

Abstract
The two-component regulatory proteins OmpR and EnvZ of Escherichia coli K-12 regulate expression of the major outer membrane porin protein, OmpF. OmpR is a DNA-binding protein that is involved in both the positive and negative control of ompF transcription. EnvZ is a histidine kinase that phosphorylates OmpR in response to environmental signals. We used DNA migration retardation analysis to examine the interactions of OmpR and the phosphorylated form of OmpR (OmpR-P) with the regulatory region immediately upstream of the ompF promoter. Our results indicate that the binding of OmpR to this regulatory region is cooperative and that phosphorylation significantly stimulates these cooperative interactions. Moreover, although phosphorylation increases the intrinsic binding of OmpR to a single OmpR-binding site, the primary role of phosphorylation in ompF regulation is to facilitate cooperative interactions between OmpR molecules bound at adjacent sites. Based on these results, we propose a model to explain how the phosphorylation of OmpR could stimulate the occupancy of specific sites in the ompF regulatory region, thereby resulting in the activation or repression of ompF transcription under the appropriate environmental conditions.
Keywords: two-component regulatory system, osmoregulation
Expression of the major outer membrane porin proteins OmpF and OmpC of Escherichia coli K-12 is regulated in response to a wide variety of environmental signals (reviewed in refs. 1–3). The most extensively studied of these signals is the osmolarity of the growth medium. At low osmolarity, the OmpF porin is preferentially expressed and only low levels of the OmpC porin are found. Conversely, at high osmolarity, OmpC is preferentially expressed and only low levels of OmpF are found. This fluctuation in porin expression is controlled at the transcriptional level by the two-component regulatory system, EnvZ and OmpR. EnvZ is a membrane-bound histidine kinase that modulates the activity of the DNA-binding protein OmpR via phosphorylation (refs. 2 and 3 and references therein). The phosphorylated form of OmpR (OmpR-P), which is the active form of the protein, is a transcriptional activator at the ompC promoter and can function as either an activator or a repressor at the ompF promoter (4).
According to the current model (3), at low osmolarity the cellular concentration of OmpR-P is low. This level is sufficient for OmpR-P to interact with the sites in the ompF regulatory region that are responsible for activating ompF transcription. At high osmolarity, the cellular concentration of OmpR-P is much greater. As a result of this increase, OmpR-P is now capable of occupying the sites in the ompC regulatory region responsible for activating ompC transcription, and the sites at ompF responsible for repressing ompF transcription. Thus, this model predicts that the regulation of ompF and ompC is a direct consequence of the level of OmpR-P in the cell and is dependent on the way in which OmpR-P interacts with sites in the ompF and ompC regulatory regions.
A number of studies have addressed the basic predictions of this model (see refs. 3 and 5–8 and references therein). The results of these studies have been taken as support for the cooperative nature of OmpR binding, the sequential occupancy of the multiple sites by OmpR in the ompF regulatory region, and the stimulatory role of phosphorylation in OmpR binding. However, these earlier studies were limited in one of two ways. Either they did not systematically compare OmpR binding in the presence and absence of kinase or they did not clearly separate the effects of phosphorylation on intrinsic binding vs. cooperative binding of OmpR to the DNA. In this paper, we examine how phosphorylation influences the ability of OmpR to bind to the OmpR-binding sites in the ompF regulatory region. Our results indicate that phosphorylation has little effect on the intrinsic binding of OmpR to the single site F1 and that the primary effect of phosphorylation at ompF is to stimulate cooperative interactions between OmpR molecules bound at adjacent sites. The fact that phosphorylation stimulates cooperativity leads us to propose a model to explain why phosphorylation is required for OmpR function in vivo.
MATERIALS AND METHODS
DNA Fragments Used in the DNA Migration Retardation Assays.
