

Abstract
The fusion-active conformation of the envelope protein gp41 of HIV-1 consists of an N-terminal trimeric α-helical coiled coil domain, and three anti-parallel C-terminal helices which fold down the grooves of the coiled coil to form a six-helix bundle. Disruption of the six-helix bundle is considered to be a key component of an effective non-peptide fusion inhibitor. In the current study, a fluorescence resonance energy transfer experiment for the detection of inhibitor binding to the gp41 N-peptide coiled coil of HIV-1 has been assessed, utilizing peptide inhibitors derived from the gp41 C-terminal helical region. The FRET acceptor is a 31-residue N-peptide containing a known deep hydrophobic pocket, stabilized into a trimer by ferrous ion ligation. The FRET donor is a selected 16-18-residue fluorophore-labeled C-peptide, designed to test the specificity of the N-C interaction. Low μM dissociation constants were observed, correlated to the correct sequence and helical propensity of the C-peptides. Competitive inhibition was demonstrated using the assay, allowing for rank ordering of peptide inhibitors according to their affinity in the 1-20μM range. The assay was conducted by measuring fluorescence intensity in 384-well plates. The rapid detection of inhibitor binding may permit identification of novel drug classes from a library.
Keywords: gp41 peptides, fusion inhibitors, fluorescence, high-throughput screening, metal ion ligation
Introduction
The gp41 envelope protein of HIV-1 is a primary candidate for drug targeting to prevent viral entry into host cells. The extracellular domain of gp41 consists of an N- and a C-terminal heptad repeat region, which adopt a six-helix bundle conformation in the isolated trimeric form of the protein. The N-heptad repeat forms a 50-residue long trimeric coiled coil, and three helical C-heptad repeats wrap in the antiparallel direction down the grooves of the coiled coil (Caffrey, M. et al., 1998). The inner coiled coil domain has been the focus of drug targeting because disruption of the six-helix bundle is known to prevent fusion (Wild et al., 1994; Chen et al., 2000; Kilgore et al., 2003; Root and Steger, 2004). In particular a deep hydrophobic pocket in the grooves of the coiled coil has been identified as an ideal site for drug binding, because it is well-defined for the association of small molecule inhibitors, and represents a region of high conservation among viral strains, both for the residues lining the pocket as well as those of the C-helix which interact in the pocket (Chan et al., 1998). Mutation of any of the key residues involved in this interaction significantly attenuates binding affinity between N- and C-helix, and the corresponding mutant viruses are unable to establish productive infection (Mo et al., 2004).
The search for small molecule inhibitors is greatly aided by the existence of a high-throughput screen for rapid searching of chemical databases. Such screens usually permit the identification of a precursor template, which would then be improved by derivatization and selection of drug-like properties. Some ideas have been proposed for establishing a high-throughput screen for the gp41 coiled coil. One involves an ELISA assay specific for the formation of the six-helix bundle (Jiang et al., 1999), although it was recently reported that small molecules can interfere with the antibody - six-helix bundle interaction, resulting in false positives (Liu et al., 2003). Recently, an assay utilizing fluorescence polarization to detect binding of a 32-residue fluorescently labeled C-peptide to N-helix fused to maltose binding protein was described (Mo et al., 2004). The wild-type C-peptide was found to bind with a Kd of 40 nM. We have been developing an assay that relies on fluorescence resonance energy transfer (FRET) to identify the interaction between N- and C-helices. The components of the assay include a 31-residue long N-peptide encompassing the hydrophobic pocket, stabilized into a trimeric structure by an N-terminal iron(II)- bipyridyl coordination complex, and a 16-18 residue C-peptide labeled with a fluorophore. The emission wavelength of the fluorophore is selected to match with the 540 nm absorption maximum of the iron(II)-bipyridyl complex resulting in fluorescence quenching on binding (Gochin et al., 2003). The small peptides involved in the assay bind with low micromolar affinity, a range that corresponds well with the concentrations used in the fluorescence experiments, as well as with the likely affinity of novel small molecule scaffolds from a chemical library. Thus, the system is designed to focus on site-specific detection of small molecule inhibitors of moderate (or higher) affinity, in this case for the hydrophobic pocket. As with the fluorescence polarization assay (Mo et al., 2004), fluorophore excitation and emission frequencies well outside the range of peptide absorption and most small molecule absorption should prevent complications due to UV absorbance by peptides or fluorescence by organic molecules. In addition, the method offers a minimalist approach in terms of composition of components of the assay.
