

Abstract
The purpose of this study was to determine the role of prostaglandin E2 (PGE2) in modulating neuronal activity of the dorsolateral periaqueductal gray (dl-PAG) through excitatory and inhibitory synaptic inputs. First, whole cell voltage-clamp recording was performed to obtain excitatory and inhibitory postsynaptic currents (EPSCs and IPSCs) of the dl-PAG neurons. Our results show that PGE2 significantly decreased the frequency of miniature EPSCs and amplitude of evoked EPSCs. The effects were mimicked by sulprostone, an agonist to PGE2 EP3 receptors. In contrast, PGE2 had no distinct effect on IPSCs. In addition, spontaneous action potential of the dl-PAG neurons was recorded using whole cell current-clamp methods. PGE2 significantly attenuated the discharge rate of the dl-PAG neurons. The decreased firing activity was abolished in the presence of glutamate NMDA and non-NMDA receptors antagonists. The results from the current study provide the first evidence indicating that PGE2 inhibits the neuronal activity of the dl-PAG via selective attenuation of glutamatergic synaptic inputs, likely due to activation of presynaptic EP3 receptors.
Keywords: Prostaglandin, synaptic transmission, glutamate, midbrain PAG
1. Introduction
Prostaglandin E2 (PGE2) has been known to contribute to inflammation and pain hypersensitivity (Minami et al., 2001; Omote et al., 2002; Samad et al., 2001). At the site of inflammation, PGE2 sensitizes peripheral nociceptors through activation of EP receptors present on the peripheral terminals of sensory neurons by reducing threshold and increasing responsiveness (Omote et al., 2002). PGE2 is also produced in the spinal cord after tissue injury, where it increases excitability of the spinal cord dorsal horn neurons that produces pain hypersensitivity (Minami et al., 2001; Samad et al., 2001).
Studies have further shown that PGE2 receptors appear in several supraspinal regions including hypothalamus, midbrain periaqueductal gray (PAG) and hippocampus (Ek et al., 2000; Nakamura et al., 2000). Among the PGE2 receptor subtypes (EP1–4), EP3 has been reported to mediate a number of the physiological functions of PGE2 in the CNS such as pain modulation and regulation of the autonomic nervous system (Kumazawa et al., 1993; Oka et al., 1997; Yokotani et al., 1995). EP3 receptors have specifically been localized in the PAG (Ek et al., 2000; Nakamura et al., 2000).
A cyclooxygenase inhibitor injected into the PAG attenuates pain response from cutaneous and visceral afferent nerves (Vanegas and Tortorici, 2002). A prior study has also shown that microinjection of PGE2 into the PAG facilitates nociception through descending activation of the rostral ventromedial medulla (Heinricher et al., 2004). Activation of EP3 receptor within the PAG increases formalin-induced nociceptive response by modulating glutamate and GABA releases (Oliva et al., 2006). However, the underlying mechanisms by which PGE2 participates in excitatory glutamatergic and inhibitory GABAergic synaptic signaling to the PAG neurons have not specifically been studied.
It has been reported that the dorsal horn of the spinal cord has neuronal terminations in the dorsolateral (dl), lateral and ventrolateral regions of the PAG (Craig, 1995; Keay et al., 1997; Wiberg and Blomqvist, 1984). Those regions of the PAG further send descending neuronal projections to the medulla (Hudson and Lumb, 1996; Odeh and Antal, 2001) in regulating pain and autonomic activity (McGaraughty et al., 2003; Tjen-A-Looi et al., 2006; Verberne and Guyenet, 1992). For example, activation of the dl-PAG contributes to an increase in arterial blood pressure and antinociception (Bandler et al., 1991; Behbehani, 1995).
Glutamate, the major excitatory neurotransmitter, appears in the dl-PAG region (Beitz and Williams, 1991). The dl-PAG also has the high density of excitatory amino acid binding sites (glutamate receptor subtypes) including a-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA)/kainate, N-methyl-d-aspartate (NMDA) and metabotropic receptors (Albin et al., 1990; Cotman et al., 1987).
In this report, therefore, we used an in vitro whole cell recording technique in the midbrain slice to determine the role of PGE2 in modulating the firing activity of the dl-PAG neurons through the excitatory glutamatergic inputs. We hypothesized that PGE2 would decrease discharge of the dl-PAG neurons via inhibition of glutamatergic synaptic inputs. Furthermore, we determined the role of EP3 receptor activation in glutamatergic synaptic signaling to the dl-PAG neurons.
In addition, GABA-mediated neuronal elements constituting ~50% of the total population of neurons play a crucial role in the intrinsic neuronal circuitry of the PAG (Mugnaini and Oertel, 1985; Reichling, 1991). The GABA synaptic inputs make up ~50% of the synaptic innervation of the PAG neurons and the majority of GABAergic neurons are tonic active interneurons (Barbaresi, 2005). The release of GABA from those neurons may play a role in modulation of the synaptic inputs to the PAG neurons. Studies have further shown that GABAA receptors are dense within the PAG (Bowery et al., 1987; Chu et al., 1990). Thus the effect of PGE2 on the inhibitory GABAergic inputs to the dl-PAG neurons was also examined in this study.
2. Results
At the end of each experiment, the location of the recording pipette in the PAG slice was visualized and identified under a microscope using differential interference contrast (DIC, x40 magnification). We have confirmed that all the cells included for data analysis in this experiment located in the dl-PAG (shown in Fig. 1) according to rat brain atlas (Swanson, 1998). Whole cell patch-clamp experiments were performed and experimental data were collected from 69 dl-PAG neurons.
Figure 1.
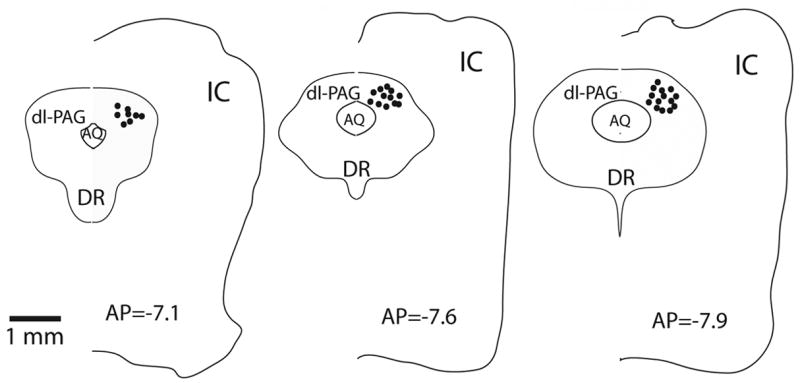
The electrophysiological activity was recorded from the dorsolateral PAG (dl-PAG). At the end of each experiment, recording sites were examined under a microscope using differential interference contrast. The representative locations (solid circles) of recorded neurons are shown with anterior-posterior (AP) coordinates of the sections using Swanson’s rat brain maps. AQ, cerebral aqueduct; DR, dorsal nucleus raphe; IC, inferior colliculus external nucleus.
