

Abstract
Failure to control oxidative stress is closely related to aging and to a diverse range of human diseases. We have reported that protein kinase C γ (PKCγ) acts as a primary oxidative stress sensor in the lens. PKCγ has a Zn-finger C1B stress switch domain, residues 101-150. Mutation, H101Y, in the C1B domain of PKCγ proteins causes a failure of the PKCγ oxidative stress response (Lin and Takemoto (2005) J. Biol. Chem. 280:13682-13693). Some human neurodegenerative spinocerebellar ataxia type 14 are caused by mutations in the PKCγ C1B domain. In the current study we have investigated the effects of these mutations on lens epithelial cell responses to oxidative stress. Results demonstrate that PKCγ C1B mutants had lower basal enzyme activities and were not activated by H2O2. Furthermore, the PKCγ mutations caused a failure of endogenous wild type PKCγ to be activated by H2O2. These PKCγ mutations abolished the effect of H2O2 on phosphorylation of Cx43 and Cx50 by H2O2—activation of PKCγ. The cells with PKCγ C1B mutations had more Cx43 and/or Cx50 gap junction plaques which were not decreased by H2O2. Since open gap junctions could have a bystander effect this could cause apoptosis to occur. H2O2 (100 μM, 3 h) activated a caspase-3 apoptotic pathway in the lens epithelial cells but was more severe in cells expressing PKCγ mutations. The presence of 18α-glycyrrhetinic acid (AGA), an inhibitor of gap junctions, decreased Cx43 and Cx50 protein levels and gap junction plaque number. This reduction in gap junctions by AGA resulted in inhibition of H2O2-induced apoptosis. Our results demonstrate that there is a dominant negative effect of PKCγ C1B mutations on endogenous PKCγ which results in loss of control of gap junctions. Modeled structures suggest that the severity of C1B mutation effects may be related to extent of loss of C1B structure. Mutations in the C1B domain of PKCγ result in increased apoptosis in lens epithelial cells. This can be prevented by a gap junction inhibitor. Thus, propagation of apoptosis from cell-to-cell in lens epithelial cells may be through open gap junctions. The control of gap junctions requires PKCγ.
Keywords: Cx43, Cx50, protein kinase C γ, caspase-3, C1B domain
Introduction
Gap junctions are hydrophilic channels and/or hemichannels that allow the passage of small molecules including necessary metabolites, second messengers, ions, and cell death signals from cell to cell (Thompson et al., 2006). Propagation or amplification of cell death signals through open gap junctions results in cell apoptosis (Cusato et al., 2003; Lin et al., 1998; Neijssen et al., 2005). Lens epithelial cells express gap junction proteins, Cx43 and Cx50, while lens fiber cells express Cx50 and Cx46. Fiber cell gap junctions provide a pathway to maintain lens homeostasis through circulating fluxes, and disruption of this gap junctional cell-to-cell communication pathway causes cataractogenesis (Goodenough et al., 1996). We have previously determined that gap junctions, Cx43 and Cx50, are the major targets of activated PKCγ in the lens epithelial cells in culture and in the whole lens (Lin and Takemoto, 2005; Lin et al., 2003a & b, 2004; Zampighi et al., 2005). Loss of control of gap junctions by PKCγ, ie., no PKCγ phosphorylation of Cx50 in PKCγ knockout lenses, causes PKCγ knockout lenses to be more susceptible to oxidative stress-induced cataracts in the mouse (Lin et al., 2006). Inhibition of gap junctions prevents cell death (Farahani et al., 2005; de Pina-Benabou et al., 2005; Krysko et al., 2005). It is apparent that control of gap junctions is essential for lens cell survival.
PKCγ is primarily found in the central and peripheral nervous systems (Shutoh et al., 2003). It is also found in the eye tissues including retina and lens epithelium and cortex (Correia et al., 2003; Saleh et al., 2001). PKCγ consists of C1 and C2 regulatory domains and C3 and C4 catalytic domains. The C1 domain contains two tandem repeat, Cys-rich subdomains, C1A and C1B. The C1 domain is a diacylglycerol (DAG) binding domain, while calcium binds to the C2 domain. Unlike other conventional PKC's, the C1B subdomain of the PKCγ protein is always exposed and calcium is not required for activation of this enzyme (Ananthanarayanan et al., 2003; Lin et al., 2003a). We have demonstrated that the PKCγ C1B subdomain is oxidized when cells are treated with H2O2 which results in formation of disulfide bonds and activation of the enzyme. This occurs without an elevation in cellular DAG levels. The activated PKCγ phosphorylates Cx43 on Ser368 and this causes disassembly of gap junction plaques and inhibition of gap junction dye transfer activity (Lin and Takemoto, 2005). PKCγ acts as an oxidative stress sensor to prevent the lens from oxidative damage through proper control of gap junctions. It has been reported recently that missense mutations in PKCγ cause the dominant spinocerebellar ataxia type 14 (SCA14), a neurodegenerative disorder with onset age as early as three years (Chen et al., 2003, 005; van de Warrenburg et al., 2003; Stevanin et al., 2004; Yabe et al., 2003, Verbeek et al., 2005; Seki et al., 2005; Alonso et al., 2005; Klebe et al., 2005; Vlak et al., 2006; Fahey et al., 2005). Most of the PKCγ SCA14 mutations occur in the C1B subdomain. Since this is a newly identified neuronal disorder, long term follow-up study is necessary to uncover what PKCγ SCA14 mutations do to cataractogenesis. We previously observed that the PKCγ H101Y mutant is not activated by oxidative stress, e.g. H2O2 (Lin and Takemoto, 2005). But its effect on gap junctions and cell survival remains unclear.
In this paper we study the structure/function effects of three PKCγ C1B mutations on PKCγ enzyme activity, lens gap junction control, and on the induction of apoptosis in N/N1003A lens epithelial cells. This cell line expresses Cx43, Cx50, and PKCγ. These all mutants are expressed against a background of endogenous wild type PKCγ. This is because humans with SCA14 are heterozygous. Thus, this more correctly models the human disease. We demonstrate that cells with PKCγ C1B mutants lacked control of Cx43 and Cx50 gap junctions when exposed to H2O2 and this caused cells to be more susceptible to H2O2 -induced, caspase-3-dependent cell apoptosis.
