

Abstract
The GIT proteins, GIT1 and GIT2, are GTPase-activating proteins for the ADP-ribosylation factor family of small GTP binding proteins, but also serve as adaptors to link signaling proteins to distinct cellular locations. One role for GIT proteins is to link the PIX family of Rho guanine nucleotide exchange factors and their binding partners, the p21-activated protein kinases, to remodeling focal adhesions by interacting with the focal adhesion adaptor protein paxillin. We here identified the C-terminal domain of GIT1 responsible for paxillin binding. Combining structural and mutational analysis, we show that this region folds into an antiparallel four-helix domain highly reminiscent to the focal adhesion targeting (FAT) domain of focal adhesion kinase (FAK). Our results suggest that the GIT1 FAT-homology (FAH) domain and FAT bind the paxillin LD4 motif quite similarly. Since only a small fraction of GIT1 is bound to paxillin under normal conditions, regulation of paxillin binding was explored. Although paxillin binding to the FAT domain of FAK is regulated by tyrosine phosphorylation within this domain, we find that tyrosine phosphorylation of the FAH domain GIT1 is not involved in regulating binding to paxillin. Instead, we find that mutations within the FAH domain may alter binding to paxillin that has been phosphorylated within the LD4 motif. Thus, despite apparent structural similarity in their FAT domains, GIT1 and FAK binding to paxillin is differentially regulated.
Keywords: GIT1 protein, paxillin, focal adhesion targeting domain, PIX protein, Arf GTPase-activating protein, guanine nucleotide exchange factor
The GIT and PIX proteins tightly associate to form an oligomeric complex that functions as a recruitable scaffold for various signaling proteins [1]. The GIT proteins, GIT1 and GIT2 (also known as p95-APP1/2, p95-PKL, and Cat-2 proteins), are GTPase-activating proteins for the Arf family of small GTP-binding proteins [2-4]. The PIX proteins, α–PIX and β–PIX (also known as Cool-1/2, p85-SPR and ARHGEF6/7 proteins), are guanine nucleotide exchange factors for the Rho family of small GTP-binding proteins [5]. In addition to functioning as regulators of small GTP-binding proteins, GIT/PIX complexes have been reported to bind a variety of signaling molecules, including GRKs, PAKs, FAK, POPX, PLC–γ, and MEK proteins [2, 5-9]. Through regulated binding to molecules having distinct cellular localizations, GIT/PIX complexes and associated signaling molecules can be transiently localized to many parts of the cell, including focal adhesions, membrane ruffles, intracellular vesicles and synapses, by binding to proteins such as paxillin/Hic-5, liprin-α, shank or scribble [10-13].
GIT proteins have been shown to bind to paxillin and the paxillin family member Hic-5 [3, 6, 10, 14]. GIT proteins act as adaptors to link GIT-associated proteins such as PIX - together with PIX-binding partners, the PAKs - to focal adhesions [6, 10, 15]. GIT proteins bind paxillin at its LD4 motif, and mutation of the paxillin LD4 motif prevents GIT protein binding and prevents GIT protein localization to focal adhesions [10, 15, 16]. Two putative paxillin binding sites (PBS) on GIT proteins were identified based on similarity to other paxillin-binding proteins, and binding was localized to a short carboxyl terminal region, PBS2 [10, 15], although this binding site has never been precisely defined through mutagenesis.
Like GIT proteins, focal adhesion kinase (FAK) is targeted to focal adhesions by binding to paxillin [17-19]. Indeed, GITs and FAK share a binding site on paxillin, the LD4 motif [20]. In FAK, the extreme carboxyl terminal region is known as the “focal adhesion targeting” (FAT) domain, since this is the paxillin and talin binding site and this domain is required for focal adhesion localization [19]. The structure of the FAT domain of FAK, both alone or bound to paxillin LD motif peptides, has been determined [21-25]. The FAT sequence folds to form a single structural domain comprised of a bundle of four anti-parallel alpha helices. The FAT domain contains two paxillin LD motif binding sites, one between helices 1 and 4, and a second on the opposite side between helices 2 and 3 [24]. In vitro, the paxillin LD2 and LD4 peptides each bind to either site, although a preference of LD2 for site 1/4 and LD4 for site 2/3 has been observed [24-26]. Thus, it is likely that one molecule of FAK binds to both the LD2 and LD4 motifs of one paxillin molecule at the same time.
Combining structural and functional analyses, we have characterized the structure of the carboxyl terminal domain of GIT1 responsible for paxillin binding. We show that the folding and paxillin LD4 binding mode of this domain are very similar to the FAT domain of FAK. The binding of LD4 to these domains however is regulated differently.
Experimental Procedures
Materials
Tissue culture media and supplements were obtained from Life Technologies (Rockville, MD). General chemicals were from Sigma (St. Louis, MO). Restriction enzymes were from New England Biolabs (Beverly, MA). Electrophoresis chemicals, protein size standards and nitrocellulose filters were from BioRad (Hercules, CA). Anti-mouse and anti-rabbit secondary antibody-horseradish peroxidase conjugates were from Amersham Biosciences (Piscataway, NJ), and anti-mouse secondary antibody-horseradish peroxidase conjugate selected to be non-crossreactive with denatured mouse IgG chains (TrueBlot) was from eBioscience (San Diego, CA). SuperSignal West Pico chemiluminescent substrate was from Pierce (Rockford, IL). OctA (Flag) probe, 6xHis probe, myc probe, HA probe, GIT1 (H-170) and PAK antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Flag M2 antibody, M2 Flag-HRP conjugate and M2 Flag-agarose were obtained from Sigma (St. Louis, MO). 12CA5 anti-HA monoclonal antibody was from Roche Applied Sciences (Indianapolis, IN), 12CA5 anti-HA-Sepharose was from Covance (Richmond, CA), and HA-HRP conjugate was from Abcam (Cambridge, MA). Anti-paxillin mouse monoclonal antibody 349 was from BD/Transduction Labs (San Diego, CA) and anti-paxillin rabbit monoclonal antibody from Epitomics (Burlingame, CA). Antisera were raised in rabbits by Cocalico Labs (Reamstown, PA) using full-length rat GIT1/6xHis or human GIT2-short/6xHis proteins purified from recombinant baculovirus-infected Sf9 cells as described [3]. Antiserum raised against β–PIX [27] was the gift of Dr. R. Cerione (Cornell University, Ithaca).
cDNA clones and constructs
pBK(Δ)-rat GIT1/Flag, pBK(Δ)Flag-GIT1, pBK(Δ)-GIT1(ΔABC)/Flag, pBK(Δ)-GIT1/6xHis, pBK(Δ)HA-α-PIX and pBK(Δ)HA-β-PIX have been described previously [2, 3]. GIT1/EGFP was prepared by replacing the Flag cassette of pBK(Δ)-GIT1/Flag with EGFP (from pEGFP-C1 (Invitrogen)) using EcoRI and XbaI sites. pCS2-myc-Hic-5 [28] was obtained from Dr. Sheila Thomas (Harvard Medical School). pcDNA3.1-chicken paxillin [10] was obtained from Dr. Chris Turner (SUNY Upstate Medical Center, Syracuse). Site-directed mutations were prepared using specific oligonucleotide primers using the QuickChange mutagenesis kit (Stratagene, La Jolla, CA). All clones were confirmed by DNA sequencing.
Cell culture and transfection
COS-7 and HEK293 cells were obtained from ATCC, and maintained in DMEM or MEM media supplemented with 10% fetal calf serum and penicillin/streptomycin at 37°C under 5% CO2. COS-7 cells at 70% confluency in 15 cm plates were transiently transfected with a total of 10 μg of plasmid DNA and 60 μl of Lipofectamine (Life Technologies, Rockville, MD).
Immunoprecipitation and Immunoblotting
Forty-eight hours after transfection, cells were rinsed with cold phosphate buffered saline and scraped into lysis buffer (20 mM Tris–HCl, 1 mM EDTA, 100 mM NaCl, 5 μg/ml aprotinin, 150 μg/ml benzamidine, 5 μg/ml leupeptin, 4 μg/ml pepstatin, and 20 mg/ml phenylmethylsulfonyl fluoride, pH 7.4). Cells were lysed by repeated passage through a 26-gauge needle, and spun at 21,000×g for 20 minutes at 4°C. The soluble fraction was removed and used for immunoprecipitation. One milliliter of lysate was transferred to a new tube and 25 μl of a 50% slurry of M2 Flag-agarose beads or HA-Sepharose beads was added, or 1 μg of primary antibody and 40 μl of Protein A/G beads (Calbiochem, San Diego, CA). The lysate was rotated at 4°C for 4 hours, and the beads washed three times with lysis buffer. Immunoprecipitated proteins were removed from beads by addition of 40 μl of SDS-sample buffer and heating at 95°C for 10 minutes. Proteins were separated on 10% polyacrylamide gels and transferred to nitrocellulose filters. Filters were incubated with 3% bovine serum albumin in Tween-Tris-buffered saline (20 mM Tris, pH 7.4, 500 mM NaCl, 0.2% Tween-20) to block non-specific binding sites, exposed to primary antibodies, washed, and exposed to secondary antibody-peroxidase conjugates. After final washes, immunoreactive bands were detected by chemiluminescence and exposure to X-ray film.
Immunocytochemistry and microscopy
A7r5 cells were grown on fibronectin-coated glass coverslips. After transfection with either wt or mutant GIT1/EGFP constructs, cells were incubated for 48 hours, washed twice with PBS and fixed with 4% paraformaldehyde in PBS for 10 min at RT. After another two washes with PBS, cells were permeabilized using 0.1% Triton X-100 in PBS for 10 min at RT. Cells were then blocked with 1% BSA in PBS for 1 hr at RT and washed twice with PBS. Endogenous paxillin and actin were detected with an anti-paxillin-TRITC labeled antibody (1:1000 in 1% BSA/PBS, BD Transduction Labs) or Alexa633-phalloidin (1:2000 in 1% BSA/PBS, Molecular Probes) incubated for 1 hour at RT followed by two washes with PBS. Cells were then air dried at RT and mounted on microscope slides using FluorSave Reagent (Calbiochem). Pictures were taken with a Zeiss LSM 510 Meta confocal microscope.