The sequences of the nontemplate strands of the DNA fragments used in this study are presented in Fig. 1B. Each DNA fragment has been assigned a descriptive name that lists the OmpR-binding sites present on the fragment and a reference name, which is given in parentheses. F1 (IKH20) carries the single OmpR-binding site F1 and contains ompF sequence from −99 to −76. F1-F2 (IKH21) carries both the F1 and the F2 sites and contains ompF sequence from −99 to −56. F1-F1 (IKH22) contains a tandem duplication of the F1 site. To construct F1-F1, the nucleotide sequence between −79 and −56 was replaced with the nucleotide sequence between −99 and −76. As a result, the phasing between the regions of contact between OmpR and the DNA in the two F1 sites is the same as the phasing observed between the F1 and F2 sites in the ompF regulatory region.
Figure 1.

The ompF regulatory region. (A) The locations of the two separate OmpR binding regions upstream of the start point of ompF transcription (arrow marked +1) and the locations of individual OmpR binding sites (F1, F2, F3, F4) within these regions are indicated. (B) The sequences of the nontemplate strands for the five double-stranded oligonucleotides used in the DNA migration retardation assays are indicated. The DNA fragments consist of two parts: DNA sequences from the ompF regulatory region (uppercase) and flanking sequences that are not present at ompF (lowercase). The underlined bases indicate the positions of the half-sites in the individual sites (6). The asterisk indicates the position of the G-to-A substitution in the mutant F2 site.
Each of these three DNA fragments was generated by first chemically synthesizing two single strands of DNA (MilliGen/Biosearch, Millipore). The single-stranded oligonucleotides were purified using a denaturing polyacrylamide gel, mixed, and allowed to anneal. The resulting double-stranded oligonucleotides were then labeled with [α-32P]dATP (specific activity = 3,000 Ci/mmol; 1 Ci = 37 GBq) using the Klenow fragment of DNA polymerase I.
For the experiments shown in Fig. 4, the DNA fragments F1-F2-F3 (IKH23) and F1-F2*-F3 (IKH24) were generated by PCR from the plasmid pKJH6 (6). The DNA fragment F1-F2-F3 contains the wild-type sequence from −99 to −35. The DNA fragment F1-F2*-F3 is identical to F1-F2-F3, except for a G-to-A substitution at position −70 in the F2 site. The PCR-generated DNA fragments were labeled with [γ-32P]ATP (spec. act. = 6,000 Ci/mmol) using T4 polynucleotide kinase and then purified using a 6% nondenaturing polyacrylamide gel.
Figure 4.

Effect of OmpR phosphorylation on the binding of OmpR to a mutant F2 site. The DNA fragments were generated as described: F1-F2-F3 carries the wild-type sites F1, F2, and F3 (A), whereas F1-F2*-F3 carries a mutated F2 site between flanking wild-type F1 and F3 sites (B). The labeled DNA fragments (≈3.3 nM) were incubated with different amounts of wild-type OmpR in the absence or presence of 0.63 μM of the kinase MBP-EnvZ, as indicated. The OmpR/DNA complexes were resolved on a nondenaturing gel containing 10% polyacrylamide (75:1; wt/wt, acrylamide to bisacrylamide) and analyzed by autoradiography. The concentrations of OmpR used in this analysis were as follows. Lanes: 1, 0; 2, 37.6 nM; 3, 75.2 nM; and 4, 150.4 nM. The OmpR/DNA complexes corresponding to one-site occupancy (I), two-site occupancy (II), and three-site occupancy (III) are indicated.
Purification of Proteins and DNA Migration Retardation Analysis.
The DNA migration retardation assays were performed as described (6) using the proteins and DNA fragments indicated in the figure legends. Wild-type OmpR and maltose binding protein (MBP)-EnvZ fusion protein were overexpressed and purified as described (6). The OmpR D55Q mutant protein was overexpressed using plasmid pLAN202 (unpublished results) and then purified using the protocol outlined previously for wild-type OmpR (6).
Estimation of Cooperativity of Binding.
