

Abstract
There are many forms of iron storage disease, some hereditary and some acquired. The most common of the hereditary forms is HFE-associated hemochromatosis, and it is this disorder that is the main focus of this presentation. The body iron content is regulated by controlling absorption, and studies in the past decade have clarified, in part, how this regulation functions. A 25 amino acid peptide hepcidin is upregulated by iron and by inflammation, and it inhibits iron absorption and traps iron in macrophages by binding to and causing degradation of the iron transport protein ferroportin. Most forms of hemochromatosis results from dysregulation of hepcidin or defects of hepcidin or ferroportin themselves.
Hereditary hemochromatosis was once considered to be very rare, but in the 1970s and 1980s, with the introduction of better diagnostic tests, it became to be considered the most common disease of Europeans. Controlled epidemiologic studies carried out in the last decade have shown, however, the disease itself actually is rare, and it is only the genotype and associated biochemical changes that are common. We do not understand why only a few homozygotes develop severe disease. It now seems unlikely that there are important modifying genes, and although alcohol is known to have some effect, excess drinking probably plays only a modest role in determining the hemochromatosis phenotype.
Hereditary hemochromatosis is readily treated by phlebotomy. Secondary forms of the disease require chelation therapy, and the recent introduction of effective oral chelating agents is an important step forward in treating patients with disorders in which iron overload often proves to be fatal, such as thalassemia, myelodysplastic anemias, and dyserythropoietic anemias.
While much has been learned about the regulation of iron homeostasis in the past decade, many mysteries remain and represent challenges that will keep us occupied for years to come.
It is a great honor for me to present this lecture in memory of my good friend Bracha Ramot. It is because this lecture is in honor of Bracha that I do not feel the least bit uncomfortable speaking about iron storage diseases in a symposium entitled “Genomics and Molecular Biology of Hematopoietic Malignancies”. I know that had it been possible to ask her whether this might be appropriate Bracha would have said: “Of course, Ernie. Talk about whatever you want to”. That was her way. And, of course, Bracha was no stranger to iron metabolism. She was scientifically broad, a Renaissance hematologist. A MEDLINE search reveals 261 publications by Bracha and eight of those deal with iron -- iron in thalassemia, iron in sideroblastic anemia, and iron in iron deficiency anemia.
The ease with which iron can shed or acquire electrons has suited it well for a variety of chemical reactions, and it is probably for this reason that iron is essential for virtually all forms of life. But because of its reactivity iron can also produce damage, and thus life forms have had to evolve mechanisms for acquiring and retaining the right amount of iron -- enough iron to perform its essential functions, but not enough to produce damage. As with all finely tuned mechanisms, things can go wrong with maintaining this delicate balance. Iron deficiency ranks, along with inflammation, as the most common cause of anemia. Iron overload is much less common, but is also clinically important, although much less so than was believed only a few years ago.
Iron storage disease may be divided into hereditary forms and acquired forms, as shown in table 1. The acquired forms of iron storage disease are generally associated with marrow hyperactivity, a finding that has suggested that there may be a direct connection between erythropoiesis and iron absorption. The mechanism of this effect remains unknown, but the possibility that a humoral factor elaborated by the marrow might modulate iron absorption is very real.
Table 1.
A Classification of Iron Storage Disease
| Hereditary |
Autosomal Dominant
|
Autosomal Recessive
|
Acquired
|
Of the hereditary forms of iron storage disease, only HFE-associated hemochromatosis is characterized by a high mutant gene frequency. The other abnormalities, which will not be discussed here, are important too, not only because they are the cause of severe diseases, but also because the discovery of the mutations that cause them have played an important role in furthering our understanding of iron homeostasis.
The Fundamentals of Iron Metabolism
Since the classical studies of McCance and Widdowson [1] 70 years ago it has been recognized that the iron content of the body is regulated through the modulation of iron absorption. Although small amounts of iron are lost from the body, chiefly through desquamation of cells, there is no regulated iron excretion. Rather, iron metabolism is a closed circuit in which the iron that the body has captured is reutilized (figure 1). Hemochromatosis is characterized by excessive accumulation of iron in the body. Thus, it is a state in which the absorption of iron is dysregulated, and more iron is absorbed than is needed. But how does the body “know” how much iron to absorb? Over the past 60 years attempts have been made to explain the regulation of iron absorption through the existence of a “mucosal block” [3], a flawed concept based on flawed data, as I have described in detail elsewhere [4]. What the experimental data actually show is not the existence of a “blocking” of iron absorption after an initial dose of iron has been given, but rather “mucosal intelligence”. Iron-deficiency results in enhanced iron absorption and iron overload decreases iron absorption. Other factors that modulate iron absorption include anemia, which increases iron absorption probably largely through the enhanced erythropoiesis that is generally present, inflammation, which decreases iron absorption, and hypoxia which increases iron absorption.
Figure 1.