GST-LD pulldown assays
GST-LD motif constructs were prepared by ligating annealed oligonucleotides encoding LD repeat sequences of paxillin, Hic-5 or leupaxin into the pGEX 4T-1 vector (Amersham). For paxillin, the LD4 construct encodes amino acid residues 263-277 from human paxillin. Paxillin LD4 S272D, S274D and S272D/S274D mutants were also prepared. For Hic-5, the LD3 construct encodes amino acid residues 154-168 from mouse Hic-5, and for leupaxin, the LD3 construct encodes amino acid residues 89-103 from human leupaxin. GST fusions were induced by treating growing bacteria with isopropyl β-d-thiogalactoside and the induced proteins were purified on glutathione-agarose beads. To 5 μg of fusion proteins on beads, crude Sf9 lysates expressing GIT1/6xHis was added in lysis buffer and incubated for 30 min at 4°C. Beads were collecting by centrifugation and washed 3 times with lysis buffer, prior to SDS-PAGE and Coomassie blue staining or immunoblotting.
Homology modeling
Homology modeling of rat GIT1(647-770) was carried out with SWISS-MODEL [30] using the unliganded FAT domain (residues 916 – 1047 of PDB entry 1K05) as a template.
E. coli protein production and purification
The rat GIT1(647-770) fragment was subcloned into pGEX-4T-1 (Novagen), and transformed into E. coli BL21(DE3) cells. Bacteria were grown in LB media at 30°C for 6 h after induction with 0.2 mM IPTG. Cells were centrifuged 20 minutes at 4000 g and the pellet was frozen in −80°C for subsequent purification. Cells were resuspended in lysis buffer (200 mM NaCl, 50 mM Tris pH 8, 5 mM β-mercaptoethanol, 0.4% Triton X-100, 5 mM EDTA), and lysed by mild sonication. Proteins and bacterial membranes were separated by centrifugation (45 minutes, at 35,000 g) and the supernatent was applied to glutathione-Sepharose 4B beads (Pharmacia). The column was washed thoroughly with 500 mM NaCl, 50 mM Tris pH 8 and 5 mM β-mercaptoethanol. Beads were resuspended in cleavage buffer (200 mM NaCl, 50 mM Tris pH 8, 5 mM β-mercaptoethanol, 5 mM EDTA), and the GST tag was cleaved by 1 h incubation with thrombin. Protease-eluted protein was further purified by size-exclusion chromatography using a Superdex 75 column (Pharmacia) in gel filtration buffer (200 mM NaCl, 20 mM Tris pH 8, 5 mM β-mercaptoethanol).
Isothermal Titration Calorimetry (ITC)
Cleaved recombinant GIT1(647-770) was dialysed in ITC buffer (150 mM NaCl, 20 mM Hepes pH 7.5, 1 mM TCEP) and placed in the 1.4 ml sample cell at a concentration of 50 μM. Peptides were dissolved directly into the dialysate at a concentration of 1 mM and placed in the injection syringe. For each titration, peptide was injected at 25°C in five minute intervals, using injection sequences of 2/10/10 μl followed by 18 injections of 15 μl. All peptides were also titrated into ITC buffer alone, and the resulting heat of dilution was subtracted from the binding curves. ITC was performed using a VP-ITC MicroCalorimeter from MicroCal Incorporated, and data were fit with Origin software.
Circular Dichroïsm (CD)
FAT(892-1052) [24] and GIT1(647-770) were dialysed in specific CD buffer (150mM NaF, 20mM NaPO4 pH 7.5, 1mM TCEP). CD spectra were performed on a Chirascan circular dichroïsm spectrometer (Applied Photophysics), using protein concentrations of 0.3 μM. CD spectra were recorded at 20°C between 180 and 250 nm with a step of 0.5 nm, a bandwidth of 1 nm and an optical path of 0.02 cm.
Small Angle X-ray Scattering (SAXS)
Data were collected at beamline X33, at DESY, EMBL, Hamburg, at 10°C, using a wavelength of λ=1.5 Å. GIT1(647-770) in gel filtration buffer was used at a concentration of 21 mg/ml (apo-GIT1(647-770)), and of 8 mg/ml in presence of 3 mg/ml chicken paxillin LD4 peptide (S262ATRELDELMMASLSDFKFMAQGK). The ratio of GIT1(647-770) to LD4 peptide was 1:2. Prior to data recording, the samples were extensively centrifuged to eliminate aggregates, and supplemented with 2 mM DTT. Diffusion spectra for buffer only were taken before and after the protein sample, averaged, and subtracted from the protein scattering curve. Data analysis and ab initio shape calculations were performed using PRIMUS, GNOM, GASBOR, DAMMIN, CREDO, CRYSOL and DAMAVER from the SAXS program suite by D. Svergun and colleagues [31].
Results
Alignment of GIT1 and GIT2 protein sequences from diverse species reveals that the extreme carboxyl terminal ~130 residues of these proteins exhibit an unusually high degree of sequence conservation (Figure 1A). While residues 643-679 had been proposed to form a paxillin binding sequence (PBS2) in chicken GIT2, and deletion of residues 643-679 in chicken GIT2 leads to a protein that no longer binds to a paxillin LD4-GST fusion protein in vitro [15], the ~75 residues beyond this PBS2 region to the end of the protein have no known function in any GIT family member. Based on the over 20 % sequence identity (50 % similarity) with the FAK focal adhesion targeting (FAT) domain, we have recently predicted that the entire 130 residue carboxyl terminal region of chicken GIT2 folds into a 4-helix bundle to form a paxillin binding domain analogous to the FAT domain of FAK [24].
Figure 1.

GIT1 has a conserved carboxyl terminal sequence that is required for paxillin binding.
A. Alignment of carboxyl terminal sequences of rat and chicken GIT1 and GIT2 proteins. Residues mutated in this study are indicated above the alignment by the amino acid that was substituted, and the location of inserted stop codons in the three truncation mutants are indicated by asterisks. Predicted alpha helical regions in the rat GIT1 model are underlined. Predicted PBS2 region in chicken GIT2 is underlined. Residue numbers in rat GIT1 are shown below the alignment.
B. Paxillin does not bind to GIT1 lacking the carboxyl terminal four-helix bundle, or any of the bundle helices. The indicated Flag-GIT1 truncation mutants were transfected into COS7 cells. Cells were scraped and lysed after 48 hours, and soluble lysates immunoprecipitated by addition of M2 Flag-agarose beads. Co-immunoprecipitated endogenous paxillin and β–PIX were measured by immunoblotting with specific antisera, and precipitated Flag-GIT1 directly visualized with M2 Flag-HRP conjugate.
We reasoned that if the entire carboxyl terminal folds to form a compact 4-helix bundle FAT Homology (FAH) domain, the residues beyond the previously described PBS2 should be critical to form the functional paxillin binding site. On the other hand, if the predicted PBS2 region alone were sufficient for paxillin binding, removal of these final 75 residues may have little or no effect on paxillin binding. We verified that the major paxillin binding site in GIT1 resides at the carboxyl terminus by truncating the protein to remove the predicted PBS2 region and all residues beyond it, GIT1(1-646). We also created truncated forms of GIT1 containing the predicted PBS2 region but lacking portions of the conserved stretch of amino acids that follows it. GIT1(1-705) contains the entire predicted PBS2, but is missing all the conserved region beyond this (i.e., predicted alpha helices 3 and 4), while GIT1(1-735) lacks the only last half of this conserved domain beyond the PBS2 (i.e., predicted alpha helix 4). These constructs, together with full length Flag-GIT1, were expressed in COS-7 cells, immunoprecipitated with M2 Flag beads, and immunoblotted for co-precipitated endogenous paxillin (Figure 1B). All four constructs were well expressed and bound endogenous β-PIX, suggesting appropriate overall folding. None of the three truncated forms of GIT1 demonstrated any binding to paxillin. Thus, the major paxillin binding site does appear to reside in the carboxyl terminal region of GIT1, as it does in chicken GIT2 [15], but sequences beyond the originally-identified PBS2 region also appear critical for paxillin binding.
We next expressed the GIT1 FAT-homology (FAH) region (residues 647-770 of rat GIT1) as a recombinant protein in E. coli. CD analysis of GIT1(647-770) revealed a spectrum characteristic for an all-alpha helix protein. Moreover, the GIT1 CD spectrum showed remarkable similarity to that of the FAT domain of FAK (Figure 2). Thus, the GIT1 FAH domain forms an independently folded domain with secondary structure features matching those of FAT.
Figure 2.
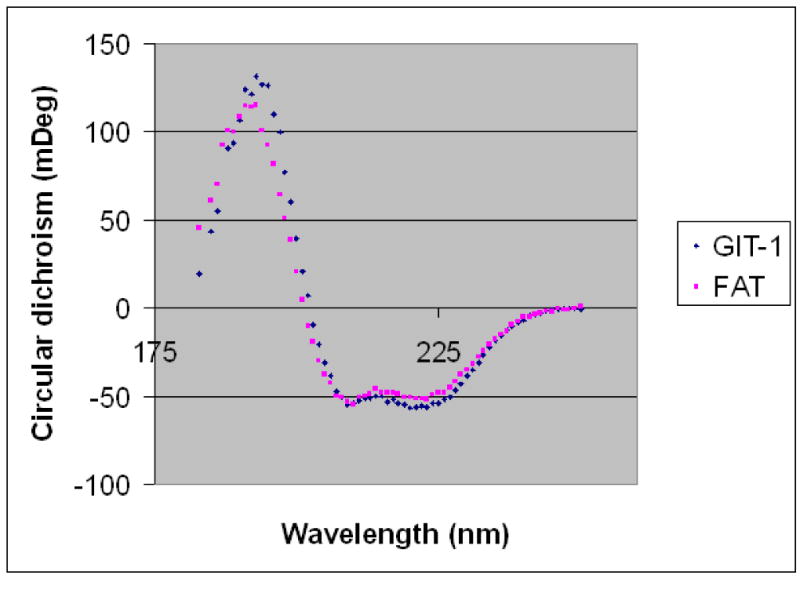
Circular dichroism comparison of the GIT1 FAH domain and the FAK FAT domain. Circular dichroism spectra of FAT(892-1052) (violet) and GIT1(647-770) (blue) at 0.3 μM concentrations are superimposed.