Materials and Methods
Materials
Antibodies against active caspase-3, PKCγ, connexin 43 (Cx43), and GFP were purchased from BD Biosciences (Palo Alto, CA). Rabbit polyclonal PKCγ phospho T514 antibody was purchased from Abcam (Cambridge, MA). Caspase-3 colorimetric assay kit, polyclonal rabbit anti-phosphoserine, and anti-phospho-S368-Cx43 were purchased from Chemicon (Temicula, CA). Protein A/G PLUS-agarose beads were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-mouse or anti-rabbit IgG conjugated with HRP, EcoRI, KpnI, and PepTag non-radioactive protein kinase C assay system were purchased from Promega (Madison, WI). Mouse anti-α-tubulin and anti-connexin 50 were purchased from Zymed Laboratories (South San Francisco, CA). Geneticin (G418), Dulbecco's Modified Eagle Medium (DMEM) (low glucose, or high glucose), trypsin-EDTA, gentamicin, penicillin/streptomycin, and lipofectamine 2000 were purchased from Invitrogen Life Technologies (Carlsbad, CA). Dimethyl sulfoxide (DMSO), dithiothreitol (DTT), H2O2, sodium fluoride (NaF), calcium chloride (CaCl2) and Takara Ex Taq DNA polymerase, Delta T dishes were purchased from Fisher Scientific (Pittsburgh, PA). Fetal bovine serum was purchased from Atlanta Biologicals (Norcross, GA). Phenylmethanesulfonyl fluoride (PMSF), 18α-glycyrrhetinic acid (AGA), and protease inhibitor cocktail were from Sigma (St Louis, MO). Protein molecular weight marker was purchased from Bio-Rad Laboratories (Hercules, CA). QuikChange site-directed mutagenesis kits were purchased from Stratagene (La Jolla, CA). Alexa fluor 568 goat anti-mouse IgM, Alexa fluor 568 goat anti-mouse IgG (H+L), Alexa fluor 568 goat anti-rabbit IgG (H+L) and SlowFade antifade were purchased from Molecular Probes (Eugene, OR). Plasmid DNA purification kits were purchased from Qiagen (Valencia, CA).
Cell cultures
The rabbit lens epithelial cells N/N1003A were cultured in low glucose DMEM supplemented with 10 % fetal bovine serum and 50 μg/ml gentamicin, 0.05 unit/ml penicillin, 50 μg/ml streptomycin, pH 7.4 at 37°C in an atmosphere of 95 % air and 5 % CO2.
PKCγ C1B mutation plasmid construction, transfection, and fluorescent confocal microscopy
Rat PKCγ:EGFP plasmid DNA (in pEGFP-N3 vector) was used as a template. SCA14 mutations have been constructed by PCR using a site-directed, QuikChange mutagenesis kit. Briefly, the two oligonucleotide primer pairs containing the desired mutations (see below for detailed sequences), each complementary to opposite strands of the PKCγ:EGFP vector, were extended during PCR cycling. Incorporation of the oligonucleotide primers generated a mutated plasmid containing staggered nicks. The products were digested with Dpn I to remove the parental DNA templates, and mutation-containing synthesized plasmid DNA was transformed into XL1-Blue competent cells which were further selected on LB+kanamycin plates. In all cases construct sequences were verified by sequencing. Forward primer sequences are shown below with highlighted mutation nucleotides.
H101Y: 5' -C GAC CCT CGC AAC AAG TAC AAG TTC CGT CTG CAC AGC- 3'
S119P: 5'-C TTC TGC GAC CAC TGT GGT CCC CTC CTC TAC GGG CGC A-3'
G128D: 5'-C TAC GGG CTG GTG CAC CAG GAC ATG AAA TGT TCC TGT TGC GAA-3'
Plasmid DNA transient transfection into N/N1003A cells was performed by Lipofectamine 2000 transfection reagent according to manufacturer's protocol. Briefly, 60-80 % confluent N/N1003A cells seeded in 6-well plates were incubated with plasmid DNA-Lipofectamine™ 2000 complexes (1μg DNA to 2.5 μL Lipofectamine™ 2000 in 125 μL serum-free DMEM) in a regular CO2 incubator for 24 hours. Expression of plasmid DNA was confirmed by Western blotting and by confocal microscopy to monitor the GFP fluorescence and plasma membrane translocation of GFP fusion proteins. The efficiency of the transient transfection as measured by GFP-confocal microscopy was between 25-35 %. Transiently-transfected N/N1003A cells were selected in DMEM (low glucose) supplemented with 500 μg/mL geneticin (G418) for at least 6 weeks to establish the stable transfection lines with greater than 95 % transfection efficiency.
Western blot and immunoprecipitation
Cells were collected and lysed on ice with cell lysis buffer followed by homogenization and sonication. The cell lysis buffer contained 20 mM Tris-HCl, pH 7.5, 0.5 mM EDTA, 0.5 mM EGTA, 0.5 % Triton X-100, 0.1 % protease inhibitor cocktail, 5 mM NaF, and 2 mM PMSF. After centrifugation at 12,000 × g for 20 min, the supernatants were collected and used as whole cell extracts. Western blotting, immunoprecipitation, and Western blot band digitalization were carried out as described previously (Lin and Takemoto, 2005).
PKCγenzyme activity assay
Specific PKCγ activity was analyzed by use of the PepTag assay kit as described (Lin and Takemoto, 2005). Equal protein amounts of whole cell extracts were immunoprecipitated with PKCγ antisera at 4 °C for 4 h. Immunoprecipitated PKCγ/agarose bead complexes were recovered and incubated with PKC reaction mixture (25 μL) according to the manufacturer's instructions. The reactions were stopped by heating at 95 °C for 10 min and fluorescent phospho-PepTag peptides (phosphorylated by PKCγ) were resolved by 0.8 % agarose gel electrophoresis and visualized under UV light. The phosphorylated peptide bands were excised and their fluorescence intensities were quantified by spectrophotometry at 570 nm. The enzyme activity was normalized by calibration of the relative level of phosphorylated substrates to the relative amount of PKCγ:EGFP or wild type PKCγ + PKCγ:EGFP in the immunoprecipitation as determined by Western blotting, and was expressed as % of nontreated specific PKCγ activity. Exogenous rat PKCγ proteins fused with EGFP tag have similar enzymatic activity compared to endogenous rabbit PKCγ enzymes in N/N1003A cells as shown previously (Lin et al., 2003b).