The degree of cooperativity exhibited by OmpR was estimated from data obtained by DNA migration retardation analysis using the DNA fragment F1-F1. The OmpR/DNA complexes were resolved and the amount of free DNA and bound DNA in the different complexes was determined using a Fuji Bas1000 phosphorImager. The fraction of DNA in the different complexes was then calculated by using the macbas2.0 software (Fujix Bas system) under the profile measurement mode. These data were used to calculate the degree of cooperativity of binding of OmpR using equations derived by Hudson and Fried (9) and by Tsai et al. (10). This method was chosen for two reasons. First, these equations made it possible to determine the degree of cooperative binding between OmpR molecules using calculations that are independent of the concentration of the OmpR protein and its affinity for its binding site. Second, this method was successfully used to estimate the degree of cooperativity exhibited by another response regulator protein NtrC (11).
The equation used to calculate the cooperativity factor (KdI/KdII) pertains to the special case in which the two binding sites on the DNA fragment are equivalent. This formula is a simplification of the Hudson–Fried equation (9), which was previously described by Tsai et al. (10).
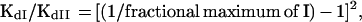 |
1 |
where KdI is the apparent dissociation constant for binding of two OmpR molecules to one site on the DNA fragment, KdII is the apparent dissociation constant for the binding of two additional OmpR molecules at the adjacent site, and the fractional maximum of I refers to the maximum fraction of the intermediate species I, which is the species with only one of the two sites occupied. Therefore, to use this equation, we will assume that the two F1 sites are equivalent and that occupancy of either site results in the formation of complex II with equal efficiency.
RESULTS
In this paper, we examined how phosphorylation influences the ability of OmpR to bind to the individual sites in the ompF regulatory region (Fig. 1A). A major difference between this study and earlier studies of the ompF regulatory region (7, 8, 12, 13) concerns the definition of the OmpR binding sites used to design the DNA fragments employed in the DNA migration retardation assays. Previously, we used dimethyl sulfate and hydroxyl radical footprinting analyses to define the regions of contact between OmpR and the DNA (6). These studies indicated that each individual OmpR binding site spans ≈18 bp and contains two half-sites oriented as asymmetric direct repeats. Analysis of the individual sites at ompF also established that only the F1 site is capable of forming a stable OmpR/DNA complex as an isolated site (6–8, 12) and that two OmpR molecules are required for stable binding to this site (6). Therefore, although OmpR exists in solution as a monomer under most conditions that have been examined (5, 14, 15), OmpR is unable to form stable protein/DNA complexes as a monomer. Using this information as a foundation, we designed a series of DNA fragments to examine how phosphorylation influences the interaction of OmpR with the sites in the ompF regulatory region. These DNA fragments (Fig. 1B) take into account the contact information obtained from our DNA footprinting experiments (6). That the DNA fragments used in this study have different endpoints when compared with the DNA fragments used in earlier studies seems the simplest explanation for any disagreement between our results and those obtained by others (7, 8, 12, 13).
Effects of OmpR Phosphorylation on Binding to a Single OmpR-Binding Site vs. Two Adjacent Sites.
To obtain a more detailed understanding of how OmpR and OmpR-P interact with the ompF regulatory region, we performed a series of DNA migration retardation assays using DNA fragments containing different portions of the ompF regulatory region (see Materials and Methods; Fig. 1B). We first examined the interaction of OmpR with the single OmpR-binding site F1. In this analysis, the DNA fragment containing the F1 site was labeled and incubated with different concentrations of OmpR in the presence or absence of a fusion protein kinase, MBP-EnvZ. The addition of the kinase to the reaction results in the phosphorylation of the OmpR protein (data not shown). As shown in Fig. 2A, the DNA fragment containing the F1 site is capable of forming stable OmpR/DNA complexes in both the absence and the presence of the kinase. The presence of the kinase results in a slight stimulation of stable complex formation, approximately 2- to 3-fold under these particular experimental conditions. We also noticed that there is increased smearing in the presence of the kinase, which is indicative of unstable complex formation. Experiments to determine the exact nature of these complexes are in progress.
Figure 2.