We then used small angle X-ray scattering (SAXS) to investigate the structure of the GIT1 FAH domain. SAXS data on GIT1 FAH were recorded to a maximum resolution of s=4.48 nm-1. Guinier plot and GNOM analysis yielded a radius of gyration (Rg) of 1.9 nm for GIT1 FAH. This Rg is in good agreement with the Rg value of 1.7 nm calculated for FAT (residues 916-1047, PDB entry 1K05), considering that the Rg values calculated from model coordinates do not include a hydration shell. The shape of the SAXS curve obtained for GIT1(647-770) is typical for a rod-shaped object, like the one formed by FAT. Indeed, the scattering curve calculated from FAT (CRYSOL) fitted the experimental data of GIT1 FAH extremely well (χ=0.28), whereas other helix-containing domains of same size and Rg gave a significantly worse fit (Figure 3A). In fact, this approach even indicates that the structure of GIT FAH more closely resembles the 4-helical structure of FAT than the 4-helix bundle of the vinculin paxillin LD motif-binding tail domain (Figure 3A).
Figure 3.
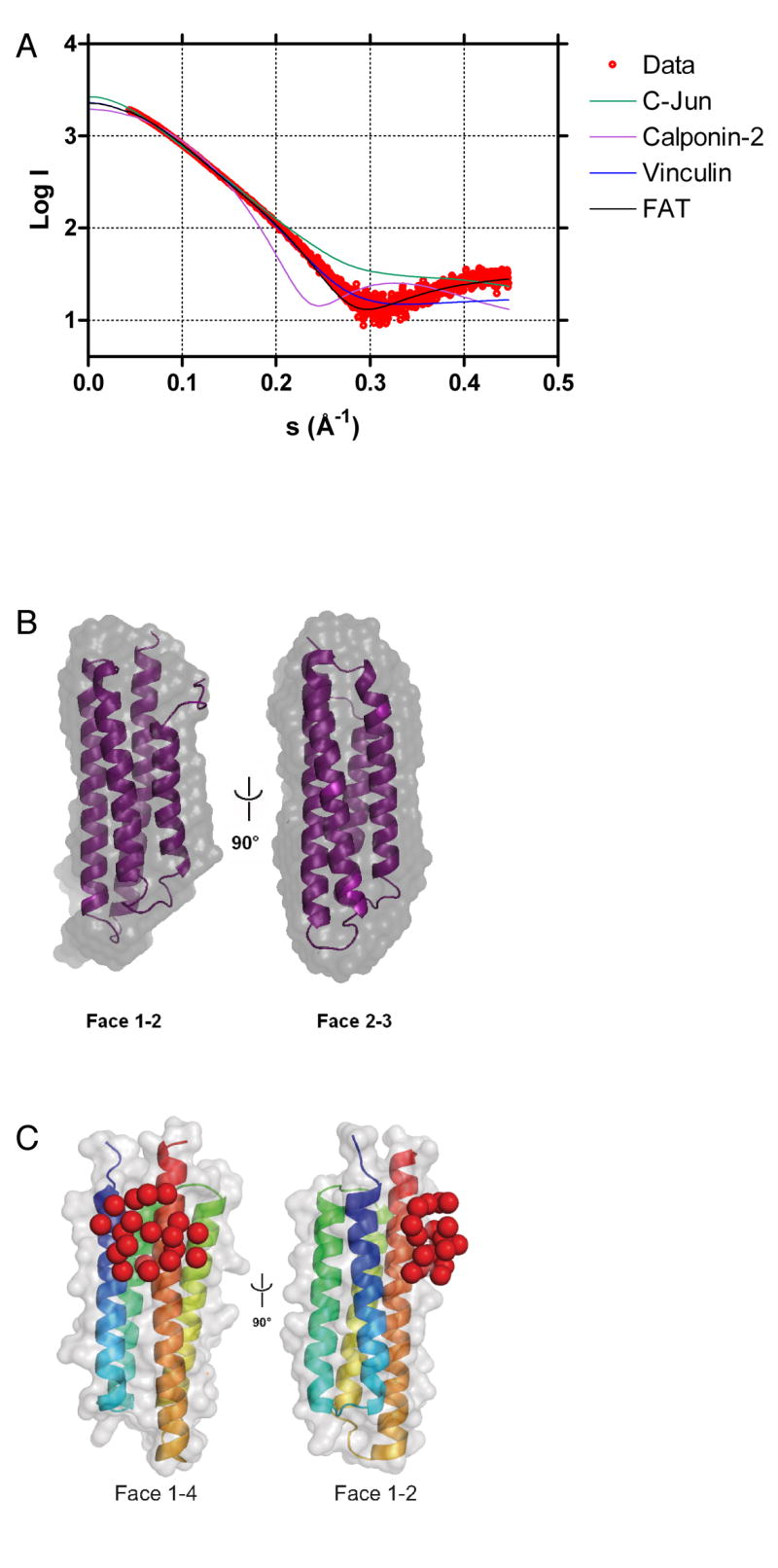
Small angle X-ray scattering.
A. SAXS versus model. GIT1(647-770) SAXS scattering curve (red) superimposed by CRYSOL to the calculated scattering pattern of the FAK FAT domain (residues 916 – 1047 of PDB entry 1K05 [21]; black), the 4-helix bundle of the vinculin tail domain (residues 917 – 1049 of PDB entry 1QKR [50]; blue), the CH domain of human calponin-2 (PDB entry 1WYN; pink) and the coiled-coil homodimer of C-Jun (PDB entry 1JUN [51]; green).
B. SAXS shape. The FAK FAT domain (residues 916 – 1047 of PDB entry 1K05 [21]) (violet) fitted into the SAXS envelope of GIT1(647-770) (grey), obtained from averaging ten individual GASBOR runs.
C. Paxillin LD4 peptide binding. Violet pseudo-residues corresponding to the LD4 peptide positioned by CREDO onto the GIT1(647-770) homology model.
Moreover, ab initio structure reconstructions (GASBOR) yielded rod-shaped structures (Figure 3B). The molecular envelope obtained after averaging and filtering ten individual GASBOR models (DAMAVER) compares very well in shape and size with the FAT domain [normalized spatial discrepancy = 0.76 (SUPCOMB).
To experimentally localize the LD4 peptide on the GIT1 FAH domain, we recorded SAXS data on GIT1 FAH in presence of two-fold excess of LD4 peptide. SAXS scattering patterns and distance distribution functions were very similar between LD4-bound and apo-GIT1, indicating that LD4 binding did not introduce gross structural changes. We then used CREDO on the LD4:GIT1 FAH SAXS data to localize the LD4 peptide on the GIT1 FAH domain. Individual CREDO runs placed the 23 pseudo-residues corresponding to the LD4 peptide either on face 1/4 or face 2/3 of the 4 helix bundle (Figure 3C). Given the moderate resolution of the SAXS data, faces 1/4 and 2/3 are very similar, both being composed of a long and a short helix. In contrast, faces 1/4 or 2/3 can be distinguished from face 1/2 (being composed of two short helices) and face 3/4 (two long helices). Therefore, SAXS suggests that LD4 binds GIT1(647-770) either on face 1/4 or 2/3, but not on faces 1/2 or 3/4.
Together, our structural data show that the last 130 amino acids of GIT family proteins folds into a compact, FAT-like anti-parallel 4-helix bundle. We have therefore used the FAT structure as a template, together with our experimental data, to produce a three-dimensional model of the FAH region of rat GIT1 (Figure 4). The GIT1 FAH sequence is very compatible with the FAT fold, yielding a model without steric clashes and a Verify3D score [32] close to the one obtained for the FAT crystal structure (0.27 and 0.23 for GIT1(647-770) and FAT, respectively). The molecular model established for GIT1 FAH reveals a potential paxillin LD binding site between helices 1 and 4, where a hydrophobic patch is surrounded by positive charges (Figure 4).
Figure 4.
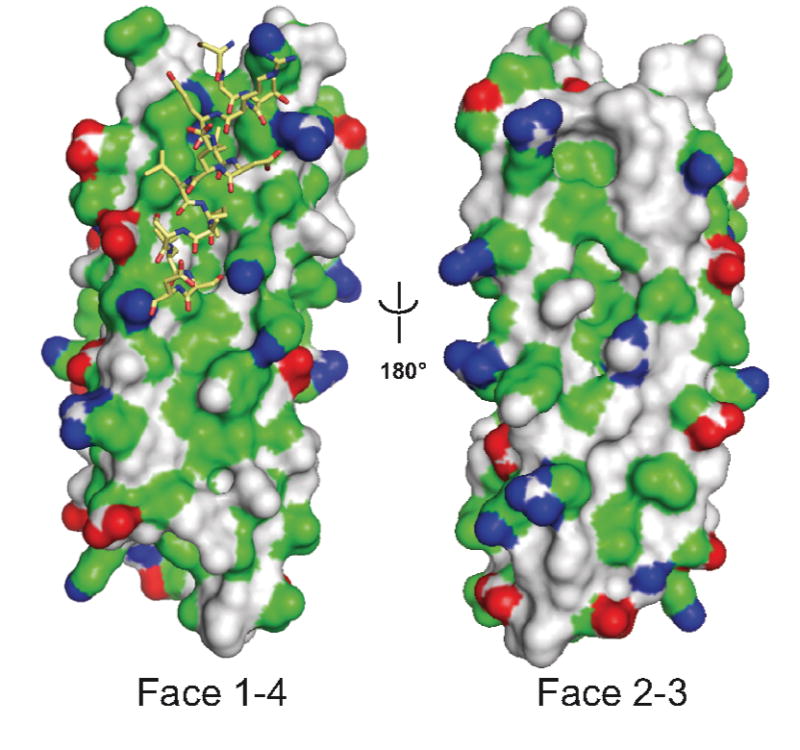
Model of rat GIT1 carboxyl terminal domain as a FAH four-helix bundle. GIT1(647-770) was modeled on the X-ray structure of the human FAK FAT domain as described in Experimental Procedures. The GIT1 FAH domain is represented by its molecular surface (red, negatively charged atoms; blue, positively charged atoms; green hydrophobic atoms, and grey polar atoms). The LD4 peptide (stick model) has been docked manually as suggested by experimental data.