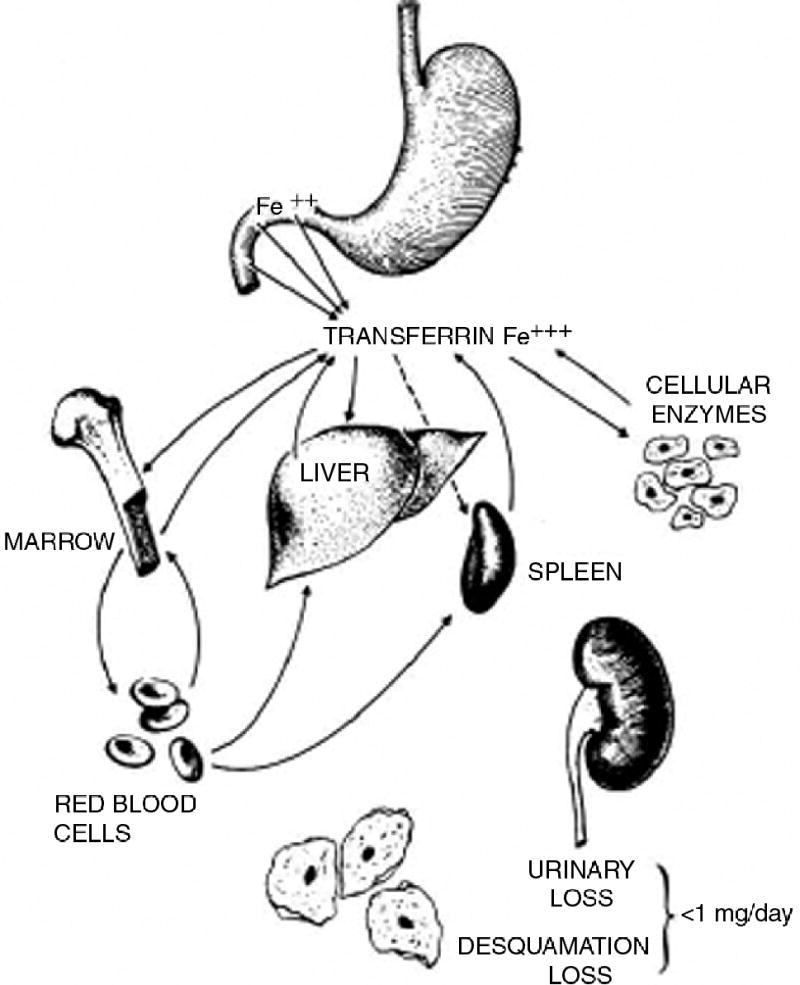
The closed-circuit of iron metabolism (From Beutler et al. Reprinted with permission from Elsevier [2])
In the past 10 years a considerable number of genes involved in the regulation of iron homeostasis has been identified. These have been generally found either through the study of mutant laboratory animals or the positional cloning of genes in humans with iron storage disease. In table 2, those genes hypomorphic mutations of which cause iron overload have classified as “down-regulators”. Conversely, those in which hypomorphs cause iron deficiency are classified as “up-regulators”.
Table 2.
Some Of The Known Participants In The Regulation of Iron Homeostasis
| Down-Regulators | Up-Regulators |
|---|---|
| Ceruloplasmin | Duodenal cytochrome b (dcytb) |
| Ferroportin (SLC1A3) | Hephaestin |
| Hemojuvelin | Nramp2 (DMT-1) |
| Hepcidin | |
| HFE | |
| IRP2 | |
| Transferrin | |
| Transferrin receptor 1 | |
| Transferrin receptor 2 | |
| β2 microglobulin |
The interaction between these participants is complex and has been very difficult to unravel. To a large extent we do not understand how the various parts fit together; it is like a difficult jigsaw puzzle (figure 2).
Figure 2.

The various participants in the regulation of iron homeostasis have been somewhat of a jigsaw puzzle. We are only now gradually beginning to put together the pieces.
The Central Role of Hepcidin
The 25 amino acid peptide, hepcidin, plays a central role in the regulation of iron homeostasis, and it is directly or indirectly involved in the development of most iron overload syndromes [5]. Hepcidin is an anti-microbial peptide that has apparently evolved into a regulator of iron transport. Ferroportin, the transport protein that moves ferrous iron from intestinal epithelial cells and macrophages into the serum serves as a receptor for hepcidin. When hepcidin binds to ferroportin it causes internalization of ferroportin and its proteolytic destruction [6]. Thus, hepcidin serves to prevent the egress of iron both from intestinal cells and from macrophages.
Hepcidin is normally upregulated by excess iron stores [7,8]. This serves to prevent further absorption of iron from the gastrointestinal tract, and its release from macrophages. It is this regulation that is impaired in several types of hemochromatosis. HFE, transferrin receptor 2, and hemojuvelin deficiency all are associated with failure of hepcidin to be appropriately upregulated. In the case of HFE and transferrin receptor-2 mutations some rise of hepcidin levels does occur, and as a consequence iron overload is relatively mild in most cases. In hemojuvelin deficiency, the mutation that causes juvenile hemochromatosis, the defect is more severe [9,10]. It is apparent that these three molecules are involved in some way in the signaling pathway from iron to hepcidin transcription. Although a number of models have been proposed, none have been established to be correct. The recent proposal [11] that a complex consisting of HFE, transferrin receptor 2, and hemojuvelin serves as a receptor for a bone morphogenetic protein (BMP) has attractive features, but does not explain our finding that the response to BMPs is undiminished in cells deficient either in HFE or transferrin receptor 2 [12] or that hepcidin-2 in mice responds to iron but not to BMPs (Truksa J, Lee P, Flanagan J, Beutler E. Different regulatory elements are required for response of hepcidin to IL-6 and BMP-9. In preparation, 2007).
The possible role of the marrow in sending signals that modulate iron absorption has been alluded to above. Such a factor might well work by signaling hepcidin, but its nature remains elusive. Recently, we were able to show that the level of plasma soluble transferrin receptor, greatly increased both in iron deficiency and in ineffective erythropoiesis, does not modulate hepcidin expression [13]. On the other hand, it has recently been suggested that plasma from thalassemic patients has a negative effect on hepcidin levels in HepG2 cells [14]. It may be that soluble hemojuvelin, which has been shown to be present in the plasma, acts as a regulator by inhibiting ligation of the receptor for BMPs [15,16].
The History of Hereditary Hemochromatosis
Described more than a hundred years ago, hereditary hemochromatosis was considered to be a rare curiosity until Sheldon [17] published his classic monograph about the disease, reviewing over 345 cases that have been described in the literature. In Sheldon’s own words:
“it may be accepted that haemochromatosis is a rare disease, although it is probably a good deal less rare then is usually believed.”
A simple treatment, viz., phlebotomy was introduced. Credit for this innovation is usually accorded to Davis and Arrowsmith [18], but the actual suggestion was first made three years earlier by Finch [19].
The view that hemochromatosis was a rare disease prevailed until the 1970s. The discovery that the disorder was HLA-linked [20,21], and the fact that serum iron determinations and methods for measuring serum ferritin were becoming commonplace changed the landscape. Quickly, and, in retrospect, uncritically, hemochromatosis was considered to be the most common genetic disease of Europeans. The gene responsible for the most common form of the disease was cloned in 1996 [22] and designated HFE. The common mutation that was strongly associated with hemochromatosis was c.845G>A (C282Y) and it was soon possible to confirm that homozygote frequency was about 5:1,000.