2.1. Effect of PGE2 on Glutamatergic Excitatory Postsynaptic Currents (EPSCs)
The spontaneous miniature EPSCs (mEPSCs) were recorded in the dl-PAG in order to determine the effects of PGE2 on synaptic glutamate release onto the neurons (Fig. 2). PGE2, in the concentration of 5μM, perfused into the recording chamber significantly decreased the frequency of mEPSCs from 3.06±0.18 to 1.39±0.14 Hz (P<0.05, n=8), but did not alter the amplitude and the decay time constant of mEPSCs (7.55±0.36 ms in control vs. 7.85±0.51 ms after PGE2, P>0.05) in all neurons tested. The mEPSCs recovered during washout of the perfusion solution and were completely abolished with 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, Fig. 2A). The cumulative probability analysis of mEPSCs shows that the distribution pattern of the inter-event interval of mEPSCs shifted toward the right but the distribution pattern of the amplitude was not altered as PGE2 was applied (Fig. 2B&C). Average data of the PGE2 effects on the frequency and amplitude of mEPSC of the dl-PAG neurons are also shown (Fig. 2D&E).
Figure 2.
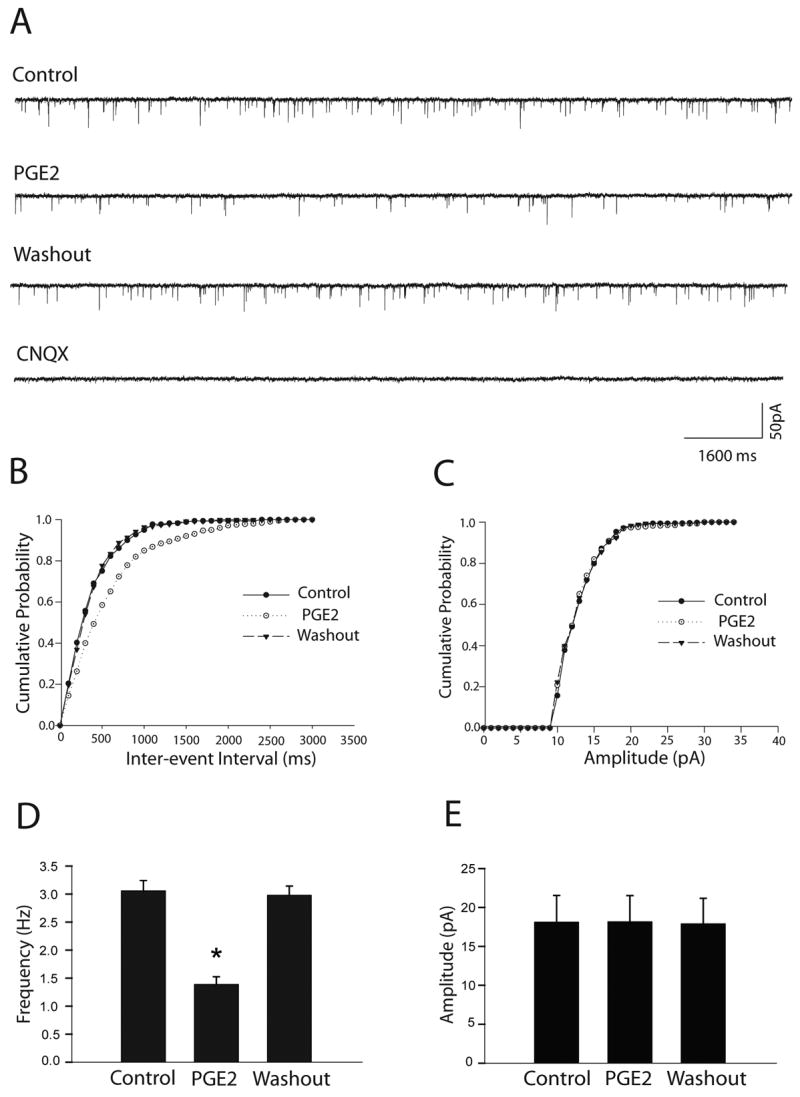
PGE2 decreased the frequency of glutamatergic mEPSCs of the dl-PAG neurons. The effect was observed in eight neurons tested. A: Representative tracings from a dl-PAG neuron show that 5μM of PGE2 attenuated the frequency of mEPSCs, and that the mEPSCs recovered during washout and completely abolished in the presence of 20 μM of CNQX. B&C: The cumulative probability analysis shows that PGE2 increased the inter-event interval of mEPSCs but did not alter the distribution pattern of the amplitude of the mEPSCs. D&E: Average data show the effects of PGE2 on the frequency and amplitude of mEPSCs of the dl-PAG neurons. *P<0.05, vs. control and washout.
Furthermore, the effects of PGE2 on mEPSCs were mimicked by EP3 activation (Fig. 3). Sulprostone, an EP3 agonist (Clarke et al., 2004), in the concentration of 5μM, significantly decreased the frequency of mEPSCs from 3.15±0.10 to 1.27±0.16 Hz (P<0.05, n=8), but did not alter the amplitude and the decay time constant of mEPSCs in all neurons tested.
Figure 3.
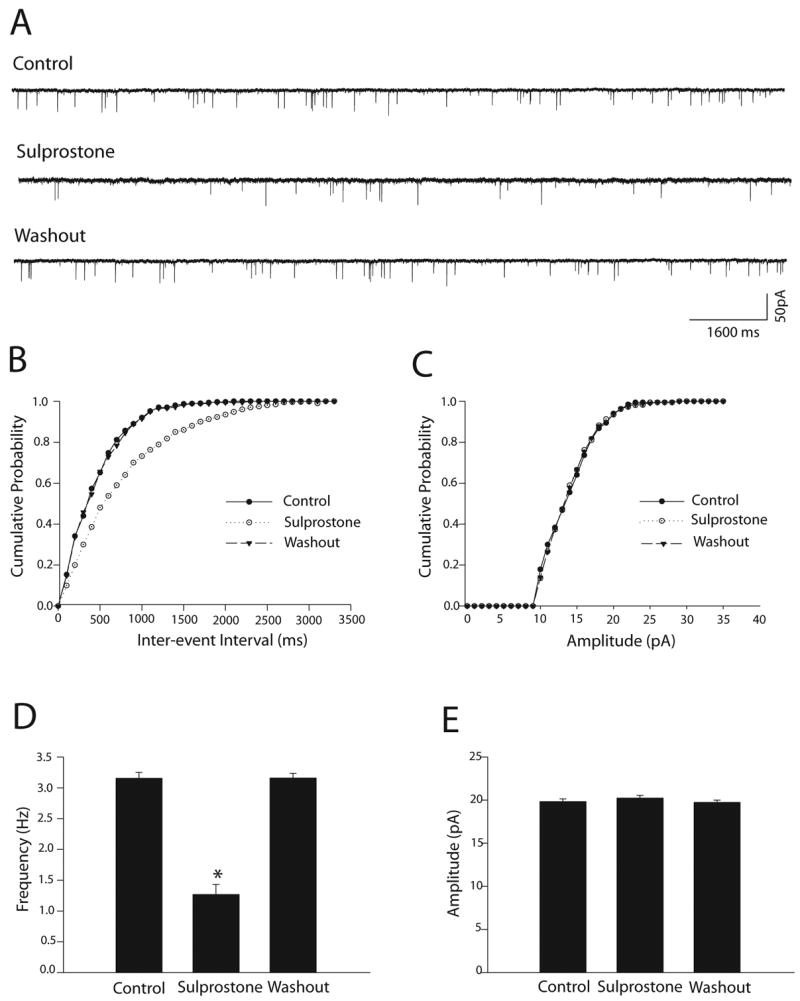
EP3 activation decreased the frequency of mEPSCs of the dl-PAG neurons. Sulprostone (5μM) was used to activate EP3 in eight neurons. A: Representative tracings from a dl-PAG neuron show that sulprostone inhibited the frequency of mEPSCs, and the mEPSCs recovery during washout. B&C: Sulprostone increased the inter-event interval of mEPSCs without altering the distribution pattern of the amplitude of the mEPSCs. D&E: Average data show the effect of sulprostone. *P<0.05, vs. control and washout.