In this study, we examined the effect of peptide sequence on the discriminatory ability of this assay. This is important in order to establish the integrity of the N-peptide coiled coil construct, and the specificity of the binding interaction with small relatively unstructured peptides. We examined two sequences for the N-peptide and several C-peptide sequences. Ideally, tight binding observed for a C-peptide of the correct sequence should be abrogated for a peptide of jumbled sequence or for a peptide in which key hydrophobic contacts have been removed. We also examined the effect of innate helical conformation on the C-peptide binding constant. We show that there is some indiscriminate hydrophobic association due to the hydrophobic nature of the peptides, but that we are able to distinguish between specific and non-specific binding. In addition, the ability of the assay to detect inhibitor binding is demonstrated using unlabeled C-peptides as inhibitors of varying affinity.
Results and Discussion
N-peptide coiled coil creation
The peptides used for construction of the gp41 coiled coil receptor were as follows:
env2.0: bpy-GQAVEAQQHLLQLATVWGIKQLQARILAVEKK-amide
env2.1: bpy-GQAVQALEKLLQLATVWGIKQLQARILAVEKK-amide
bpy is 5-carboxy-2,2′-bipyridine. Residues in bold form the deep hydrophobic pocket identified in the crystal structure of the gp41 core (Eckert et al., 1999). Underlined residues constitute a continuous region of wild-type sequence. The two peptides differ in the composition of the first heptad repeat, which is modified from the native sequence in env2.0 to a sequence calculated to promote three-helix bundle formation (Ogihara et al., 1997) in env2.1.
Formation of the three-helix bundle structure upon metal binding was assessed indirectly by UV, NMR and CD studies. Figures 1 and 2 show the CD and aromatic NMR spectra of env2.0 and env2.1 and Table 1 lists the observed helicity as a function of metal ion. Both env2.0 and env2.1 demonstrate intrinsic helicity, but differ in degree of helicity and residual aggregation. Aggregation can be assessed from the appearance of the Trp resonances in the aromatic NMR spectrum. env2.0 shows virtually no aggregation in the NMR spectrum, but env2.1 does aggregate in the absence of metal ion, causing broadening of the aromatic region of the spectrum. Metal binding is clearly accompanied by an increase in helicity, shifting of Trp resonances and, in the case of env2.1, dissipation of aggregation. FeII has the most pronounced stabilizing effect of the three metal ions tested, due to the fact that binding of the three bidentate ligands to FeII is cooperative. Cooperativity does not occur with CoII, which behaves more as a probe of the stability of the coiled coil structure. CoII can readily form mixed bpy and H2O ligand complexes. It appears that env2.1 has higher trimeric stability, as expected from the inclusion of a trimer-stabilizing heptad repeat at the N-terminus.
Figure 1.
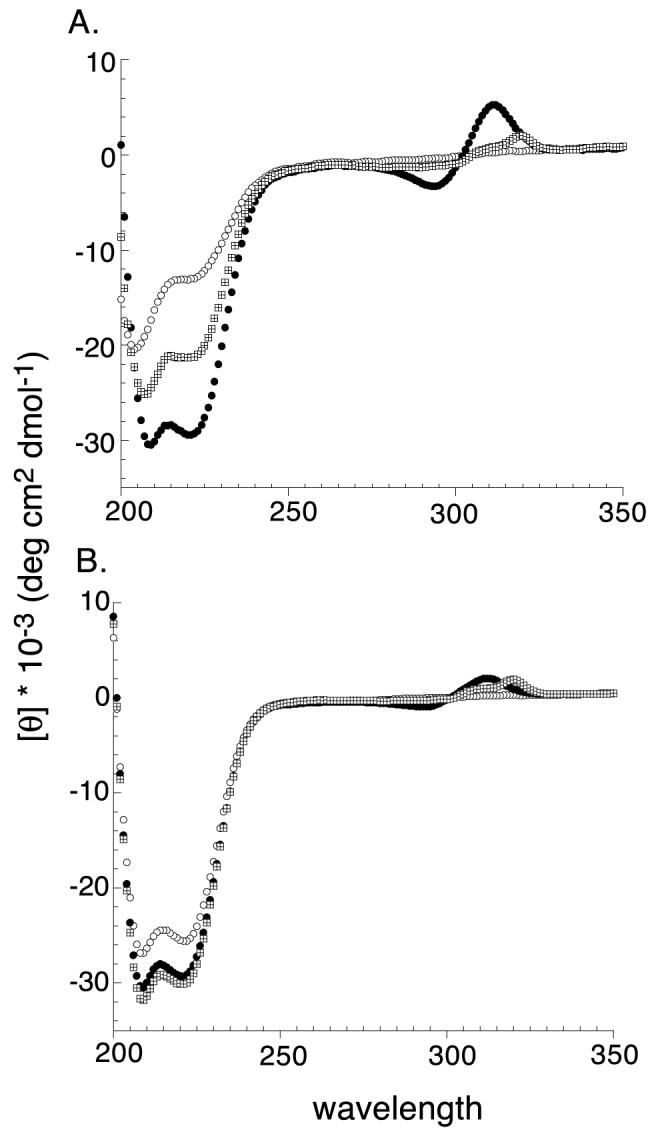
CD spectra of 10μM env2.0 (A) and 20μM env2.1 (B) in the absence(○) and presence of equimolar amounts of NiCl2 (⊞) and FeNH4SO4 (●).