Screening for Hemochromatosis
With the cloning of the HFE gene there was strong advocacy for universal screening for hemochromatosis. The belief that screening for hemochromatosis would be useful was driven by the perception that the disorder was very common and by the availability of a treatment, phlebotomy. Thus, Adams et al. [23] wrote “Screening blood donors for hemochromatosis has the potential to improve overall societal health status and decrease third-party payer health care costs over the long-term”; Yapp and Powell [24] averred “Our message is simple: there should be general population screening for HFE-associated haemochromatosis”; and Allen and Williamson [25] stated “We believe that to benefit all those at risk there is an ethical imperative to implement screening for the major mutation causing haemochromatosis now, rather than wait years for confirmation of what is currently known that at least half of those with the predisposing genotype will develop some form of the disease”. But the fact was that, Allen and Williamson’s statement notwithstanding, we did not know the actual penetrance of the homozygous state. The entire advocacy for screening was based on the softest type of data -- clinical impressions. Indeed, a consensus conference held in the year 2000 [26] posed the question
“May clinically asymptomatic patients never present life threatening complications?”
and provided the following response:
“The answer is yes: However, this seems to occur in a low proportion of subjects: it is estimated that only 5% of C282Y/C282Y men over the age of 40 years will not express an HC phenotype.”
Thus, in the year 2000 a group of experts concluded that 95% of males with a homozygous genotype manifested the phenotype. No definition of the phenotype was specified. The biochemical changes in hemochromatosis—the high transferrin saturation or increased serum ferritin levels—are, indeed, common although they do not achieve a 95% penetrance. But the meaningful clinical phenotype, illness as a result of iron overload is actually rare (see below). Clearly it is the penetrance this phenotype, the percentage of homozygotes manifesting actually clinical morbidity, not merely elevation of serum iron, transferrin saturation, or ferritin levels that is relevant. Since so many of the putative symptoms of hemochromatosis, fatigue, impotence, and arthropathy are highly subjective and are common in older patients whether or not they have hemochromatosis, this question could only be answered in a well-controlled study.
The Kaiser-Scripps Study of Hemochromatosis Penetrance
We have had the opportunity of performing a study which, for the first time, has made it possible to objectively estimate the clinical penetrance of HFE hemochromatosis. The subjects of the study were attendees at a “Health Appraisal Clinic”, at the Kaiser Permanente in Southern California in the San Diego area. Over a period of three years we were able to obtain DNA samples from 41,702 individuals. Because of the demography of the San Diego area the ancestry of 32,820 of these patients was principally European, the population in which the C282Y mutation is by far the most common. Each volunteer completed a questionnaire consisting of 400 questions covering a variety of health topics and detailed information about family ancestry. Importantly, this information was obtained before genetic or laboratory information was known. The median age of participants was approximately 56 years, a time at which most patients with hemochromatosis who develop clinical disease are already symptomatic. Details about this population have been published elsewhere [27–30].
We found that about two-thirds of the males and one -half of the females homozygous for the C282Y mutation manifested an increased transferrin saturation and/or increased serum ferritin levels. But this was not the purpose of our study. What we had set out to ascertain is what proportion of the patients would have symptoms of hemochromatosis. Previous studies had concluded, as pointed out above, that many or most would have such symptoms. But these studies did not take into account the symptoms of matched controls. Such comparisons are critical because the symptoms that were evaluated are the very symptoms that are very common in older people, symptoms such as fatigue, arthritis, and impotence. Our findings are summarized in figure 3.
Figure 3.

Findings in 156 homozygotes for the C282Y HFE mutation characteristic of hereditary hemochromatosis and wild type (wt) controls (Reprinted with permission from McGraw-Hill Companies [31]).
It should not come as a surprise that results so much at variance with the conventional wisdom of the day were received with considerable skepticism. Legitimate questions were raised. Was our population biased by the fact that our studies were conducted in a “health appraisal clinic”? Ironically, one group of skeptics suggested our population was too sickly [32]; another group suggested that they were too healthy [33]. They couldn’t both be right! But the question of whether there was selection bias in the population studied is a legitimate one that deserves a clear answer, and there is one. Suppose that we were just examining the survivors, that a sizable number of seriously ill patients suffering from hemochromatosis had either died or were being treated by a hepatologist, and therefore had no reason or inclination to attend a “health appraisal clinic”? The consequence of this circumstance would be that the percentage of homozygotes in the population would decrease with progressive age. It does not. A second consequence would be that the number of homozygotes would fall short of the number predicted by the Hardy-Weinberg equilibrium. They do not. Gradually, the results of our study with Kaiser have been accepted as representing the true state of affairs, and two large studies [34,35] and several smaller ones have confirmed our results.
What Causes the Variable Penetrance of HFE-Hemochromatosis?
Over the past several decades we learned that there really are no single gene diseases. Whether we are dealing with a dominant disorder like factor V Leiden, a sex-linked defect like G6PD deficiency, or homozygotes for recessive disorders such as those that cause sickle cell disease, Gaucher disease, or hereditary hemochromatosis, individuals with the same genotype have very different clinical phenotypes [36]. This variation can be due to: 1) environmental factors; 2) genetic factors; or 3) epigenetic factors. The role of the last of these, epigenetic factors, may well be the most important. The power of this form of modification is dramatically shown by the fact that littermates in inbred strains of mice may show marked differences in the expression of a single mutant gene [37]. Unfortunately, we do not have the tools to study epigenetic modification of internal organs in man, and circulating blood cells may be poor surrogates for the epigenetic pattern of the patient, since epigenetic activation and inactivation of genes is usually tissue-specific. Accordingly, at the present time we can only limit our attention to environmental and genetic factors.
Environmental Modifie rs of Hereditary Hemochromatosis
Alcohol
The association between the ingestion of alcohol and severe hemochromatosis is so strong clinically that it was believed at one time that hemochromatosis was due to alcoholism [17,38–40]. The anecdotal evidence supporting the view that alcohol intake exacerbates the clinical manifestations of hemochromatosis is so strong as to be very convincing. Interestingly, the controlled clinical evidence is considerably weaker (fig 4). However, in some large studies there is a tendency for serum ferritin levels to be somewhat higher and liver function tests to be more abnormal in patients who ingest 60 g or more of alcohol when compared to homozygotes ingesting less alcohol [41] and in one study the incidence of cirrhosis was 9 times as high in such patients [42]. Hepcidin mRNA is decreased [43,44] and iron absorption is increased [43] by alcohol in normal 129x1/SvJ mice, but, somewhat surprisingly, the already elevated iron absorption in hfe knockout mice is not increased further by alcohol ingestion [43]. The effect of alcohol in mice is highly strain specific [43], and therefore caution must be exercised in extrapolating the murine findings to man.