In the next group of experiments, the effects of PGE2 and sulprostone on evoked EPSCs (eEPSCs) were examined in the dl-PAG neurons (Fig. 4). PGE2 (Fig. 4A&B) and sulprostone (Fig. 4C&D) significantly inhibited the peak amplitude of eEPSCs by 40% and by 42%, respectively. In order to determine whether the effects of PGE2 and sulprostone were via presynaptic sites, we further examined the paired-pulse ratio (PPR) of eEPSCs when PGE2 (Fig. 4A&B) and sulprostone (Fig. 4C&D) were perfused into the recording chamber. The PPR increased by 102% in control vs. 143% after PGE2 (n=8, P<0.05), and by 109% in control vs. 162% after sulprostone (n=8, P<0.05).
Figure 4.
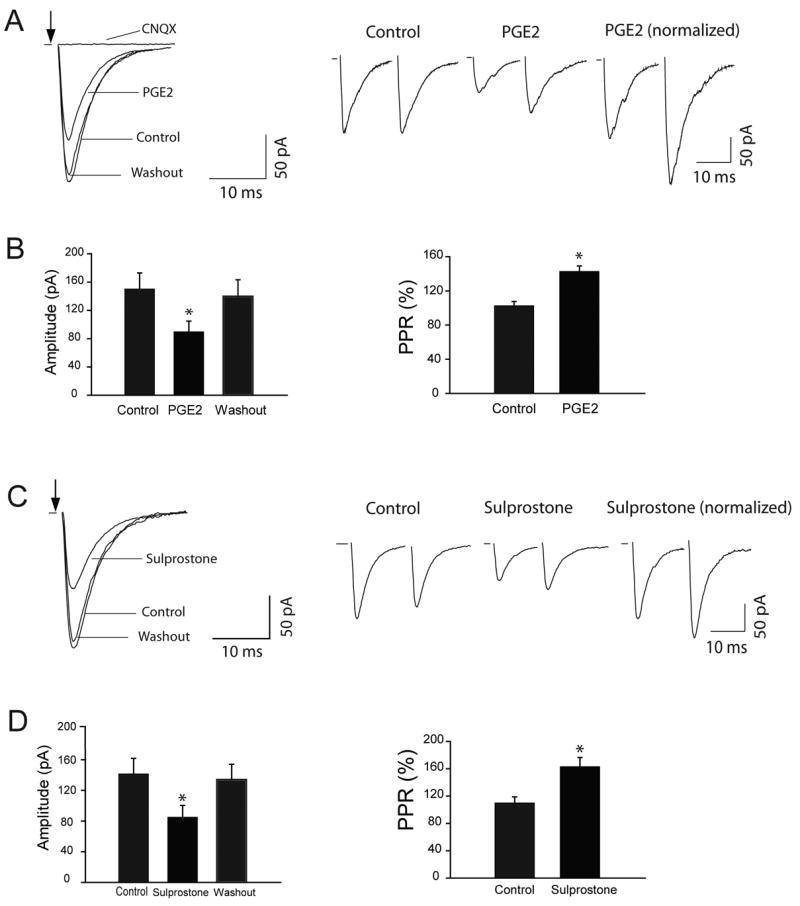
PGE2 and sulprostone attenuated the peak amplitude of eEPSCs of the dl-PAG neurons and increased the PPR of eEPSCs. A&B: Typical traces from a dl-PAG neuron and summarized data (n=8) showing the peak amplitude of eEPSCs during control, PGE2 and washout; and the PPR of eEPSCs. C&D: Typical traces from a dl-PAG neuron and summarized data (n=8) showing the peak amplitude of eEPSCs during control, sulprostone and washout; and the PPR of eEPSCs. *P<0.05, vs. control and washout for the amplitude; and vs. control for the PPR. The traces are average of 10 consecutive responses. Stimulation artifacts are removed and indicated by arrows.
2.2. Effect of PGE2 on GABAergic Inhibitory Postsynaptic Currents (IPSCs)
The spontaneous miniature IPSCs (mIPSCs) were also examined in the dl-PAG neurons in order to determine the effects of PGE2 on synaptic GABA release onto neurons (Fig. 5). PGE2, in the concentration of 5μM, did not produce a significant effect on the frequency and amplitude of mIPSCs in ten dl-PAG neurons. The mIPSCs were completely eliminated in the presence of 20 μM of bicuculline (Fig. 5A). Average data further show that PGE2 had no effect on the frequency and amplitude of mIPSCs of the dl-PAG neurons (Fig. 5B).
Figure 5.
