

Abstract
Cytoplasmic ATP inhibits human erythrocyte glucose transport protein (GLUT1)–mediated glucose transport in human red blood cells by reducing net glucose transport but not exchange glucose transport (Cloherty, E.K., D.L. Diamond, K.S. Heard, and A. Carruthers. 1996. Biochemistry. 35:13231–13239). We investigated the mechanism of ATP regulation of GLUT1 by identifying GLUT1 domains that undergo significant conformational change upon GLUT1–ATP interaction. ATP (but not GTP) protects GLUT1 against tryptic digestion. Immunoblot analysis indicates that ATP protection extends across multiple GLUT1 domains. Peptide-directed antibody binding to full-length GLUT1 is reduced by ATP at two specific locations: exofacial loop 7–8 and the cytoplasmic C terminus. C-terminal antibody binding to wild-type GLUT1 expressed in HEK cells is inhibited by ATP but binding of the same antibody to a GLUT1–GLUT4 chimera in which loop 6–7 of GLUT1 is substituted with loop 6–7 of GLUT4 is unaffected. ATP reduces GLUT1 lysine covalent modification by sulfo-NHS-LC-biotin by 40%. AMP is without effect on lysine accessibility but antagonizes ATP inhibition of lysine modification. Tandem electrospray ionization mass spectrometry analysis indicates that ATP reduces covalent modification of lysine residues 245, 255, 256, and 477, whereas labeling at lysine residues 225, 229, and 230 is unchanged. Exogenous, intracellular GLUT1 C-terminal peptide mimics ATP modulation of transport whereas C-terminal peptide-directed IgGs inhibit ATP modulation of glucose transport. These findings suggest that transport regulation involves ATP-dependent conformational changes in (or interactions between) the GLUT1 C terminus and the C-terminal half of GLUT1 cytoplasmic loop 6–7.
INTRODUCTION
Blood–tissue barriers, which comprise endothelial cells connected by tight junctions (Takata et al., 1997; Mann et al., 2003), protect the brain, peripheral nerve, myocardium, retina, olfactory epithelium, and the inner ear from the external environment. Metabolism in these tissues is fueled by glucose that is transported across the endothelial barrier via a transcellular mechanism mediated by the type I facilitative glucose transport protein, GLUT1 (Takata et al., 1997; Mann et al., 2003; Leybaert, 2005). GLUT1 is a prototypic member of the facilitative glucose transporter family (Joost et al., 2002) and of the wider major facilitator superfamily (MFS) of structurally and functionally related transport proteins (Saier et al., 1999; Saier, 2000).
GLUT1 is expressed at very high levels in endothelial cells and erythrocytes (Takata et al., 1997) where it displays substrate affinities, kinetic properties, and inhibitor pharmacodynamics that distinguish it from other GLUT facilitative sugar transporter members (Takakura et al., 1991). Glucose metabolism in erythrocytes and endothelial cells is not rate limited by transport because the glucose transport capacities of these cells greatly exceeds their glycolytic capacities (Jacquez, 1984; Gerritsen et al., 1988). In spite of this, GLUT1-mediated sugar transport displays acute and adaptive regulation in endothelial cells (Takata et al., 1997; Loaiza et al., 2003; Mann et al., 2003) and acute regulation in erythrocytes (Jung et al., 1971; Taverna and Langdon, 1973; Jacquez, 1983; Weiser et al., 1983; Levine et al., 2005) where cellular ATP depletion enhances GLUT1-mediated sugar import capacity (Carruthers and Zottola, 1996; Heard et al., 2000; Levine et al., 2002; Levine et al., 2005; Leitch and Carruthers, 2007). Acute responses occur within seconds to minutes and involve stimulation of existing cell surface glucose transporters (Diamond and Carruthers, 1993; Shetty et al., 1993; Cloherty et al., 1996). Adaptive responses occur over several hours in response to hypoxia and hypoglycemia and involve changes in glucose transporter expression (Mann et al., 2003).
GLUT1 is a nucleotide binding protein that, when complexed with ATP, displays reduced glucose import capacity but increased affinity for sugar (Carruthers and Helgerson, 1989; Levine et al., 1998; Levine et al., 2002). ATP modulation of GLUT1-mediated transport is competitively inhibited by AMP and ADP, but does not require ATP hydrolysis (Heard et al., 2000). Peptide mapping studies of azidoATP-labeled GLUT1 demonstrate that ATP interacts with GLUT1 residues 301–364 (Levine et al., 1998). This sequence spans transmembrane helices 8 and 9 (TM8 and TM9) and cytoplasmic loop 8–9. Residues 332–343 of this region display 50% sequence identity with a component of the adenylate kinase ATP binding pocket (Levine et al., 1998) and mutagenesis of key residues within this subdomain abolishes ATP modulation of transport (Levine et al., 2002). These observations suggest that nucleotide binding pocket minimally consists of L8–9 and a portion of TM9. Competitive antagonism of ATP regulation of transport by AMP eliminates the possibility that transport regulation is a simple consequence of nucleotide binding to GLUT1. Rather, regulation must involve nucleotide- induced GLUT1 conformational changes but the nature of these changes is unknown. Neither is it known whether the details of GLUT1 regulation are isoform specific or reflect a mechanism fundamental to all structurally related GLUT family members that extends to transport catalyzed by other structurally and functionally related MFS proteins. GLUT1 regulation appears to involve rapid changes in GLUT1 intrinsic activity while regulation of the insulin-sensitive transporter GLUT4 involves rapid redistributions of GLUT4 proteins between intracellular and cell surface membranes in addition to GLUT4 activation (Joost et al., 1986; Simpson and Cushman, 1986).
The results of the present study suggest that ATP binding to GLUT1 causes the GLUT1 carboxyl terminus to interact with GLUT1 cytoplasmic loop 6–7 in a sequence-specific fashion to inhibit transport.
MATERIALS AND METHODS
Materials
Fresh, de-identified human blood was purchased from Biological Specialties Corporation. Protein assays, Pro Blue coomassie stain, and Supersignal chemiluminescence kits were from Pierce Chemical Co. Nitrocellulose and Immobilon-P were purchased from Fisher Scientific. Purified rabbit IgGs raised against synthetic peptides corresponding to GLUT1 subdomains were obtained from Animal Pharm Services, Inc. These are N-Ab (GLUT1 residues 1–13); L2–3-Ab (GLUT1 residues 85–95); L6–7-Ab (GLUT1 residues 217–231); L7–8-Ab (GLUT1 residues 299–311); C-Ab (GLUT1 residues 480–492). All other reagents were purchased from Sigma-Aldrich.
Solutions
Saline comprises 150 mM NaCl, 10 mM Tris-HCl, and 0.5 mM EDTA, pH 7.4. Lysis medium contained 10 mM Tris-HCl and 0.2 mM EDTA, pH 7.2. Stripping solution contained 2 mM EDTA, 15.2 mM NaOH, pH 12. Tris medium contained 50 mM Tris-HCl, pH 7.4. Kaline consisted of 150 mM KCl, 5 mM HEPES, 4 mM EGTA, and 5 mM MgCl2. Ammonium bicarbonate was 0.5% (63 mM), pH 9.0. PBS containing Tween (PBS-T) comprised 140 mM NaCl, 10 mM Na2HPO4, 3.4 mM KCl, 1.84 mM KH2PO4, 0.1% Tween, pH 7.3. Stop solution comprises ice-cold Kaline plus cytochalasin B (CB) (10 μM) and phloretin (100 μM).
Red Cells and Red Cell Ghosts
Red cells were isolated by as described previously (Leitch and Carruthers, 2007). Red cell ghosts were formed by reversible hypotonic lysis of washed red cells (Leitch and Carruthers, 2007).