Figure 4.

The relationship between alcohol intake and serum ferritin levels of HFE C282Y homozygotes and wildtype (wt) controls in the Scripps-Kaiser study. Although the ferritin levels are, as expected, higher in the homozygotes, the upward trend is found only in the controls. The error bars ± 1 SEM.
Diet
Diet seems to have a relatively modest influence on iron stores as judged by serum ferritin levels. There is a weak, albeit significant correlation between serum ferritin levels and heme, but not non-heme dietary intake [45,46]. It seems reasonable to suppose that the dietary intake may have some small effect on the penetrance of the disorder, but there is no compelling evidence that would lead one to believe that it is a major factor in determining the degree of iron overload that occurs in patients homozygous for the C282Y mutation.
Inflammation
It seemed possible that inflammation might be a modifying factor in the hemochromatosis phenotype. Inflammation up-regulates the levels of hepcidin and does so independently of HFE [47]. We measured the levels of C-reactive protein and of IL-6 in homozygous patients, but found no relationship between the size of the iron burden as measured by phlebotomy and the levels of these inflammatory markers [48].
H. pylori infection
H. pylori infection is associated with iron deficiency, and this infection also impairs the response to iron therapy [49,50]. It seemed reasonable that by blocking iron absorption this common infestation might protect against iron overload. However, we found that the iron burden of patients with antibodies against H. pylori was no greater than those without serologic evidence of infection [51].
Genetic Modifiers of Hereditary Hemochromatosis
Considerable effort has been expended, particularly by our laboratory [52–58] but also by others [59–67] to find mutations that interact with HFE mutations to increase or decrease the penetrance. These efforts have been virtually entirely fruitless. Only in the case of hepcidin mutations is there some evidence of modification of the expression of the C282Y homozygous state. Co-inheritance of hepcidin mutations appears to increase iron loading in homozygotes for the C282Y mutation [60,66,68]. Such hepcidin mutations are excessively rare; we have never encountered one in our sizeable experience. Perhaps we should not be too surprised that no genetic modifiers have emerged. The evidence that the expression of hemochromatosis is genetically determined is very modest [69]. Because of the fact that there was a modest sib-to-sib correlation in serum ferritin levels in fraternal and identical twins homozygous for the C282Y HFE mutation, it was concluded that genetic factors were operative. But the correlation was no greater in monozygotic twins than in sibs. This is actually more suggestive of environment than genetic factors.
Treatment of Iron Overload
The standard treatment for hereditary hemochromatosis is the removal of iron by phlebotomy. The evidence that the iron excess can be removed by this means is quite straightforward. It is more difficult to prove rigorously that the treatment is clinically effective since a controlled trial would be ethically unacceptable. The treatment is usually justified by citing studies that show that the lifespan of noncirrhotic patients who are treated by phlebotomy is normal [70,71]. But these studies were not controlled with patients who had not been phlebotomized. We now have come to recognize that the vast majority of patients with hemochromatosis are never diagnosed and never treated, and their lifespan appears to be essentially normal. This can be deduced from the fact that the proportion of homozygotes in the population does not decrease with increasing age [72]. Thus, the fact that phlebotomized patients also have a normal lifespan hardly constitutes proof of efficacy.
To determine the effectiveness of phlebotomy therapy we must go back to the pre-phlebotomy era. On this basis, it seems very likely that phlebotomy is useful. The lifespan of patients who are diagnosed after the introduction of phlebotomy was considerably longer than those diagnosed in the earlier decades [73]. This could, of course, be merely due to the fact that the disorder was diagnosed at earlier and earlier stages or that better supportive treatment became available for diabetes, cirrhosis, and infections. More compelling is the fact that cirrhosis of the liver seems, on serial biopsy, to improve after phlebotomy treatment is instituted [74–76].
But when should patients with hereditary hemochromatosis be treated? Any patient with clear clinical manifestations of hemochromatosis such as cirrhosis of the liver, cardiomyopathy or diabetes mellitus should be treated with phlebotomy. The risk of cirrhosis seems to increase sharply when the serum ferritin level reaches 1000 μg/L [77–79] and patients with such levels should be phlebotomized. On the other hand, joint symptoms in such patients, often ascribed to iron overload, are usually due to other causes and do not respond to phlebotomy. Similarly, the fatigue that is ascribed to the disease, no more common in such patients than in the general population does not seem to be a reason to start therapy.
Based on earlier misconceptions regarding the progressive nature of the disease and of the severity of its clinical manifestations, much of the “educational material” to which patients are subjected on the internet and elsewhere is quite misleading and creates anxiety in patients regarding the necessity for treatment.
It is not uncommon to encounter patients who expect to be phlebotomized, even though none of the indications enumerated above are present. Since the treatment itself, viz., phlebotomy, is essentially harmless, there is no need to resist the desire of such misinformed patients for treatment. Even if the response is only a placebo effect, if the patient feels that his or her energy level is improved, all the better. On the other hand, patients who have no such preconceived ideas can be reassured that they do not need any treatment, not at the moment and probably never. Although it was formerly believed that patients with the hemochromatosis genotype need to be monitored for an increasing iron burden, more recent studies have shown that there is no tendency for the iron burden to increase [80–82]. Based on the available evidence, reassessing patients’ iron status every 5 years or so should be more than adequate.
There are potential complications that should be taken into account in the care of the patient with hereditary hemochromatosis. In the case of those patients who have developed cirrhosis of the liver, the risk of developing a hepatoma is high. Bomford and Williams [83] found that 29% of treated patients and 19% of untreated patients developed this tumor. (Most of the untreated patients were from an era before the introduction of phlebotomy therapy; some were patients who refused treatment or were diagnosed at autopsy). In two recent studies 20% [84] and 21% [85] of cirrhotic patients with hemochromatosis developed this tumor. Non-cirrhotic patients also appear to be at risk. There are a number of anecdotal case-reports of such patients with hepatoma [86–88]. Recently, Dale et al reported (in abstract [89]) that the results of a 492 patient-year study of hemochromatosis patients with and without cirrhosis showed a relative risk of 58.99 [95% CI 7.70–453.27] (p=0.0004) without cirrhosis and with a relative risk of 464.78 [103.49~987.3] (p <0.0001). Moreover, Willis et al [90] estimated that 1.31 to 2.1% of all males with the homozygous genotype develop hepatoma. Since the studies of Willis et al. included all homozygotes and only a small percentage of homozygotes develop cirrhosis (recently estimated at 5.6% and 1.9% for males and females respectively [91]), the finding by Willis et al. implies a considerable percenta ge of non-cirrhotic patients are at risk for hepatoma. More data regarding the risk of developing hepatoma in non-cirrhotic patients are needed, and we are undertaking such studies in the Kaiser population.