Figure 2.

Aromatic region of the 600 MHz NMR spectrum of 400μM env2.0 (A,C) and env2.1 (B,D) in the presence (A,B) or absence (C,D) of 33μM NiCl2.
Table 1.
Percent helical content measured by CD†
| Peptide |
No metal |
CoII |
NiII |
FeII |
| env2.0 | 40 | 51 | 64 | 89 |
| env2.1 | 70 | 85 | 91 | 91 |
assuming 100% helix at a mean residue ellipticity θ222=33,000 deg.cm2.dmol-1
NiII was specifically chosen for the NMR experiments because the paramagnetic effect shifts bipyridine resonances downfield and out of the aromatic region of the NMR spectrum. This reveals apparent multiplicity for the single His resonance at position 9 of the sequence of env2.0. This may be associated with the existence of up to 8 possible stereoisomers of the NiII-bipyridyl complex, which causes heterogeneity up to 10 residues away from the metal (Gochin et al., 2002). The multiplicity could also arise from a mixed monomer-trimer equilibrium, which cannot be distinguished from conformational heterogeneity of the trimer from CD or NMR results.
FeII was selected for the fluorescence experiments, due to the presence of a chargetransfer band at 540 nm, a region of the spectrum in which peptide absorption does not occur. 540nm is close to the wavelength of emission of the fluorophores dansyl aziridine and Lucifer Yellow (LY). The latter was selected for use based on its environmental insensitivity, higher quantum yield and superior overlap with the FeII-bpy absorption band compared to dansyl. Both env2.0 and env2.1 are 90% helical in the presence of ferrous ion, and stable to air oxidation over long periods.
C-peptide selection
In order to characterize the specificity of the binding event between the metallopeptide construct and C-peptides, a series of C-peptides with various residue modifications was examined. A correctly folded metallopeptide should contain the correct hydrophobic pocket within the grooves of the coiled coil and should bind a C-peptide displaying the correct orientation of side chains more tightly. The assay should be able to discriminate between C-peptides with modified sequence and/or helicity. The series of C-peptides that were examined is shown in Table 2. The natural helical propensity of the C-peptides was enhanced by the following increasingly constraining factors: (1) WT-peptide (C18-WT), (2) Salt bridges introducing an i, i+4 constraint (C18-SB) (Otaka et al., 2002), and (3) α-amino-isobutyric acid substitution (C18-Aib) (Sia et al., 2002). A more tightly constrained α-helical peptide should reduce the entropic penalty associated with binding, resulting in a lower dissociation constant Kd. In addition a C-peptide with a scrambled sequence (C16-Scr), and a peptide with all hydrophobic residues replaced with alanine (C16-Ala) were tested as negative controls. Table 2 lists the C-peptides tested using the fluorescence assay for their binding affinity to FeII complexes of env2.0 and env2.1. In each case, the C-peptide was labeled with LY for the measurement of affinity.
Table 2.