GLUT1 Purification
Glucose transporter (plus endogenous lipids) was purified from human erythrocyte membranes in the absence of reductant as described previously (Hebert and Carruthers, 1992). The resulting GLUT1 proteoliposomes contain (by protein mass) 90% GLUT1, 8% RhD protein, 2% nucleoside transporter (ENT1) and have a lipid:total protein mass ratio of 1:1 (Zottola et al., 1995). Experiments were restricted to the use of GLUT1 preparations in which the stoichiometry of proteoliposomal cytochalasin B binding is 0.48 ± 0.07 mol CB per mol nonreduced GLUT1 and 0.9 ± 0.1 mol CB per mol reduced GLUT1.
Sugar Transport Determinations
Transport in red cell ghosts was measured as described previously (Leitch and Carruthers, 2007).
Limited Proteolytic Digestion of Purified GLUT1
Purified GLUT1 (10 μg) was digested with a 20:1 (protein: enzyme) ratio of purified porcine trypsin (Princeton Separations) in 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 4 mM ATP (pH 7.5). Digestions were performed at 4°C for 60 min, or the indicated time period. Reactions were immediately loaded onto 15% SDS-PAGE.
Western Blotting
After SDS-PAGE, samples were transferred to nitrocellulose and blocked overnight in 25% nonfat dry milk/PBS-T. Primary antibody (N-Ab [1:200], L2–3-Ab [1:200], L6–7-Ab [1:500], L7–8-Ab [1:200], L8–9 [1:200], C-Ab [1:15,000], δ-Ab [1:1,000]) was incubated in 3% nonfat dry milk/PBS-T for 1 h at room temperature. Blots were washed three times in PBS-T and incubated with horseradish peroxidase (HRP)–conjugated goat anti-rabbit secondary antibody (1:5,000 dilution) at room temperature for 1 h. Blots were washed three times in PBS-T, developed using Pierce SuperSignal West Pico Chemiluminescent substrate and visualized by autoradiography.
ELISA
Purified GLUT1 (200 ng) in PBS was adsorbed to each well of the ELISA plate for 2 h at 37°C. Plates were blocked with PBS + 3% BSA for 2 h at 37°C. Primary antibody (N-Ab [1:200], L2–3-Ab [1:200], L6–7-Ab [1:500], L7–8-Ab [1:200], L8–9 [1:100 to 1:500], C-Ab [1:15,000], δ-Ab [1:1,000]) was added to each well in PBS + 0.1% BSA ± ATP and binding was allowed to proceed for 2 h. The plate was washed five times with PBS, and then each well was incubated with HRP-conjugated goat anti-rabbit secondary antibody in PBS + 0.1% BSA (1:5,000 dilution) for 1 h at 37°C. The plate was washed five times with PBS and wells were developed using 100 μl of 1-Step ABTS solution (Pierce Chemical Co.). Primary IgG binding was quantitated as absorbance at 415 nm.
Construction of the GLUT1–L6-7–GLUT4 Chimera
This chimera substitutes the middle loop (L6–7) with that of its rat GLUT4 counterpart and was constructed using a six-step PCR protocol. In PCR 1A, a HindIII primer complimentary to the 5′ end of human GLUT1 and a reverse primer complimentary to nucleotides 600–619 of GLUT1 and 667–685 of GLUT4 was used to generate a fragment containing TM 1–6 (nucleotides 1–620) of GLUT1. In a separate reaction, PCR1B, primer (TGCATCGTGCTGCCCTTCTGTCCTGAGAGCCCCCGA) containing sequence complimentary to nucleotides 600–619 of GLUT1 and 667–685 of GLUT4 (5′end of L6–7) and a reverse primer (containing a NotI restriction site) complimentary to the 3′ end of GLUT4 was used to generate a fragment containing the middle loop and TM 7–12 (nucleotides 667–1531) of GLUT4. In PCR2, the HindIII primer complimentary to the 5′ end of human GLUT1 and the reverse primer (containing a NotI restriction site) complimentary to the 3′ end of GLUT4 were used along with the products from PCR1A and B to generate an intermediate chimera containing sequence from nucleotides 1–620 of GLUT1 and 667–1531 of GLUT4. This intermediate chimera contained TM 1–6 of GLUT1 in frame with L6–7 and TM 7–12 of GLUT4. In PCR 3A, the HindIII primer complimentary to the 5′ end of human GLUT1 and a primer (CGCACCCAGGGGCAGCCTATCCTCATCGCTGTGGTC) complimentary to nucleotides 844–862 of GLUT4 and 815–829 of GLUT1 were used along with the product from PCR2 to generate a fragment containing TM 1–6 of GLUT1 (nucleotides 1–619) in frame with L6–7 (nucleotides 667–862) of GLUT4. In PCR 3B, a primer (CGCACCCAGGGGCAGCCTATCCTCATCGCTGTGGTC) complimentary to nucleotides 844–862 of GLUT4 and nucleotides 815–829 of GLUT1 were used with wild-type GLUT1 plasmid as a template to generate a fragment containing TM 7–12 (nucleotides 815–1480) of GLUT1. In the final reaction, PCR 4, HindIII primer complimentary to the 5′ end of human GLUT1 and the NotI primer complimentary to the 3′ end of GLUT1 were used with the products of PCR 3A and B as a template generating the final product, which contained TM 1–6 (nucleotides 1–619) of GLUT1, followed by L6–7 of GLUT4 (nucleotides 667–862), followed by TM 7–12 (nucleotides 815–1480) of GLUT1. This PCR product was digested with HindIII and NotI and ligated into the mammalian expression vector pcDNA 3.1+, cut with the same enzymes. Sequences were confirmed by sequencing and the chimera was transiently expressed in HEK 293 cells as described previously (Levine et al., 2005).
Modification of GLUT1 Lysine Residues by Sulfo-NHS-LC-Biotin
GLUT1 was covalently modified at accessible lysine residues (±ATP) using sulfosuccinimidyl-6-(biotinamido) hexanoate (sulfo-NHS- LC-biotin) as described previously (unpublished data). Detection of modified residues was achieved by limited proteolysis of labeled GLUT1 followed by RP-HPLC separation of fragments and ESI-MS/MS identification of peptides as described previously (unpublished data).
RESULTS
Human GLUT1 copurifies with red cell membrane lipids and is reconstituted into unsealed proteoliposomes upon detergent removal (Baldwin et al., 1979; Appleman and Lienhard, 1985; Sultzman and Carruthers, 1999). Although sealed proteoliposomes may be formed if exogenous lipid is added before detergent dialysis (Baldwin et al., 1982; Carruthers and Melchior, 1984; Zeidel et al., 1992), we employed purified human GLUT1 in unsealed proteoliposomes to ensure that added reagent has access to both surfaces of the lipid bilayer and thus full access to exo- and endofacial GLUT1 domains.
ATP Protects GLUT1 against Tryptic Digestion
Exposure of GLUT1 proteoliposomes to trypsin (62.5:1 by mass) at 37°C results in the rapid loss of intact GLUT1 as judged by silver stain detection of peptides resolved by 10% SDS-PAGE. The time course of GLUT1 proteolysis is characterized by a rapid burst phase of proteolysis in which ∼30% of GLUT1 is hydrolyzed with a τ of 4.5 s (Fig. 1 A). The remaining intact GLUT1 is proteolyzed with a τ of 2,500 s (see Fig. 1 B). Addition of ATP (0.1 to 4 mM always in the presence of 5 mM MgCl2) to the reaction reduces the size and rate of the proteolysis burst phase significantly (extent = 22%, τ = 56 s; Fig. 1 B). AMP, ADP, GTP, and CTP do not affect the rate or extent of trypsin-catalyzed GLUT1 proteolysis. ATP inhibits GLUT1 digestion at 60 s by 50% at 627 ± 268 μM ATP (Fig. 1 C).