It is obvious that phlebotomy cannot be used to treat secondary hemochromatosis. These patients are usually anemic and other means must be used to mobilize the excess iron. The gold standard for chelation therapy is desferrioxamine, but the fact that this drug must we given parenterally, either by intravenous or subcutaneous route, over a period of many hours has spurred the search for oral agents capable of mobilizing iron safely. Two such drugs have emerged, deferiprone (L1) and deferasirox (ICL670; exJade). Deferiprone has been in use for nearly 20 years, and was slow in gaining acceptance because a relatively high incidence of agranulocytosis was reported in some studies, although this seems to be less of a risk than originally thought. Deferasirox has been introduced relatively recently and appears to have a favorable toxicity profile, although skin rashes, gastrointestinal disturbances, and occasionally abnormal liver function tests have been encountered. It seems to be roughly equivalent in effectiveness to desferrioxamine, and may well become the treatment of choice for patients with secondary hemochromatosis [92].
Conclusions
Much has been learned about the regulation during homeostasis in the past few years. We are beginning to understand how iron absorption is regulated from the gastrointestinal tract, but our understanding is still incomplete. Our approach to hereditary hemochromatosis has been revolutionized by appreciation of the fact that the clinical penetrance of the C282Y HFE mutation is very low. We still do not understand why some patients have fully expressed disease, while most do not. Nor do we know just how iron stimulates hepcidin, the central regulator of iron homeostasis. While much as been learned, there are still many challenges ahead.
Acknowledgments
This is manuscript number 18813-MEM. Supported by the National Institutes of Health grant DK53505 and the Stein Endowment Fund.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McCance RA, Widdowson EM. Absorption and excretion of iron. Lancet. 1937;2:680–684. doi: 10.1042/bj0312029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beutler E, Fairbanks VF, Fahey JL. Clinical Disorders of Iron Metabolism. Grune & Stratton, Inc; New York: 1963. [Google Scholar]
- 3.Hahn PF, Bale WF, Ross JF, et al. Radioactive iron absorption by gastro-intestinal tract: Influence of anemia, anoxia, and antecedent feeding. J Exp Med. 1943;78:169–188. doi: 10.1084/jem.78.3.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beutler E. History of iron in Medicine. Blood Cells Mol Dis. 2002;29:297–308. doi: 10.1006/bcmd.2002.0560. [DOI] [PubMed] [Google Scholar]
- 5.Nemeth E, Ganz T. Regulation of iron metabolism by hepcidin. Annu Rev Nutr. 2006;26:323–342. doi: 10.1146/annurev.nutr.26.061505.111303. [DOI] [PubMed] [Google Scholar]
- 6.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin Regulates Iron Efflux by Binding to Ferroportin and Inducing Its Internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 7.Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 9.Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 10.Nemeth E, Roetto A, Garozzo G, et al. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105:1803–1806. doi: 10.1182/blood-2004-08-3042. [DOI] [PubMed] [Google Scholar]
- 11.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281:28494–28498. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 12.Truksa J, Peng H, Gelbart T, et al. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc Natl Acad Sci USA. 2006;103:10289–10293. doi: 10.1073/pnas.0603124103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flanagan JM, Peng H, Wang L, et al. Soluble transferrin receptor-1 levels in mice do not affect iron absorption. Acta Haematol (Basel) 2006;116:249–254. doi: 10.1159/000095875. [DOI] [PubMed] [Google Scholar]
- 14.Weizer-Stern O, Adamsky K, Amariglio N, et al. Down regulation of hepcidin and haemojuvelin expression in the hepatocyte cell-line HepG2 induced by thalassaemic sera. Blood. 2006;108:447a. doi: 10.1111/j.1365-2141.2006.06258.x. [DOI] [PubMed] [Google Scholar]
- 15.Lin L, Goldberg YP, Ganz T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood. 2005;106:2884–2889. doi: 10.1182/blood-2005-05-1845. [DOI] [PubMed] [Google Scholar]
- 16.Zhang AS, Anderson SA, Meyers KR, et al. Evidence that inhibition of hemojuvelin shedding in response to iron is mediated through neogenin. J Biol Chem. 2007:M608788200. doi: 10.1074/jbc.M608788200. [DOI] [PubMed] [Google Scholar]
- 17.Sheldon JH. Haemochromatosis. Oxford University Press; London: 1935. [Google Scholar]
- 18.Davis WD, Arrowsmith WR. The effect of repeated phlebotomies in hemochromatosis. J Lab Clin Med. 1952;39:526–532. [PubMed] [Google Scholar]
- 19.Finch C. Iron metabolism in hemochromatosis. J Clin Invest. 1949;28:780. [Google Scholar]
- 20.Simon M, Pawlotsky Y, Bourel M, et al. Hémochromatose idiopathique: Maladie associée à l’antigène tissulaire. Nouv Presse Med. 1975;4:1432. [PubMed] [Google Scholar]