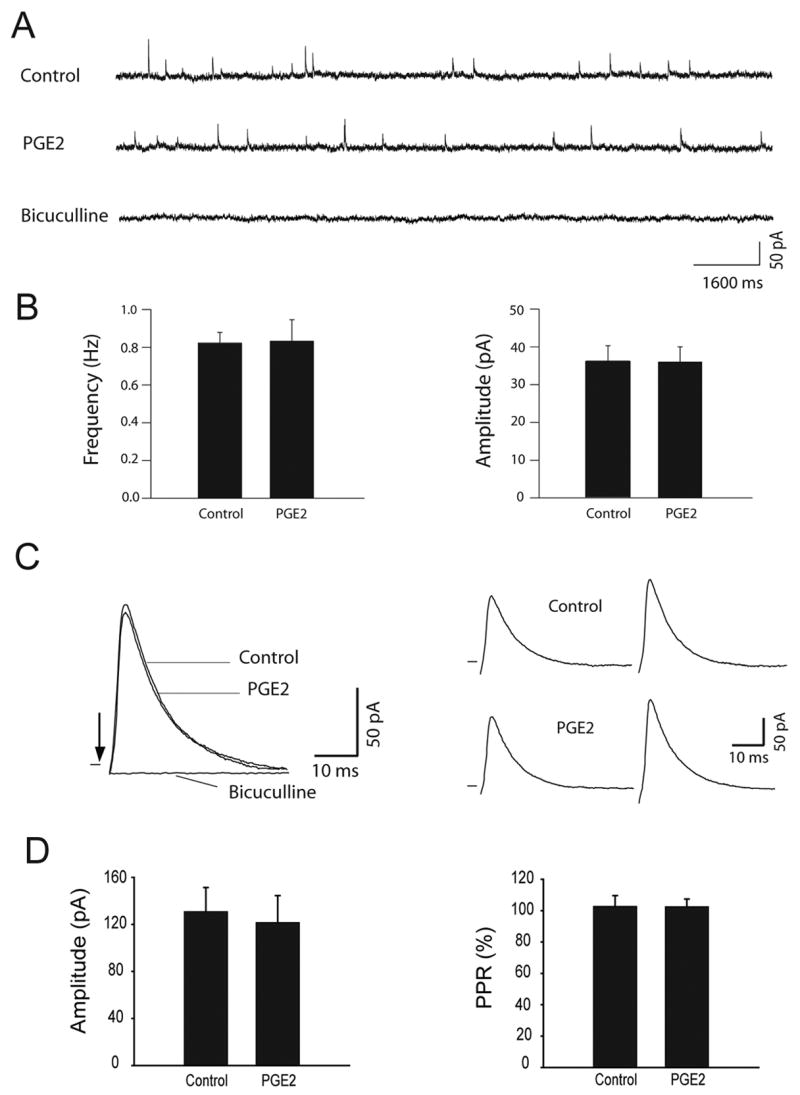
PGE2 had no distinct effects on GABAergic IPSCs of the dl-PAG neurons. Representative tracings from a dl-PAG neuron (A) and average data (B) show that the frequency and amplitude of spontaneous mIPSCs were not altered by bath application of 5μM of PGE2. The results were seen in ten neurons tested. Effects of PGE2 on eIPSCs were further examined in the dl-PAG neurons. Averaged traces of 10 consecutive responses from a dl-PAG neuron (C) and average data of eight neurons (D) show that 5μM of PGE2 did not significantly alter the peak amplitude and PPR of eIPSCs. The eIPSCs were completely abolished with bicuculline.
In another group of experiments, the effects of PGE2 on evoked IPSCs (eIPSCs) were examined in eight dl-PAG neurons (Fig. 5C&D). PGE2 had no distinct effect on the peak amplitude and PPR of eIPSCs of the dl-PAG neurons.
2.3. Effect of PGE2 on Discharge of dl-PAG Neurons
Since our results have shown that PGE2 decreased the excitatory glutamatergic inputs to the dl-PAG neurons without altering the inhibitory GABAergic synaptic activity, it was likely that PGE2 inhibited the activity of the dl-PAG neurons. To test this hypothesis, the effects of PGE2 on the discharge of the dl-PAG neurons were examined using whole cell current-clamp recordings (Fig. 6A&B). PGE2 (5 μM) significantly decreased the discharge rate of the dl-PAG neurons from 4.07±0.61 to 1.96±0.34Hz (P<0.05, n=8). Application of PGE2 did not significantly alter the resting membrane potential of the dl-PAG neurons (−66.4±1.96 mV vs. 65.0±1.16 mV, P> 0.05 n=10). Note that the resting membrane potential was measured from the cells without the firing activity.
Figure 6.
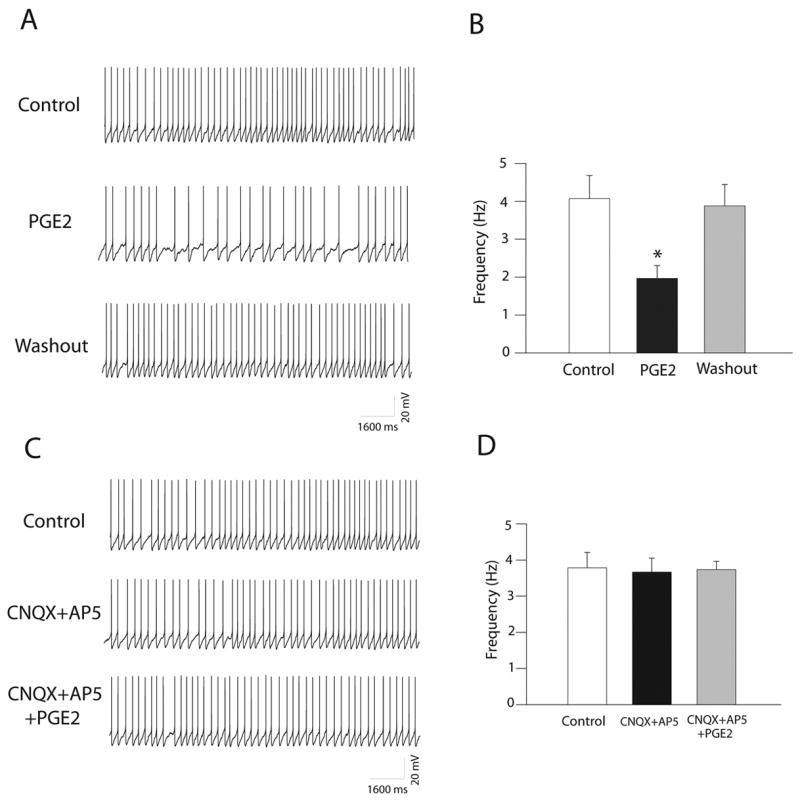
PGE2 had an inhibitory effect on the firing activity of the dl-PAG neurons. A: Original tracings from a dl-PAG neuron show the spontaneous discharge activity during control, PGE2 (5 μM) perfusion and washout. B: Average data (n=8). *P<0.05, vs. control and washout. C&D: Original tracings from a dl-PAG neuron and average data (n=9) show the spontaneous discharge activity during control, CNQX (20 μM) plus AP-5 (50 μM), and PGE2 perfusion in the presence of the glutamate receptors antagonists. The effect of PGE2 was abolished after CNQX and AP-5 application.
In addition, the role of the glutamatergic synaptic inputs and glutamate receptors in PGE2 attenuation of the dl-PAG neurons was determined (Fig. 6C&D). The firing activities of the dl-PAG neurons were examined in the presence of glutamate NMDA and non-NMDA antagonists, 2-amino-5- phosphonopentanoic acid (AP-5) and CNQX, following application of PGE2. The spontaneous discharge activities of the PAG neurons were slightly decreased following perfusion of 20 μM of CNQX and 50 μM of AP-5 (3.78±0.43 vs. 3.66±0.39 Hz, P>0.05 n=9). Subsequent application of 5 μM of PGE2 failed to decrease the spontaneous neuronal activities in the presence of CNQX and AP-5.