Comparison of OmpR binding to a single site vs. two adjacent sites. (A) Binding of OmpR to the single site F1. Approximately 0.23 nM of the labeled DNA fragment (F1) was incubated with different amounts of OmpR in the absence or presence of 0.63 μM of the kinase MBP-EnvZ. The concentrations of OmpR used in this analysis were as follows. Lanes: 1, 9.4 nM; 2, 18.8 nM; 3, 37.6 nM; and 4, 75.2 nM. (B) Binding of OmpR to the two adjacent sites, F1 and F2. Approximately 0.23 nM of the labeled DNA fragment (F1-F2) was incubated with different amounts of OmpR in the absence or presence of 0.63 μM of the kinase MBP-EnvZ. The concentrations of OmpR used in this analysis were as follows. Lanes: 1, 9.4 nM; 2, 18.8 nM; 3, 37.6 nM; and 4, 75.2 nM. The OmpR/DNA complexes were resolved on a nondenaturing gel containing 10% polyacrylamide (30:1; wt/wt, acrylamide to bisacrylamide) followed by autoradiography. The positions of the OmpR/DNA complexes containing one site (I) or two sites (II) occupied are indicated.
Phosphorylation had a much greater impact on the binding of OmpR to the DNA fragment containing the two adjacent OmpR-binding sites F1 and F2. As shown in Fig. 2B, in the absence of the kinase, two stable OmpR/DNA complexes were observed. Based on their mobility, complex I corresponds to a DNA fragment with one site occupied, whereas complex II corresponds to a DNA fragment with two sites occupied. The predominant complex in the absence of kinase is complex I. However, in the presence of kinase, most of the DNA is associated with complex II. Complex II also forms at much lower OmpR concentrations (compare Fig. 2B, complex II, lane 2 in the absence and presence of kinase). Based on these results, we conclude that the addition of kinase to the reaction greatly stimulates the binding of OmpR to DNA containing the two adjacent sites F1 and F2.
To confirm that this stimulation is due to phosphorylation of OmpR, we repeated the above experiments using a mutant form of OmpR, OmpR D55Q. This mutation changes the amino acid at position 55, the site of OmpR phosphorylation, from an aspartic acid residue to glutamine (16, 17). Biochemical characterization of this mutant indicates that this alteration of the phosphorylation site prevents the phosphorylation of OmpR by the EnvZ kinase. As expected, the stimulatory effect of the kinase on OmpR binding is not observed with this mutant form of OmpR (data not shown). It was also possible to eliminate the stimulation by performing the experiment using wild-type OmpR and the MBP-EnvZ kinase, but in the absence of ATP (data not shown). These experiments support the hypothesis that the stimulatory effect of the EnvZ kinase is due to its ability to phosphorylate OmpR.
Phosphorylation Stimulates Cooperative Interactions Between OmpR Molecules Bound at Neighboring Sites.
The DNA fragments used in the above experiments (Fig. 2B) contained two OmpR binding sites, the F1 site and the F2 site. Since the F2 site is not capable of independently binding OmpR even in the presence of the kinase, these experiments also suggest that cooperative interactions play an important role in the binding of OmpR to these sites and that phosphorylation stimulates this cooperativity. To examine cooperative binding more quantitatively, we generated the double-stranded oligonucleotide IKH22, which contains two F1 sites (see Fig. 1B). The F1 sites on this DNA fragment are oriented as direct repeats and are precisely spaced to maintain the normal phasing of the OmpR/DNA contacts found in the ompF regulatory region. Therefore, both sites on the DNA fragment are capable of independent OmpR binding and should have the same affinity for OmpR.
