ.), HA-tagged protein kinase A (catalytic subunit) from Susan Taylor (Univ. of California San Diego) and CaMKII from Angus Nairn (Yale Univ.).
Purified proteins
Full length OCRL and INPP5B were generated in insect cells as GST fusion proteins using the bacculovirus vector pAcGHLT-A (Pharmingen). cDNA contructs encoding fragments of OCRL, INPP5B, APPL1, and megalin were subcloned into the pGEX-6P-1 expression vector (Amersham) to obtain GST-fusion proteins or the pET30 vector (Qiagen) to obtain His-tagged proteins. Fusion proteins were purified on glutathione sepharose or nickel beads according to standard protocols.
Pulldown and coimmunoprecipitation experiments
Extracts from adult rat tissues (lysates) were prepared by homogenization in lysis buffer [PBS, 0.5% Triton (v/v), protease inhibitor mixture (Roche)] followed by ultracentrifugation (100,000g, 45 min, 4°C) to remove insoluble material. GST-pull downs from the extracts were performed by standard protocols. For immunoprecipitation, cell extracts (2–3 mg) were first incubated with antibodies (3 h) and then with protein A–Sepharose beads (Amersham Biosciences) for 2 h at 4°C followed by centrifugation and several washes in lysis buffer. Proteins recovered on beads in either procedure were eluted using SDS sample buffer and separated by SDS-PAGE.
Cell culture, transfection and microscopy
Cos-7 cells or HEK 293 cells (ATCC, Rockville, MD) were cultured at 37 °C and 10% CO2 in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum. Transfections were carried out using Fugene (Roche) or Lipofectamine 2000 (Life Technologies), and cells were observed after 16–24 hours. Still fluorescence microscopy was performed by standard procedures.
Total internal reflection microscopy was performed as described in (Zoncu et al., 2007). For details see supplemental material.
Isothermal Titration Calorimetry (ITC)
ITC measurements were performed as described (Lee et al., 2005) using a Microcal VP-ITC isothermal titration calorimeter equipped with a PC running VPViewer software(http://www.info.med.yale.edu/wmkeck/prochem/)]. Dissociation constant values were obtained with Origin software. See supplementary material for details.
In vitro phosphorylation
GST-fusions (each 2.5–5 μg) were mixed with purified kinases (150–200 ng)(kind gift of Dr. Angus Nairn, Yale University) in the presence of 10 μCi γ-[32P]ATP (1 Ci = 37 GBq) and 100 μM ATP for 1 h at 32°C. Protein kinase A phosphorylation reactions were performed in buffer A [50 mM Hepes (pH 7.4), 10 mM MgCl2, 1 mM EGTA, and 1 mM β-mercaptoethanol]. Reactions were quenched by addition of SDS-PAGE sample buffer, and samples were analyzed by SDS-PAGE and autoradiography.
Protein crystallization, data collection and structure determination
A selenomethionine-substituted GST fusion of the COOH-terminal region of human OCRL1 (residues 564–901), was prepared and crystallized as described in Supplementary Methods. X-ray data were acquired at the APS NE-CAT 24ID beamline (Argonne, IL). All diffraction data were processed using HKL 2000 (Otwinowski and Minor, 1997) and the structure of the crystal was determined by single anomalous dispersion (SAD) phasing with CNS (Brunger et al., 1998). The resulting experimental electron density map was displayed and an initial model was built with Xtalview (McRee, 1999). The structure was refined against the 2.4 Å refinement data (Supplemental Table 1) by simulated annealing, conjugate gradient minimization, and restrained isotropic B-factor refinement. The final model has R and Rfree values of 24.7% and 28.8% respectively and has no residues in the disallowed region of the Ramachandran plot. Other details of structure determination are given in the Supplementary Methods.
Miscellaneous Procedures
SDS-PAGE and Western Blotting were performed by standard procedures. The APPL1 band was identified by LC MS/MS at the Keck Research Facility at Yale University. The malachite green assay was performed as described (Maehama et al., 2000) using 20–50ng GST-OCRL or GST-INPP5B purified from baculovirus infected SF9 cells. The RhoGAP assay was performed using the Biochem kit (Cytoskeleton). Mutations were introduced into plasmids using the QuikChange Site-Directed Mutagenesis Kit (Stratagene).