3. Discussion
In the present study, regulatory effects of PGE2 on excitatory glutamatergic and inhibitory GABAergic synaptic activity in the dl-PAG were determined using in vitro PAG slice preparation. Our results have demonstrated that PGE2 significantly attenuated the frequency of mEPSCs of the dl-PAG neurons, but had no distinct effect on the amplitude of mEPSCs (Fig. 2). The similar effects were seen after EP3 receptor was activated with sulprostone, an EP3 agonist (Fig. 3). Moreover, both PGE2 and sulprostone significantly decreased the peak amplitude of eEPSCs with increasing the PPR (Fig. 4). These data suggest that EP3 activation inhibited the synaptic glutamate release in the PAG and the site of the action was likely at the presynaptic glutamatergic terminals (Sulzer and Pothos, 2000).
In contrast, PGE2 had no distinct effects on the frequency and amplitude of GABAergic mIPSCs, and amplitude of eIPSC recorded from the dl-PAG neurons (Fig. 5). This suggests the lack of PGE2 effects on the synaptic GABAergic terminals in the dl-PAG.
The mEPSCs represent the synaptic quanta release of glutamate that plays a role in modulating the activity of the postsynaptic neuron. Therefore, on the basis of the data showing that PGE2 had an inhibitory effect on the EPSCs of the dl-PAG neurons (Fig. 2–4), we have further determined the effects of PGE2 on the firing activity of the dl-PAG neurons in this report. The data have shown that PGE2 significantly inhibited the discharge frequency of the dl-PAG neurons (Fig. 6). To further support this notion, blocking glutamate receptors with CNQX and AP-5 abolished PGE2-induced decrease in firing activity of the dl-PAG neurons (Fig. 6). Thus our results suggest that PGE2 suppresses neuronal activity of the dl-PAG through attenuation of the excitatory glutamatergic synaptic inputs.
Previous studies have shown that EP3 receptors on presynaptic nerve terminals regulate the release of neurotransmitters (Exner and Schlicker, 1995; Nakamura et al., 1998; Schlicker and Marr, 1997). Whether EP3 receptors are present on glutamatergic terminals of presynaptic sites of the dl-PAG has not, to our knowledge, been reported although EP3 immunoreactivity has been identified in the dl-PAG (Ek et al., 2000; Nakamura et al., 2000). Our data from the current experiment demonstrated that activation of EP3 receptors decreased glutamate release from presynaptic sites. This provides electrophysiological evidence that PGE2 receptor EP3 is likely to appear on presynaptic nerve terminals in the dl-PAG.
A prior study has shown that microinjection of PGE2 into the PAG facilitates nociception through descending activation of the rostral ventromedial medulla (Heinricher et al., 2004). However, a mechanism by which PGE2 within the PAG induces hyperalgesia is unclear. Intra-PAG perfusion with misoprostol, a synthetic PGE2 analogue, increases glutamate release and formalin-induced nociceptive response (Oliva et al., 2006). Misoprostol also produces a biphasic effect on GABA release (Oliva et al., 2006). In this previous study, microdialysis methods were employed to collect extracellular samples from the PAG tissues. It was unlikely to determine whether the effect of misoprostol on the releases of glutamate and GABA was via presynaptic terminals within the PAG. Furthermore, an interaction between GABAB receptor and presynaptic glutamate release (Lei and McBain, 2003) wasn’t ruled out in the previous report. Thus PAG glutamate and GABA concentrations that were measured using microdialysis methods (Oliva et al., 2006) might be difficult to account for the effects of PGE2 on pain modulation in this brain region.
Activation of glutamate receptors in the PAG has been reported to produce analgesia and this is dependent on activated subtypes of glutamate receptors (Maione et al., 1998; Maione et al., 2000). On the other hand, the effects of PGE2 on glutamate release are not precisely determined. It is noted that activation of subtypes of PGE2 receptor differentially modulates glutamate release in the CNS. For example, activation of presynaptic EP2 receptors in the hippocampus has been reported to facilitate synaptic glutamate release via increasing cAMP pathway (Sang et al., 2005). However, activation of EP3 receptors mainly inhibits cAMP generation via Gi coupled mechanisms (Hatae et al., 2002). A large population of EP3 receptors has been identified in the PAG (Ek et al., 2000; Nakamura et al., 2000). The present experiment provides additional evidence suggesting that EP3 may appear at the presynaptic glutamatergic terminals in the PAG. Thus it is reasoned that the glutamate release is decreased after increasing PGE2 in the PAG. PGE2 receptors activation in the dl-PAG has been reported to facilitate nociception by affecting descending neuronal activity of the rostral ventromedial medulla (Heinricher et al., 2004). Nevertheless, those data suggest that PGE2 within the PAG plays a role in the processing of nociception. Overall effects of PGE2 on pain modulation may be dependent on activated subtypes of PGE2 receptor and/or glutamate receptors. The results from our current study provide, for the first time, electrophysiological evidence that 1) PGE2 within the dl-PAG neurons decreases the spontaneous firing rate of the PAG cells; and 2) the inhibitory effects of PGE2 on the neuronal activity are likely mediated via presynaptic EP3 modulation of the glutamate release.
Four subtypes of PGE2 receptors (EP1–4) have been classified (Boie et al., 1997; Narumiya et al., 1999). EP1 receptors couple with the Gq-phospholipase C-IP3 pathway. EP2 and EP4 receptors couple with the Gs-adenylyl cyclase-cAMP pathway and activation of those receptors increases cAMP levels (Hatae et al., 2002). Elevated cAMP enhances glutamatergic transmission in the PAG as well as in other brain regions (Huang and Hsu, 2006; Kaneko and Takahashi, 2004; Marabese et al., 2005). Activation of presynaptic EP2 receptors in the hippocampus has been reported to facilitate synaptic glutamate release via increasing cAMP pathway (Sang et al., 2005). In contrast, activation of EP3 receptors mainly inhibits cAMP generation via Gi coupled mechanisms (Hatae et al., 2002). EP3 is widely distributed in the CNS and plays a role in mediating a number of the physiological functions of PGE2 (Kumazawa et al., 1993; Oka et al., 1997; Yokotani et al., 1995). In the current study, sulprostone, an EP3 receptors agonist (Clarke et al., 2004) has been seen to inhibit the synaptic glutamate release in the PAG, which mimics the PGE2 effects. This suggests that PGE2 receptors EP3 may play a dominant role in attenuation of the glutamatergic synaptic inputs to dl-PAG neurons. However, one must consider the possibility that the effects seen with PGE2 could be due to activation of other subtypes of PGE2 receptors until specific PGE2 subtype blockers become available.
In summary, PGE2 significantly decreases the frequency of glutamatergic mEPSCs as well as the amplitude of eEPSCs but not GABAergic IPSCs of the dl-PAG neurons. The effect is likely mediated via activation of EP3 receptors. The inhibited glutamatergic synaptic inputs attenuate neuronal activity of the dl-PAG because the effect of PGE2 on the firing activity is blocked in the presence of glutamate NMDA and non-NMDA receptor antagonists. Our data suggest a mechanism by which PGE2 modulates neuronal activity in the dl-PAG via synaptic glutamate. The current study provides new information that the dl-PAG could be an important supraspinal site to be involved in PGE2-related modulation of physiological functions.