To test this proposed structure experimentally, we mutated individual residues throughout the entire carboxyl terminal region of GIT1 based on the FAH model (see Figure 1A). One set of mutations is within the originally proposed PBS2 region, residues 654-690 in rat GIT1. This region corresponds to helix 1 and the first half of helix 2 in the FAH domain model, and residues predicted to contribute to the LD4 binding surface in helix 1 were particularly targeted. Another set of mutations is of residues predicted to form part of the LD4 binding surface on predicted helix 4. These are quite distal from the originally proposed PBS2, but closely apposed to helix 1 in the model. A third class of mutations is in regions of GIT1 predicted to be on the surface of predicted helices 2 and 3, in positions equivalent to the second LD binding site in FAK.
GIT1/Flag was mutated, expressed in COS-7 cells, and immunoprecipiated with M2 Flag-beads to measure the co-immunoprecipitation of endogenous paxillin (Figure 5A). In every case, the mutant GIT1 expressed well and co-immunoprecipitated normally with endogenous β-PIX. Within putative helix 1, residues I655, T662, K663 and Q666 are proposed to be located on the surface of the helix 1/4 interface. By analogy to basic residues on the FAT LD binding sites, GIT1 residue K663 could contribute to LD4 motif binding by forming a salt bridge with the acidic residues of the paxillin LD4 motif, or by forming hydrogen bonds with peptide backbone carbonyls. The GIT1/Flag K663E mutation results in substantially decreased paxillin co-immunoprecipitation. Among the helix 1 residues predicted to participate in mainly hydrophobic and/or polar interactions with LD4 (I655, T662 and Q666), both T662E and Q666E mutants had reduced paxillin binding, while the I655E mutant seemed largely unaffected. The nearby conserved residue E667 is not predicted to be on the surface of the helix 1/4 interface, but the E667R charge swap mutation resulted in increased interaction of paxillin with GIT1, presumeably by influencing the conformation of K663. We also performed a charge swap mutation between another two conserved nearby residues, K676 and D678, which are proposed to form part of the loop between helices 1 and 2. The K676D/D678K double mutant of GIT1 had an unchanged interaction with paxillin (data not shown). Within predicted helices 2 and 3, V681, H688, T692, A718, and Q722 are suggested to form the mainly hydrophobic surface of the helix 2/3 interface. The V681E, T692E, A718K and Q722E mutations had little or no effect on paxillin interaction with GIT1, while H688A increased binding. The only mutation on the 2/3 interface that affected paxillin binding, H688A, probably causes an allosteric change, such as stabilization of the GIT1 FAH fold. Like residue K663, K758 within helix 4 is predicted to either form a salt bridge to negatively charged side chains of the LD4 motif, or to form a hydrogen bond with its backbone. The K758E mutant bound essentially no paxillin above background, in accord with its important role in the model. Residue I747 is also proposed to be located on the surface of the helix 1/4 interface and to participate in nonpolar interactions with LD4, and mutation I747E also strongly reduced paxillin association. However, from its position in the GIT1 model, it can not be excluded that the effect of I747E is due to a destabilization of the hydrophobic core of the domain. We combined the two most effective point mutations to create a GIT1 protein with essentially no ability to bind to paxillin. This GIT1/Flag K663E/K758E double mutant binds paxillin at background levels only (data not shown). In agreement, purified recombinant GIT1(647-770) K663E/K758E failed to show a significant affinity for LD4 peptide in isothermal titration calorimetry experiments (Table I).
Figure 5.

Paxillin binding to GIT1 FAH domain mutants.
A. Point mutants within the GIT1 carboxyl terminal four-helix bundle identify the paxillin site as the helix 1/4 interface. The indicated GIT1/Flag mutants were transfected into COS7 cells. Cells were scraped and lysed after 48 hours, and soluble lysates immunoprecipitated by addition of M2 Flag-agarose beads. Co-immunoprecipitated endogenous paxillin and β–PIX were measured by immunoblotting with specific antisera, and precipitated GIT1/Flag directly visualized with M2 Flag-HRP conjugate.
B. Mutational analysis mapped onto the surface of the GIT1 model. Mutated residues shown in Figure 1A and analyzed in Figure 3B (and Figure 7 and Figure 9) are colored according to their effect on paxillin binding: red: reduced LD4 binding, green: no influence on LD4 binding, blue: enhanced LD4 binding, grey: not mutated. Together, these data suggest that GIT1 binds LD4 between helices 1 and 4, but not helices 2 and 3.
TABLE I.
Isothermal calorimetry data
| N | Kd (μM) | ΔH(kJ/mol) | TΔS (kJ/mol) | ΔG(kJ/mol) | |
|---|---|---|---|---|---|
| Git1647-770 : LD4 | 0.8 | 10 | -29.7 | -1.3 | -23.2 |
| Git1647-770 : LD4-p273 | 1* | 123 | -16.0 | 6.3 | -22.3 |
| Git1647-770 : LD4-p275 | 1.2 | 18 | -19.0 | 7.8 | -26.8 |
| Git1647-770 : LD4 (K663E,K758E) | N.D. | > 500 | N.D. | N.D. | N.D. |
N.D., not determined.
, value imposed and fixed in fitting procedure.
Thus, we have demonstrated that mutation of several individual residues within the predicted helix 1/4 interface, including residues well outside the originally proposed PBS2 region, are able to alter binding to paxillin (summarized in Figure 5B). On the other hand, several residues on another predicted surface of the 4-helix bundle had little or no effect on paxillin binding, including mutation of several highly conserved residues within the originally described PBS2 region. Overall, these mutation and earlier truncation analyses support the contention that the entire carboxyl terminal region of GIT proteins participates in paxillin binding by folding into a compact 4-helix bundle, creating a paxillin binding domain analogous to the focal adhesion targeting domain of focal adhesion kinase. Within this FAH domain, paxillin LD4 binds in the cleft between helices 1 and 4.
The paxillin family member Hic-5 has been reported to bind to rat GIT1 and chicken GIT2 [10, 14]. To determine whether GIT1 binding site for Hic-5 is similar to the binding site for paxillin, we first verified that Hic-5 binds to the carboxyl terminal FAH domain using GIT1 truncation mutants (Figure 6A). Transfected mouse Hic-5 binds tightly to Flag-GIT1, but neither GIT1(1-646) lacking then entire FAH domain, GIT1(1-705) lacking helices 3 and 4, nor GIT1(1-735) lacking just helix 4, were able to bind Hic-5. To ascertain whether Hic-5 and paxillin bind to similar or distinct sites on the GIT1 carboxyl terminal FAH domain, the Hic-5 binding ability of a mutant of GIT1 (K663E + K758E) that no longer binds to paxillin was assessed (Figure 6B). Mutation of residue K663 in helix 1 and K758 in helix 4 ablates binding to Hic-5, as it does for paxillin.
Figure 6.
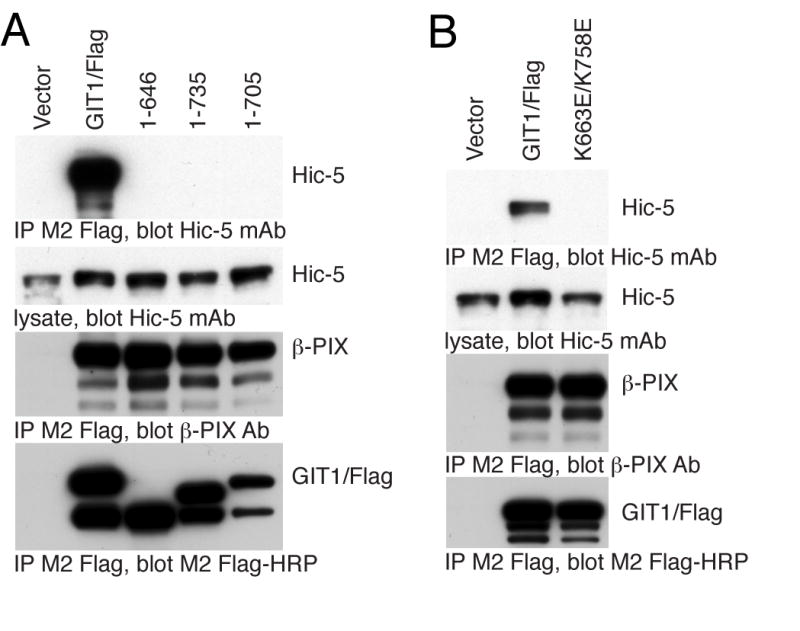
Hic-5 binding to the GIT1 FAH domain.
A. GIT1 truncations that fail to bind paxillin also fail to bind Hic-5. Myc-Hic-5 was transfected together with Flag-GIT1 or the 1-646 (helix 1-4), 1-705(helix 3 and 4) or 1-735 (helix 4 only) truncation mutants into COS7 cells. Cells were scraped and lysed after 48 hours, and soluble lysates immunoprecipitated by addition of M2 Flag-agarose beads. Co-immunoprecipitated Hic-5 was measured by immunoblotting with specific monoclonal antibody, and precipitated GIT1/Flag directly visualized with M2 Flag-HRP conjugate.
B. A GIT1 point mutant that does not bind paxillin also fails to bind Hic-5. Myc-Hic-5 was transfected together with GIT1/Flag or the GIT1(K663E,K758E)/Flag mutant into COS7 cells. Cells were scraped and lysed after 48 hours, and soluble lysates immunoprecipitated by addition of M2 Flag-agarose beads. Co-immunoprecipitated Hic-5 was measured by immunoblotting with specific monoclonal antibody, and precipitated GIT1/Flag directly visualized with M2 Flag-HRP conjugate.