This DNA fragment (termed the F1-F1 fragment) was then used to analyze the degree of cooperativity exhibited by OmpR and by OmpR-P. In this experiment, DNA migration retardation assays were performed and the amount of free DNA and bound DNA in the different complexes was determined using a phosphorImager, as described in Materials and Methods. In Fig. 3A, the fraction of free DNA was measured and then used to calculate the total fraction of bound DNA. Therefore, this fraction takes into account both the stable and unstable OmpR/DNA complexes present in the reaction. The total fraction of bound DNA was then plotted as a function of OmpR concentration. Fig. 3A shows the results from two experiments, one performed using OmpR alone and the other performed using both OmpR and MBP-EnvZ. One feature of the resulting graphs is that the OmpR binding to the F1-F1 fragment shows a classical sigmoidal titration curve, indicative of a cooperative binding reaction. The sigmoidal nature of the curve is particularly apparent in the presence of the kinase. This calculation may be an underestimate of the degree of cooperativity. If only stable OmpR/DNA complexes were used in this analysis, the sigmoidal nature of the curve was even more pronounced (data not shown). Based on these results, we conclude that the binding of OmpR to the ompF regulatory region is highly cooperative and that the role of OmpR phosphorylation is to enhance the cooperative interactions between OmpR molecules bound at adjacent sites.
Figure 3.

Quantitation of OmpR binding to the F1-F1 fragment. A DNA fragment (F1-F1) containing a tandem duplication of the F1 site was labeled using [α-32P]dATP. The labeled fragments (≈0.13 nM) were incubated with increasing amounts of OmpR in the absence (•) or presence (○) of the kinase (0.63 μM), and the resulting OmpR/DNA complexes were resolved on a nondenaturing gel containing 10% polyacrylamide (30:1; wt/wt, acrylamide to bisacrylamide) followed by autoradiography. The amount of free DNA and bound DNA in the different complexes was determined as described. In A, the fraction of free DNA was measured and then used to calculated the total fraction of bound DNA. The total percentage of bound DNA observed was then plotted as a function of OmpR concentration. In B, the fraction of the intermediate species (I), which corresponds to occupancy of only one site, was plotted as a function of OmpR concentration. The fractional maximum of species I was then determined and used to calculate the cooperativity factor (kdI/kdII), as described.
It is also possible to calculate the degree of cooperativity using the F1-F1 fragment. For this analysis, OmpR was titrated using the F1-F1 fragment in the absence or presence of the kinase. The fraction of the DNA associated with the intermediate species (OmpR/DNA complexes with one site occupied) was then determined as a function of OmpR concentration (Fig. 3B). The fractional maximum of the intermediate species was ≈0.11 in the absence of kinase and ≈0.03 in the presence of kinase. The determination of the fractional maximum of the intermediate species makes it possible to estimate the degree of cooperative binding between OmpR molecules using equations derived by Hudson and Fried (9) and by Tsai et al. (10). This method of calculating the degree of cooperativity uses equations that are independent of the concentration of OmpR and its affinity for a binding site. Based on these calculations, the occupancy of one OmpR binding site stimulates the occupancy of the adjacent site by ≈60-fold. Further stimulation is observed when the kinase is added to the reaction. Under these conditions, the occupancy of one OmpR binding site stimulates the occupancy of the adjacent site by ≈1,000 fold. Because the F1-F1 fragment does not carry the configuration of sites normally found at the ompF regulatory region, these values are only valid with this DNA fragment and under the conditions used in our DNA migration retardation assays. However, these experiments establish two basic aspects of the interaction of OmpR with its target sites: the binding of the unphosphorylated form of OmpR can be cooperative and these cooperative interactions can be further stimulated by phosphorylation.
Phosphorylation Stimulates Cooperative Interactions in the ompF Regulatory Region.
The importance of cooperative interactions in ompF regulation and the stimulatory effect of phosphorylation on these interactions is further supported by the experiment presented in Fig. 4. For this experiment, we compared the effects of phosphorylation on the binding of OmpR to a DNA fragment containing the wild-type F1, F2, and F3 sites vs. a DNA fragment containing a mutant F2 site (F2*) between flanking wild-type F1 and F3 sites. Our strategy for inactivating the F2 site was derived from our DNA footprinting analysis of the ompF regulatory region (6). These studies identified a G residue in the left half-site that was highly conserved among all OmpR-binding sites. Indeed, mutant variants of the OmpR binding site C1 in the ompC regulatory region indicate that a G residue at this position is essential for independent OmpR binding (unpublished data). Therefore, we synthesized the DNA fragment F1-F2*-F3, which carries a single G-to-A substitution at position −70 in the F2 site. As a control, we also synthesized the DNA fragment F1-F2-F3, which contains the wild-type ompF sequence.