Measurement of cell surface Cx43 or Cx50 gap junction plaques
Stably transfected N/N1003A cells, with or without 18α-glycyrrhetinic acid (AGA, 200 μM, 3 h) (28), were treated with 100 μM H2O2 for 20 min. Determination of endogenous Cx43 or Cx50 gap junctions was performed as described previously (Lin and Takemoto, 2005). Briefly, the cells were fixed with 2.5 % paraformaldehyde for 5 min and labeled with anti-Cx43 or Cx50 for 2 h at room temperature. After washing, the fixed cells were incubated with the secondary antisera which were attached to a fluorochrome and had specific excitation and emission wavelength. The cells were then mounted on slides and examined using a Nikon scanning confocal microscope. We photographed ten points per slide, three slides for each treatment, and examples are shown. For quantitation, the cell surface Cx43 or Cx50 gap junctions from single cells in single sections in each image were counted. The number of gap junction plaques (larger than 1 μm in length) was expressed as mean ± SEM. Values of p ≤ 0.05 were considered to be statistically significant (*).
Caspase-3 colorimetric assay
H2O2-activation of caspase-3 in stably transfected N/N1003A cells was determined by caspase-3 colorimetric assay. Cells with or without 100 μM H2O2 treatments were lysed and the whole cell extracts were used for caspase-3 assay following the protocol provided by the manufacturer. Caspase-3 activity is expressed as the percentages of that in the untreated wild type PKCγ stably transfected cells shown as mean ± SEM. Values of p ≤ 0.05 were considered to be statistically significant (*).
To examine the effect of gap junction inhibition on H2O2-activation of caspase-3, cells were pre-incubated with 200 μM AGA for 30 min, then additionally with 100 μM H2O2 for 3 h co-incubation. Cellular caspase-3 activity was measured. AGA is a gap junction inhibitor which can also prevents the synthesis of Cx43 (El-Sabban et al., 2003). Cellular Cx43 and/or Cx50 protein levels were determined by Western blotting.
C1B domain modeling
Structural models of three PKCγ C1B mutants were generated via homology modeling. The target structures were predicted via alignment to a set of the C1B domain of PKCγ for which experimental structural characterization was available. Using the computational informatics software SYBYL 7.0, three single amino acid mutate monomer processes were performed on the PDB file (1TBN) of the C1B domain of PKCγ attained from RCSB Protein Data Bank. After the mutation, a standard side chain torsion scan was administered for all rotateable side chains to minimize Van der Waals contacts. A dynamic simulation was ran to allow the chain to overcome low kinetic barriers and to cover a wider section of conformational space followed by an energy minimization (Tripos force field ) on the modified sequence using SYBYL biopolymer modeling program from Tripos Inc. (St. Louis, MO, USA and http://www.tripos.com). Lastly, a subset energy minimization was performed to adjust the geometry of the protein and to find the lowest total energy. Analysis of the peptide secondary structure and surface characteristics was carried out on the resulting structures via SYBYL 7.0. The figures were created using MolMol 2K.2- a molecular graphics program (Institute for Molecular Biology and Biophysics, ETH, Zurich, Switzerland)
Statistical analysis
All analyses represent at least triplicate experiments. The statistical analysis employed in this paper is the Student's t-test. The level of significance (*) was considered at p ≤ 0.05. All data are mean ± SEM.
Results
Modeled structures of the C1B mutants
We chose three mutations in the PKCγ C1B domain (Fig. 1A) to test the structure/function relationship of the PKCγ C1B domain. The predicted three-dimensional structure of PKCγ mutants (H101Y, S119P, and G128D) were modeled based on the secondary structure of PKCγ C1 domain for which NMR experimental structural characterization was available (Xu et. al., 1997). WT displays a short helix (V142-V146) and five β-strands (H101-S107, T112-C114, S119-L121, G128-C131, and M136-H139). The three-dimensional structure models of H101Y and G128D reveal conformational changes in the zinc-finger region (H101, C131, C134, and C150 for site 1 and H139, C114, C117, and C142 for site 2). These findings are in agreement with our previous report showing altered DAG/phorbol ester activation character in the PKCγ H101Y mutant (Lin and Takemoto, 2005). However, the modeled structure of the S119P mutant shows loosening of the global fold and overall conformational changes in the C1B domain structure, indicating disruption of the C1B Zn-finger stress switch and a collapse in the DAG/Phorbol ester binding loop (S110, T112, F113, L121, Y122, and G123). This is in contrast to previous models which did not confirm C1B folding changes for this mutant (Chen et al. 2003)
Figure 1. Domain structure of PKCγ showing C1B mutations.
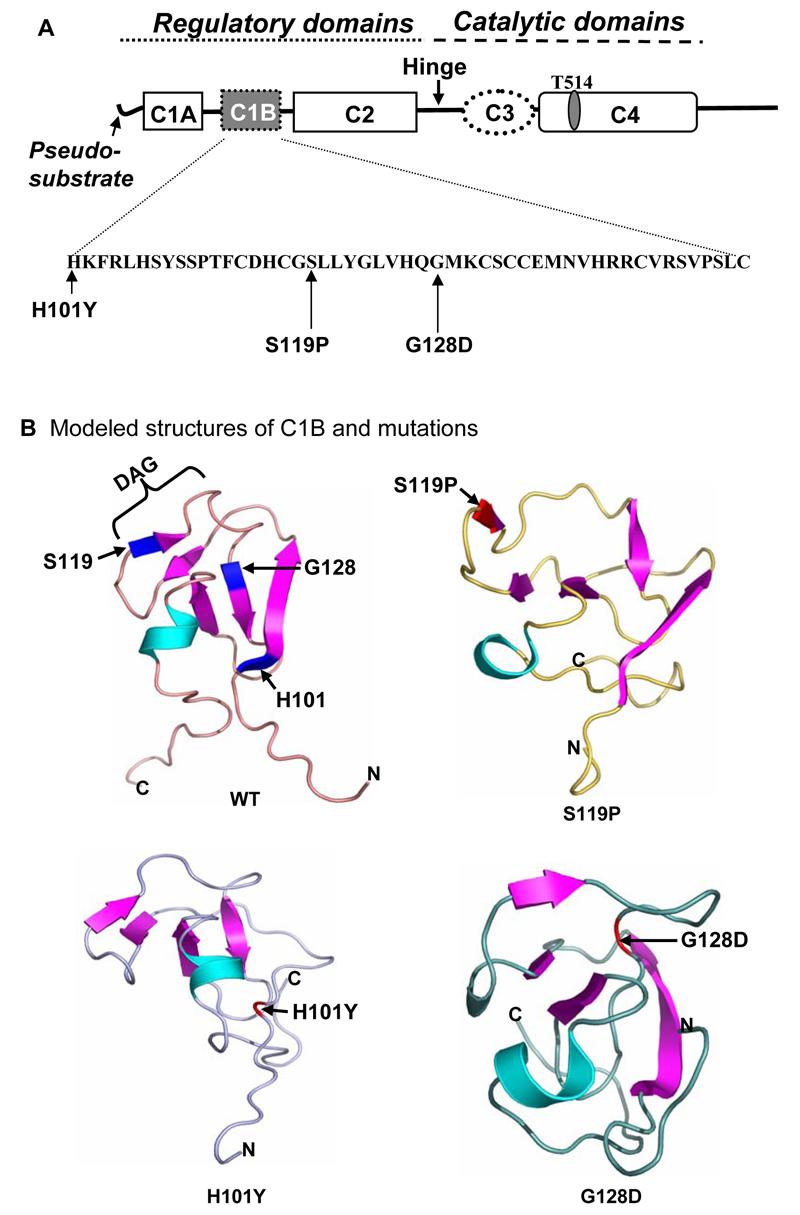
A. The amino acid sequence of the C1B domain is shown in the insert below. PKCγ SCA-14 missense mutations used in the current study are indicated. One of the autophosphorylation sites Thr514 is revealed above the diagram. B. Molecular computation models of the wild type C1B domain and the three mutations showing disruption of the C1B Zn-finger stress switch and a collapse in the DAG/Phorbol ester binding loop (DAG) in the C1B mutants, H101Y, S119P, and G128D.