C-peptide dissociation constants (mM) obtained using the FRET assay*

|
std. dev. is shown in brackets; key residues in the binding interaction are shown in bold;
position of salt bridges in gray
Binding assay
The affinity (Kd) for the interaction of the metallopeptide coiled coil with C-peptide was determined using the binding assay described in Materials and Methods. Binding experiments were repeated several times and the results are given in Table 2 and illustrated in Figure 3. Peptide mixtures were left for 1-3 hours to establish an equilibrium prior to measurement. Differences in Kd’s between env2.0 and env2.1 are small, generally within the range of experimental error, indicating that the first heptad repeat is not playing a substantial role in the binding. Dissociation constants are reported separately and as a combined average for env2.0 and env2.1.
Figure 3.

Normalized fluorescence intensity as a function of FeII-env2.0 concentration measured in 96-well or 384-well plate format, for the series of peptides in Table 2. The data are fit to a standard binding curve. The curves correspond to C18-Aib (--■--), C18-WT (--○--), C18-SB (...□ ...), C16-Scr (--●--) and C16-Ala (-- --).
Overall, the highest binding affinity was observed for C18-Aib, where substitution of two α-amino-isobutyric acid residues constrains the peptide in a helical conformation. A Kd of 1.8 μM was observed for binding to env2.0, comparable to that obtained for a similarly constrained peptide in the literature (Sia et al., 2002). The binding affinity of C18-Aib for env2.0 is over 12 times stronger than for the scrambled peptide C16-Scr. The WT-sequence C18-WT shows the next highest affinity to C18-Aib, and has a slightly higher affinity than a similar peptide with salt bridges incorporated for helical stability. The peptides are most likely not long enough for the salt-bridges to contribute to any significant stabilization of the helical conformation, and indeed show little helical propensity in the CD. The peptide C16-Ala, with every hydrophobic residue replaced by alanine and with salt bridges maintained, binds very weakly. In the final analysis, a discrimination factor between a positive control (C18-Aib, C18-WT) and a negative control (C16-Scr, C16-Ala), binding to env2.0, is of the order of 10 or higher, making the assay feasible for the discovery of competitive inhibitors.
Competitive Inhibition Assay
In a first test of the potential of the fluorescence assay to select for inhibitors of the gp41 N-peptide C-peptide interaction, a well plate was prepared containing selected amounts of FeII-env2.0, LY probe peptide and an unlabeled peptide from the list in Table 2. A tightly binding unlabeled peptide should displace LY probe peptide from the coiled coil, resulting in fluorescence enhancement. The extent of fluorescence enhancement should correlate with the affinity of the inhibitor peptide. This is crucial for examining the potential scope of the method for detecting and ranking binding of non-fluorescent small molecules to the hydrophobic pocket. The small size of the peptides tested, and consequently their moderate affinity for the binding site and only a 20-fold difference between the strongest and weakest binders places a stringent test on the ability of the assay to discriminate between them.
FeII-coiled coil concentration was selected to provide ≤ 30% residual fluorescence in the absence of inhibitor peptide. Solutions were mixed in 384 well plates with 5 μM probe peptide in the presence or absence of 100 μM of each of the inhibitor peptides from Table 2. The experiment was conducted using the probe peptides C18-WT-LY, C18-Aib-LY and C18-SB-LY, and the results are shown in Table 3.
Table 3.
Observed percent fluorescence intensity compared to control in the absence and presence of 100mM of inhibitor peptides from Table 2†
| [FeII-env-2.0] (μM) | LY probe peptide | C18-Aib | C18-WT | C18-SB | C16-Scr | C16-Ala | blank* |
| C18-Aib-LY | 65 | 55 | 24 | 21 | 20 | 22 | |
| 16.7 | C18-WT-LY | 85 | 70 | 32 | 26 | 21 | 21 |
| C18-SB-LY | 86 | 68 | 42 | 34 | 25 | 24 | |
| C18-Aib-LY | 32 | 27 | 21 | 22 | 14 | 30 | |
| 50 | C18-WT-LY | 46 | 35 | 20 | 17 | 22 | 22 |
| C18-SB-LY | 59 | 48 | 30 | 18 | 19 | 22 |
all LY-labeled probe peptides at 5mM, all inhibitors at 100mM, FeII-env2.0 concentration based on peptide monomer. Control is 5mM LY-peptide in the absence of FeII-env2.0;
no inhibitor peptide. Error estimated at ±5 percentage points.