Fig. 4A shows the binding of OmpR to the F1-F2-F3 fragment containing the wild-type sequence in the region from −99 to −35. In the absence of the kinase, three OmpR/DNA complexes exhibiting different migration properties were observed. The two predominant complexes exhibit the mobility predicted for occupancy of one site (complex I) and of two sites (complex II), with the third, less predominant form (complex III) exhibiting the mobility characteristic of occupancy of three sites. However, in the presence of the kinase, the predominant complexes exhibit the mobility corresponding to two-site occupancy (complex II) and three-site occupancy (complex III).
A very different pattern was observed using the F1-F2*-F3 fragment. In the absence of the kinase, the mutant F2 site permitted only one OmpR/DNA complex to form (Fig. 4B). The simplest explanation for this result is that OmpR is occupying the F1 site and that occupancy of the F3 site is dependent on occupancy of F2. Thus, in the absence of the kinase, the mutation in the F2 binding site is sufficient to eliminate occupancy of the F2 site. However, in the presence of the kinase, all three sites were occupied in spite of the presence of the mutated F2 site on the DNA fragment. Moreover, the complex corresponding to two-site occupancy (complex II) is absent. This would suggest that the cooperative interactions between OmpR molecules are strong enough to overcome the mutation in the F2 site and that occupancy of the F3 site plays an important role in occupancy of the F2 site. We tested this hypothesis by examining the binding pattern of a DNA fragment that is mutant for both the F2 and F3 sites. This oligonucleotide showed only one-site occupancy (data not shown). We interpret these results to mean that binding of OmpR to the mutant F2 site is dependent on its ability to interact cooperatively with OmpR molecules bound at the flanking F1 and F3 sites. The results with the F1, F2, and F3 sites in their normal configuration (Fig. 4) parallel the results with the artificial F1-F1 construct (Fig. 3). Thus, both sets of experiments underscore the importance of the stimulatory effect of phosphorylation on cooperative interactions between OmpR molecules.
DISCUSSION
In this paper, we examined the effects of phosphorylation of the transcriptional regulator OmpR on its binding to single and multiple DNA-binding sites. A prerequisite of this study was the careful definition of the sites in the ompF regulatory region using DNA footprinting analysis (6). These footprinting studies revealed the location of the individual single sites at ompF, thereby allowing the design of DNA fragments appropriate for this work. Although previous studies have examined the binding of OmpR to DNA, the impact of OmpR phosphorylation on this binding has never been fully evaluated. In one study, what was thought to be a single OmpR-binding site was, in fact, an incomplete site (7). Hence, the apparent increase in binding to this “single site” upon phosphorylation was most likely due to the stimulation of cooperative interactions between OmpR molecules bound at this incomplete site rather than due to an increased affinity of OmpR for the DNA. In other studies, although the binding of OmpR-P was examined using DNA fragments containing intact sites, this binding was not compared with the binding of the unphosphorylated form of OmpR (5, 8). Therefore, it was not possible to draw firm conclusions concerning the effects of phosphorylation on the intrinsic binding of OmpR to a single site or on the cooperative binding of OmpR to multiple sites.
The major conclusions from our studies are that binding of OmpR to the ompF regulatory region is cooperative and that phosphorylation stimulates these cooperative interactions. Furthermore, although phosphorylation results in a slight increase in OmpR binding to a single site, the primary role of phosphorylation in ompF regulation is to facilitate cooperative interactions between OmpR molecules bound at adjacent sites. The importance of cooperative interactions in the binding of OmpR to the ompF regulatory region is supported by two lines of evidence.