PKCγ C1B mutants are not activated by H2O2 and prevent activation of endogenous wild type PKCγ by H2O2
C1B mutants and endogenous wild type PKCγ (WT) were analyzed by PKCγ enzyme activity assays and autophosphorylation of PKCγ on Thr514. The results demonstrated that PKCγ H101Y enzyme had a similar basal enzyme activity level and was much less activated by H2O2 as that of wild type PKCγ, but PKCγ S119P and G128D had lower basal enzyme activities and were not activated by H2O2, compared to the wild type PKCγ (Fig. 2A). We also measured total PKCγ enzyme activities using anti-PKCγ antibodies to immunoprecipitate both endogenous wild type PKCγ and exogenous wild type PKCγ or C1B mutants with EGFP tags. Western blotting results demonstrated the wild type PKCγ and mutant protein levels (Fig. 2B, insert right), one lower band corresponding to the endogenous PKCγ and one upper band corresponding to the PKCγ with EGFP tag at higher migration. Expression of wild type PKCγ did not affect total PKCγ enzyme activation by H2O2 (Fig. 2B, graph, WT+WT). However, expression of C1B mutants caused lowered basal level of PKCγ enzyme activities and failure to be activated by H2O2 even while endogenous PKCγ levels were not changed as shown by immunoprecipitation and Western blot (Fig. 2B, insert right), indicating that the presence of the exogenous C1B PKCγ mutant, but not the wild type PKCγ, prevented normal function and activation of endogenous wild type PKCγ.
Figure 2. PKCγ C1B mutations alter endogenous PKCγ activity.


Stably-transfected N/N1003A lens epithelial cells were treated with 100 μM H2O2 for 20 min. After that cells were harvested. The whole cell extracts were used to immunoprecipitate either the EGFP-tagged wild type PKCγ or C1B mutants by anti-GFP antibodies (A), or both endogenous PKCγ and EGFP tagged PKCγ or mutants by anti-PKCγ antibodies (B). The precipitates were used for the enzyme sources to determine enzyme activity as described. As a loading control, total immunoprecipitated wild type PKCγ (endogenous and/or EGFP-tagged) or C1B mutant proteins were shown by Western blot (inserted panels). PKCγ enzyme activity was expressed as % of the activity of untreated wild type PKCγ with GFP tags. The enzyme activity was normalized by calibration of the relative level of phosphorylated substrates to the relative amount of PKCγ:EGFP or wild type PKCγ + PKCγ:EGFP in the immunoprecipitation as determined by Western blotting. * indicates significant increases in PKCγ enzyme activities compared to levels in control cells with overexpression of wild type PKCγ only (A) or with both endogenous and exogenous wild type PKCγ (B). Same whole cell extracts were used to detect the phosphorylation of PKCγ on Thr514 by Western blot using anti-phospho-Thr514 PKCγ as a probe. Phospho-Thr514 PKCγ:EGFP or mutants are shown (C). Total PKCγ:EGFP protein levels were revealed as loading controls (C, lower panel). H2O2-induced autophosphorylation of endogenous PKCγ on Thr514 in cells overexpressing EGFP tag, wild type PKCγ, or PKCγ C1B mutants are shown (D). Endogenous wild type PKCγ protein levels were revealed as loading controls (D, lower panel).
Autophosphorylation of PKCγ on Thr514 is necessary for the full activation of PKCγ. We determined the phosphorylation profiles of Thr514 in the PKCγ C1B mutants by immunoblotting with anti-phospho-Thr514 PKCγ specific antibodies. As shown in Figure 2C, Thr514 phosphorylation in the C1B mutants was not observed with or without H2O2. In separate experiments, endogenous PKCγ phosphorylation on Thr514 was also determined (Fig. 2D). Transfection of EGFP only or wild type PKCγ did not alter endogenous PKCγ autophosphorylation on Thr514 after H2O2. Expression of PKCγ H101Y mutant significantly reduced endogenous PKCγ autophosphorylation after H2O2 (H101Y column). Expression of PKCγ S119P or G128D mutant completely abolished the effect of H2O2 on endogenous PKCγ autophosphorylation (S119P and G128D columns). Taken together, this suggests that PKCγ C1B mutants, especially S119P and G128D, lack the responses to H2O2-induced oxidative stress. Furthermore, expression of C1B mutants abolished endogenous PKCγ responses to H2O2.
AGA decreases Cx50 levels in lens epithelial cells
To determine if AGA decreased Cx43 or Cx50 levels, Western blotting was done using α-tubulin levels as equal loading controls. Figure 3 demonstrates that, although AGA is reported to alter Cx43 levels (el-Sabban et al., 2003), in lens epithelial cells Cx50 protein levels were much more decreased than that of Cx43. When N/N1003A cells were stably transfected with PKCγ C1B mutants, no significant differences in AGA effects, among each of the mutants, were observed (Fig. 3). This is consistent with the previous report (El-Sabban et al., 2003).
Figure 3. Decreased Cx protein levels after AGA treatment.

Cells were pre-incubated with or without 200 μM AGA for 30 min followed by addition of 100 μM H2O2 for additional 3 h (total AGA treatment for 3 h 30 min). Whole cell extracts were isolated from PKCγ or C1B mutants (EGFP-tagged). Total protein levels of Cx43 and Cx50 are determined by Western blotting. α-tubulin is used as a loading control. The Western blot bands were digitized and relative Cx protein levels compared to α-tubulin were graphed. * indicates significant changes compared to levels in control cells with overexpression of wild type PKCγ.