Several characteristics are immediately apparent. The assay is able to detect the highest affinity binders quite readily, with a significant increase in fluorescence intensity. Fluorescence recovery correlates with the affinity of the peptide, leading to an observed ranking of the tested peptides in the order C18-Aib > C18-WT > C18-SB > C16-Scr > C16-Ala, covering a range of 1-100 μM Kd. This corresponds with the observed relative binding affinities in Table 2. The assay could be made more selective by increasing the concentration of FeII-env2.0 to 50 μM, resulting in detection of inhibitors with a Kd ≤ 5 μM (Table 3).
Calculations reveal a general discrepancy between observed and expected fluorescence recovery, as shown in Figure 4A,C, with the observed fluorescence recovery in general 10-20 percentage points below the expected value, with the exception of C18-SB for which recovery is up to 50% below the expected value. Lower than expected values may be attributed to effects such as light scattering, bimolecular quenching (Lakowitz,1986) or inhibitor peptide aggregation (by cysteine oxidation or hydrophobic association). C18-SB is the least soluble of the peptides (Materials and Methods), and displays the most pronounced quenching, in accordance with an aggregation and light scattering effect. The baseline fluorescence of LY-labeled peptides shows a clear dependence on sequence and composition, with C16-Ala having a 3-fold higher intrinsic fluorescence due to the absence of Trp residues. Both intra- and intermolecular quenching by tryptophan is likely to be occurring (Marme et al., 2003).
Figure 4.

Percentage fluorescence recovery as a function of inhibitor peptide, probe peptide and inhibitor concentration, compared with calculated values from the Kd’s in Table 2. Percent fluorescence is calculated as a fraction of the total fluorescence observed for free probe. Calculation of fluorescence recovery is described in Materials and Methods. Observed values are shown in gray, calculated values in white. A. probe peptide C18-Aib, inhibitor concentration 100μM; B. probe peptide C18-Aib, inhibitor concentration 33μM; C. probe peptide C18-SB, inhibitor concentration 100μM; D. probe peptide C18-SB, inhibitor concentration 33μM. 17-20μM FeII-env2.0 was used.
The intermolecular quenching/aggregation effect could be tested by carrying out the competitive inhibition experiment with a lower concentration of inhibitory peptide. Results in the presence or absence of 33 μM inhibitory peptides are shown in Figure 4B,D. An improved correlation between observed and calculated fluorescence recovery indicates that intermolecular quenching effects and/or peptide aggregation play a role at higher inhibitor peptide concentrations, and that the effects are alleviated by reducing the concentration, without compromising the ability of the assay to detect and rank inhibitors.
At 33 μM inhibitor concentration, the use of C18-Aib-LY as a probe permits the detection of peptide inhibitors with Kd’s ≤ 10 μM, using 20 μM FeII-env2.0 receptor peptide (Figure 4B). At 50 μM FeII-env2.0, no inhibition can be detected (Figure 5). The weaker C18-SB-LY probe appears to detect inhibitors ≤ 20 μM in Kd using 20 μM FeII-env2.0, and < 10 μM using 50 μM FeII-env2.0 (Figure 4D, 5). Thus the assay can be tuned to select inhibitors of greater or lesser affinity. The sensitivity of the assay to lower concentrations of inhibitor is important for the selection of inhibitors that may be only marginally soluble or available in small quantities. The assay has been tested using the D-peptide D10-p1-2K (Eckert et al., 1999), a known but relatively weak fusion inhibitor with an EC50 of 46 μM. In the presence of 5 μM C18-SB-LY as a probe and 17 μM env2.1 as a receptor, 32 μM D-peptide yielded a 44% fluorescence recovery of the probe.
Figure 5.