Supplementary Material
Acknowledgments
We thank Drs. K. Rajashankar and I. Kourinov of the APS (Advanced Photon Source, Argonne, Il) for assistance in x-ray data collection, Dr. Angus Nairn for critical reagents, and D. Balkin, S.A. Mentone, and M. Pypaert for help with microscopy. The work was supported by the following sources: NIH (NS36251, CA46128), The G. Harold and Leila Y. Mathers Charitable Foundation, the Kavli Foundation, the Yale/NIDA Neuroproteomic Center (DA018343), the Yale DERC (DK45735), and the Yale Center for Genomics and Proteomics (PDC); Feodor Lynen Fellowship of the Alexander von Humboldt Foundation (KSE); Howard Hughes Medical Institute Fellowship of the Life Sciences Research Foundation (YM); NIH (MSTP TG 5T32GM07205) (HJM); Boehringer Ingelheim Fonds PhD Scholarship (RZ).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Coordinates of the ASH-RhoGAP like domains of OCRL have been deposited in the Protein Data Bank at the RCSB with the accession code: 2QV2.
References
- Astle MV, Seaton G, Davies EM, Fedele CG, Rahman P, Arsala L, Mitchell CA. Regulation of phosphoinositide signaling by the inositol polyphosphate 5-phosphatases. IUBMB Life. 2006;58:451–456. doi: 10.1080/15216540600871159. [DOI] [PubMed] [Google Scholar]
- Attree O, Olivos IM, Okabe I, Bailey LC, Nelson DL, Lewis RA, McInnes RR, Nussbaum RL. The Lowe’s oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature. 1992;358:239–242. doi: 10.1038/358239a0. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Rojas R. Retrograde transport from endosomes to the trans-Golgi network. Nat Rev Mol Cell Biol. 2006;7:568–579. doi: 10.1038/nrm1985. [DOI] [PubMed] [Google Scholar]
- Bork P, Holm L, Sander C. The immunoglobulin fold. Structural classification, sequence patterns and common core. J Mol Biol. 1994;242:309–320. doi: 10.1006/jmbi.1994.1582. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Caruso-Neves C, Pinheiro AA, Cai H, Souza-Menezes J, Guggino WB. PKB and megalin determine the survival or death of renal proximal tubule cells. Proc Natl Acad Sci U S A. 2006;103:18810–18815. doi: 10.1073/pnas.0605029103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury R, Diao A, Zhang F, Eisenberg E, Saint-Pol A, Williams C, Konstantakopoulos A, Lucocq J, Johannes L, Rabouille C, et al. Lowe syndrome protein OCRL1 interacts with clathrin and regulates protein trafficking between endosomes and the trans-Golgi network. Mol Biol Cell. 2005;16:3467–3479. doi: 10.1091/mbc.E05-02-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O, Di Paolo G, Wenk MR, Luthi A, Kim WT, Takei K, Daniell L, Nemoto Y, Shears SB, Flavell RA, et al. Essential role of phosphoinositide metabolism in synaptic vesicle recycling. Cell. 1999;99:179–188. doi: 10.1016/s0092-8674(00)81649-9. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, De Camilli P. Phosphoinositids in cell regulation and membrane dynamics. Nature. 2006 doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- Dressman MA, Olivos-Glander IM, Nussbaum RL, Suchy SF. Ocrl1, a PtdIns(4,5)P(2) 5-phosphatase, is localized to the trans-Golgi network of fibroblasts and epithelial cells. J Histochem Cytochem. 2000;48:179–190. doi: 10.1177/002215540004800203. [DOI] [PubMed] [Google Scholar]
- Faucherre A, Desbois P, Nagano F, Satre V, Lunardi J, Gacon G, Dorseuil O. Lowe syndrome protein Ocrl1 is translocated to membrane ruffles upon Rac GTPase activation: a new perspective on Lowe syndrome pathophysiology. Hum Mol Genet. 2005;14:1441–1448. doi: 10.1093/hmg/ddi153. [DOI] [PubMed] [Google Scholar]