Having defined the paxillin/Hic-5 LD motif binding site on GIT1 as consistent with a FAH 4-helix bundle structure, we sought to understand how paxillin binding to the GIT1 FAH domain might be regulated. Focal adhesion kinase binding to paxillin through its four-helix bundle FAT domain, and therefore localization to focal adhesions, is regulated in part through phosphorylation of FAK on tyrosine 925, a residue in helix 1 of the FAT domain itself [33]. When phosphorylated at tyrosine 925 by Src family members, FAK is released from focal complexes [33]. We sought to determine whether GIT1 association with paxillin might also be regulated through tyrosine phosphorylation. While no tyrosine residue is found in GIT1 in a position directly analogous to Y925 in helix 1 of the FAT domain of focal adhesion kinase, GIT1 and GIT2 share conserved tyrosine residues at positions 719 (in helix 3) and 751 (in helix 4). GIT1 is known to be phosphorylated on tyrosine [9, 27, 34-38], and is a substrate for phosphorylation by Src and FAK [27] and for dephosphorylation by PTPζ and PTP-PEST [39-41], although tyrosine residues 719 and 751 have not been reported to phosphorylated We therefore examined whether GIT1 mutants lacking specific potential tyrosine phosphorylation sites still bind normally to paxillin. GIT1/Flag proteins with mutations in tyrosine residues in the FAT domain were expressed in COS-7 cells, immunoprecipitated, and immunoblotted with anti-paxillin antibody (Figure 7). Neither of the tyrosine residues within the FAH domain, when mutated, had any effect on the amount of paxillin associated with immunoprecipitated GIT1. Thus tyrosine phosphorylation within the FAH domain appears to play no role in regulating GIT - paxillin association. However, tyrosine phosphorylation elsewhere in GIT1 might contribute to regulating paxillin binding. We have tested the paxillin binding of mutant GIT1 molecules in which each tyrosine has been mutated to phenylalanine. Most mutants had no change in paxillin binding (data not shown), but the mutant Y563F had enhanced interaction with paxillin (Figure 7). The mechanism by which this mutant enhances paxillin interaction is under investigation. Another group has recently reported a mutant of chicken GIT2/PKL in which three tyrosine residues are mutated to phenylanaline, and which cannot bind to paxillin or localize to focal adhesions [42]. These authors also reported that GIT2 association with paxillin in focal adhesions is ablated in cells lacking Src family kinases, suggesting that Src-mediated phosphorylation of these tyrosine residues is critical for allowing paxillin binding, whereas GIT1 was still found to be associated normally with paxillin-containing focal adhesions in cells lacking Src family members [42]. None of these tyrosine residues are within the carboxyl terminal FAH domain, but all are conserved in GIT1. We created an equivalent mutant in GIT1, Y293F/Y392F/Y607F, and tested the ability of this GIT1(3YF) mutant and its three component point mutations to co-immunoprecipitate with paxillin. In agreement with Turner and colleagues [42], we find that the GIT1(3YF) mutant does still bind to paxillin, albeit more weakly than the wildtype GIT1 (Figure 7). Nevertheless, we conclude that paxillin binding by GIT1 is not regulated by tyrosine phosphorylation within the FAH domain itself. Tyrosine mutations elsewhere, shown to be critical in regulating GIT2, do not prevent GIT1 binding to paxillin. However, GIT1 does appear to be regulated in part by tyrosine phosphorylation at Y563. Intriguingly, tyrosine phosphorylation-dependent regulation of GIT protein paxillin binding activity appears to be distinctly regulated in GIT1 and GIT2.
Figure 7.
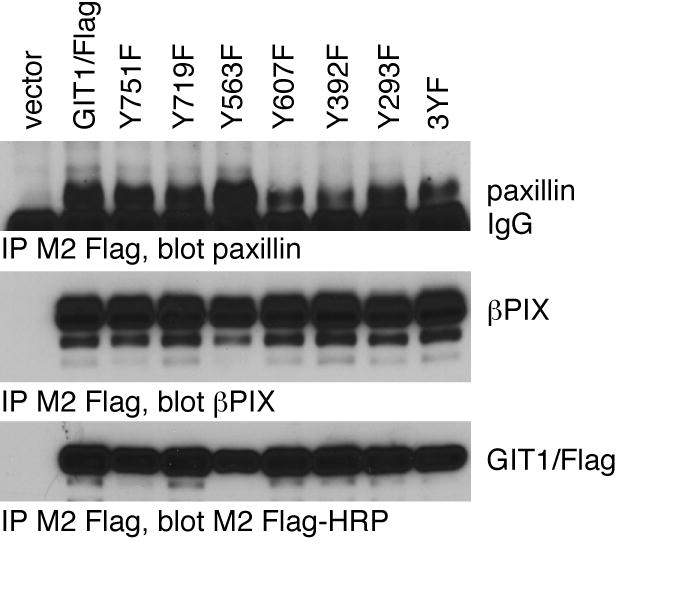
Role of tyrosine phosphorylation of GIT1 on paxillin binding.
The indicated GIT1/Flag tyrosine-to-phenylalanine mutants were transfected into COS7 cells together with chicken paxillin. Cells were scraped and lysed after 48 hours, and soluble lysates immunoprecipitated by addition of M2 Flag-agarose beads. The 3YF mutant is Y293F/Y392F/Y607F. Co-immunoprecipitated paxillin and endogenous β–PIX were measured by immunoblotting with specific antisera, and precipitated Flag-GIT1 directly visualized with M2 Flag-HRP conjugate.
Horwitz and colleagues recently presented evidence that chicken paxillin is phosphorylated at serine 273 within the LD4 motif, and that serine 273 phosphorylation may be an important regulator of the GIT1 - paxillin interaction [43, 44]. Further, serine 275 within the LD4 motif was also found to be phosphorylated [43]. It was reported that the paxillin S273D mutant, a mimic of the phosphorylation of this serine residue, had increased association with GIT1 [44].
In human paxillin, these two serines are conserved, and are located at positions 272 and 274, respectively. Human Hic-5 LD3 also contains serines at residues 164 and 166 that are cognate to human paxillin serines 272 and 274. The third paxillin family member, leupaxin, has neither of these serine residues conserved within its LD3 motif, which is the one most similar to the paxillin LD4 and Hic-5 LD3 sequences. We prepared GST fusions with human paxillin LD4, human Hic-5 LD3 and human leupaxin LD3 peptides, as well as human paxillin LD4 in which serine 272 and/or serine 274 were mutated to aspartate to mimic a phosphorylated serine, or human Hic-5 in which serine 164 was mutated to aspartate. Since the bacteria that are used to prepare the GST fusions contain no serine kinases, the wildtype peptides lack serine phosphorylation. To each of these GST fusion proteins, we added Sf9 cell lysate overexpressing GIT1/6xHis protein, and washed the beads extensively, then detected GIT1 association with the LD repeats by staining with Coomassie blue (Figure 8A). While the paxillin LD4 and Hic-5 LD3 fusions all bound to GIT1, the paxillin S272D, S274D and S272D/S274D LD4 fusions all bound less GIT1 than their wildtype counterparts. The paxillin LD4 S272D/S274D double mutant bound the least GIT1, the S272D mutant had intermediate binding to GIT1, while the S274D mutant had the least effect. Similarly, the Hic5 S164D mutant bound less GIT1 than the wildtype Hic-5 LD3 fusion. While we also tested the leupaxin LD3 fusion in the same assay, we saw no GIT1 pulldown; but the small size difference between GST alone and the GST-LD3 fusion left us unable to verify that the purified fusion protein indeed had an intact LD3 motif. GIT1 association with leupaxin or the leupaxin LD3 peptide has not been demonstrated elsewhere, although GIT2 was reported to co-immunoprecipitate with leupaxin [45].
Figure 8.
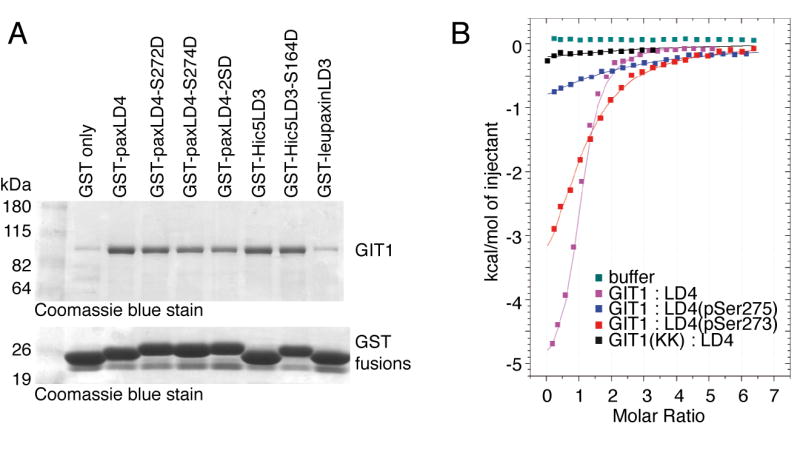
GIT1 binding to paxillin LD peptides in vitro.
A. Pull-down of GIT1 by GST-LD mutants. GST fusions of the human paxillin LD4 motif, mouse Hic-5 LD3 motif and human leupaxin LD3 motif (or the indicated mutants thereof) were expressed in BL21 strain E. coli, and purified on glutathione agarose beads. Fusions were normalized based on Coomassie blue-stained SDS gels, and the beads mixed with Sf9 cell lysates expressing recombinant GIT1. Beads were washed, and bound GIT1 visualized by SDS-PAGE and staining with Coomassie blue.
B. Isothermal titration calorimetry of GIT1 FAH domain and paxillin LD4 peptides. Data (squares) are fitted by Microcal Origin 7 software (lines). GIT1(647-770) was tested with several chicken LD4 phosphopeptides: violet, unphosphorylated LD4; red, LD4 phosphorylated on serine 275; blue, LD4 phosphorylated on serine 273. The K663E/K758E mutant of GIT1(647-770):LD4 data is shown in black, and heats obtained by titrating LD4 into buffer alone are displayed in green.