Figure 1.

Kinetics of GLUT1 digestion by trypsin at 37°C. GLUT1 (10 μg) was incubated with trypsin (0.16 μg) ± 4 mM ATP (plus 5 mM MgCl2) for 60 s (A and B) or with 0 ATP, 4 mM ATP, or 4 mM GTP for the times indicated (B). Peptides were separated by SDS-PAGE and the fraction of intact GLUT1 remaining quantitated by densitometry of silver-stained gels. The bars to the left of the gel in A indicate the mobility of molecular weight standards (113, 92, 50.1, 35.4, 29, 21.5 kD from top to bottom). The curves drawn through the data points in B assume two exponential phases of proteolysis. The fast phase accounts for 33 ± 2% of GLUT1 proteolysis and has a first order rate constant of 0.055 ± 0.008 per second (control, •) or 0.049 ± 0.026 per second (GTP, ▵). The slow phase accounts for 66% of GLUT1 digestion and has a first order rate constant of 0.00027 ± 0.00007 per second (control, •) or 0.00026 ± 0.00022 per second (GTP, ▵). ATP (○) reduces the size of the fast phase to 22 ± 2% and slows the fast rate constant to 0.017 ± 0.003 per second. Insufficient data exists to analyze slow phase kinetics in the presence of ATP. Data are shown as mean ± SEM of five separate experiments. (C) ATP protection of GLUT1 during fast phase proteolysis is half-maximal at 627 ± 268 μM ATP. Data are shown as mean ± SEM of 3 separate experiments.
To evaluate which GLUT1 domains were most susceptible to proteolysis, we examined GLUT1 fragmentation patterns by immunoblot analysis of GLUT1 digests using a series of GLUT1 peptide-directed IgGs. These included N-Ab (directed against GLUT1 amino acids 1–13), L6–7-Ab (directed against GLUT1 residues 217– 231), L7–8-Ab (GLUT1 residues 299–311), and C-Ab (residues 480–492). ATP (4 mM) reduces the efficiency of GLUT1 digestion (100:1 GLUT1:trypsin by mass, 20°C, 30 min reaction) as detected by immunoblot using IgGs specific to each of these GLUT1 subdomains (Fig. 2 A). For example, the C-Ab panel indicates the absence of higher molecular weight C-Ab–reactive fragments when GLUT1 proteolysis proceeds in the absence of ATP. When ATP is included, however, both high and low molecular weight C-Ab–reactive fragments are visible and more abundant, suggesting a general slowing of proteolysis of both intact and partially cleaved GLUT1. To identify protected peptides, we aligned each GLUT1 peptide detected by immunoblot analysis to GLUT1 sequence using two criteria: (1) the fragment must contain the reactive epitope, and (2) the mass (electrophoretic mobility) of the fragment must be consistent with previously detected GLUT1 tryptic cleavage sites (unpublished data). The trypsin-accessible sites observed here are generally consistent with the surface accessibility maps produced by systematic cysteine scanning mutagenesis of GLUT1 (Mueckler and Makepeace, 2006).
Figure 2.

Immunoblot analysis of trypsin-digested, purified GLUT1 ± 4 mM ATP. The digest was analyzed by Western blot analysis using a panel of antibodies directed against specific GLUT1 domains: N-Ab (GLUT1 residues 1–13); L6–7-Ab (GLUT1 residues 217–231); L7–8-Ab (GLUT1 residues 299–311); and C-Ab (GLUT1 residues 480–492). (A) Immunoblot analysis of tryptic digests. The key indicates the presence (+) or absence (−) of trypsin (tryp) or ATP (4 mM). The bars to the left of each blot show the mobility of molecular weight standards (kD). The arrows to the right of each blot indicate peptides whose intensity is greater when digests are performed in the presence of ATP. (B) Linear representation of GLUT1. The open boxes show the locations of N-Ab, L6–7-Ab, L7–8-Ab, and C-Ab directed IgG binding domains. The vertical arrows indicate GLUT1 tryptic cleavage sites determined by MS/MS analysis of GLUT1 tryptic digests. The horizontal bars below indicate putative assignments of ATP-sensitive GLUT1 peptides detected by peptide-directed IgGs and their theoretical molecular weight (kD). (C) [ATP] dose response to trypsin digestion. The relative intensities of C-Ab–reactive 20 kD (•) and 25–27 kD (○) peptides increase with [ATP] during proteolysis. Peptide intensity (volume) was quantitated by densitometry of immunoblots using the Image J software package and plotted as a function of [ATP]. Curves were computed by nonlinear regression assuming that intensity increases with [ATP] in a simple, saturable fashion. 20 kD and 25–27 kD peptides are half maximally protected by ATP at (366 ± 202) and (440 ± 211) μM, respectively. This figure represents a single dose–response experiment.
Fig. 2 B summarizes potential alignments of protected peptides. GLUT1 is most susceptible to tryptic digestion at cytoplasmic loop 6–7 and at the C terminus. ATP protection is distributed across GLUT1 primary structure. ATP inhibition of trypsin-catalyzed GLUT1 proteolysis increases in a saturable manner with ATP concentration and is half maximal at ∼400 μM ATP (n = 1; Fig. 2 C).
ATP-dependent GLUT1 Conformational Changes
The results of the proteolysis experiments suggest that ATP promotes significant GLUT1 conformational changes. To determine whether this also occurs in intact GLUT1, we analyzed ATP-dependent changes in GLUT1 epitope accessibility by measuring peptide-directed IgG binding to membrane-resident GLUT1. GLUT1 proteoliposomes were adsorbed to ELISA wells and then incubated ± ATP and peptide-directed IgGs. As reported previously (Carruthers and Helgerson, 1989), ATP inhibits C-Ab binding to GLUT1 proteoliposomes (Fig. 3 A; Table I). This is also observed with C-Ab binding to unsealed red cell membranes (Fig. 3 B) and in membranes isolated from HEK cells heterologously expressing wild-type human GLUT1 (Fig. 3 C). When HEK293 cells are transfected with a GLUT1–GLUT4 chimera in which the GLUT1 large middle loop (L6–7) is replaced by equivalent GLUT4 sequence, ATP inhibition of C-Ab binding to the GLUT1 C terminus of the chimera is lost (Fig. 3 D; Table I). Analysis of equilibrium C-Ab binding to these membranes indicates that ATP significantly reduces C-Ab binding to purified GLUT1, red cell–resident GLUT1 and wtGLUT1 (P < 0.001) but not to the GLUT1–GLUT4 loop 6 chimera (P > 0.1). This chimera is expressed efficiently (Fig. 3 D) and reaches the cell surface where it facilitates 2-deoxy-d-glucose transport. Untransfected HEK cells are characterized by Vmax and Km(app) for 2-deoxy-d-glucose uptake at 30°C of 1.2 ± 0.1 pmol/μg cell protein/min and 3.6 ± 1.4 mM, respectively. HEK cells transfected with wild-type GLUT1 (1.6 μg DNA per 106 cells) show significantly greater 2-deoxy-d-glucose uptake and are characterized by Vmax and Km(app) of 29.3 ± 9.4 pmol/μg cell protein/min and 3.6 ± 1.4 mM, respectively. Cells transfected with the loop 6–7 GLUT1–GLUT4 chimera (1.6 μg DNA per 106 cells) are characterized by Vmax and Km(app) for 2- deoxy-d-glucose uptake of 21.6 ± 2.6 μmol/106 cells/min and 1.7 ± 0.7 mM, respectively.
Figure 3.