- Gekle M, Mildenberger S, Freudinger R, Schwerdt G, Silbernagl S. Albumin endocytosis in OK cells: dependence on actin and microtubules and regulation by protein kinases. Am J Physiol. 1997;272:F668–677. doi: 10.1152/ajprenal.1997.272.5.F668. [DOI] [PubMed] [Google Scholar]
- Hellsten E, Evans JP, Bernard DJ, Janne PA, Nussbaum RL. Disrupted sperm function and fertilin beta processing in mice deficient in the inositol polyphosphate 5-phosphatase Inpp5b. Dev Biol. 2001;240:641–653. doi: 10.1006/dbio.2001.0476. [DOI] [PubMed] [Google Scholar]
- Hoopes RR, Jr, Shrimpton AE, Knohl SJ, Hueber P, Hoppe B, Matyus J, Simckes A, Tasic V, Toenshoff B, Suchy SF, et al. Dent Disease with mutations in OCRL1. Am J Hum Genet. 2005;76:260–267. doi: 10.1086/427887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyvola N, Diao A, McKenzie E, Skippen A, Cockcroft S, Lowe M. Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. Embo J. 2006;25:3750–3761. doi: 10.1038/sj.emboj.7601274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janne PA, Suchy SF, Bernard D, MacDonald M, Crawley J, Grinberg A, Wynshaw-Boris A, Westphal H, Nussbaum RL. Functional overlap between murine Inpp5b and Ocrl1 may explain why deficiency of the murine ortholog for OCRL1 does not cause Lowe syndrome in mice. J Clin Invest. 1998;101:2042–2053. doi: 10.1172/JCI2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferson AB, Majerus PW. Properties of type II inositol polyphosphate 5-phosphatase. J Biol Chem. 1995;270:9370–9377. doi: 10.1074/jbc.270.16.9370. [DOI] [PubMed] [Google Scholar]
- Kagiwada S, Hosaka K, Murata M, Nikawa J, Takatsuki A. The Saccharomyces cerevisiae SCS2 gene product, a homolog of a synaptobrevin-associated protein, is an integral membrane protein of the endoplasmic reticulum and is required for inositol metabolism. J Bacteriol. 1998;180:1700–1708. doi: 10.1128/jb.180.7.1700-1708.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser SE, Brickner JH, Reilein AR, Fenn TD, Walter P, Brunger AT. Structural basis of FFAT motif-mediated ER targeting. Structure. 2005;13:1035–1045. doi: 10.1016/j.str.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Lee SY, Voronov S, Letinic K, Nairn AC, Di Paolo G, De Camilli P. Regulation of the interaction between PIPKI gamma and talin by proline-directed protein kinases. J Cell Biol. 2005;168:789–799. doi: 10.1083/jcb.200409028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leheste JR, Rolinski B, Vorum H, Hilpert J, Nykjaer A, Jacobsen C, Aucouturier P, Moskaug JO, Otto A, Christensen EI, Willnow TE. Megalin knockout mice as an animal model of low molecular weight proteinuria. Am J Pathol. 1999;155:1361–1370. doi: 10.1016/S0002-9440(10)65238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichter-Konecki U, Farber LW, Cronin JS, Suchy SF, Nussbaum RL. The effect of missense mutations in the RhoGAP-homology domain on ocrl1 function. Mol Genet Metab. 2006;89:121–128. doi: 10.1016/j.ymgme.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Lin DC, Quevedo C, Brewer NE, Bell A, Testa J, Grimes ML, Miller FD, Kaplan DR. APPL1 associates with TrkA and GIPC1, and is required for NGF-mediated signal transduction. Mol Cell Biol. 2006 doi: 10.1128/MCB.00228-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yao F, Wu R, Morgan M, Thorburn A, Finley RL, Jr, Chen YQ. Mediation of the DCC apoptotic signal by DIP13 alpha. J Biol Chem. 2002;277:26281–26285. doi: 10.1074/jbc.M204679200. [DOI] [PubMed] [Google Scholar]
- Loewen CJ, Roy A, Levine TP. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. Embo J. 2003;22:2025–2035. doi: 10.1093/emboj/cdg201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, McQuistan T, Orlando RA, Farquhar MG. GAIP, GIPC and Galphai3 are concentrated in endocytic compartments of proximal tubule cells: putative role in regulating megalin’s function. J Am Soc Nephrol. 2002;13:918–927. doi: 10.1681/ASN.V134918. [DOI] [PubMed] [Google Scholar]