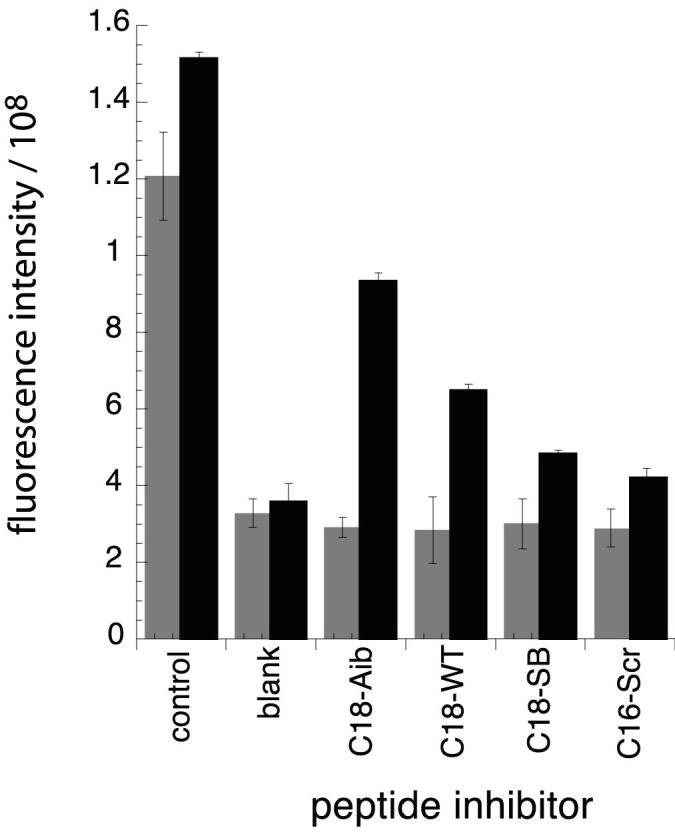
Raw fluorescence intensity as a function of peptide inhibitor for a mixture containing 33μM inhibitor peptide, 5μM probe peptide (C18-Aib-LY in gray; C18-SB-LY in black) and 50μM FeII-env2.0. Experiments were repeated in duplicate.
The results obtained here affirm the basic tenet of the assay; that it is able to selectively detect and rank inhibitors in the low μM range by their effect on fluorescence intensity. Problems associated with high concentrations of hydrophobic peptides should not be a factor in tests for small molecule inhibitors, although self-association of small molecules, should it occur, could affect fluorescence readings.
Conclusion
The deep hydrophobic pocket of the coiled coil domain has been identified as an important target for anti-HIV fusion inhibitors. We have developed a FRET assay to detect peptide binding to this region of the gp41 coiled coil. By specific selection of small peptides comprising the hydrophobic pocket region, and small probe peptides which bind to it, we have demonstrated the validity of the FRET assay for the determination of binding to the hydrophobic pocket, opening up the possibility of searching compound libraries for hits. Three characteristics of the system were assessed: (a) the integrity of the metallopeptide construct as a suitable receptor for displaying the hydrophobic pocket, (b) the specificity of binding of the correct peptide sequence to the target receptor, and (c) the sensitivity of the assay for detecting competitive inhibition by a potential drug candidate. We observed a strong correlation between helix formation and metal ion binding, consistent with hydrophobic collapse of the heptad repeat peptides into a helix bundle, and a highly structured domain using FeII, the metal of choice for the fluorescence assay. C-peptide binding assays revealed a minimum ten-fold enhancement of binding for the peptide of correct sequence and helical conformation, over a scrambled sequence. Finally, the assay was able to detect competitive inhibition by peptides with KI’s on the order of 1-10 μM. The range of the assay can be adjusted by varying the composition and concentration of assay components. Fluorescence recovery depends, as expected, on the concentration of FeII-coiled coil and the affinity of the LY probe peptide. The use of 50 μM FeII-coiled coil limits observation of inhibitors to those with KI’s less than 5 μM. The range of detectable binding affinities is adjustable by the selection of probe peptide. C18-Aib-LY, C18-WT-LY and C18-SB-LY permit the observation of inhibitors in the approximate range < 5 μM, < 10 μM and < 20 μM respectively, as shown, for example, by the highlighted area in Table 3. Further data collection and analysis are underway for statistical validation of the screening assay.
The range of concentrations and binding affinities of components permit the detection of compounds with binding affinities in the low micromolar range. This is appropriate for the selection of precursor molecules from a library. The assay is run in high-throughput mode, using 384 well-plates in a fluorescence plate reader. It is hoped that novel compound scaffolds may be discovered by this method.