- 21.Simon M, Bourel R, Fauchet R, Genetet B. Association of HLA-A3 and HLA-B14 antigens with idiopathic hemochromatosis. Gut. 1976;17:332–334. doi: 10.1136/gut.17.5.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 23.Adams PC, Gregor JC, Kertesz AE, Valberg LS. Screening blood donors for hereditary hemochromatosis: Decision analysis model based on a 30-year database. Gastroenterology. 1995;109:177–188. doi: 10.1016/0016-5085(95)90283-x. [DOI] [PubMed] [Google Scholar]
- 24.Yapp TR, Eijkelkamp EJ, Powell LW. Population screening for HFE-associated haemochromatosis: should we have to pay for our genes? Internal Medicine Journal. 2001;31:48–52. doi: 10.1046/j.1445-5994.2001.00005.x. [DOI] [PubMed] [Google Scholar]
- 25.Allen K, Williamson R. Screening for hereditary haemochromatosis should be implemented now. BMJ. 2000;320:183–184. [PMC free article] [PubMed] [Google Scholar]
- 26.Adams P, Brissot P, Powell L. EASL International Consensus Conference on Haemochromatosis - Part II. Expert document. J Hepatol. 2000;33:487–496. doi: 10.1016/s0168-8278(01)80874-6. [DOI] [PubMed] [Google Scholar]
- 27.Beutler E, Felitti V, Gelbart T, Ho N. The effect of HFE genotypes in patients attending a health appraisal clinic. Ann Intern Med. 2000;133:329–337. doi: 10.7326/0003-4819-133-5-200009050-00008. [DOI] [PubMed] [Google Scholar]
- 28.Beutler E, Felitti VJ, Koziol JA, et al. Penetrance of the 845G->A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet. 2002;359:211–218. doi: 10.1016/S0140-6736(02)07447-0. [DOI] [PubMed] [Google Scholar]
- 29.Waalen J, Felitti V, Gelbart T, et al. Prevalence of hemochromatosis-related symptoms in homozygotes for the C282Y mutation of the HFE gene. Mayo Clin Proc. 2002;77:522–530. doi: 10.4065/77.6.522. [DOI] [PubMed] [Google Scholar]
- 30.Waalen J, Felitti VJ, Gelbart T, et al. Penetrance of hemochromatosis. Blood Cells Mol Dis. 2002;29:418–432. doi: 10.1006/bcmd.2002.0596. [DOI] [PubMed] [Google Scholar]
- 31.Beutler E. Disorders of iron metabolism. In: Lichtman MA, Beutler E, Kipps TJ, Seligsohn U, Kaushansky K, Prchal J, editors. Williams Hematology. McGraw-Hill; New York: 2006. pp. 511–553. [Google Scholar]
- 32.Cox T, Rochette J, Camaschella C, et al. Clinical haemochromatosis in HFE carriers. Lancet. 2002;360:412. doi: 10.1016/S0140-6736(02)09582-X. [DOI] [PubMed] [Google Scholar]
- 33.Allen KJ, Warner B, Delatycki MB. Clinical haemochromatosis in HFE carriers. Lancet. 2002;360:412–413. doi: 10.1016/s0140-6736(02)09583-1. [DOI] [PubMed] [Google Scholar]
- 34.Åsberg A, Hveem K, Thorstensen K, et al. Screening for hemochromatosis: High prevalence and low morbidity in an unselected population of 65,238 persons. Scand J Gastroenterol. 2001;36:1108–1115. doi: 10.1080/003655201750422747. [DOI] [PubMed] [Google Scholar]
- 35.Adams PC, Reboussin DM, Barton JC, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769–1778. doi: 10.1056/NEJMoa041534. [DOI] [PubMed] [Google Scholar]
- 36.Beutler E. Discrepancies between genotype and phenotype in hematology: an important frontier. Blood. 2001;98:2597–2602. doi: 10.1182/blood.v98.9.2597. [DOI] [PubMed] [Google Scholar]
- 37.Whitelaw E, Martin DI. Retrotransposons as epigenetic mediators of phenotypic variation in mammals. Nat Genet. 2001;27:361–365. doi: 10.1038/86850. [DOI] [PubMed] [Google Scholar]
- 38.MacDonald RA. Idiopathic hemochromatosis: A variant of portal cirrhosis and idiopathic hemosiderosis. Arch Intern Med. 1961;107:606–616. doi: 10.1001/archinte.1961.03620040132015. [DOI] [PubMed] [Google Scholar]
- 39.MacDonald RA. Idiopathic hemochromatosis. Genetic or acquired? Arch Intern Med. 1963;112:82–88. doi: 10.1001/archinte.1963.03860020082010. [DOI] [PubMed] [Google Scholar]
- 40.Milder MS, Cook JD, Stray S, Finch CA. Idiopathic hemochromatosis, an interim report. Medicine. 1980;59:34–49. doi: 10.1097/00005792-198001000-00002. [DOI] [PubMed] [Google Scholar]
- 41.Scotet V, Merour MC, Mercier AY, et al. Hereditary hemochromatosis: Effect of excessive alcohol consumption on disease expression in patients homozygous for the C282Y mutation. Am J Epidemiol. 2003;158:129–134. doi: 10.1093/aje/kwg123. [DOI] [PubMed] [Google Scholar]
- 42.Fletcher LM, Dixon JL, Purdie DM, et al. Excess alcohol greatly increases the prevalence of cirrhosis in hereditary hemochromatosis. Gastroenterology. 2002;122:281–289. doi: 10.1053/gast.2002.30992. [DOI] [PubMed] [Google Scholar]
- 43.Flanagan JM, Peng H, Beutler E. Effects of alcohol consumption on iron metabolism in mice with hemochromatosis mutations. Alcohol Clin Exp Res. 2006;31:138–143. doi: 10.1111/j.1530-0277.2006.00275.x. [DOI] [PubMed] [Google Scholar]
- 44.Harrison-Findik DD, Schafer D, Klein E, et al. Alcohol Metabolism-mediated Oxidative Stress Down-regulates Hepcidin Transcription and Leads to Increased Duodenal Iron Transporter Expression. J Biol Chem. 2006;281:22974–22982. doi: 10.1074/jbc.M602098200. [DOI] [PubMed] [Google Scholar]
- 45.Cade JE, Moreton JA, O’hara B, et al. Diet and genetic factors associated with iron status in middle-aged women. Am J Clin Nutr. 2005;82:813–820. doi: 10.1093/ajcn/82.4.813. [DOI] [PubMed] [Google Scholar]