PKCγ C1B mutants do not phosphorylate Cx43 or Cx50
PKCγ catalyzed phosphorylation of Cx43 or Cx50 is shown in Fig. 4. H2O2 stimulated PKCγ activation and subsequent phosphorylation of both Cx43 on Ser368 and Cx50 on serines (unidentified at present) in the cells with expressed wild type PKCγ (WT). However, phosphorylation of Cx43 or Cx50 did not occur in cells expressing C1B mutations before or after H2O2 treatment (Fig. 4), even though endogenous wild type PKCγ is also present in these cells. Data suggest the loss of function of endogenous wild type PKCγ and loss of control of gap junctions in the cells expressing exogenous PKCγ C1B mutations. This suggests that humans with SCA14 which are heterozygous for C1B mutations may also shown such a dominant negative effect even as carriers.
Figure 4. Lack of phosphorylation of Cx43 on Ser368 or Cx50 on serines by PKCγ C1B mutants.

A. Stably-transfected cells were treated with 100 μM H2O2 for 20 min. Phosphorylation of Cx43 on Ser368 was determined by Western blot using anti-phosphoSer368 Cx43 antibodies. α-tubulin levels were shown as controls. B. Cx50 was immunoprecipitated by anti-Cx50 antibodies. The immunoprecipitates were resolved by SDS-PAGE and immunoblotted with anti-phospho-serines antibodies (i.e., site of phosphorylation not presently identified). Cx50 levels were shown as controls.
Expression of PKCγ C1B mutants cause increased Cx43 and Cx50 plaques: Disassembly does not occur after H2O2
To test if the altered Cx phosphorylation levels altered Cx43 or Cx50 plaque formation, we used anti-Cx43 or anti-Cx50 antibodies to immunolabel cell surface Cx43 or Cx50 gap junction plaques in the stably transfected cells with greater than 95 % transfection efficiency. The gap junction plaque number was graphed and sample images of Cx43 plaques are shown (Fig. 5A-C). The experimental results showed that expression of exogenous PKCγ C1B mutations caused significant increases in cell surface gap junction plaques (Fig. 5A and B, respectively). H2O2 treatment significantly decreased both Cx43 and Cx50 gap junction plaque numbers in the cells with overexpressed wild type PKCγ (WT), but not with C1B mutant PKCγ's. As a control, pre-incubation with 200 μM AGA for 3h followed by 100 μM H2O2 for 20 min (AGA treatment, totally 3 h 20 min) caused significant decreases in the number of Cx43 and Cx50 gap junction plaques (Fig. 5A and B, AGA+H2O2). We conclude that PKCγ C1B mutants have a dominant negative effect on endogenous PKCγ which results in increases in cell surface gap junction plaques, and in failure to respond to H2O2 by stress-induced decreases in gap junction plaques. Although cell surface plaques decreased with AGA, results from Fig. 3 suggest that Cx50 protein levels were much more decreased. Thus, Cx43 plaque reduction does not appear to reflect a decrease in total Cx43 protein levels.
Figure 5. PKCγ C1B mutations cause increased Cx43 and Cx50 gap junction plaques.

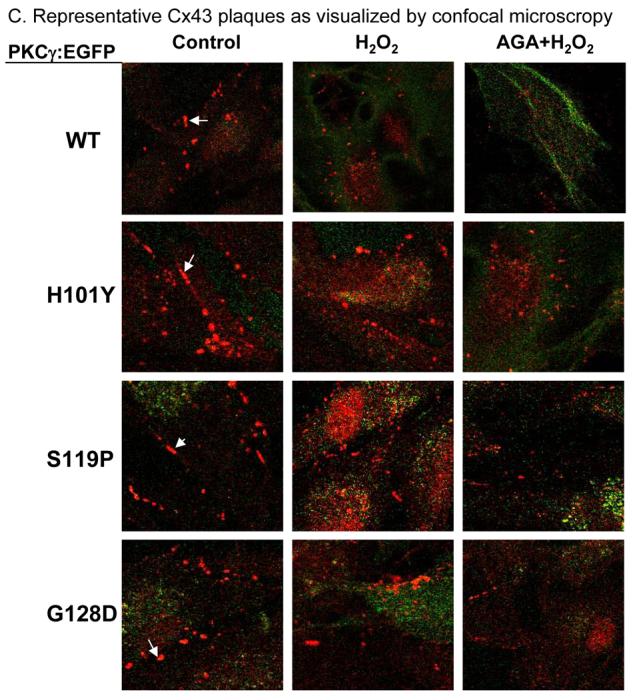
A. Stably-transfected cells, with or without AGA (200 μM, 3 h), were treated with 100 μM H2O2 for 20 min. Cell surface Cx43 gap junction plaques were visualized by anti-Cx43 immunolabeling and counted and graphed (in red, ≥ 1 μm in length, white arrows indicate representative plaques) and expressed as Cx43 plaques per cell per section. Green label revealed the EGFP fusion proteins of the wild type PKCγ or C1B mutants. The example images are shown in C. B. Cell surface Cx50 gap junction plaques were revealed using anti-Cx50 immunolabeling. Cx50 gap junction plaques were counted and graphed and expressed as Cx50 plaques per cell per section.
Expression of PKCγC1B mutations increased N/N1003A cell susceptibility to oxidative stress and cell death, even when endogenous wild type PKCγ was present
To determine if PKCγ C1B mutations caused increased response to oxidative stress we measured the activation of caspase-3 in stably transfected cells (Fig. 6). Initially caspase-3 enzyme activity was low in both untreated and stably transfected cells expressing either the wild type PKCγ or C1B mutants. However, H2O2 induced activation of caspase-3 in a time-dependent manner. After 3 h of H2O2 greater increases in caspase-3 enzyme activity were observed in cells expressing C1B mutants when compared to that of the WT cells. Data indicate that overexpression of exogenous PKCγ C1B mutants caused N/N1003A lens epithelial cells to be more sensitive to H2O2 via a caspase-3 activation pathway.
Figure 6. PKCγ C1B mutations cause N/N1003A cells to be more susceptible to H2O2-induced apoptosis (caspase-3 activity assay): prevention using a gap junction inhibitor.