Time course of C-Ab binding to ELISA dish–immobilized GLUT1 proteoliposomes (A), red cell membranes (B), HEK cell membranes expressing GLUT (C), and HEK cell membranes expressing the GLUT1–GLUT4 loop 6 chimera in which GLUT1 L6–7 is substituted by GLUT4 L6–7 (D). Ordinate, extent of C-Ab binding (OD415); Abscissa, duration of C-Ab exposure to membranes (min). Filled circles (•) show C-Ab binding in the presence of ATP (4 mM), and open circles (○) show C-Ab binding in the absence of ATP. Results are the mean ± SEM of quadruplicate measurements. Each experiment was repeated three or more times (A–C) or twice (D). Open triangles show C-Ab binding to membranes isolated from untransfected HEK cells. Curves were calculated assuming a single exponential phase of IgG binding described by B (1 − e−kt), where B is equilibrium binding, k is the first order rate constant for binding, and t is time. The results are summarized in Table I. The inset of D shows a C-Ab immunoblot of HEK membranes (20 μg) isolated from untransfected cells (lane 1), cells transfected with wt GLUT1 (lane 3), and cells transfected with the GLUT1–GLUT4 loop 6 chimera (lane 2). The bars to the left of the blot indicate the mobility (top to bottom) of 108-, 90-, and 51-kD molecular weight standards.
TABLE I.
Effects of ATP on C-Ab Binding to GLUT1
| Equilibrium binding
|
k per min
|
||||
|---|---|---|---|---|---|
| [ATP] mM
|
[ATP] mM
|
||||
| Membranes | 0 | 4 | P | 0 | 4 |
| GLUT1 | 0.65 ± 0.012 | 0.46 ± 0.02 | <0.001 | 0.13 ± 0.06 | 0.12 ± 0.01 |
| RBC | 0.84 ± 0.13 | 0.49 ± 0.05 | <0.001 | 0.10 ± 0.03 | 0.17 ± 0.05 |
| wtGLUT1 | 0.67 ± 0.02 | 0.47 ± 0.03 | <0.001 | 0.11 ± 0.01 | 0.11 ± 0.01 |
| GLUT1–GLUT4 (L6) chimera | 0.63 ± 0.02 | 0.56 ± 0.04 | >0.10 | 0.14 ± 0.05 | 0.12 ± 0.01 |
Results from the curve fits of Fig. 3. C-Ab binding to GLUT1 is a simple exponential process characterized by a constant, B, that describes the extent of binding at equilibrium and by a rate constant k (per min). Binding was measured in the absence and presence of 4 mM ATP. Binding to GLUT1 was measured in GLUT1 proteoliposomes (GLUT1), in red cell membranes (RBC), in membranes isolated from HEK cells expressing wild-type GLUT1 (wtGLUT1), and in membranes isolated from HEK cells expressing the GLUT1–GLUT4 loop 6 chimera (GLUT1–GLUT4 (L6) chimera). P is a test of the hypothesis that binding in the absence of ATP is identical to binding in the presence of 4 mM ATP (t test of equilibrium binding obtained in three or more experiments). k is unaffected by ATP.
To understand whether this response is restricted to the GLUT1 C terminus or more widespread, we examined the available peptide-directed IgGs for ability to bind to intact GLUT1 and for sensitivity of binding to ATP (Table II). ATP does not affect binding of ∂-Ab, loop 2–3-Ab or loop 6–7-Ab to membrane-resident GLUT1 but does reduce loop 7–8-Ab and C-Ab binding to GLUT1 proteoliposomes. N-Ab and loop 8–9-Ab binding to native GLUT1 structure are undetectable, indicating that these epitopes are inaccessible in membrane-resident GLUT1.
TABLE II.
Effects of ATP on Peptide-directed IgG Binding to GLUT1
| GLUT1 IgG and domain | IgG binding by Western blot | IgG binding to proteoliposomes | Effect of ATP on IgG binding to proteoliposomes |
|---|---|---|---|
| N-Ab; 1–13 | Yes (1:200) | ND | NA |
| L2–3-Ab; 84–96 | Yes (1:200) | Yes | No effect |
| L6–7-Ab; 217–231 | Yes (1:500) | Yes | No effect |
| L7–8-Ab; 299–311 | Yes (1:200) | Yes | Inhibits |
| L8–9-Ab; 329–241 | Yes (1:200) | ND | NA |
| C-Ab; 480–492 | Yes (1:15,000) | Yes | Inhibits |
| Exofacial (δ-Ab) | Yes (1:1,000) | Yes | No effect |
Immune sera generated against synthetic peptides corresponding to the indicated GLUT1 amino acid residues were raised in rabbits (e.g., N-Ab is immune serum raised against GLUT1 amino acids 1–13). IgG binding was measured by immunoblot analysis of proteins resolved by SDS-PAGE (see Fig. 1) or by ELISA analysis of binding to GLUT1 proteoliposomes (see Fig. 2). The serum dilution used in immunoblot analyses is indicated in parenthesis. Yes indicates specific binding is detectable. ND indicates that binding is not detectable. The effect of 4 mM ATP on IgG binding was measured. Inhibits indicates that equilibrium binding is reduced by >25% (see Fig. 2). NA indicates that the experiment is not applicable because IgG binding to proteoliposomes is undetectable. Exofacial GLUT1 domain-reactive polyclonal IgGs (δ-Ab) were raised against nonreduced, native GLUT1. The reactive epitope is not known.
ATP-dependent Changes in Amino Acid Side Chain Accessibility
We assessed whether ATP-dependent conformational changes in loop 6–7 and the C terminus are reflected at the level of specific amino acid side chains by analysis of ATP modulation of sulfo-NHS-LC-biotin covalent modification of loop 6–7 and C terminus lysine residues. GLUT1 proteoliposomes were preincubated in the presence or absence of 4 mM ATP before addition of a 20-fold molar excess of sulfo-NHS-LC-biotin to initiate GLUT1 labeling. This ratio of probe to GLUT1 produces significant labeling of GLUT1 cytoplasmic loop lysine residues without affecting the ability of GLUT1 to bind cytochalasin B (unpublished data). ATP reduces the extent of GLUT1 modification by ∼40%, without slowing the reaction rate (Fig. 4 A). ATP inhibition of labeling is half maximal at 2 mM ATP (Fig. 4 B). AMP (0 to 4 mM) alters neither the rate nor the extent of GLUT1 modification but increases the amount of ATP required to inhibit GLUT1 modification. When the concentration dependence of ATP inhibition of GLUT1 modification is measured in the presence of 2 mM AMP, the effects of ATP are half maximal at 3.8 mM. Assuming AMP competitively antagonizes ATP inhibition of modification, Ki(app) for AMP antagonism of ATP modulation of GLUT1 is 2.2 mM (Fig. 4 B).
Figure 4.