4. Experimental Procedure
4.1. Brain Slice Preparations
All procedures outlined in this study were approved by the Animal Care Committee of this institution. Sprague Dawley rats of either gender (4–6 weeks old) were anesthetized by inhalation of isoflurane oxygen mixture (5% isoflurane in 100% oxygen), and then were decapitated. Briefly, the brain was quickly removed and placed in ice-cold artificial cerebral spinal fluid (aCSF) perfusion solution. A tissue block containing the midbrain PAG was cut from the brain and glued onto the stage of the vibratome (Technical Product International, St. Louis, MO). Coronal slices (300 μm) containing the midbrain PAG were dissected from the tissue block in ice-cold aCSF solution. An equilibrium period of 60 min was required to incubate the slices in the aCSF at 34°C before they were transferred to the recording chamber. During the procedures described above, aCSF were saturated with 95% O2 - 5% CO2. The aCSF perfusion solution contained (in mM) 124.0 NaCl, 3.0 KCl, 1.3 MgSO4, 2.4 CaCl2, 1.4 NaH2 PO4, 10.0 glucose, and 26.0 NaHCO3 (Li et al., 2004).
4.2. Electrophysiological Recordings
4.2.1. Postsynaptic currents of dl-PAG neurons
A whole cell voltage-clamp technique was used to record postsynaptic currents in the dl-PAG neurons. Borosilicate glass capillaries (1.2 mm OD, 0.69 mm ID; Harvard Apparatus) were pulled to make the recording pipettes using a puller (Sutter Instrument, Novato, CA). The resistance of the pipette was 4–6 MΩ when it was filled with the internal solution (contained in mM: 130.0 potassium gluconate, 1.0 MgCl2, 10.0 HEPES, 10.0 EGTA, 1.0 CaCl2, and 4.0 ATP-Mg) (Li et al., 2004). The solution was adjusted to pH 7.25 with 1 M of KOH and osmolarity of 280–300 mOsm. The slice was placed in a recording chamber (Warner Instruments, Hamden, CT) and fixed with a grid of parallel nylon threads supported by a U-shaped stainless steel weight. The aCSF saturated with 95% O2 - 5% CO2 was perfused into the chamber at 3.0 ml/min. The temperature of the perfusion solution was maintained at 34°C by an in-line solution heater with a temperature controller (Model TC-324; Warner Instruments). Whole cell recordings from the dl-PAG neurons were performed visually using DIC optics on an upright microscope (BX50WI, Olympus, Tokyo, Japan). The tissue image was captured and enhanced through a camera and displayed on a video monitor. A tight giga-ohm seal was subsequently obtained in the dl-PAG neuron viewed using DIC optics. An equilibration period of 5–10 min was allowed after whole cell access was established and the recording reached a steady state. The recording was abandoned if the monitored input resistance changed >15%.
The mEPSCs were recorded in the presence of 1 μM of TTX and 20 μM of bicuculline at a holding potential of −70 mV. The mIPSCs were recorded in the presence of 1 μM of tetrodotoxin (TTX) and 20 μM of CNQX at a holding potential of 0 mV. QX-314 (10 mM) and GDP-β-s (1 mM) were contained in the pipette solution to block sodium current and possible postsynaptic effects mediated through G proteins in this experiment.
In order to examine the eEPSCs and eIPSCs in the dl-PAG neurons, electrical stimulation (0.1 ms, 0.4–0.8 mA, and 0.2 Hz) was induced using a bipolar tungsten electrode connected to a stimulator (Grass Instruments, Quincy, MA). The tip of the stimulating electrode was placed 200-500 μm away from the recorded neuron. The eEPSCs was determined at a holding potential of −70 mV in the presence of bicuculline (20 μM), and eIPSCs at 0 mV in the presence of CNQX (20 μM), respectively. QX-314 and GDP-β-s were also contained in the pipette solution in this experiment.
Single stimuli and paired stimuli at short intervals (40 ms for eEPSCs and 50 ms for eIPSCs) were applied. PPR of eEPSCs and eIPSCs was expressed as percentage (%) of the amplitude of the second synaptic response/the first synaptic response. Ten consecutive responses were averaged for subsequent analysis.
4.2.2. Spontaneous action potentials of dl-PAG neurons
A whole cell current-clamp technique was used to record the spontaneous firing activity of the dl-PAG neurons (under holding current = 0). The recording procedures were described above. The whole cell access was first established. For the cells that did not display firing activity, resting membrane potential was measured when it became stable. For those cells showing activity, discharge rate of the dl-PAG neurons was examined 5-10 min after the firing activity reached a steady state. The action potential with amplitude >60 mV was included for data analysis in this experiment.
4.2.3. Drugs and their application
TTX, bicuculline, CNQX, and AP-5 were obtained from Sigma Co. PGE2 and sulprostone were obtained from Cayman Chemical Co. All drugs were dissolved in the aCSF solution immediately before they were used. According to experimental protocol, the drugs were delivered into the recording chamber at final concentrations using syringe pumps during the experiment (Xing and Li, 2007). The responses of EPSCs, IPSCs and firing activity of the dl-PAG to application of drugs were recorded after control data were collected.
4.3. Data Acquisition and Analysis
Signals were recorded with a MultiClamp 700B amplifier (Axon Instruments, Foster City, CA), digitized at 10 kHz with a DigiData 1440A, and filtered at 1–2 kHz and saved in a PC-based computer using pClamp 10.1 software (Axon Instruments). A liquid junction potential of −15.0 mV (for the potassium gluconate pipette solution) was corrected during off-line analysis (Li et al., 2002; Li et al., 2004). The mEPSCs, mIPSCs, and firing activities of the PAG neurons were analyzed off-line with a peak detection program (MiniAnalysis, Synaptosoft, Leonia, NJ). Detection of events was accomplished by setting a threshold above the noise level. The distribution of cumulative probability of the inter-event interval and amplitude of mEPSCs and mIPSCs was estimated using the Komogorov–Smirnov test (Li et al., 2002; Li et al., 2004). The amplitude of eEPSCs and eIPSCs, and PPR were analyzed using Clampfit 10.1 (Axon Instruments). Experimental data (frequency, amplitude and decay time of mEPSCs and mIPSCs, the firing rate of dl-PAG neurons and the PPR of evoked currents) were analyzed with one-way ANOVA. Tukey’s post hoc analyses were utilized to determine differences between groups, as appropriate. Paired t test was used to analyze amplitude of the eEPSCs and eIPSCs. All values were expressed mean ± SE. For all analyses, differences were considered significant at P<0.05. All statistical analyses were performed using SPSS for windows version 15.0.
Acknowledgments
Drs. Jihong Xing and Jian Lu are visiting scholar from The First Hospital of Jilin University Changchun 130021, Jilin Province of PR China.