Cells were treated with 100 μM H2O2 for different time periods. Cell apoptosis was determined by caspase-3 activity assay. In addition, cells were pre-incubated with 200 μM AGA for 30 min followed by addition of 100 μM H2O2 for additional 3 h and caspase-3 activation was analyzed (AGA+H2O2 (3h) columns) . * indicates significant changes compared to levels in control cells with overexpression of wild type PKCγ.
Finally, we used AGA to confirm that the increased gap junction plaques (Fig. 5) could account for increased cell susceptibility to H2O2-induced apoptosis. Cells were pre-incubated with 200 μM AGA for 30 min followed by addition of 100 μM H2O2 for additional 3 h (AGA treatment, totally 3 h 30 min) and caspase-3 activation was measured. The results demonstrated that gap junction inhibition (or decreased protein levels) by AGA prevented activation of caspase-3 in cells with wild type PKCγ or C1B mutants (Fig. 6, far right four columns, AGA+H2O2). Thus, AGA can lower Cx50 levels (Fig. 3) and both Cx43 and Cx50 plaque number (Fig. 5) and this is correlated with protection from apoptosis caused by H2O2.
Discussion
We have previously demonstrated that PKCγ serves as an oxidative stress sensor through proper control of gap junctions in the lens (Lin et al., 2003a & b, 2004; Lin and Takemoto, 2005; Zampighi et al., 2005; Lin et al., 2006). We have also observed that the PKCγ H101Y C1B mutant is not activated by oxidative stress, e.g. H2O2 (Lin and Takemoto, 2005). In this current study we have tested three SCA14 PKCγ mutants which have modeled structures indicating disruption of the C1B Zn-finger stress switch. We have further provided an additional link between PKCγ C1B domain stress switch → gap junction control → caspase activation. Our data conclusively demonstrate that defective PKCγ C1B domains can cause 1) dysfunction of endogenous PKCγ (i.e., a dominant effect), 2) loss of gap junction control in lens epithelial cells, and 3) increased susceptibility to caspase-3-linked apoptosis.
PKCγ is a unique isoform of classical PKC which has an exposed C1B domain which binds diacylglycerol (DAG) and is oxidized to form disulfide bonds when the cells are exposed to hydrogen peroxide or other oxidative stress (Lin and Takemoto 2005; Lin et al., 2003a; Ananthanarayanan et al., 2003). Inactive PKCγ is always associated with 14-3-3 protein in the cytosol by binding to the PKCγ C1B domain where most SCA14 mutations occur (Nguyen et al., 2004). Release of 14-3-3 and oxidation within the C1B domain of the PKCγ lead to membrane translocation and activation of PKCγ (Nguyen et al., 2004; Lin and Takemoto 2005). Structural modeling results predict that H101Y and G128D SCA14 mutations caused Zinc-finger conformational changes (Chen et al., 2003; Xu et al., 1997, and Fig. 1). Our predicted model of the S119P mutation also suggests a dramatically altered C1B domain structure (Fig. 1B). We previously demonstrated the expression of exogenous wild type rat PKCγ with full enzyme activity (Lin et al., 2003b). In this report we demonstrate that PKCγ S119P and G128D mutations have lower basal enzyme activities when compared to the wild type PKCγ. We further demonstrate that all three of the PKCγ C1B mutants lacked responses to H2O2 (Fig. 2). Expression of the PKCγ C1B mutants caused a lack of oxidative stress responses of the remaining endogenous wild type PKCγ (Fig. 2B and D). The data suggest that C1B mutations might disrupt the association of endogenous PKCγ with other associated proteins, such as 14-3-3 proteins, which, in turn, causes inactivation of wild type endogenous PKCγ. This dysfunction of the endogenous PKCγ could lead to alteration of cell signaling pathways, such as responses of gap junctions to stress.
The passage of apoptotic signals through open gap junctions is linked to oxidative stress-induced cell death (Naus et al., 2001; Frantseva, 2002). Inhibition of gap junctions prevents cell death (Farahani et al., 2005; de Pina-Benabou et al., 2005; Krysko et al., 2005). We, for the first time, show that PKCγ C1B mutations have a negative effect on endogenous wild type PKCγ. The consequence of these mutations are: 1) altered control of gap junctions as determined by Cx43 and Cx50 phosphorylation and gap junction plaques (Fig. 4 and 5), and 2) increased caspase-3 linked apoptosis (Fig. 6). Gap junction inhibition experiments with AGA further confirmed that PKCγ C1B domain mutations induced increased cell susceptibility to H2O2-linked caspase-3 apoptosis through improper control of gap junctions.
Gap junction plaque formation is a dynamic process caused by multiple factors (Gaietta et al., 2002). Gap junction plaques assemble and disassemble at a rate of about every 2-5hr (Lauf et al., 2002; Berthoud et al., 2004; Musil et al., 2000), but little is known about what forces disassembly during this dynamic process. We have previously shown that activation of endogenous PKCγ results in disassembly of Cx43 and Cx50 gap junction plaques (Lin and Takemoto 2005). We show here that overexpression of PKCγ C1B mutants results in failure of endogenous PKCγ to respond to oxidative stress (e.g. H2O2) (Fig. 2). The dysfunction of endogenous PKCγ caused by the mutant PKCγ prevents the dynamic process of gap junction plaque disassembly before or after oxidative stress. This would result in increases in functional cell surface gap junction plaques in the cells with the PKCγ C1B mutants.
Apoptotic caspase-3 activation is a common event in cell death. During ischemia or stroke, cell death signals may pass through gap junction channels to adjacent cells (Velaquez et al., 2003; Contreras et al., 2004; Thompson et al., 2006). This gap junction “Bystander effect” accounts for cell death (Farahani et al., 2005). In our study, overexpression of PKCγ C1B mutations induced cells to be more susceptible to apoptosis by H2O2 as determined by caspase-3 activation. This effect was abolished by AGA inhibition of gap junctions. Since AGA caused a greater decrease in Cx50 (Fig. 3), we suggest that altered Cx50 gap junctions in N/N1003A cells contribute to passage of apoptotic cell signals to adjacent cells which, in turn, stimulates activation of caspase-3 and causes cells to be more susceptible to oxidative stress. Gap junction inhibition results suggest that protection of cells from oxidative stress may be restored by lowering Cx50 gap junction levels in lens epithelial cells.
In summary, PKCγ C1B mutants lack PKCγ stress sensing activity. Overexpression of these mutants results in dysfunction of endogenous PKCγ, which, in turn, leads to altered control of gap junctions by PKCγ during oxidative stress. Altered gap junctions subsequently result in failure to be protected from further exposure to damaged signals which, in turn, causes cellular caspase-3 activation and subsequent cell apoptosis.