- Lou X, Yano H, Lee F, Chao MV, Farquhar MG. GIPC and GAIP form a complex with TrkA: a putative link between G protein and receptor tyrosine kinase pathways. Mol Biol Cell. 2001;12:615–627. doi: 10.1091/mbc.12.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M. Structure and function of the Lowe syndrome protein OCRL1. Traffic. 2005;6:711–719. doi: 10.1111/j.1600-0854.2005.00311.x. [DOI] [PubMed] [Google Scholar]
- Maehama T, Taylor GS, Slama JT, Dixon JE. A sensitive assay for phosphoinositide phosphatases. Anal Biochem. 2000;279:248–250. doi: 10.1006/abio.2000.4497. [DOI] [PubMed] [Google Scholar]
- Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8:516–523. doi: 10.1038/ncb1404. [DOI] [PubMed] [Google Scholar]
- Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383–397. doi: 10.1016/j.cell.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRee DE. XtalView/Xfit--A versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG, Zerial M. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004;116:445–456. doi: 10.1016/s0092-8674(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Mitsuuchi Y, Johnson SW, Sonoda G, Tanno S, Golemis EA, Testa JR. Identification of a chromosome 3p14.3-21.1 gene, APPL, encoding an adaptor molecule that interacts with the oncoprotein-serine/threonine kinase AKT2. Oncogene. 1999;18:4891–4898. doi: 10.1038/sj.onc.1203080. [DOI] [PubMed] [Google Scholar]
- Monnier N, Satre V, Lerouge E, Berthoin F, Lunardi J. OCRL1 mutation analysis in French Lowe syndrome patients: implications for molecular diagnosis strategy and genetic counseling. Hum Mutat. 2000;16:157–165. doi: 10.1002/1098-1004(200008)16:2<157::AID-HUMU8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Naccache SN, Hasson T, Horowitz A. Binding of internalized receptors to the PDZ domain of GIPC/synectin recruits myosin VI to endocytic vesicles. Proc Natl Acad Sci U S A. 2006;103:12735–12740. doi: 10.1073/pnas.0605317103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai J, Christensen EI, Morris SM, Willnow TE, Cooper JA, Nielsen R. Mutually dependent localization of megalin and Dab2 in the renal proximal tubule. Am J Physiol Renal Physiol. 2005;289:F569–576. doi: 10.1152/ajprenal.00292.2004. [DOI] [PubMed] [Google Scholar]
- Nagai M, Meerloo T, Takeda T, Farquhar MG. The adaptor protein ARH escorts megalin to and through endosomes. Mol Biol Cell. 2003;14:4984–4996. doi: 10.1091/mbc.E03-06-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechamen CA, Thomas RM, Cohen BD, Acevedo G, Poulikakos PI, Testa JR, Dias JA. Human follicle-stimulating hormone (FSH) receptor interacts with the adaptor protein APPL1 in HEK 293 cells: potential involvement of the PI3K pathway in FSH signaling. Biol Reprod. 2004;71:629–636. doi: 10.1095/biolreprod.103.025833. [DOI] [PubMed] [Google Scholar]
- Norden AG, Lapsley M, Igarashi T, Kelleher CL, Lee PJ, Matsuyama T, Scheinman SJ, Shiraga H, Sundin DP, Thakker RV, et al. Urinary megalin deficiency implicates abnormal tubular endocytic function in Fanconi syndrome. J Am Soc Nephrol. 2002;13:125–133. doi: 10.1681/ASN.V131125. [DOI] [PubMed] [Google Scholar]
- Olivos-Glander IM, Janne PA, Nussbaum RL. The oculocerebrorenal syndrome gene product is a 105-kD protein localized to the Golgi complex. Am J Hum Genet. 1995;57:817–823. [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Peck J, Douglas Gt, Wu CH, Burbelo PD. Human RhoGAP domain-containing proteins: structure, function and evolutionary relationships. FEBS Lett. 2002;528:27–34. doi: 10.1016/s0014-5793(02)03331-8. [DOI] [PubMed] [Google Scholar]