- 46.van der DL, Peeters APH, Grobbee DE, et al. HFE genotypes and dietary heme iron: No evidence of strong gene-nutrient interaction on serum ferritin concentrations in middle -aged women. Nutr Metab Cardiovasc Dis. 2006;16:60–68. doi: 10.1016/j.numecd.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 47.Lee PL, Peng H, Gelbart T, Beutler E. The IL-6 and lipopolysaccharide-induced transcription of hepcidin in HFE, transferrin receptor-2, and β2 microglobulin deficient hepatocytes. Proc Natl Acad Sci USA. 2004;101:9263–9265. doi: 10.1073/pnas.0403108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beutler E, Waalen J, Gelbart T. Chronic inflammation does not appear to modify the homozygous hereditary hemochromatosis phenotype. Blood Cells Mol Dis. 2005;35:326–327. doi: 10.1016/j.bcmd.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 49.Dubois S, Kearney DJ. Iron-Deficiency Anemia and Helicobacter pylori Infection: A Review of the Evidence. Am J Gastroe nterol. 2005;100:453–459. doi: 10.1111/j.1572-0241.2005.30252.x. [DOI] [PubMed] [Google Scholar]
- 50.Hershko C. A hematologist’s view of unexplained iron deficiency anemia in males: impact of Helicobacter pylori eradication. Blood cells, molecules & diseases. 2007;38:45–53. doi: 10.1016/j.bcmd.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 51.Beutler E, Gelbart T. Helicobacter pylori infection and HFE hemochromatosis. Blood Cells Mol Dis. 2006;37:188–191. doi: 10.1016/j.bcmd.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 52.Beutler E, Gelbart T, Lee P. Haptoglobin polymorphism and iron homeostasis. Clin Chem. 2002;48:2232–2235. [PubMed] [Google Scholar]
- 53.Beutler E, Beutler L, Lee PL, Barton JC. The mitochondrial NT 16189 polymorphism and hereditary hemochromatosis. Blood Cells Mol Dis. 2004;33:344–345. doi: 10.1016/j.bcmd.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 54.Lee PL, Ho NJ, Olson R, Beutler E. The effect of transferrin polymorphisms on iron metabolism. Blood Cells Mol Dis. 1999;25:374–379. doi: 10.1006/bcmd.1999.0267. [DOI] [PubMed] [Google Scholar]
- 55.Lee PL, Halloran C, Beutler E. Polymorphisms in the transferrin 5′ flanking region associated with differences in total iron binding capacity: Possible implications in iron homeostasis. Blood Cells Mol Dis. 2001;27:539–548. doi: 10.1006/bcmd.2001.0418. [DOI] [PubMed] [Google Scholar]
- 56.Lee PL, Gelbart T, West C, et al. A study of genes that may modulate the expression of hereditary hemochromatosis: Transferrin receptor-1, ferroportin, ceruloplasmin, ferritin light and heavy chains, iron regulatory proteins (IRP)-1 and -2, and hepcidin. Blood Cells Mol Dis. 2001;27:783–802. doi: 10.1006/bcmd.2001.0445. [DOI] [PubMed] [Google Scholar]
- 57.Lee PL, Halloran C, West C, Beutler E. Mutation analysis of the transferrin receptor-2 gene in patients with iron overload. Blood Cells Mol Dis. 2001;27:285–289. doi: 10.1006/bcmd.2001.0381. [DOI] [PubMed] [Google Scholar]
- 58.Lee PL, Gelbart T, West C, et al. Seeking candidate mutations that affect iron homeostasis. Blood Cells Mol Dis. 2002;29:471–487. doi: 10.1006/bcmd.2002.0586. [DOI] [PubMed] [Google Scholar]
- 59.Krayenbuehl PA, Maly FE, Hersberger M, et al. Tumor Necrosis Factor-{alpha}-308G>A Polymorphism Modulates Iron Accumulation in Patients with Hereditary Hemochromatosis. Clin Chem. 2006 doi: 10.1373/clinchem.2005.065417. [DOI] [PubMed] [Google Scholar]
- 60.Jacolot S, Le Gac G, Scotet V, et al. HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype. Blood. 2004;103:2835–2840. doi: 10.1182/blood-2003-10-3366. [DOI] [PubMed] [Google Scholar]
- 61.Le Gac G, Scotet V, Ka C, et al. The recently identified type 2A juvenile haemochromatosis gene (HJV), a second candidate modifier of the C282Y homozygous phenotype. Hum Mol Genet. 2004;13:1913–1918. doi: 10.1093/hmg/ddh206. [DOI] [PubMed] [Google Scholar]
- 62.Livesey KJ, Wimhurst VL, Carter K, et al. The 16189 variant of mitochondrial DNA occurs more frequently in C282Y homozygotes with haemochromatosis than those without iron loading. J Med Genet. 2004;41:6–10. doi: 10.1136/jmg.2003.008805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valenti L, Conte D, Piperno A, et al. The mitochondrial superoxide dismutase A16V polymorphism in the cardiomyopathy associated with hereditary haemochromatosis. J Med Genet. 2004;41:946–950. doi: 10.1136/jmg.2004.019588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carter K, Bowen DJ, McCune CA, Worwood M. Haptoglobin type neither influences iron accumulation in normal subjects nor predicts clinical presentation in HFE C282Y haemochromatosis: phenotype and genotype analysis. Br J Haematol. 2003;122:326–332. doi: 10.1046/j.1365-2141.2003.04436.x. [DOI] [PubMed] [Google Scholar]
- 65.Distante S, Elmberg M, Haug KBF, et al. Tumour necrosis factor alpha and its promoter polymorphisms’ role in the phenotypic expression of hemochromatosis. Scand J Gastroenterol. 2003;38:871–877. doi: 10.1080/00365520310003444. [DOI] [PubMed] [Google Scholar]
- 66.Merryweather-Clarke AT, Cadet E, Bomford A, et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum Mol Genet. 2003;12:2241–2247. doi: 10.1093/hmg/ddg225. [DOI] [PubMed] [Google Scholar]
- 67.Delanghe JR, Langlois MR. Haptoglobin polymorphism and body iron stores. Clin Chem Lab Med. 2002;40:212–216. doi: 10.1515/CCLM.2002.035. [DOI] [PubMed] [Google Scholar]
- 68.Nicolas G, Andrews NC, Kahn A, Vaulont S. Hepcidin, a candidate modifier of the hemochromatosis phenotype in mice. Blood. 2004;103:2841–2843. doi: 10.1182/blood-2003-09-3358. [DOI] [PubMed] [Google Scholar]