Materials and Methods
N- and C-peptides were purchased from Biosynthesis Inc (Lewisville, Texas). N-terminal bipyridylation of N-peptide was achieved by attachment of 2,2′-bipyridyl-5-carboxylic acid on the resin prior to cleavage, as previously described (Case and McLendon, 2000). C-peptides are either 16 residues long or 18 residues long with an additional (N-terminal) Met-Thr, added for comparison with a previously prepared α-amino-isobutyric acid substituted peptide (Sia et al., 2002). Each of the C-peptides was labeled at a C-terminal cysteine with Lucifer Yellow (LY) (Invitrogen-Molecular Probes, Carlsbad, CA) by stirring a 200 μM solution of peptide with 5 equivalents of LY under argon at room temperature in 100 mM phosphate buffer, pH 7.2, for 2 hours, followed by HPLC purification. Peptide concentrations were measured using an extinction coefficient of 17,300 M-1 cm-1 at 291 nm for bipyridylated N-peptide, 12,660 M-1 cm-1 at 280 nm for unlabeled C-peptides (with the exception of C16-Ala, which was measured by weight), and 10,800 M-1 cm-1 at 425 nm for Lucifer-Yellow labeled C-peptides. NMR spectra were recorded on a Varian 600 MHz spectrometer equipped with a cryoprobe. CD experiments were recorded on a Jasco 715 spectropolarimeter using 10-20 μM solutions. Fluorescence experiments were carried out on a plate reader using 96-well plates in a Perkin-Elmer LS55 luminescence spectrometer, or 384-well plates in an LJL Biosystems Analyst Plate Reader (Molecular Devices, Sunnyvale, CA). All biophysical experiments were carried out in Tris-acetate buffer, pH 7. NMR experiments were conducted using stoichiometric amounts of NiCl2 (1/3 of peptide concentration). CD and fluorescence experiments were typically run with a 1:1 metal: peptide ratio. Fluorescence plates were prepared using 500 μM stock peptide solutions, except for C18-WT (320 μM) and C18-SB (200 μM) due to the limited solubility of these peptides.
Fluorescence assay
Lucifer Yellow (LY) labeled C-peptide binding to the FeII-bound N-heptad domain was observed by excitation of the LY fluorophore at 425 nm and measurement of the intensity of emission at 525 nm. Excitation and emission slits of 2.5 nm and a cutoff dichroic mirror at 515 nm were used. 10 μM LY-peptide was used as the probe, and the concentration of FeII complex varied from 3 nM to 500 μM. Data were fit to a standard binding curve using Kaleidagraph®. Competitive inhibition studies were carried out using 5 μM LY-peptide, 20 μM or 50 μM FeII-env2.0 and varying amounts of unlabeled peptide as the competitive inhibitor.
Calculations
Dissociation constants for peptide binding to the coiled coil were determined using Kaleidagraph by fitting the data to the equation
, where Fmax is the maximum fluorescence of free probe and Fobs is the observed fluorescence for a given concentration Pt of env2.0. c1 and c2 are constants. Fluorescence recovery in the presence of a competitive inhibitor peptide was calculated numerically using Mathcad (Mathsoft), assuming standard competitive binding with a 1:1 binding stoichiometry. It was assumed that there are three equivalent binding sites for the three peptides comprising the coiled coil. Fluorescence of bound probe peptide was assumed to be zero, and that of free peptide to be 100%.
Acknowledgements
The authors are grateful to the Bay Area Screening Center at the UCSF Institute for Quantitative Biomedical Research, for the use of equipment and training. NMR experiments were conducted at the UCSF NMR facility. Use of the CD spectropolarimeter was kindly provided by Prof. Dave Agard at UCSF. Funding for this study was provided by the American Foundation for AIDS Research Grant # 106388-33-RGGN to M.G. and NIH AI060361 to M.G. Additional resources were provided by Research Pilot Award activity DRES-029 to R.S. and DRES-033 to S.H. from the Arthur A. Dugoni School of Dentistry, University of the Pacific.