The authors thank Dr. Lawrence Sinoway for his support and scientific input. This study was supported by NIH R01 HL075533 (Li), R01 HL078866 (Li) and R01 HL060800 (Sinoway).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature References
- Albin RL, Makowiec RL, Hollingsworth Z, Dure LS, Penney JB, Young AB. Excitatory amino acid receptors in the periaqueductal gray of the rat. Neurosci Lett. 1990;118:112–115. doi: 10.1016/0304-3940(90)90261-7. [DOI] [PubMed] [Google Scholar]
- Bandler R, Carrive P, Zhang SH. Integration of somatic and autonomic reactions within the midbrain periaqueductal gray: Viscerotopic, somatotopic and functional organization. Prog In Brain Res. 1991;87:269–305. doi: 10.1016/s0079-6123(08)63056-3. [DOI] [PubMed] [Google Scholar]
- Barbaresi P. GABA-immunoreactive neurons and terminals in the cat periaqueductal gray matter: a light and electron microscopic study. Journal of Neurocytology. 2005;34:471–87. doi: 10.1007/s11068-006-9440-7. [DOI] [PubMed] [Google Scholar]
- Behbehani MM. Functional characteristics of the midbrain periaqueductal gray. Prog Neurobiol. 1995;46:575–605. doi: 10.1016/0301-0082(95)00009-k. [DOI] [PubMed] [Google Scholar]
- Beitz AJ, Williams FG. Localization of putative amino acid transmitters in the PAG and their relationship to the PAG–raphe magnus pathway. In: Depaulis A, Bandler R, editors. The Midbrain Periaqueductal Gray Matter. Plenum; New York: 1991. [Google Scholar]
- Boie Y, Stocco R, Sawyer N, Slipetz DM, Ungrin MD, Neuschafer-Rube F, Puschel GP, Metters KM, Abramovitz M. Molecular cloning and characterization of the four prostaglandin E2 prostanoid receptor subtypes. Eur J Pharmacol. 1997;340:227–241. doi: 10.1016/s0014-2999(97)01383-6. [DOI] [PubMed] [Google Scholar]
- Bowery NG, Hudson AL, Price GW. GABAA and GABAB receptor site distribution in the rat central nervous system. Neurosci. 1987;20:365–383. doi: 10.1016/0306-4522(87)90098-4. [DOI] [PubMed] [Google Scholar]
- Chu CDM, Albin RL, Young AB, Penney JB. Distribution and kinetics of GABAB binding sites in rat central nervous system: a quantitative autoradiographic study. Neurosci. 1990;34:341–357. doi: 10.1016/0306-4522(90)90144-s. [DOI] [PubMed] [Google Scholar]
- Clarke DL, Giembycz MA, Patel HJ, Belvisi MG. E-ring 8-isoprostanes inhibit Ach release from parasympathetic nerves innervating guinea-pig trachea through agonism of prostanoid receptors of the EP3-subtype . Br J Pharmacol. 2004;141:600–609. doi: 10.1038/sj.bjp.0705648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman CW, Monaghan DT, Ottersen OP, Storm-Mathisen J. Anatomical organization of excitatory amino acid receptors and their pathways. Trends Neurosci. 1987;10:273–280. [Google Scholar]
- Craig AD. Distribution of brainstem projections from spinal lamina I neurons in the cat and the monkey. J Comp Neurol. 1995;361:225–248. doi: 10.1002/cne.903610204. [DOI] [PubMed] [Google Scholar]
- Ek M, Arias C, Sawchenko P, Ericsson-Dahlstrand A. Distribution of the EP3 prostaglandin E2 receptor subtype in the rat brain: Relationship to sites of interleukin-1 induced cellular responsiveness. J Comp Neurol. 2000;428:5–20. doi: 10.1002/1096-9861(20001204)428:1<5::aid-cne2>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Exner J, Schlicker E. Prostanoid receptors of the EP3 subtype mediate the inhibitory effect of prostaglandin E2 on noradrenaline release in the mouse brain cortex. Naunyn Schmiedebergs Arch Phamracol. 1995;351:46–52. doi: 10.1007/BF00169063. [DOI] [PubMed] [Google Scholar]
- Hatae N, Sugimoto Y, Ichikawa A. Prostaglandin receptors: advanced in the study of EP3 receptor signaling. J Biochem. 2002;131:781–784. doi: 10.1093/oxfordjournals.jbchem.a003165. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Martenson ME, Neubert MJ. Prostaglandin E2 in the midbrain periaqueductal gray produces hyperalgesia and activates pain-modulating circuitry in the rostral ventromedial medulla. Pain. 2004;110:419–426. doi: 10.1016/j.pain.2004.04.026. [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Presynaptic mechanism underlying cAMP-induced synaptic potentiation in medial prefrontal cortex pyramidal neurons. Mol Pharmacol. 2006;69:846–856. doi: 10.1124/mol.105.018093. [DOI] [PubMed] [Google Scholar]
- Hudson PM, Lumb BM. Neurones in the midbrain periaqueductal grey send collateral projections to nucleus raphe magnus and the rostral ventrolateral medulla in the rat. Brain Res. 1996;733:138–141. doi: 10.1016/0006-8993(96)00784-6. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Takahashi T. Presynaptic mechanism underlying cAMP-dependent synaptic potentiation. J Neurosci. 2004;24:5202–5208. doi: 10.1523/JNEUROSCI.0999-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keay KA, Feil K, Gordon BD, Herbert H, Bandler R. Spinal afferents to functionally distinct periaqueductal gray columns in the rat - An anterograde and retrograde tracing study. J Comp Neurol. 1997;385:207–229. doi: 10.1002/(sici)1096-9861(19970825)385:2<207::aid-cne3>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Kumazawa T, Mizumura K, Koda H. Involvement of EP3 subtype of prostaglandin E receptors in PGE2-induced enhancement of the bradykinin response of nociceptors. Brain Res. 1993;632:321–324. doi: 10.1016/0006-8993(93)91169-s. [DOI] [PubMed] [Google Scholar]
- Lei S, McBain CJ. GABAB receptor modulation synaptic transmission interneurons. J Physiol. 2003;546:439–453. doi: 10.1113/jphysiol.2002.034017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan HL. Nitric oxide inhibits spinally projecting paraventricular neurons through potentiation of presynaptic GABA release. J Neurophysiol. 2002;88:2664–2674. doi: 10.1152/jn.00540.2002. [DOI] [PubMed] [Google Scholar]
- Li DP, Chen SR, Pan HL. VR1 receptor activation induces glutamate release and postsynaptic firing in the paraventricular nucleus. J Neurophysiol. 2004;92:1807–1816. doi: 10.1152/jn.00171.2004. [DOI] [PubMed] [Google Scholar]
- Maione S, Marabese I, Leyva J, Palazzo E, de Novellis V, Rossi F. Characterization of mGluRs which modulate nociception in the PAG of mouse. Neuropharmacol. 1998;37:1475–1483. doi: 10.1016/s0028-3908(98)00126-9. [DOI] [PubMed] [Google Scholar]