Acknowledgements
The authors are grateful to K. H. Fischbeck of NINDS/NIH for helpful discussion. We thank Dr. John Reddan of Oakland University for the N/N1003A cells, Dr. Peggy Zelenka of National Eye Institute for the PKCγ:EGFP vector. This work was supported by National Institutes of Health Grant EY 13421 (to D. J. T.) and by a grant from the National Organization for Rare Disorders (to D. L.). This is publication 06-13-5 from the Kansas Agricultural Experiment Station.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alonso I, Costa C, Gomes A, Ferro A, Seixas AI, Silva S, Cruz VT, Coutinho P, Sequeiros J, Silveira I. A novel H101Q mutation causes PKCgamma loss in spinocerebellar ataxia type 14. J Human Genet. 2005;50:523–529. doi: 10.1007/s10038-005-0287-z. [DOI] [PubMed] [Google Scholar]
- Ananthanarayanan B, Stahelin RV, Digman MA, Cho W. Activation mechanisms of conventional protein kinase C isoforms are determined by the ligand affinity and conformational flexibility of their C1 domains. J Biol Chem. 2003;278:46886–46894. doi: 10.1074/jbc.M307853200. [DOI] [PubMed] [Google Scholar]
- Berthoud V, Tadros P, Beyer E. Connexin and gap junction degradation. Methods. 2000;20:180–187. doi: 10.1006/meth.1999.0935. [DOI] [PubMed] [Google Scholar]
- Chen DH, Brkanac Z, Verlinde CL, Tan XJ, Bylenok L, Nochlin D, Matsushita M, Lipe H, Wolff J, Fernandez M, Cimino PJ, Bird TD, Raskind WH. Missense mutations in the regulatory domain of PKC gamma: a new mechanism for dominant nonepisodic cerebellar ataxia. Am J Hum Genet. 2003;72:839–849. doi: 10.1086/373883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DH, Cimino PJ, Ranum LP, Zoghbi HY, Yabe I, Schut L, Margolis RL, Lipe HP, Feleke A, Matsushita M, Wolff J, Morgan C, Lau D, Fernandez M, Sasaki H, Raskind WH, Bird TD. The clinical and genetic spectrum of spinocerebellar ataxia 14. Neurol. 2005;64:1258–1260. doi: 10.1212/01.WNL.0000156801.64549.6B. [DOI] [PubMed] [Google Scholar]
- Contreras J, Sanchez H, Veliz L, Bukauskas F, Bennett M, Saez J. Role of connexin-based gap junctional channels and hemichannels in ischemia-induced cell death in nervous tissue. Brain Res Rev. 2004;47:290–303. doi: 10.1016/j.brainresrev.2004.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia SS, Duarte CB, Faro CJ, Pires EV, Carvalho AL. Protein kinase C gamma associates directly with the GluR4 alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor subunit. Effect on receptor phosphorylation. J Biol Chem. 2003;278:6307–6313. doi: 10.1074/jbc.M205587200. [DOI] [PubMed] [Google Scholar]
- Cusato K, Bosco A, Rozental R, Guimaraes CA, Reese BE, Linden R, Spray DC. Gap junctions mediate bystander cell death in developing retina. J Neurosci. 2003;23:6413–6422. doi: 10.1523/JNEUROSCI.23-16-06413.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pina-Benabou MH, Szostak V, Kyrozis A, Rempe D, Uziel D, Urban-Maldonado M, Benabou S, Spray DC, Federoff HJ, Stanton PK, Rozental R. Blockade of gap junctions in vivo provides neuroprotection after perinatal global ischemia. Stroke. 2005;36:2232–2237. doi: 10.1161/01.STR.0000182239.75969.d8. [DOI] [PubMed] [Google Scholar]
- El-Sabban ME, Sfeir AJ, Daher MH, Kalaany NY, Bassam RA, Talhouk RS. ECM-induced gap junctional communication enhances mammary epithelial cell differentiation. J Cell Sci. 2003;116:3531–3541. doi: 10.1242/jcs.00656. [DOI] [PubMed] [Google Scholar]
- Fahey MC, Knight MA, Shaw JH, Gardner RJ, du Sart D, Lockhart PJ, Delatycki MB, Gates PC, Storey E. Spinocerebellar ataxia type 14: study of a family with an exon 5 mutation in the PRKCG gene. J Neurol Neurosurg Psychiatry. 2005;76:1720–1722. doi: 10.1136/jnnp.2004.044115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farahani R, Pina-Benabou MH, Kyrozis A, Siddiq A, Barradas PC, Chiu FC, Cavalcante LA, Lai JC, Stanton PK, Rozental R. Alterations in metabolism and gap junction expression may determine the role of astrocytes as “good samaritans” or executioners. Glia. 2005;50:351–361. doi: 10.1002/glia.20213. [DOI] [PubMed] [Google Scholar]
- Frantseva J. Ischemia-induced brain damage depends on specific gap-junctional coupling. J. Cerebral. Blood Flow & Metab. 2002;22:453–462. doi: 10.1097/00004647-200204000-00009. [DOI] [PubMed] [Google Scholar]
- Goodenough DA, Goliger JA, Paul DL. Connexins, Connexons, and Intercellular Communication. Ann Rev Biochem. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- Klebe S, Durr A, Rentschler A, Hahn-Barma V, Abele M, Bouslam N, Schols L, Jedynak P, Forlani S, Denis E, Dussert C, Agid Y, Bauer P, Globas C, Wullner U, Brice A, Riess O, Stevanin G. New mutations in protein kinase Cgamma associated with spinocerebellar ataxia type 14. Ann Neurol. 2005;58:720–729. doi: 10.1002/ana.20628. [DOI] [PubMed] [Google Scholar]
- Krysko DV, Leybaert L, Vandenabeele P, D'Herde K. Gap junctions and the propagation of cell survival and cell death signals. Apoptosis. 2005;10:459–469. doi: 10.1007/s10495-005-1875-2. [DOI] [PubMed] [Google Scholar]
- Lauf U, Giepmans B, Lopez P, Braconnot S, Chen S, Falk M. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci. 2002;99:10446–10451. doi: 10.1073/pnas.162055899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Boyle DL, Takemoto DJ. IGF-I-induced phosphorylation of connexin 43 by PKCgamma: regulation of gap junctions in rabbit lens epithelial cells. Investig Ophthalmol Vis Sci. 2003a;44:5259–5268. doi: 10.1167/iovs.02-0737. [DOI] [PubMed] [Google Scholar]