References
- Caffrey M, Cai M, Kaufman J, Stahl SJ, Wingfield PT, Covell DG, Gronenborn AM, Clore GM. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. Embo Journal. 1998;17(16):4572–84. doi: 10.1093/emboj/17.16.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case MA, McLendon GL. A Virtual Library Approach To Investigate Protein Folding nd Internal Packing. J. Am. Chem. Soc. 2000;122:8089–90. [Google Scholar]
- Chan DC, Chutkowski CT, Kim PS. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:15613–7. doi: 10.1073/pnas.95.26.15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y-H, Xiao Y, Dierich MP. HIV-1 gp41: role in HIV entry and prevention. Immunobiology. 2000;201:308–16. doi: 10.1016/S0171-2985(00)80085-6. [DOI] [PubMed] [Google Scholar]
- Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103–15. doi: 10.1016/s0092-8674(00)80066-5. [DOI] [PubMed] [Google Scholar]
- Gochin M, Guy RK, Case MA. A metallopeptide assembly of the HIV-1 gp41 coiled coil is an ideal receptor in fluorescence detection of ligand binding. Angew. Chem. Int. Ed. 2003;42:5325–8. doi: 10.1002/anie.200352006. [DOI] [PubMed] [Google Scholar]
- Gochin M, Khorosheva V, Case MA. Structural Characterization of a Paramagnetic Metal-Ion-Assembled Three-Stranded alpha-Helical Coiled Coil. J. Am. Chem. Soc. 2002;124:11018–11028. doi: 10.1021/ja020431c. [DOI] [PubMed] [Google Scholar]
- Jiang S, Lin K, Zhang L, Debnath AK. A screening assay for antiviral compounds targeted to the HIV-1 gp41 core structure using a conformation-specific monoclonal antibody. J Virol Methods. 1999;80:85–96. doi: 10.1016/s0166-0934(99)00041-5. [DOI] [PubMed] [Google Scholar]
- Kilgore NR, Salzwedel K, Reddick M, Allaway GP, Wild CT. Direct evidence that C-peptide inhibitors of human immunodeficiency virus type 1 entry bind to the gp41 N-helical domain in receptor-activated viral envelope. J. Virol. 2003;77:7669–72. doi: 10.1128/JVI.77.13.7669-7672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowitz JR. In Principles of Fluorescence Spectroscopy. Plenum Press; New York and London: 1986. Chapter 8. [Google Scholar]
- Liu S, Zhao Q, Jiang S. Determination of the HIV-1 gp41 fusogenic core conformation modeled by synthetic peptides: applicable for identification of HIV-1 fusion inhibitors. Peptides. 2003;24:1303–13. doi: 10.1016/j.peptides.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Marme N, Knemeyer J-P, Sauer M, Wolfrum J. Inter- and Intramolecular Fluorescence Quenching of Organic Dyes by Tryptophan. Bioconjugate Chem. 2003;14:1133–39. doi: 10.1021/bc0341324. [DOI] [PubMed] [Google Scholar]
- Mo H, Konstantinidis AK, Stewart KD, Dekhtyar T, Ng T, Swift K, Matayoshi ED, Kati W, Kohlbrenner W, Molla A. Conserved residues in the coiled-coil pocket of human immunodeficiency virus type 1 gp41 are essential for viral replication and interhelical interaction. Virology. 2004;329:319–27. doi: 10.1016/j.virol.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Ogihara NL, Weiss MS, Degrado WF, Eisenberg D. The crystal structure of the designed trimeric coiled coil coil-VaLd: implications for engineering crystals and supramolecular assemblies. Protein Science. 1997;6:80–8. doi: 10.1002/pro.5560060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otaka A, Nakamura M, Daisuke N, Kodama E, Uchiyama S, Nakamura H, Kobayashi Y, Matsuoka M, Fujii N. Remodelling of gp41-C34 Peptide Leads to Highly Effective Inhibitors of the Fusion of HIV-1 with Target Cells. Angew. Chem. Int. Ed. 2002;41:2938–9. doi: 10.1002/1521-3773(20020816)41:16<2937::AID-ANIE2937>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Root M, Steger H. HIV-1 gp41 as a target for viral entry inhibition. Curr. Pharm. Des. 2004;10:1805–25. doi: 10.2174/1381612043384448. [DOI] [PubMed] [Google Scholar]
- Sia SK, Carr PA, Cochran AG, Malashkevich VN, Kim PS. Short constrained peptides that inhibit HIV-1 entry. Proc. Natl. Acad. Sci. (USA) 2002;99:14664–9. doi: 10.1073/pnas.232566599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. 1994;91:9770–4. doi: 10.1073/pnas.91.21.9770. [DOI] [PMC free article] [PubMed] [Google Scholar]