- Maione S, Oliva P, Marabese I, Palazzo E, Rossi F, Berrino L, Rossi F, Filippelli A. Periaqueductal grey matter metabotropic glutamate receptors modulate formalin-induced nociception. Pain. 2000;85:183–189. doi: 10.1016/s0304-3959(99)00269-9. [DOI] [PubMed] [Google Scholar]
- Marabese I, de Novellis V, Palazzo E, Mariani L, Siniscalco D, Rodella L, Rossi F, Maione S. Differential roles of mGlu8 receptors in the regulation of glutamate and r-aminobutyric acid release at periaqueductal grey level. Neuropharmacol. 2005;49:157–166. doi: 10.1016/j.neuropharm.2005.02.006. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Chu KL, Bitner RS, Martino B, El Kouhen R, Han P, Nikkel AL, Burgard EC, Faltynek CR, Jarvis F. Capsaicin infused into the PAG affects rat tail flick responses to noxious heat and alters neuronal firing in the RVM. J Neurophysiol. 2003;90:2702–2710. doi: 10.1152/jn.00433.2003. [DOI] [PubMed] [Google Scholar]
- Minami T, Nakano H, Kobayashi T, Sugimoto Y, Ushikubi F, Ishikawa A, Narumiya S, Ito S. Characterization of EP receptor subtypes responsible for prostaglandin E2-induced pain responses by use of EP1 and EP3 receptor knockout mice. Br J Pharmacol. 2001;133:438–444. doi: 10.1038/sj.bjp.0704092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugnaini E, Oertel WH. An atlas of the distribution of GABAergic neurons and terminals in the rat CNS as revealed by GAD immunohistochemistry. In: Bjorklund A, Hokfelt T, editors. Handbook of Chemical Neuroanatomy 4: GABA and Neuropeptides in the CNS. Elsevier; Amsterdam: 1985. [Google Scholar]
- Nakamura K, Kaneko T, Yamashita Y, Hasegawa H, Katoh H, Negishi M. Immunohistochemical localization of prostaglandin EP3 receptor in the rat nervous system. J Comp Neurol. 2000;421:543–569. doi: 10.1002/(sici)1096-9861(20000612)421:4<543::aid-cne6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Katoh H, Ichikawa A, Negishi M. Inhibition of dopamine release by prostaglandin EP3 receptor via pertussis toxin-sensitive and -insensitive pathways in PC12 cells. J Neurochem. 1998;71:646–652. doi: 10.1046/j.1471-4159.1998.71020646.x. [DOI] [PubMed] [Google Scholar]
- Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors, structures, properties and functions. Parmacol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- Odeh F, Antal M. The projections of the midbrain periaqueductal grey to the pons and medulla oblongata in rats. Eur J of Neurosci. 2001;14:1275–1286. doi: 10.1046/j.0953-816x.2001.01760.x. [DOI] [PubMed] [Google Scholar]
- Oka T, Hori T, Hosoi M, Oka K, Abe M, Kubo C. Biphasic modulation in the trigeminal nociceptive neuronal responses by the intracerebroventricular prostaglandin E2 may be mediated through different EP receptors subtypes in rats. Brain Res. 1997;771:278–284. doi: 10.1016/s0006-8993(97)00802-0. [DOI] [PubMed] [Google Scholar]
- Oliva P, Berrino L, de Novellis V, Palazzo E, Marabese I, Siniscalco D, Scafuro M, Mariani L, Rossi F, Maione S. Role of periaqueductal grey prostaglandin receptors in formalin-induced hyperalgesia. Eur J Pharmacol. 2006;530:40–47. doi: 10.1016/j.ejphar.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Omote K, Kawamata T, Nakayama Y, Yamamoto H, Kawamata M, Namiki A. Effects of a novel selective agonist for prostaglandin receptor subtype EP4 on hyperalgesia and inflammation in monoarthritic model. Anesthesiology. 2002;97:170–176. doi: 10.1097/00000542-200207000-00024. [DOI] [PubMed] [Google Scholar]
- Reichling DB. GABAergic neuronal circuitry in the periaqueductal gray matter. In: Depaulis A, Bandler R, editors. The Midbrain Periaqueductal Gray Matter. Plenum; New York: 1991. [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1 beta-mediated induction of Cox-2 in hte CNS contrubutes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Sang N, Zhang J, Marcheselli V, Bazan NG, Chen C. Postsynaptically synthesized prostaglandin E2 (PGE2) modulates hipocampal synaptic transmission via a presynaptic PGE2 EP2 receptor. J Neurosci. 2005;25:9858–9870. doi: 10.1523/JNEUROSCI.2392-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker E, Marr I. Mutual interactions of the presynaptic histamine H3 and prostaglandin EP3 receptors on the noradrendergic terminals in the mouse brain. Neurosci. 1997;79:247–254. doi: 10.1016/s0306-4522(96)00685-9. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Pothos EN. Regulation of quantal size by presynaptic mechanisms. Rev Neurosci. 2000;11:159–212. doi: 10.1515/revneuro.2000.11.2-3.159. [DOI] [PubMed] [Google Scholar]
- Swanson LW. Brain Maps: Structure of the rat brain. 2. Elsevier; New York: 1998. [Google Scholar]
- Tjen-A-Looi SC, Li P, Longhurst JC. Midbrain vlPAG inhibits rVLM cardiovascular sympathoexcitatory responses during electroacupuncture. Am J Physiol. 2006;290:H2543–H2553. doi: 10.1152/ajpheart.01329.2005. [DOI] [PubMed] [Google Scholar]
- Vanegas H, Tortorici V. Opioidergic effects of nonopioid analgesics on the central nervous system. Cell Mol Neurobiol. 2002;22:655–661. doi: 10.1023/A:1021896622089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verberne AJM, Guyenet PG. Midbrain central gray: Influence on medullary sympathoexcitatory neurons and the baroreflex in rats. Am J Physiol. 1992;263:R24–R33. doi: 10.1152/ajpregu.1992.263.1.R24. [DOI] [PubMed] [Google Scholar]
- Wiberg M, Blomqvist A. The spinomesencephalic tract in the cat: its cells of origin and termination pattern as demonstrated by the intraaxonal transport method. Brain Res. 1984;291:1–18. doi: 10.1016/0006-8993(84)90645-0. [DOI] [PubMed] [Google Scholar]
- Xing J, Li J. TRPV1 receptor mediates glutamatergic synaptic input to dorsolateral periaqueductal gray (dl-PAG) neurons. J Neurophysiol. 2007;97:503–511. doi: 10.1152/jn.01023.2006. [DOI] [PubMed] [Google Scholar]
- Yokotani K, Nishihara M, Murakami Y, Hasegawa T, Okuma Y, Osumi Y. Elevation of plasma noradrenaline levels in urethane-anaesthetized rats by activation of central prostanoid EP3 receptors. Br J Pharmacol. 1995;115:672–676. doi: 10.1111/j.1476-5381.1995.tb14985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]