Analysis of ATP modulation of lysine modification. (A) Time course of GLUT1 labeling by sulfo-NHS-LC-biotin. GLUT1 proteoliposomes were preincubated in buffer lacking (○) or containing 4 mM AMP (•) or 4 mM ATP (▪) before addition of sulfo-NHS-LC-biotin. Ordinate, extent of labeling (measured as OD415); abscissa, time in minutes. Results are shown as mean ± SEM of at least four determinations. Biotinylation kinetics follow pseudo-first order kinetics where labeling equals B0 + B∞(1 − e−kt). The curves have the following constants: buffer (○), B0 = 0.15 ± 0.02, B∞ = 1.09 ± 0.05, k = 0.0023 ± 0.003 per minute; AMP (•), B0 = 0.16 ± 0.01, B∞ = 1.20 ± 0.03, k = 0.0019 ± 0.0001 per minute; ATP (▪), B0 = 0.098 ± 0.009, B∞ = 0.66 ± 0.02, k = 0.0022 ± 0.0002 per minute. (A, inset) Average B∞ (±SEM) of at least three labeling experiments made in triplicate for control, 4 mM AMP, and 4 mM ATP conditions. (B) Effects of nucleotides on equilibrium GLUT1 biotinylation. Ordinate, extent of total biotin incorporation (shown as mean ± SEM; n = 3 or greater); abscissa, [AMP] or [ATP] (mM) present during labeling. The pseudo-first-order rate constant describing GLUT1 labeling by sulfo-NHS-LC-biotin is unaffected by nucleotides. The extent of labeling is not significantly affected by AMP alone (•). Assuming labeling is described by BC − BN[nucleotide]/(Ki + [nucleotide]), nonlinear regression analysis indicates that for labeling in the presence of ATP (▪), BC = 1.210 ± 0.007, BN = 0.72 ± 0.04, and Ki = 2.1 ± 0.1 mM. ATP inhibition of labeling was also measured in the presence of 2 mM AMP (▵), where BC = 0.9, BN = 0.6, and Ki = 3.8 ± 1.5 mM. AMP therefore anatagonizes ATP modulation of biotinylation with Ki(app) for AMP = 2.2 mM.
GLUT1 lysine residues whose accessibility to sulfo-NHS-LC-biotin is specifically affected by ATP were identified by ESI-MS/MS analysis of labeled GLUT1. After GLUT1 biotinylation in the presence of 4 mM AMP (control) or ATP and subsequent tryptic digestion/ESI-MS-MS, we identified all peptides originating from a specific GLUT1 region and quantitated each peak area. The peak areas of biotinylated peptides in a denoted region were summed and expressed as the fraction of all peptides (labeled and unlabeled) in that region. Fig. 5 shows a representative analysis of lysine 245 modification.
Figure 5.

K245 accessibility to biotin labeling is modified by ATP. GLUT1 was labeled to equilibrium by sulfo-NHS-LC-biotin in the presence of 4 mM AMP (A) or ATP (B), trypsin-digested, and soluble peptides examined by RP-HPLC-ESI-MS/MS. Two peptides containing K245 were isolated. The first (m/z = 1444.7 D; sequence R(232).GTADVTHDLQEMK(245).E) was unlabeled and cleaved at K245. The second (m/z = 2285.1 D; R(232).GTADVTHDLQEMKEESR(249).Q) was labeled at K245 (339.2 D adduct covalently attached to K245) and cleaved at R249. Peak areas were calculated for each peptide. (A) Labeling conducted in the presence of 4 mM AMP. Unlabeled peptide area (•) = 5.54 × 108; labeled peptide area (□) = 1.15 × 108; % modified peptide = 17.24%. (B) Labeling conducted in the presence of 4 mM ATP. Unlabeled peptide area (•) = 5.09 × 108, labeled peptide area (□) = 5.73 × 107, % modified peptide = 10.12%. Labeling of K245 is reduced by 41% in the presence of ATP. This is one of three representative experiments. Fig. 6 summarizes the extent of ATP inhibition of labeling of this and three additional peptides.
To simplify the analysis, cytoplasmic GLUT1 lysine residues were grouped into four regions based on sequence location (see Fig. 6): region 1 contains K225, K229, and K230; region 2 contains K245; region 3 comprises K255 and K256; region 4 contains K477. Regions 1, 2, and 3 are located in cytoplasmic loop-6–7. Region 4 lies in the GLUT1-C terminus.
Figure 6.

ATP protection of GLUT1 lysine residues. GLUT1 putative membrane-spanning topography (Salas-Burgos et al., 2004) is illustrated. Lysine residues modified by sulfo-NHS-LC-biotin (○) are indicated. This is representative of three separate experiments. Rectangles indicate cytoplasmic regions 1 (K225, K229, and K 230), 2 (K245), 3 (K255 and K 256), and 4 (K477). The extent (%) of ATP protection against lysine modification is indicated as a percentage (mean ± SEM of three separate experiments) adjacent to each region.
Region 1 shows no discernible change in the quantity of labeled peptide in the presence of ATP (Fig. 6). Regions 2, 3, and 4 show decreased lysine modification in the presence of ATP (Fig. 6). ATP reduces labeling of K245 of region 2 by 38% (see Fig. 5 A [AMP] and Fig. 5 B [ATP]). Similarly, region 3 displays a 24% reduction and labeling of K477 of region 4 shows a 26% decrease in the presence of ATP (Fig. 6). Overall, ATP reduces labeling of lysine residues found within the C-terminal half of L6–7 and the C terminus of GLUT1 by ∼30%.
Effects of the GLUT1 C Terminus on Sugar Transport
These findings reinforce the hypothesis that the GLUT1 C terminus and loop 6–7 undergo significant conformational change in the presence of cytoplasmic ATP. We asked, therefore, how complexation of the C terminus using intracellular C-Ab might impact ATP modulation of sugar transport. ATP increases the rate of sugar transport at subsaturating 3MG because at these sugar concentrations the rate of transport, v, is given by:
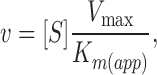 |
where [S] is the sugar concentration. ATP reduces Vmax and Km (app) for net 3MG uptake by red cell ghosts at 4°C by 8- and 18-fold, respectively (Helgerson et al., 1989), thereby increasing the ratio Vmax/ Km (app) and the rate of subsaturating transport by 2.3-fold (Table III).
TABLE III.
Effects of Intracellular C-terminal Peptide and Cytoplasmic Loop-directed IgGs on Red Cell Sugar Transport
| Experiment | Vmax/Km |
|---|---|
| No addition | 0.26 ± 0.02 |
| ATP | 0.53 ± 0.03 |
| 13 μM Ctp | 0.62 ± 0.05 |
| 13 μM Ctp + ATP | 1.04 ± 0.07 |
| 25 μM 1D4 | 0.30 ± 0.09 |
| 25 μM 1D4 + ATP | 0.64 ± 0.11 |
| Preimmune serum | 0.23 ± 0.03 |
| Preimmune serum + ATP | 0.60 ± 0.08 |
| C-Ab | 0.25 ± 0.04 |
| C-Ab + ATP | 0.38 ± 0.06 |
| L6–7-Ab | 0.20 ± 0.02 |
| L6–7-Ab + ATP | 0.55 ± 0.02 |
Experiments were made using resealed human erythrocyte ghosts containing or lacking 4 mM ATP and various other additions which include GLUT1-C-terminal peptide (Ctp-GLUT1 residues 480–492 at 13 μM); 1D4 (rhodopsin peptide ETQSVAPA at 25 μM); preimmune rabbit serum (4 mg/ml); C-Ab (20 μg/ml affinity purified IgG raised against GLUT1 Ctp) or L6 rabbit serum (immune serum obtained from rabbits immunized with GLUT1 peptide 217-231; 4 mg/ml). 3MG transport was measured at 4°C at 100 μM 3MG. The rate constant (per min) is equivalent to Vmax/Km. Results are shown as the mean ± SEM of two or four separate measurements made in duplicate or triplicate.
Incorporation of GLUT1 C-Ab into resealed erythrocyte ghosts severely blunts ATP stimulation of 3MG uptake at 4°C (Table III). Preimmune serum and loop 6–7-Ab are without effect on ATP action.
Exogenous, intracellular GLUT1 C-terminal peptide (EELFHPLGADSQV), but not 1D4 peptide (ETQSVAPA, a rhodopsin peptide), mimics the ability of ATP to stimulate sugar transport and acts synergistically with ATP (Table III). In the absence of ATP, C-terminal peptide triples the rate of 3MG uptake and is half maximally active at 9 μM.