The first line of evidence comes from our experiments using a DNA fragment containing a tandem duplication of the F1 site. This analysis showed the cooperative nature of OmpR binding and allowed us to estimate the degree of cooperativity for the F1-F1 construct. For the unphosphorylated form of OmpR, the occupancy of one site stimulated the occupancy of the second equivalent site by ≈60-fold. Significantly, when the kinase was also present in the reaction, this occupancy was further stimulated, resulting in a total increase of ≈1,000-fold. Although the F1-F1 fragment does not carry the configuration of sites normally found at the ompF regulatory region, these experiments established that the unphosphorylated form of OmpR can manifest cooperative interactions and that these interactions can be further stimulated by phosphorylation.
The second line of evidence, which highlights the importance of OmpR phosphorylation and cooperativity in the binding of OmpR to the ompF regulatory region, comes from our site-inactivation experiments. In the absence of the kinase, a single base substitution in the F2 site is sufficient to eliminate the binding of the unphosphorylated form of OmpR to the mutated F2 site and the adjacent F3 site. This substitution also eliminates the formation of complex II by OmpR-P, the complex in which both sites F1 and F2 are occupied. Together, these results indicate that a single base pair substitution is sufficient to eliminate OmpR binding to F2. The unexpected result from this analysis was the ability of OmpR-P to form complex III. Based on the mobility of this complex, all three sites, including the mutated F2 site, are occupied. This would suggest that the presence of OmpR bound at the flanking sites F1 and F3 is sufficient to stabilize the interaction of OmpR molecules with the mutated F2 site, and furthermore, that this stabilization only occurs under conditions where OmpR can be phosphorylated. That this mutated F2 site can still bind OmpR in the presence of kinase could explain why it has been difficult to obtain single mutations in the ompF regulatory region that affect activation or repression by OmpR in vivo. These results also imply that the occupancy of the different sites in the ompF regulatory region can be influenced by the phosphorylation state of OmpR.
How might the effect of phosphorylation on the cooperative DNA-binding properties of OmpR explain the observed regulation of ompF transcription in vivo? Our results are consistent with the model diagrammed in Fig. 5. The ompF regulatory region contains four binding sites (Fig. 1A) that have different affinities for OmpR. The site with the highest affinity for OmpR is the F1 site (6–8, 12). In the absence of OmpR phosphorylation, OmpR may be able to bind F1, but it is unable to bind F2 in sufficient quantities to influence ompF transcription (Fig. 5A). However, under conditions where OmpR is phosphorylated, the F2 site is also occupied due to cooperative interactions between OmpR-P molecules bound at the F1 site and the weaker F2 site. Based on the position of the F2 site relative to the start point of ompF transcription, we postulate that occupancy of the F2 site allows OmpR to activate transcription from the ompF promoter under low osmolarity conditions. This could occur either by facilitating RNA polymerase (RNAP) binding and/or by allowing productive contacts between OmpR and RNAP (Fig. 5B). Finally, it seems likely that the stimulatory effect of phosphorylation on cooperative interactions could also be used to explain ompF repression under high osmolarity conditions. Previous studies have suggested that the repression of ompF transcription involves the formation of a DNA loop (8, 18, 19). One intriguing possibility is that the stimulation of cooperative interaction by high cellular levels of OmpR-P could result in the occupancy of the F3 and F4 sites. This increase in cooperative interactions, together with additional proteins such as integration host factor, could lead to the formation of a DNA loop that represses ompF transcription under high osmolarity conditions.
Figure 5.

Model for the regulation of ompF by OmpR-P. According to this model, the unphosphorylated form of OmpR binds primarily to the F1 site. (A) Although occupancy of the F2 site may occur in the absence of OmpR phosphorylation, this occupancy does not seem to be sufficient to activate ompF transcription. (B) In contrast, phosphorylation of OmpR results in increased occupancy of the F2 site due to the stimulation of cooperative interactions. We proposed that the increased occupancy of the F2 site is responsible for the observed activation of ompF transcription under low osmolarity conditions.