- Piwon N, Gunther W, Schwake M, Bosl MR, Jentsch TJ. ClC-5 Cl- -channel disruption impairs endocytosis in a mouse model for Dent’s disease. Nature. 2000;408:369–373. doi: 10.1038/35042597. [DOI] [PubMed] [Google Scholar]
- Ponting CP. A novel domain suggests a ciliary function for ASPM, a brain size determining gene. Bioinformatics. 2006 doi: 10.1093/bioinformatics/btl022. [DOI] [PubMed] [Google Scholar]
- Rodman JS, Kerjaschki D, Merisko E, Farquhar MG. Presence of an extensive clathrin coat on the apical plasmalemma of the rat kidney proximal tubule cell. J Cell Biol. 1984;98:1630–1636. doi: 10.1083/jcb.98.5.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid AC, Wise HM, Mitchell CA, Nussbaum R, Woscholski R. Type II phosphoinositide 5-phosphatases have unique sensitivities towards fatty acid composition and head group phosphorylation. FEBS Lett. 2004;576:9–13. doi: 10.1016/j.febslet.2004.08.052. [DOI] [PubMed] [Google Scholar]
- Shin HW, Hayashi M, Christoforidis S, Lacas-Gervais S, Hoepfner S, Wenk MR, Modregger J, Uttenweiler-Joseph S, Wilm M, Nystuen A, et al. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J Cell Biol. 2005;170:607–618. doi: 10.1083/jcb.200505128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stauffer TP, Ahn S, Meyer T. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr Biol. 1998;8:343–346. doi: 10.1016/s0960-9822(98)70135-6. [DOI] [PubMed] [Google Scholar]
- ter Haar E, Harrison SC, Kirchhausen T. Peptide-in-groove interactions link target proteins to the beta-propeller of clathrin. Proc Natl Acad Sci U S A. 2000;97:1096–1100. doi: 10.1073/pnas.97.3.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima JY, Toshima J, Kaksonen M, Martin AC, King DS, Drubin DG. Spatial dynamics of receptor-mediated endocytic trafficking in budding yeast revealed by using fluorescent alpha-factor derivatives. Proc Natl Acad Sci U S A. 2006;103:5793–5798. doi: 10.1073/pnas.0601042103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujishita Y, Guo S, Stolz LE, York JD, Hurley JH. Specificity determinants in phosphoinositide dephosphorylation: crystal structure of an archetypal inositol polyphosphate 5-phosphatase. Cell. 2001;105:379–389. doi: 10.1016/s0092-8674(01)00326-9. [DOI] [PubMed] [Google Scholar]
- Ungewickell A, Ward ME, Ungewickell E, Majerus PW. The inositol polyphosphate 5-phosphatase Ocrl associates with endosomes that are partially coated with clathrin. Proc Natl Acad Sci U S A. 2004;101:13501–13506. doi: 10.1073/pnas.0405664101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varsano T, Dong MQ, Niesman I, Gacula H, Lou X, Ma T, Testa JR, Yates JR, 3rd, Farquhar MG. GIPC is recruited by APPL to Peripheral TrkA Endosomes and Regulates TrkA Trafficking and Signaling. Mol Cell Biol. 2006 doi: 10.1128/MCB.00305-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Jefferson AB, Auethavekiat V, Majerus PW. The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc Natl Acad Sci U S A. 1995;92:4853–4856. doi: 10.1073/pnas.92.11.4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Perera RM, Sebastian R, Nakatsu F, Chen H, Balla T, Ayala G, Toomre D, De Camilli PV. Loss of endocytic clathrin-coated pits upon acute depletion of phosphatidylinositol 4,5-bisphosphate. Proc Natl Acad Sci U S A. 2007;104:3793–3798. doi: 10.1073/pnas.0611733104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Z, Chung B, Nguyen T, Mentone S, Thomson B, Biemesderfer D. Linking receptor-mediated endocytosis and cell signaling: evidence for regulated intramembrane proteolysis of megalin in proximal tubule. J Biol Chem. 2004;279:34302–34310. doi: 10.1074/jbc.M405608200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.