To further validate the ability of LD4 motif serine phosphorylation to reduce rather than increase paxillin - GIT1 association, we used isothermal calorimetry to directly measure the binding affinity of purified GIT1(647-770) to the chicken paxillin LD4 peptide, as well as chicken LD4 peptides synthesized with phospho-serine at residues 273 or 275 (Figure 8B). Phosphorylation of either serine residue within the paxillin LD4 peptide led to reduced binding to the GIT1 FAH domain, with phospho-serine 273 having the lowest binding. Analysis of the binding curves shows that wildtype paxillin LD4 peptide bound to the GIT1 FAH domain with an affinity of 10 μM, while the phospho-S273 LD4 peptide had an affinity of 123 μM and phospho-S275 LD4 peptide 18 μM (Table I). Thus, by two independent measures, phosphorylation (or glutamate phosphorylation mimic) at the two serines within the paxillin LD4 motif reduces GIT1 association.
Having verified that GIT does bind less tightly to paxillin with phosphorylation at serine 272 and serine 274 in the LD4 motif, we therefore explored whether we might construct a mutant of GIT1 that is insensitive to the phosphorylation status of paxillin at serines 272 and 274. Because in our hands GIT1 appears to prefer dephosphorylated to phosphorylated paxillin, this suggests that the GIT1 FAH domain binding pocket poorly accommodates the bulky, charged phosphate group. This could be either due to steric factors or charge incompatibility. By docking the paxillin LD4 peptide onto the GIT1 model, we predicted that conserved glutamate residues E652, E659 or E674 in helix 1 might electro-statically repulse phospho-LD4 binding. Indeed, these negatively charged residues are absent in the FAK FAT LD binding sites. Mutations of these residues that make the binding phospho-insensitive should therefore increase the extent of GIT1 - paxillin association in cells. Accordingly, we mutated E652N, E659T and E674A in GIT1/Flag, expressed these mutants in COS-7 cells, and immunoprecipiated each with M2 Flag-beads to measure the co-immunoprecipitation of endogenous paxillin (Figure 9). The E659T mutant co-immunoprecipitates a greatly increased quantity of paxillin, while the E652N and E674A mutants are similar to wildtype GIT1. Thus, E659 appears to contribute to limiting GIT1 binding to the major form of paxillin in the cell, and removal of this limitation increases GIT1 - paxillin interaction. Since this mutation is predicted to alter phospho-recognition of the paxillin LD4 peptide, the effect of this mutation to enhance paxillin association provides some evidence that phosphorylation of serines 272 and/or 274 in paxillin’s LD4 motif is indeed a major control mechanism for GIT1 - paxillin binding.
Figure 9.
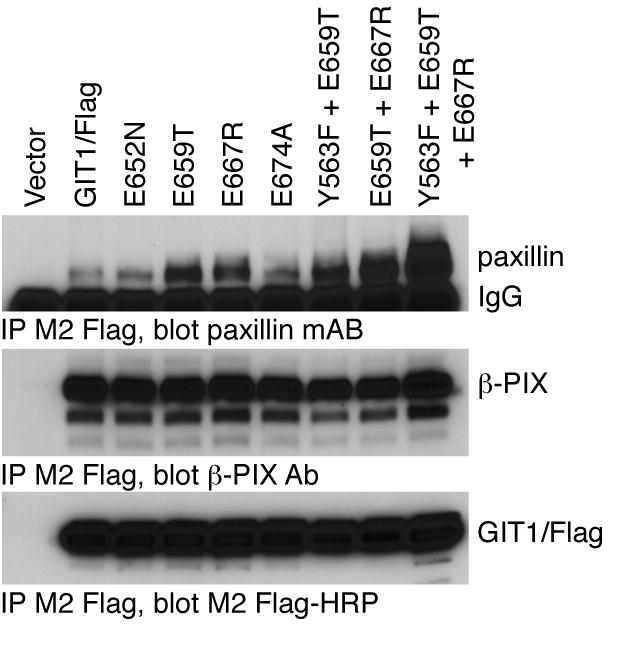
Role of GIT1 phosphate-sensing residues on paxillin binding.
The indicated GIT1/Flag glutamate residue mutants were transfected into COS7 cells together with chicken paxillin. Cells were scraped and lysed after 48 hours, and soluble lysates immunoprecipitated by addition of M2 Flag-agarose beads. Co-immunoprecipitated paxillin and endogenous β–PIX were measured by immunoblotting with specific antisera, and precipitated Flag-GIT1 directly visualized with M2 Flag-HRP conjugate.
During the course of these studies, we identified two other GIT1 point mutants that increased the binding of paxillin, E667R (Figure 5A and Figure 9) and Y563F (Figure 7). Since the E659T, E667R and Y563F mutants each seemed to have altered binding for apparently independent reasons, we asked whether a combination of these mutants might exhibit even greater paxillin binding. While the Y563F/E659T mutant appeared similar to the two individual mutants, the GIT1/Flag E659T/E667R double mutant and the particularly the Y563F/E659T/E667R triple mutant each co-immunoprecipitated with even more paxillin than their component mutants (Figure 9). GIT1/Flag always co-immunoprecipitates with much less paxillin than is present in the cell lysate control sample (1% of the immunoprecipitation initial amount). In contrast, GIT1(Y563F/E659T/E667R)/Flag pulls down more paxillin than is in this lysate control. This Y563F/E659T/E667R triple mutant appears to elude several distinct regulatory mechanisms to constitutively associate with paxillin.
To address whether GIT1 mutants with altered binding to paxillin also have altered ability to localize to focal adhesions, we expressed GIT1/EGFP fusions. Wildtype GIT1/EGFP, GIT1(K758E)/EGFP, GIT1(K663E/K758E)/EGFP, GIT1(E659T/E667R)/EGFP and GIT1(Y563F/E659T/E667R)/EGFP were transfected into A7r5 smooth muscle cells, and the cellular localization of the GIT1/EGFP fusion protein was determined using confocal microscopy (Figure 10). As has been described by others [6, 46], GIT1/EGFP was localized to peripheral focal adhesions along with paxillin, at the end of actin stress fibers (as visualized with phalloidin, not shown), as well as in a general cytoplasmic distribution. In contrast, GIT1(K758E)/EGFP was found throughout the cytoplasm, but with no accumulation at focal adhesions, which were revealed to be present by staining for paxillin. Thus, as expected, this GIT1 mutant incapable of binding to paxillin is indeed unable to localize to paxillin-containing focal adhesions, and overexpressing this GIT1 mutant does not prevent paxillin association with focal adhesions. GIT1(K663E/K758E)/EGFP behaved indistinguishably (data not shown). On the other hand, the GIT1(E659T/E667R)/EGFP mutant showed strong co-localization with paxillin at focal adhesions, as did GIT1(Y563F/E659T/E667R)/EGFP (data not shown).
Figure 10.
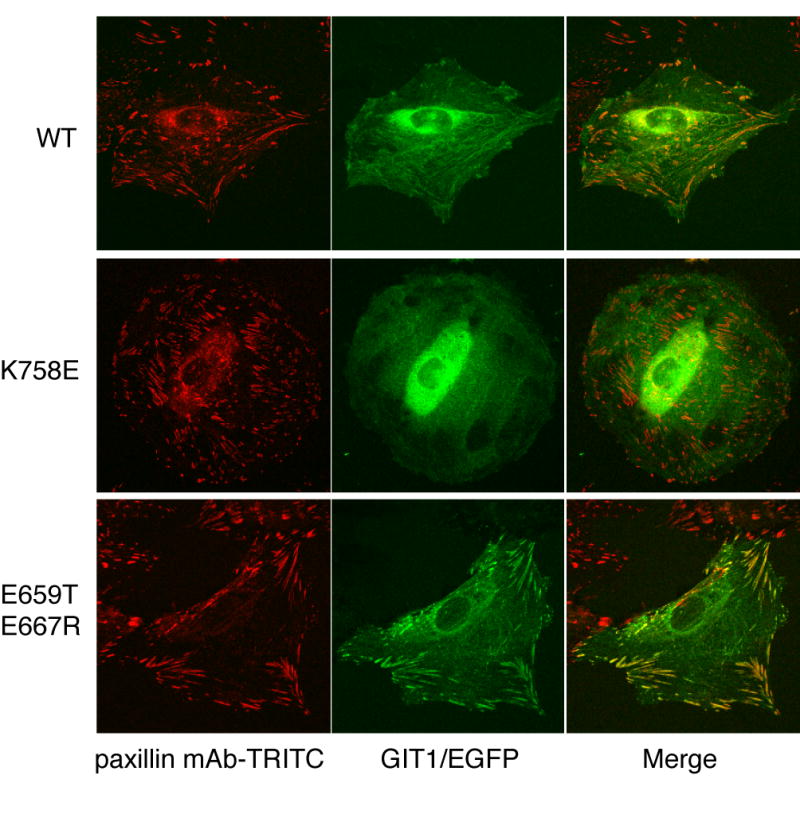
Cellular localization of GIT1 mutants with altered paxillin binding.
GIT1/EGFP, GIT1(K758E)/EGFP or GIT1(E659T+E667R)/EGFP were transfected into A7r5 cells grown on coverslips. Cells were fixed and stained with paxillin mAb conjugated to TRITC (BD Transduction labs), and viewed on a confocal microscope to image EGFP and TRITC fluorescence.
Finally, we refined our molecular model of LD4 bound to the FAH GIT1 carboxy-terminal domain. Together, our data suggest that the GIT1 FAH domain binds one LD4 motif between helices 1 and 4; in contrast to FAT, which appears to bind preferentially LD4 on site 2/3. Both LD binding sites on FAT are indeed very similar, but owing to the distribution of the four positive charges around the hydrophobic groove between the helices, the upper half of the GIT1(647-770) homology model (oriented with the amino-terminus of helix 1 upwards) displays a surface region most resembling the FAT helix 2/3 LD4 binding site (Figure 4). Most mutations that affect LD4 binding, or its phospho-regulation, are located within this upper half of site 1/4. Since SAXS located the LD4 peptide on an exocentric position either on site 1/4 or on site 2/3, we positioned the LD4 peptide on the GIT1(647-770) homology model on face 1/4 as shown in Figure 1B. In this position, S273 of the LD peptide is located in proximity of GIT1 E659, which could explain the effect of phospho-regulation within the paxillin LD4 motif. Further higher resolution structural assays will be required to refine our understanding of this binding interface.