Acknowledgments
We thank S. Kowalczykowski, S. Kustu, M. Privalsky, and the members of our laboratory for helpful discussions. We are grateful to C. Price and M. Singer for their critical comments on the manuscript. This research was supported in part by Public Service Grant GM48591 to M.I. from the National Institutes of Health.
ABBREVIATIONS
- OmpR-P
phosphorylated form of OmpR
- MBP
maltose binding protein
References
- 1.Forst S, Inouye M. Annu Rev Cell Biol. 1988;4:21–42. doi: 10.1146/annurev.cb.04.110188.000321. [DOI] [PubMed] [Google Scholar]
- 2.Mizuno T, Mizushima S. Mol Microbiol. 1990;4:1077–1082. doi: 10.1111/j.1365-2958.1990.tb00681.x. [DOI] [PubMed] [Google Scholar]
- 3.Pratt L A, Silhavy T J. In: Two-Component Signal Transduction. Hoch J A, Silhavy T J, editors. Washington, DC: Am. Soc. Microbiol.; 1995. pp. 105–127. [Google Scholar]
- 4.Slauch J M, Silhavy T J. J Mol Biol. 1989;210:281–292. doi: 10.1016/0022-2836(89)90330-6. [DOI] [PubMed] [Google Scholar]
- 5.Harlocker S L, Bergstrom L, Inouye M. J Biol Chem. 1995;270:26849–26856. doi: 10.1074/jbc.270.45.26849. [DOI] [PubMed] [Google Scholar]
- 6.Huang K-J, Igo M M. J Mol Biol. 1996;262:615–628. doi: 10.1006/jmbi.1996.0540. [DOI] [PubMed] [Google Scholar]
- 7.Maeda S, Takayanagi K, Nishimura Y, Maruyama T, Sato K, Mizuno T. J Biochem. 1991;110:324–327. doi: 10.1093/oxfordjournals.jbchem.a123579. [DOI] [PubMed] [Google Scholar]
- 8.Rampersaud A, Harlocker S L, Inouye M. J Biol Chem. 1994;269:12559–12566. [PubMed] [Google Scholar]
- 9.Hudson J M, Fried M G. J Mol Biol. 1990;214:381–396. doi: 10.1016/0022-2836(90)90188-R. [DOI] [PubMed] [Google Scholar]
- 10.Tsai S Y, Tsai M J, O’Malley B W. Cell. 1989;57:443–448. doi: 10.1016/0092-8674(89)90919-7. [DOI] [PubMed] [Google Scholar]
- 11.Porter S C, North A K, Wedel A B, Kustu S. Genes Dev. 1993;7:2258–2273. doi: 10.1101/gad.7.11.2258. [DOI] [PubMed] [Google Scholar]
- 12.Forst S, Kalve I, Durski W. FEMS Microbiol Lett. 1995;131:147–151. doi: 10.1111/j.1574-6968.1995.tb07769.x. [DOI] [PubMed] [Google Scholar]
- 13.Rampersaud A, Norioka S, Inouye M. J Biol Chem. 1989;264:18693–18700. [PubMed] [Google Scholar]
- 14.Jo Y-K, Nara F, Ichihara S, Mizuno T, Mizushima S. J Biol Chem. 1986;261:15252–15256. [PubMed] [Google Scholar]
- 15.Norioka S, Ramakrishnan G, Ikenaka K, Inouye M. J Biol Chem. 1986;261:17113–17119. [PubMed] [Google Scholar]
- 16.Brissette R, Tsung K, Inouye M. J Bacteriol. 1991;173:601–608. doi: 10.1128/jb.173.2.601-608.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanamaru K, Aiba H, Mizuno T. J Biochem (Tokyo) 1990;108:483–487. doi: 10.1093/oxfordjournals.jbchem.a123225. [DOI] [PubMed] [Google Scholar]
- 18.Huang K J, Schieberl J L, Igo M M. J Bacteriol. 1994;176:1309–1315. doi: 10.1128/jb.176.5.1309-1315.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slauch J M, Silhavy T J. J Bacteriol. 1991;173:4039–4048. doi: 10.1128/jb.173.13.4039-4048.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]