- 69.Whiting P, Fletcher L, Dixon J, et al. Concordance of iron indices in homozygote and heterozygote sibling pairs in hemochromatosis families: implications for family screening. J Hepatol. 2002;37:309. doi: 10.1016/s0168-8278(02)00216-7. [DOI] [PubMed] [Google Scholar]
- 70.Niederau C, Fischer R, Sonnenberg A, et al. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N Engl J Med. 1985;313:1256–1262. doi: 10.1056/NEJM198511143132004. [DOI] [PubMed] [Google Scholar]
- 71.Niederau C, Fischer R, Puerschel A, et al. Long-term survival in patients with hereditary hemochromatosis. Gastroenterology. 1996;110:1107–1119. doi: 10.1053/gast.1996.v110.pm8613000. [DOI] [PubMed] [Google Scholar]
- 72.Waalen J, Nordestgaard BG, Beutler E. The penetrance of hereditary hemochromatosis. Best Pract Res Clin Haematol. 2005;18:203–220. doi: 10.1016/j.beha.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 73.Williams R, Smith PM, Spicer EJF, et al. Venesection therapy in idiopathic haemochromatosis. Q J Med. 1969;38:1–16. [PubMed] [Google Scholar]
- 74.Fischer R. Klinik und Therapie der idiopathischen Hämochromatose. Erfahrungen bei 51 eigenen F„llen. Leber Magen Darm. 1976;6:316–329. [PubMed] [Google Scholar]
- 75.Knauer CM, Gamble CN, Monroe LS. The reversal of hemochromatotic cirrhosis by multiple phlebotomies. Report of a case. Gastroenterology. 1965;49:667–671. [PubMed] [Google Scholar]
- 76.Falize L, Guillygomarc’h A, Perrin M, et al. Reversibility of hepatic fibrosis in treated genetic hemochromatosis: A study of 36 cases. Hepatology. 2006;44:472–477. doi: 10.1002/hep.21260. [DOI] [PubMed] [Google Scholar]
- 77.Morrison ED, Brandhagen DJ, Phatak PD, et al. Serum ferritin level predicts advanced hepatic fibrosis among U.S. patients with phenotypic hemochromatosis. Ann Intern Med. 2003;138:627–633. doi: 10.7326/0003-4819-138-8-200304150-00008. [DOI] [PubMed] [Google Scholar]
- 78.Beaton M, Guyader D, Deugnier Y, et al. Noninvasive prediction of cirrhosis in C282Y-linked hemochromatosis. Hepatology. 2002;36:673–678. doi: 10.1053/jhep.2002.35343. [DOI] [PubMed] [Google Scholar]
- 79.Guyader D, Jacquelinet C, Moirand R, et al. Noninvasive prediction of fibrosis in C282Y homozygous hemochromatosis. Gastroenterology. 1998;115:929–936. doi: 10.1016/s0016-5085(98)70265-3. [DOI] [PubMed] [Google Scholar]
- 80.Andersen RV, Tybjaerg-Hansen A, Appleyard M, et al. Hemochromatosis mutations in the general population: iron overload progression rate. Blood. 2004;103:2914–2919. doi: 10.1182/blood-2003-10-3564. [DOI] [PubMed] [Google Scholar]
- 81.Beutler E. Natural history of hemochromatosis. Mayo Clin Proc. 2004;79:305–306. doi: 10.4065/79.3.305. [DOI] [PubMed] [Google Scholar]
- 82.Olynyk JK, Hagan SE, Cullen DJ, et al. Evolution of untreated hereditary hemochromatosis in the Busselton population: a 17-year study. Mayo Clin Proc. 2004;79:309–313. doi: 10.4065/79.3.309. [DOI] [PubMed] [Google Scholar]
- 83.Bomford A, Williams R. Long term results of venesection therapy in idiopathic haemochromatosis. Q J Med. 1976;180:611–623. [PubMed] [Google Scholar]
- 84.Beaton M, Adams PC. Prognostic factors and survival in patients with hereditary hemochromatosis and cirrhosis. Can J Gastroenterol. 2006;20:257–260. doi: 10.1155/2006/428048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fracanzani AL, Conte D, Fraquelli M, et al. Increased cancer risk in a cohort of 230 patients with hereditary hemochromatosis in comparison to matched control patients with non-iron-related chronic liver disease. Hepatology. 2001;33:647–651. doi: 10.1053/jhep.2001.22506. [DOI] [PubMed] [Google Scholar]
- 86.Blumberg RS, Chopra S, Ibrahim R, et al. Primary hepatocellular carcinoma in idiopathic hemochromatosis after reversal of cirrhosis. Gastroenterology. 1988;95:1399–1402. doi: 10.1016/0016-5085(88)90379-4. [DOI] [PubMed] [Google Scholar]
- 87.Britto MRC, Thomas LA, Balaratnam N, et al. Hepatocellular carcinoma arising in non-cirrhotic liver in genetic haemochromatosis. Scand J Gastroenterol. 2000;35:889–893. doi: 10.1080/003655200750023282. [DOI] [PubMed] [Google Scholar]
- 88.Brage A, Tome S, Figueruela B, et al. Hepatoma in a 40-year old male with hereditary hemochromatosis in the absence of cirrhosis. Implications of molecular diagnosis. Rev Esp Enferm Dig. 2002;94:493–499. [PubMed] [Google Scholar]
- 89.Dale SP, Aithal GP, Thomas JA, et al. Risk of hepatocellular carcinoma associated with genetic haemochromatosis in the absence of cirrhosis: A retrospective cohort study. J Hepatol. 2006;44:S88. [Google Scholar]
- 90.Willis G, Bardsley V, Fellows IW, et al. Hepatocellular carcinoma and the penetrance of HFE C282Y mutations: a cross sectional study. BMC Gastroenterol. 2005;5:17. doi: 10.1186/1471-230X-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Powell LW, Dixon JL, Ramm GA, et al. Screening for hemochromatosis in asymptomatic subjects with or without a family history. Arch Intern Med. 2006;166:294–301. doi: 10.1001/archinte.166.3.294. [DOI] [PubMed] [Google Scholar]
- 92.Neufeld EJ. Oral chelators deferasirox and deferiprone for transfusional iron overload in thalassen a major: New data, new questions. Blood. 2006;107:3436–3441. doi: 10.1182/blood-2006-02-002394. [DOI] [PMC free article] [PubMed] [Google Scholar]