Discussion
The GIT1 carboxyl terminal appears to fold into a single defined structural domain directly analogous to the FAT domain of focal adhesion kinase, which we term the FAT-homology (FAH) domain. In each protein, the extreme carboxyl terminal 130 residues fold to form a 4-helix bundle that binds paxillin LD motifs. We present both structural and functional evidence supporting this structure in GIT1.
We purified the carboxyl terminal paxillin binding domain of GIT1, and used circular dichroism to directly compare the overall helical content of this fragment of GIT1 with the focal adhesion targeting domain of FAK. We also have used small angle X-ray scattering to derive an overall shape of the GIT1 carboxyl terminal alone or with bound paxillin LD4 peptide, and compared this shape to the predicted shape of the FAK-based model for GIT1 carboxyl terminal. The paxillin LD4 peptide appears to bind on a face of the bundle formed by adjacent helices, presumeably the helix 1/4 interface.
Functional evidence for a FAT-like paxillin binding domain comes from two types of mutants that no longer bind paxillin: carboxyl terminal truncations of putative α-helices beyond the predicted PBS2 region, and point mutants predicted to be on the surface of the helix 1/4 interface. Truncations of GIT1 that remove one or more of the predicted α-helices that comprise this 4-helix bundle disrupt paxillin binding, despite leaving intact the predicted paxillin binding site (PBS2), suggesting that this PBS2 region is not sufficient to form a functional paxillin LD4 binding site. Mutagenesis of residues on the surface of this 4-helix bundle supports an overall model where the extreme carboxyl terminal of GIT1 (predicted helix 4) closely abuts the amino terminal end of this 120-residue domain (predicted helix 1), since mutations in each of helices 1 and 4 disrupt paxillin binding. Effective mutations cluster along a region predicted to be on the surface of the helix 1/4 interface, defining the paxillin LD4 peptide binding site on GIT1, which is reminiscent of the LD4 site on focal adhesion kinase. These mutants have the same effect on paxillin and Hic-5, suggesting that GIT1 binds these two paxillin family members through quite similar interactions. These point mutants are summarized in Figure 5B.
Although GIT1 appears to share a paxillin binding FAH domain with focal adhesion kinase, there are clear differences between these two domains. First, GIT1 and FAK bind in related but distinct ways to paxillin. GIT1 binds only to the paxillin LD4 motif, and through only one surface (the helix 1/4 interface) of the 4-helix bundle. Focal adhesion kinase binds to two paxillin LD motifs, LD2 and LD4, through the FAT domain helix 1/4 interface and the helix 2/3 interface on the opposite side of the 4-helix bundle. Second, our modeling and small-angle X-ray scattering studies suggest that the paxillin LD4 peptide binds in an opposite orientation in GIT1 than it does in FAK due to an altered charge orientation in the two binding sites. Nevertheless, key residues in the paxillin LD4 motif, glutamate at the -1 position and aspartate at the +1 position (relative to the leucine in the LD sequence), appear the same for both binding interactions. Third, while paxillin binding to FAK is prominently regulated by tyrosine phosphorylation within the FAT domain at tyrosine 925, we find no evidence that tyrosine phosphorylation within the FAH domain of GIT1 contributes to regulating paxillin association.
In the original analysis to identify potential paxillin binding sites in GIT proteins, two regions were suggested, PBS1 in the amino terminal between the Arf GAP domain and the ankyrin repeat region and PBS2 in the carboxyl terminal [10]. Deletion analysis suggested that PBS2 was the active site for binding [10, 15], and is consistent with our structural and functional analyses here. However, others have reported that a splice variant of GIT2 (GIT2-short) lacking this carboxyl terminal FAH domain still retains paxillin binding ability, mediated through this PBS1 region [47]. GIT2-short was reported to bind more weakly to paxillin than the GIT2-long splice variant that contains the FAH domain [47], just as we have noted previously that GIT2 binds paxillin more weakly than GIT1 [3]. In our hands, we are unable to detect binding of paxillin to GIT2-short by co-immunoprecipitation [3], nor residual binding of paxillin to GIT1 truncations lacking all or part of the FAH carboxyl terminal domain or bearing mutations within this domain (Figure 1B and Figure 5A). Nevertheless, while low affinity interaction of GIT proteins with paxillin through this amino terminal PBS1 region may play a role in further crosslinking GITs with paxillin, the carboxyl terminal FAH domain is clearly the primary high affinity binding site required to target GIT/PIX complexes to paxillin on focal complexes.
The regulation of GIT/PIX complex binding to paxillin remains mysterious, although some details are now apparent. Most GIT/PIX in an adherent cell, as well as most paxillin, appears cytosolic, and only a small fraction of these proteins is bound to focal adhesions. Little soluble GIT/PIX is bound to paxillin. When adhesions are altered (migration, detachment, replating), GIT/PIX and paxillin associate and are found at focal adhesions. The presence of paxillin on focal complexes in cells expressing mutants of GITs that cannot bind paxillin (Figure 10) [15] indicates that paxillin is recruited to focal adhesions prior to binding to GIT/PIX complexes. Phosphorylation of paxillin on various tyrosines [48], as well as on serine residues within the GIT-binding LD4 motif [43, 44], is one mechanism altering GIT binding. While it was reported that phosphorylation of paxillin within LD4 increased GIT1 association [44], we find reduced binding in a pulldown assay (Figure 8A) and reduced affinity for peptide binding to the isolated FAH domain (Figure 8B, Table I). One reason for this difference may be that we have analyzed this interaction with isolated proteins, whereas in living cells other regulatory modes might compensate for changes due to aspartate mutation of the phosphorylated serines, or the aspartate mutation itself might not truly mimic a phosphate in this instance. Nevertheless, phosphorylation of paxillin within the LD4 motif clearly does play a role in regulating GIT protein binding to paxillin, and bears further investigation.
While tyrosine phosphorylation of GIT1 residues within the FAH domain does not appear to be an important contributor to binding, as it is in FAK [33], tyrosine phosphorylation elsewhere in GIT proteins does appear important. First, we identified increased binding of paxillin to the Y563F mutant (Figure 7). Phosphoproteomic studies have identified phosphorylation of this site [35, 38], although the identity of any phosphotyrosine binding partners for phospho-Y563 remain unknown. Second, in GIT2, three tyrosine residues were reported to be the major sites for Src-mediated phosphorylation, and GIT2 bearing Y-F mutations at these three sites, or wildtype GIT2 in cells lacking Src family members, was unable to binding to paxillin or associate with focal adhesions [42]. How Src phosphorylation and phosphotyrosine binding partners like Nck at these sites actually contribute to regulating paxillin binding remains unknown. On the other hand, GIT1 was still able to associate with focal adhesions in cells lacking Src kinases [42]. These three tyrosine residues are located outside the FAH domain but are conserved in GIT1, and we verified that GIT1 bearing these Y-F mutations is still able to bind paxillin, albeit more weakly than wildtype GIT1 (Figure 7). It appears that mechanisms controlling GIT1 and GIT2 association with paxillin may differ. Finally, it was recently reported that phosphorylation of human GIT1 by PAK1 at serine 709 within the FAH domain also regulates paxillin binding [49]. This residue is located on the surface of our model of the GIT1 FAH domain, at the N-terminus of helix 3. The presence of S709, S710 and P713 is likely to destabilize this region, allowing structural transitions necessary to phosphorylate serine 709. Since S709 is on the opposite face of the domain from the paxillin binding site, we predict that this phosphorylation would not directly impinge the paxillin binding surface, but could alter the conformation of the overall domain to affect binding. Alternatively, phosphorylation of serine 709 could influence (intra)molecular associations that affect the accessibility of the paxillin binding site of GIT1.
Our structural and functional characterization of the paxillin binding region of GIT1 as a FAH domain provides a molecular framework allowing more detailed studies to understand the mechanisms regulating GIT1 binding to paxillin.
Conclusions
Structural analyses of the GIT1 conserved carboxyl terminal region reveal a rod shaped structure with high alpha-helical content consistent with an antiparallel 4-helix bundle highly similar to the FAT domain of focal adhesion kinase.
Structure-based mutagenesis defines the paxillin LD4 and Hic-5 LD3 binding site of this GIT1 FAT-Homology (FAH) domain at the helix 1/helix 4 interface.
Paxillin LD4 peptides phosphorylated on serine residues within the LD4 motif bind less well to the GIT1 FAH domain.
Mutations within the paxillin binding site of the GIT1 FAH domain that can accommodate LD4 phosphorylation increase binding of paxillin.
GIT1 FAH domain mutants unable to bind to paxillin no longer localize to focal adhesions.
Acknowledgments
We thank D. Svergun and M. Petoukhov from DESY beamline X33 (EMBL, Hamburg) for their assistance with data recording, and advice throughout data analysis, and Dr. Erin Whalen for helpful comments on the manuscript. This work was supported by the NIH grants GM59989 and DA016347 (RTP), and by Agence National de la Recherche, by the Agence Nationale de Recherches sur le SIDA et les Hépatites Virales, the Association pour la Recherche sur le Cancer and the Ministère de l’Education Nationale, de l’Enseignement Supérieur et de la Recherche (STA). The latter also provides support for MLG.