DISCUSSION
This study examines the structural basis of GLUT1 modulation by cytoplasmic ATP and AMP. Our findings suggest that the GLUT1 cytoplasmic C terminus and the cytoplasmic loop linking TMs 6 and 7 interact in an ATP- dependent fashion to modulate transport.
ATP-dependent GLUT1 Functional Changes
Human GLUT1 responds acutely to cellular ATP depletion with enhanced sugar transport capacity (Jung et al., 1971; Taverna and Langdon, 1973; Weiser et al., 1983; Cloherty et al., 1996; Levine et al., 2002). This effect is direct (Hebert and Carruthers, 1986; Carruthers and Helgerson, 1989), does not require ATP hydrolysis, and is antagonized by intracellular H+, AMP, and ADP (Carruthers and Helgerson, 1989; Helgerson et al., 1989). GLUT1 residues 301–364 (TM8 through TM 9) form at least one element of the nucleotide binding domain (Levine et al., 1998) and site-directed mutagenesis of loop 8–9 residues (E329A or R332A/R333A) produces an ATP-insensitive, dominant-negative transporter in HEK cells (Levine et al., 2002). The molecular mechanism of GLUT1 modulation by ATP is unknown but may involve GLUT1 C terminus conformational change (Carruthers and Helgerson, 1989).
GLUT1 Structural Changes
Our results demonstrate that ATP–GLUT1 interactions promote global changes in GLUT1 conformation. ATP-dependent GLUT1 protection against trypsin digestion, ATP inhibition of IgG binding to GLUT1 subdomains, and ATP protection of specific cytoplasmic lysine residues against modification by NHS-biotin confirm that the ATP–GLUT1 complex is conformationally distinct from its ATP-free counterpart. ATP inhibits IgG binding to the C terminus and to exofacial loop 7–8 of membrane-resident transporter. ATP does not affect the extent of covalent modification of loop 6–7 lysine residues 225, 229, and 230 or binding of an antibody directed against residues 217–231, indicating that exposure of this flexible loop subdomain to cytoplasmic water is unchanged by ATP. This ATP-insensitive region contains a putative ATP binding motif (KSVLK; residues 225–229) that is also present in GLUT4 loop 6–7 (KSLK), lending further support to the hypothesis that this domain is not involved in ATP regulation of GLUT1. Labeling of loop 6–7 lysine residues 245, 255, and 256 is inhibited by ATP, and substitution of GLUT1 loop 6–7 with the equivalent GLUT4 domain greatly diminishes ATP inhibition of IgG binding to the chimera's GLUT1 C terminus. These observations suggest that GLUT1 requires sequence present in GLUT1 loop 6–7 in order to occlude the C terminus from exogenous IgG. Two of the three ATP-sensitive lysine residues of loop 6–7 are retained in GLUT4 loop 6–7 primary structure. This suggests that loop 6–7 ATP-protected lysine residues do not play a direct role in ATP regulation of transport (e.g., ATP binding or salt bridge formation with C-terminal residues). Rather, these residues may reflect altered conformational accessibility of loop 6–7.
AMP is without effect on GLUT1 loop 6–7 NHS-biotinylation but antagonizes ATP inhibition of labeling. AMP also antagonizes ATP inhibition of C-Ab binding to GLUT1 (Carruthers and Helgerson, 1989). This behavior confirms that the functional antagonism between ATP and AMP is replicated at the molecular level and that ATP modulation of GLUT1 conformation results in modulated transport function.
How Do ATP-dependent Structural Changes Influence Sugar Transport?
We propose that GLUT1–ATP association promotes conformational change within the cytoplasmic GLUT1 C terminus and loop 6–7, resulting in their interaction and alignment with sugar translocation pathway (Fig. 7). This alignment/interaction limits sugar release into cytoplasm and is supported by several observations. (a) Sequestration of the GLUT1 C terminus by exogenous intracellular C-terminal IgGs blunts ATP modulation of sugar transport. (b) Exogenous, intracellular C-terminal peptide mimics ATP action of sugar transport. (c) Substitution of GLUT1 loop 6–7 with GLUT4 loop 6–7 (40% sequence identity with GLUT1 over residues 249–259) prevents ATP inhibition of IgG binding to the C terminus.
Figure 7.

Model for ATP regulation of GLUT1. GLUT1 putative membrane-spanning topography (Salas-Burgos et al., 2004) is illustrated. The leftmost topography summarizes findings in the presence of AMP. Trypsin cleavage sites (K, ○; R, gray circle), sites of antibody recognition (gray rectangles), and sites where IgG binding is not detected (white rectangles) are indicated. In the presence of ATP (rightmost topography), ATP-sensitive (crosshatched rectangles) and insensitive (gray rectangles) IgG binding domains are indicated. The circles show ATP-insensitive tryptic cleavage sites (○), ATP-protected tryptic cleavage sites (gray circle), and ATP-protected sites of covalent modification by sulfo-NHS-LC-biotin (•). We propose that the GLUT1 C terminus and the C-terminal half of L6–7 respond to ATP binding by undergoing a conformational change that reduces their respective accessibility to polar reagents. This interaction restricts glucose release from the translocation pathway.
The action of ATP is thus extrinsic to the membrane-spanning translocation pathway and intrinsic to cytoplasmic domains. This hypothesis is further supported by rapid quench flow studies of erythrocyte glucose transport, demonstrating that rapid translocation of sugar through the sugar translocation pathway is unaffected by ATP, but sugar release from the pathway into cytosol is regulated by ATP (Blodgett and Carruthers, 2005; Leitch and Carruthers, 2007). Loop 6–7 and the C terminus may be necessary but not sufficient for GLUT1 regulation because GLUT1 loop 8–9 mutagenesis causes the loss of GLUT1 ATP sensitivity but retention of GLUT1 ATP binding (Levine et al., 2002).
Do Other Transporters Use a Similar Regulatory Mechanism?
The parallels between channel inactivation and inhibition of GLUT1 via cytosolic “inactivation domains” are striking. Inactivation of multisubunit, voltage-dependent K+ channels and large-conductance, Ca2+-activated K+ channels involves insertion of a cytosolic N-terminal peptide segment into the ion permeation pathway, thereby obstructing ion flux (Kobertz et al., 2000).
Less is known about the intrinsic regulation of other members of the GLUT family of proteins or whether the broader family of MFS proteins coopt a similar regulatory mechanism. MFS proteins contain loop 6–7 domains ranging from 30 to 65 amino acids (Weinglass and Kaback, 2000). The LacY and GlpT structures indicate that loop 6–7 and the C terminus cytoplasmic domains do not impede access to the substrate export site (Abramson et al., 2003; Huang et al., 2003). However, loop 6–7 and the C termini of these structures are considerably shorter than equivalent domains in the mammalian sugar porters. Progressive truncation of GLUT1 loop 6–7 results in loss of transport function followed by loss of GLUT1 expression (Monden et al., 2001). The former result may reflect the loss of residues crucial to transport. The latter may reflect impaired cotranslational insertion/folding (Weinglass and Kaback, 2000).
The available evidence suggests that the GLUT1 and GLUT4 C termini are essential for transport but contain isoform-specific subsequence that suppresses intrinsic GLUT activity. Truncation of the GLUT1 C terminus by 12 amino acids is without effect on GLUT1 activity (Lin et al., 1992), whereas removal of 37 residues abolishes GLUT1 intrinsic activity (Oka et al., 1990). Substitution of the GLUT1 C-terminal tail with the equivalent GLUT4 sequence stimulates GLUT1 intrinsic activity twofold but abolishes accelerated exchange transport (Dauterive et al., 1996). The C-terminal 20 residues of GLUT4 suppress GLUT4 intrinsic activity and deletion or replacement of this region with equivalent GLUT1 sequence increases GLUT4 activity fourfold (Dauterive et al., 1996). C-terminal truncation of the organic anion transporter OAT1 reduces OAT1 intrinsic activity (Xu et al., 2006). These observations support the hypothesis that the C-terminal domains of the GLUT sugar porter and the organo anion porter MFS subfamilies are essential for transport function and that GLUT1 and GLUT4 C termini contain isoform-specific subsequence that suppresses intrinsic GLUT activity. Insulin unmasks the GLUT4 C terminus in L6 muscle cells (Ishiki et al., 2005), adipocytes (Smith et al., 1991), and muscle (Wang et al., 1996) and desuppresses GLUT4 intrinsic activity (Michelle Furtado et al., 2003). The present study demonstrates that ATP depletion unmasks the GLUT1 C terminus, resulting in desuppression of GLUT1 intrinsic activity.