- Lin D, Zhou J, Zelenka PS, Takemoto DJ. Protein kinase Cgamma regulation of gap junction activity through caveolin-1-containing lipid rafts. Investig Ophthalmol Vis Sci. 2003b;44:5259–5268. doi: 10.1167/iovs.03-0296. [DOI] [PubMed] [Google Scholar]
- Lin D, Lobell S, Jewell A, Takemoto DJ. Differential phosphorylation of connexin46 and connexin50 by H2O2 activation of protein kinase Cgamma. Mol Vis. 2004;10:688–695. [PubMed] [Google Scholar]
- Lin D, Takemoto DJ. Oxidative activation of protein kinase Cgamma through the C1 domain. Effects on gap junctions. J Biol Chem. 2005;280:13682–13693. doi: 10.1074/jbc.M407762200. [DOI] [PubMed] [Google Scholar]
- Lin D, Barnett M, Lobell S, Madgwick D, Zampighi GA, Takemoto DJ. PKC γ knockout mouse lenses are more susceptible to oxidative stress damage. J Exp Biol. 2006;209:4371–4378. doi: 10.1242/jeb.02524. [DOI] [PubMed] [Google Scholar]
- Lin JH, Weigel H, Cotrina ML, Liu S, Bueno E, Hansen AJ, Hansen TW, Goldman S, Nedergaard M. Gap-junction-mediated propagation and amplification of cell injury. Nat Neurosci. 1998;1:494–500. doi: 10.1038/2210. [DOI] [PubMed] [Google Scholar]
- Musil LS, Le AC, VanSlyke JK, Roberts LM. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem. 2000;275:25207–25215. doi: 10.1074/jbc.275.33.25207. [DOI] [PubMed] [Google Scholar]
- Naus C, Ozog M, Bechberger J, Nakase T. A neuroprotective role for gap junctions. Cell Commun Adhes. 2001;8:325–328. doi: 10.3109/15419060109080747. [DOI] [PubMed] [Google Scholar]
- Neijssen J, Herberts C, Drijfhout JW, Reits E, Janssen L, Neefjes J. Cross-presentation by intercellular peptide transfer through gap junctions. Nature. 2005;434:83–88. doi: 10.1038/nature03290. [DOI] [PubMed] [Google Scholar]
- Nguyen TA, Takemoto LJ, Takemoto DJ. Inhibition of gap junction activity through the release of the C1B domain of protein kinase Cgamma (PKCgamma) from 14-3-3: identification of PKCgamma-binding sites. J Biol Chem. 2004;279:52714–52725. doi: 10.1074/jbc.M403040200. [DOI] [PubMed] [Google Scholar]
- Saleh SM, Takemoto LJ, Zoukhri D, Takemoto DJ. PKC-gamma phosphorylation of connexin 46 in the lens cortex. Mol Vis. 2001;7:240–246. [PubMed] [Google Scholar]
- Seki T, Adachi N, Ono Y, Mochizuki H, Hiramoto K, Amano T, Matsubayashi H, Matsumoto M, Kawakami H, Saito N, Sakai N. Mutant protein kinase Cgamma found in spinocerebellar ataxia type 14 is susceptible to aggregation and causes cell death. J Biol Chem. 2005;280:29096–29106. doi: 10.1074/jbc.M501716200. [DOI] [PubMed] [Google Scholar]
- Shutoh F, Katoh A, Ohki M, Itohara S, Tonegawa S, Nagao S. Role of protein kinase C family in the cerebellum-dependent adaptive learning of horizontal optokinetic response eye movements in mice. Euro J Neurosci. 2003;18:134–142. doi: 10.1046/j.1460-9568.2003.02717.x. [DOI] [PubMed] [Google Scholar]
- Stevanin G, Hahn V, Lohmann E, Bouslam N, Gouttard M, Soumphonphakdy C, Welter ML, Ollagnon-Roman E, Lemainque A, Ruberg M, Brice A, Durr A. Mutation in the catalytic domain of protein kinase C gamma and extension of the phenotype associated with spinocerebellar ataxia type 14. Arch Neurol. 2004;61:1242–1248. doi: 10.1001/archneur.61.8.1242. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–927. doi: 10.1126/science.1126241. [DOI] [PubMed] [Google Scholar]
- van de Warrenburg BP, Verbeek DS, Piersma SJ, Hennekam FA, Pearson PL, Knoers NV, Kremer HP, Sinke RJ. Identification of a novel SCA14 mutation in a Dutch autosomal dominant cerebellar ataxia family. Neurol. 2003;61:1760–1765. doi: 10.1212/01.wnl.0000098883.79421.73. [DOI] [PubMed] [Google Scholar]
- Velaquez J, Frantseva M, Naus C. Gap junctions and neuronal injury: Protectants or executioners. Neuroscientist. 2003;9:5–9. doi: 10.1177/1073858402239586. [DOI] [PubMed] [Google Scholar]
- Verbeek DS, Knight MA, Harmison GG, Fischbeck KH, Howell BW. Protein kinase C gamma mutations in spinocerebellar ataxia 14 increase kinase activity and alter membrane targeting. Brain. 2005;128:436–442. doi: 10.1093/brain/awh378. [DOI] [PubMed] [Google Scholar]
- Vlak MH, Sinke RJ, Rabelink GM, Kremer BP, van de Warrenburg BP. Novel PRKCG/SCA14 mutation in a Dutch spinocerebellar ataxia family: Expanding the phenotype. Mov Disord. 2006 doi: 10.1002/mds.20851. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Xu RX, Pawelczyk T, Xia TH, Brown SC. NMR structure of a protein kinase C-gamma phorbol-binding domain and study of protein-lipid micelle interactions. Biochem. 1997;36:10709–10717. doi: 10.1021/bi970833a. [DOI] [PubMed] [Google Scholar]
- Yabe I, Sasaki H, Chen DH, Raskind WH, Bird TD, Yamashita I, Tsuji S, Kikuchi S, Tashiro K. Spinocerebellar ataxia type 14 caused by a mutation in protein kinase C gamma. Arch Neurol. 2003;60:1749–1751. doi: 10.1001/archneur.60.12.1749. [DOI] [PubMed] [Google Scholar]
- Zampighi GA, Planells AM, Lin D, Takemoto D. Regulation of lens cell-to-cell communication by activation of PKCgamma and disassembly of Cx50 channels. Investig Ophthalmol Vis Sci. 2005;46:3247–3255. doi: 10.1167/iovs.04-1504. [DOI] [PubMed] [Google Scholar]