Abbreviations
- ARF
ADP-ribosylation factor
- FAK
focal adhesion kinase
- FAT
focal adhesion targeting
- GAP
GTPase-activating protein
- GRK
G protein-coupled receptor kinase
- GIT
GRK-interacting ARF GAP
- PAK
p21-activated protein kinase
- PIX
PAK-interacting exchange factor
- PKL
paxillin-kinase linker protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Premont RT, Perry SJ, Schmalzigaug R, Roseman JT, Xing Y, Claing A. Cell Signal. 2004;16:1001–1011. doi: 10.1016/j.cellsig.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Premont RT, Claing A, Vitale N, Freeman JL, Pitcher JA, Patton WA, Moss J, Vaughan M, Lefkowitz RJ. Proc Natl Acad Sci U S A. 1998;95:14082–14087. doi: 10.1073/pnas.95.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Premont RT, Claing A, Vitale N, Perry SJ, Lefkowitz RJ. J Biol Chem. 2000;275:22373–22380. doi: 10.1074/jbc.275.29.22373. [DOI] [PubMed] [Google Scholar]
- 4.Vitale N, Patton WA, Moss J, Vaughan M, Lefkowitz RJ, Premont RT. J Biol Chem. 2000;275:13901–13906. doi: 10.1074/jbc.275.18.13901. [DOI] [PubMed] [Google Scholar]
- 5.Manser E, Loo TH, Koh CG, Zhao ZS, Chen XQ, Tan L, Tan I, Leung T, Lim L. Mol Cell. 1998;1:183–192. doi: 10.1016/s1097-2765(00)80019-2. [DOI] [PubMed] [Google Scholar]
- 6.Zhao ZS, Manser E, Loo TH, Lim L. Mol Cell Biol. 2000;20:6354–6363. doi: 10.1128/mcb.20.17.6354-6363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koh CG, Tan EJ, Manser E, Lim L. Curr Biol. 2002;12:317–321. doi: 10.1016/s0960-9822(02)00652-8. [DOI] [PubMed] [Google Scholar]
- 8.Haendeler J, Yin G, Hojo Y, Saito Y, Melaragno M, Yan C, Sharma VK, Heller M, Aebersold R, Berk BC. J Biol Chem. 2003;278:49936–49944. doi: 10.1074/jbc.M307317200. [DOI] [PubMed] [Google Scholar]
- 9.Yin G, Haendeler J, Yan C, Berk BC. Mol Cell Biol. 2004;24:875–885. doi: 10.1128/MCB.24.2.875-885.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, Bagrodia S, Thomas S, Leventhal PS. J Cell Biol. 1999;145:851–863. doi: 10.1083/jcb.145.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ko J, Kim S, Valtschanoff JG, Shin H, Lee JR, Sheng M, Premont RT, Weinberg RJ, Kim E. J Neurosci. 2003;23:1667–1677. doi: 10.1523/JNEUROSCI.23-05-01667.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim S, Ko J, Shin H, Lee JR, Lim C, Han JH, Altrock WD, Garner CC, Gundelfinger ED, Premont RT, Kaang BK, Kim E. J Biol Chem. 2003;278:6291–6300. doi: 10.1074/jbc.M212287200. [DOI] [PubMed] [Google Scholar]
- 13.Audebert S, Navarro C, Nourry C, Chasserot-Golaz S, Lecine P, Bellaiche Y, Dupont JL, Premont RT, Sempere C, Strub JM, Van Dorsselaer A, Vitale N, Borg JP. Curr Biol. 2004;14:987–995. doi: 10.1016/j.cub.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 14.Nishiya N, Shirai T, Suzuki W, Nose K. J Biochem (Tokyo) 2002;132:279–289. doi: 10.1093/oxfordjournals.jbchem.a003222. [DOI] [PubMed] [Google Scholar]
- 15.West KA, Zhang H, Brown MC, Nikolopoulos SN, Riedy MC, Horwitz AF, Turner CE. J Cell Biol. 2001;154:161–176. doi: 10.1083/jcb.200101039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown MC, West KA, Turner CE. Mol Biol Cell. 2002;13:1550–1565. doi: 10.1091/mbc.02-02-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaller MD. Oncogene. 2001;20:6459–6472. doi: 10.1038/sj.onc.1204786. [DOI] [PubMed] [Google Scholar]
- 18.Turner CE. Nat Cell Biol. 2000;2:E231–236. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- 19.Mitra SK, Hanson DA, Schlaepfer DD. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 20.Tumbarello DA, Brown MC, Turner CE. FEBS Lett. 2002;513:114–118. doi: 10.1016/s0014-5793(01)03244-6. [DOI] [PubMed] [Google Scholar]
- 21.Arold ST, Hoellerer MK, Noble ME. Structure (Camb) 2002;10:319–327. doi: 10.1016/s0969-2126(02)00717-7. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi I, Vuori K, Liddington RC. Nat Struct Biol. 2002;9:101–106. doi: 10.1038/nsb755. [DOI] [PubMed] [Google Scholar]
- 23.Liu G, Guibao CD, Zheng J. Mol Cell Biol. 2002;22:2751–2760. doi: 10.1128/MCB.22.8.2751-2760.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoellerer MK, Noble ME, Labesse G, Campbell ID, Werner JM, Arold ST. Structure (Camb) 2003;11:1207–1217. doi: 10.1016/j.str.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 25.Gao G, Prutzman KC, King ML, Scheswohl DM, DeRose EF, London RE, Schaller MD, Campbell SL. J Biol Chem. 2004;279:8441–8451. doi: 10.1074/jbc.M309808200. [DOI] [PubMed] [Google Scholar]
- 26.Bertolucci CM, Guibao CD, Zheng J. Protein Sci. 2005;14:644–652. doi: 10.1110/ps.041107205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagrodia S, Bailey D, Lenard Z, Hart M, Guan JL, Premont RT, Taylor SJ, Cerione RA. J Biol Chem. 1999;274:22393–22400. doi: 10.1074/jbc.274.32.22393. [DOI] [PubMed] [Google Scholar]
- 28.Thomas SM, Hagel M, Turner CE. J Cell Sci. 1999;112(Pt 2):181–190. doi: 10.1242/jcs.112.2.181. [DOI] [PubMed] [Google Scholar]
- 29.Kondo A, Hashimoto S, Yano H, Nagayama K, Mazaki Y, Sabe H. Mol Biol Cell. 2000;11:1315–1327. doi: 10.1091/mbc.11.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schwede T, Kopp J, Guex N, Peitsch MC. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Svergun DI, Koch MH. Curr Opin Struct Biol. 2002;12:654–660. doi: 10.1016/s0959-440x(02)00363-9. [DOI] [PubMed] [Google Scholar]
- 32.Luthy R, Bowie JU, Eisenberg D. Nature. 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 33.Katz BZ, Romer L, Miyamoto S, Volberg T, Matsumoto K, Cukierman E, Geiger B, Yamada KM. J Biol Chem. 2003;278:29115–29120. doi: 10.1074/jbc.M212396200. [DOI] [PubMed] [Google Scholar]
- 34.van Nieuw Amerongen GP, Natarajan K, Yin G, Hoefen RJ, Osawa M, Haendeler J, Ridley AJ, Fujiwara K, van Hinsbergh VW, Berk BC. Circ Res. 2004;94:1041–1049. doi: 10.1161/01.RES.0000125627.77235.0C. [DOI] [PubMed] [Google Scholar]
- 35.Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, Comb MJ. Nat Biotechnol. 2005;23:94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Wolf-Yadlin A, Ross PL, Pappin DJ, Rush J, Lauffenburger DA, White FM. Mol Cell Proteomics. 2005;4:1240–1250. doi: 10.1074/mcp.M500089-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Hinsby AM, Olsen JV, Mann M. J Biol Chem. 2004;279:46438–46447. doi: 10.1074/jbc.M404537200. [DOI] [PubMed] [Google Scholar]
- 38.Webb DJ, Mayhew MW, Kovalenko M, Schroeder MJ, Jeffery ED, Whitmore L, Shabanowitz J, Hunt DF, Horwitz AF. J Cell Sci. 2006;119:2847–2850. doi: 10.1242/jcs.03044. [DOI] [PubMed] [Google Scholar]
- 39.Kawachi H, Fujikawa A, Maeda N, Noda M. Proc Natl Acad Sci U S A. 2001;98:6593–6598. doi: 10.1073/pnas.041608698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujikawa A, Shirasaka D, Yamamoto S, Ota H, Yahiro K, Fukada M, Shintani T, Wada A, Aoyama N, Hirayama T, Fukamachi H, Noda M. Nat Genet. 2003;33:375–381. doi: 10.1038/ng1112. [DOI] [PubMed] [Google Scholar]
- 41.Jamieson JS, Tumbarello DA, Halle M, Brown MC, Tremblay ML, Turner CE. J Cell Sci. 2005;118:5835–5847. doi: 10.1242/jcs.02693. [DOI] [PubMed] [Google Scholar]
- 42.Brown MC, Cary LA, Jamieson JS, Cooper JA, Turner CE. Mol Biol Cell. 2005;16:4316–4328. doi: 10.1091/mbc.E05-02-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Webb DJ, Schroeder MJ, Brame CJ, Whitmore L, Shabanowitz J, Hunt DF, Horwitz AR. J Cell Sci. 2005;118:4925–4929. doi: 10.1242/jcs.02563. [DOI] [PubMed] [Google Scholar]
- 44.Nayal A, Webb DJ, Brown CM, Schaefer EM, Vicente-Manzanares M, Horwitz AR. J Cell Biol. 2006;173:587–589. doi: 10.1083/jcb.200509075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupta A, Lee BS, Khadeer MA, Tang Z, Chellaiah M, Abu-Amer Y, Goldknopf J, Hruska KA. J Bone Miner Res. 2003;18:669–685. doi: 10.1359/jbmr.2003.18.4.669. [DOI] [PubMed] [Google Scholar]
- 46.Manabe Ri R, Kovalenko M, Webb DJ, Horwitz AR. J Cell Sci. 2002;115:1497–1510. doi: 10.1242/jcs.115.7.1497. [DOI] [PubMed] [Google Scholar]
- 47.Mazaki Y, Hashimoto S, Okawa K, Tsubouchi A, Nakamura K, Yagi R, Yano H, Kondo A, Iwamatsu A, Mizoguchi A, Sabe H. Mol Biol Cell. 2001;12:645–662. doi: 10.1091/mbc.12.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown MC, Turner CE. Physiol Rev. 2004;84:1315–1339. doi: 10.1152/physrev.00002.2004. [DOI] [PubMed] [Google Scholar]
- 49.Webb DJ, Kovalenko M, Whitmore L, Horwitz AF. Biochem Biophys Res Commun. 2006;346:1284–1288. doi: 10.1016/j.bbrc.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 50.Bakolitsa C, de Pereda JM, Bagshaw CR, Critchley DR, Liddington RC. Cell. 1999;99:603–613. doi: 10.1016/s0092-8674(00)81549-4. [DOI] [PubMed] [Google Scholar]
- 51.Junius FK, O’Donoghue SI, Nilges M, Weiss AS, King GF. J Biol Chem. 1996;271:13663–13667. doi: 10.1074/jbc.271.23.13663. [DOI] [PubMed] [Google Scholar]