Relevance to GLUT1 Physiology
Sugar transport in astrocytes, vascular smooth muscle cells, basal cardiomyocytes, and cells of the reticuloendothelial system is mediated by GLUT1 (Hruz and Mueckler, 2001; Mann et al., 2003; Simpson et al., 2007). This may result in ATP-sensitive glucose transport in these tissues whereby cellular sugar transport capacity is increased in response to ATP depletion. What advantage is obtained by GLUT1 regulation in erythrocytes and endothelial cells where transport exceeds metabolic demand? The benefit may not be realized by the transporting cell but rather by the cells to which glucose is subsequently transferred.
Glucose transfer from blood to brain proceeds across capillary endothelial cells that form the blood–brain barrier (Takata et al., 1990). Endothelial cells constitute only 0.1% of brain mass yet almost all glucose used by cerebral neurons and astrocytes enters the brain by GLUT1-mediated, transendothelial cell transport (Harik, 1992). Cerebral endothelial cell GLUT1 content is adaptively up-regulated during long-term hypoglycemia and hypoxia to meet increased astrocytic and neuronal demand for glycolytic ATP (Mann et al., 2003). Blood–brain barrier glucose transport is acutely stimulated during seizure-induced stimulation of neuronal and astrocytic glycolysis (Cornford et al., 2000). Regulation of barrier GLUT1 is, therefore, an important physiologic adaptation and deficits in GLUT1 expression or hypoglycemia inevitably impact human cognitive function and development as observed in GLUT1-deficiency syndrome (Pascual et al., 2004) and hypoglycemia (Guettier and Gorden, 2006).
The very high GLUT1 content of higher primate and odontocete erythrocytes may contribute to glucose transfer from blood to tissue (Craik et al., 1998) by allowing human red cells to exchange upwards of 82–98% of intracellular glucose with serum in the 2–4 s required for an erythrocyte to transit the capillary bed of peripheral tissues (Honig et al., 1977; Regittnig et al., 2003). Regulation of erythrocyte glucose transport therefore may permit controlled expansion/contraction of the blood glucose space available for exchange with the interstitium of glycolytically active tissues.
Conclusions
GLUT1–ATP interaction restructures GLUT1 cytoplasmic loop 6–7 and C-terminal domains, resulting in their interaction and selective inhibition of sugar release from the transmembrane sugar translocation pathway into cytosol. This enables GLUT1 to respond to cytoplasmic energy charge with low or high capacity glucose transport. Interactions between cytoplasmic domains may be important in the regulation of other mammalian MFS transport proteins.
Acknowledgments
This work was supported by National Institutes of Health grants DK 36081 and DK 44888 and by ADA 1-06-IN-04 (Gail Patrick Innovation Award supported by a generous gift from the estate of Gail Patrick).
Lawrence G. Palmer served as editor.
D.M. Blodgett and J.K. De Zutter contributed equally to this work.
Abbreviations used in this paper: CB, cytochalasin B; GLUT1, human erythrocyte glucose transport protein; ESI-MS, electrospray ionization mass spectrometry; HRP, horseradish peroxidase; MFS, major facilitator superfamily; PBS-T, PBS containing Tween; sulfo-NHS-LC-biotin, sulfosuccinimidyl-6-(biotinamido) hexanoate.
References
- Abramson, J., I. Smirnova, V. Kasho, G. Verner, H.R. Kaback, and S. Iwata. 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science. 301:610–615. [DOI] [PubMed] [Google Scholar]
- Appleman, J.R., and G.E. Lienhard. 1985. Rapid kinetics of the glucose transporter from human erythrocytes. Detection and measurement of a half-turnover of the purified transporter. J. Biol. Chem. 260:4575–4578. [PubMed] [Google Scholar]
- Baldwin, S.A., J.M. Baldwin, F.R. Gorga, and G.E. Lienhard. 1979. Purification of the cytochalasin B binding component of the human erythrocyte monosaccharide transport system. Biochim. Biophys. Acta. 552:183–188. [DOI] [PubMed] [Google Scholar]
- Baldwin, S.A., J.M. Baldwin, and G.E. Lienhard. 1982. The monosaccharide transporter of the human erythrocyte. Characterization of an improved preparation. Biochemistry. 21:3836–3842. [DOI] [PubMed] [Google Scholar]
- Blodgett, D.M., and A. Carruthers. 2005. Quench-flow analysis reveals multiple phases of GluT1-mediated sugar transport. Biochemistry. 44:2650–2660. [DOI] [PubMed] [Google Scholar]
- Carruthers, A., and A.L. Helgerson. 1989. The human erythrocyte sugar transporter is also a nucleotide binding protein. Biochemistry. 28:8337–8346. [DOI] [PubMed] [Google Scholar]
- Carruthers, A., and D.L. Melchior. 1984. A rapid method of reconstituting human erythrocyte sugar transport proteins. Biochemistry. 23:2712–2718. [DOI] [PubMed] [Google Scholar]
- Carruthers, A., and R.J. Zottola. 1996. Erythrocyte sugar transport. In Handbook of Biological Physics. Transport Processes in Eukaryotic and Prokaryotic Organisms. Volume 2. W.N. Konings, H.R. Kaback, and J.S. Lolkema, editors. Elsevier, Amsterdam, Netherlands. 311–342.
- Cloherty, E.K., D.L. Diamond, K.S. Heard, and A. Carruthers. 1996. Regulation of GLUT1-mediated sugar transport by an antiport/uniport switch mechanism. Biochemistry. 35:13231–13239. [DOI] [PubMed] [Google Scholar]
- Cornford, E.M., E.V. Nguyen, and E.M. Landaw. 2000. Acute upregulation of blood-brain barrier glucose transporter activity in seizures. Am. J. Physiol. Heart Circ. Physiol. 279:H1346–H1354. [DOI] [PubMed] [Google Scholar]
- Craik, J.D., J.D. Young, and C.I. Cheeseman. 1998. GLUT-1 mediation of rapid glucose transport in dolphin (Tursiops truncatus) red blood cells. Am. J. Physiol. 274:R112–R119. [DOI] [PubMed] [Google Scholar]
- Dauterive, R., S. Laroux, R.C. Bunn, A. Chaisson, T. Sanson, and B.C. Reed. 1996. C-terminal mutations that alter the turnover number for 3-O-methylglucose transport by GLUT1 and GLUT4. J. Biol. Chem. 271:11414–11421. [DOI] [PubMed] [Google Scholar]
- Diamond, D., and A. Carruthers. 1993. Metabolic control of sugar transport by derepression of cell surface glucose transporters: an insulin-independent, recruitment-independent mechanism of regulation. J. Biol. Chem. 268:6437–6444. [PubMed] [Google Scholar]
- Gerritsen, M.E., T.M. Burke, and L.A. Allen. 1988. Glucose starvation is required for insulin stimulation of glucose uptake and metabolism in cultured microvascular endothelial cells. Microvasc. Res. 35:153–166. [DOI] [PubMed] [Google Scholar]
- Guettier, J.M., and P. Gorden. 2006. Hypoglycemia. Endocrinol. Metab. Clin. North Am. 35:753–766, viii–ix. [DOI] [PubMed] [Google Scholar]
- Harik, S.I. 1992. Changes in the glucose transporter of brain capillaries. Can. J. Physiol. Pharmacol. 70:S113–S117. [DOI] [PubMed] [Google Scholar]
- Heard, K.S., N. Fidyk, and A. Carruthers. 2000. ATP-dependent substrate occlusion by the human erythrocyte sugar transporter. Biochemistry. 39:3005–3014. [DOI] [PubMed] [Google Scholar]
- Hebert, D.N., and A. Carruthers. 1986. Direct evidence for ATP modulation of sugar transport in human erythrocyte ghosts. J. Biol. Chem. 261:10093–10099. [PubMed] [Google Scholar]
- Hebert, D.N., and A. Carruthers. 1992. Glucose transporter oligomeric structure determines transporter function. Reversible redox-dependent interconversions of tetrameric and dimeric GLUT1. J. Biol. Chem. 267:23829–23838. [PubMed] [Google Scholar]
- Helgerson, A.L., D.N. Hebert, S. Naderi, and A. Carruthers. 1989. Characterization of two independent modes of action of ATP on human erythrocyte sugar transport. Biochemistry. 28:6410–6417. [DOI] [PubMed] [Google Scholar]
- Honig, C.R., M.L. Feldstein, and J.L. Frierson. 1977. Capillary lengths, anastomoses, and estimated capillary transit times in skeletal muscle. Am. J. Physiol. 233:H122–H129. [DOI] [PubMed] [Google Scholar]
- Hruz, P.W., and M.M. Mueckler. 2001. Structural analysis of the GLUT1 facilitative glucose transporter (review). Mol. Membr. Biol. 18:183–193. [DOI] [PubMed] [Google Scholar]
- Huang, Y., M.J. Lemieux, J. Song, M. Auer, and D.N. Wang. 2003. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 301:616–620. [DOI] [PubMed] [Google Scholar]
- Ishiki, M., V.K. Randhawa, V. Poon, L. Jebailey, and A. Klip. 2005. Insulin regulates the membrane arrival, fusion, and C-terminal unmasking of glucose transporter-4 via distinct phosphoinositides. J. Biol. Chem. 280:28792–28802. [DOI] [PubMed] [Google Scholar]
- Jacquez, J.A. 1983. Modulation of glucose transport in human red blood cells by ATP. Biochim. Biophys. Acta. 727:367–378. [DOI] [PubMed] [Google Scholar]
- Jacquez, J.A. 1984. Red blood cell as glucose carrier: significance for placental and cerebral glucose transfer. Am. J. Physiol. 246:R289–R298. [DOI] [PubMed] [Google Scholar]
- Joost, H.G., T.M. Weber, S.W. Cushman, and I.A. Simpson. 1986. Insulin-stimulated glucose transport in rat adipose cells: modulation of transporter intrinsic activity by isoproterenol and adenosine. J. Biol. Chem. 261:10033–10036. [PubMed] [Google Scholar]
- Joost, H.G., G.I. Bell, J.D. Best, M.J. Birnbaum, M.J. Charron, Y.T. Chen, H. Doege, D.E. James, H.F. Lodish, K.H. Moley, et al. 2002. Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am. J. Physiol. Endocrinol. Metab. 282:E974–E976. [DOI] [PubMed] [Google Scholar]
- Jung, C.Y., L.M. Carlson, and D.A. Whaley. 1971. Glucose transport carrier activities in extensively washed human red cell ghosts. Biochim. Biophys. Acta. 241:613–627. [DOI] [PubMed] [Google Scholar]
- Kobertz, W.R., C. Williams, and C. Miller. 2000. Hanging gondola structure of the T1 domain in a voltage-gated K+ channel. Biochemistry. 39:10347–10352. [DOI] [PubMed] [Google Scholar]
- Leitch, J.M., and A. Carruthers. 2007. ATP-dependent sugar transport complexity in human erythrocytes. Am. J. Physiol. Cell Physiol. 292:C974–C986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, K.B., E.K. Cloherty, N.J. Fidyk, and A. Carruthers. 1998. Structural and physiologic determinants of human erythrocyte sugar transport regulation by adenosine triphosphate. Biochemistry. 37:12221–12232. [DOI] [PubMed] [Google Scholar]
- Levine, K.B., E.K. Cloherty, S. Hamill, and A. Carruthers. 2002. Molecular determinants of sugar transport regulation by ATP. Biochemistry. 41:12629–12638. [DOI] [PubMed] [Google Scholar]
- Levine, K.B., T.K. Robichaud, S. Hamill, L.A. Sultzman, and A. Carruthers. 2005. Properties of the human erythrocyte glucose transport protein are determined by cellular context. Biochemistry. 44:5606–5616. [DOI] [PubMed] [Google Scholar]
- Leybaert, L. 2005. Neurobarrier coupling in the brain: a partner of neurovascular and neurometabolic coupling? J. Cereb. Blood Flow Metab. 25:2–16. [DOI] [PubMed] [Google Scholar]
- Lin, J.L., T. Asano, H. Katagiri, K. Tsukuda, H. Ishihara, K. Inukai, Y. Yazaki, and Y. Oka. 1992. Deletion of C-terminal 12 amino acids of GLUT1 protein does not abolish the transport activity. Biochem. Biophys. Res. Commun. 184:865–870. [DOI] [PubMed] [Google Scholar]
- Loaiza, A., O.H. Porras, and L.F. Barros. 2003. Glutamate triggers rapid glucose transport stimulation in astrocytes as evidenced by real-time confocal microscopy. J. Neurosci. 23:7337–7342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann, G.E., D.L. Yudilevich, and L. Sobrevia. 2003. Regulation of amino acid and glucose transporters in endothelial and smooth muscle cells. Physiol. Rev. 83:183–252. [DOI] [PubMed] [Google Scholar]
- Michelle Furtado, L., V. Poon, and A. Klip. 2003. GLUT4 activation: thoughts on possible mechanisms. Acta Physiol. Scand. 178:287–296. [DOI] [PubMed] [Google Scholar]
- Monden, I., A. Olsowski, G. Krause, and K. Keller. 2001. The large cytoplasmic loop of the glucose transporter GLUT1 is an essential structural element for function. Biol. Chem. 382:1551–1558. [DOI] [PubMed] [Google Scholar]
- Mueckler, M., and C. Makepeace. 2006. Transmembrane segment 12 of the glut1 glucose transporter is an outer helix and is not directly involved in the transport mechanism. J. Biol. Chem. 281:36993–36998. [DOI] [PubMed] [Google Scholar]
- Oka, Y., T. Asano, Y. Shibasaki, J.L. Lin, K. Tsukuda, H. Katagiri, Y. Akanuma, and F. Takaku. 1990. C-terminal truncated glucose transporter is locked into an inward-facing form without transport activity. Nature. 345:550–553. [DOI] [PubMed] [Google Scholar]
- Pascual, J.M., D. Wang, B. Lecumberri, H. Yang, X. Mao, R. Yang, and D.C. De Vivo. 2004. GLUT1 deficiency and other glucose transporter diseases. Eur. J. Endocrinol. 150:627–633. [DOI] [PubMed] [Google Scholar]
- Regittnig, W., M. Ellmerer, G. Fauler, G. Sendlhofer, Z. Trajanoski, H.J. Leis, L. Schaupp, P. Wach, and T.R. Pieber. 2003. Assessment of transcapillary glucose exchange in human skeletal muscle and adipose tissue. Am. J. Physiol. Endocrinol. Metab. 285:E241–E251. [DOI] [PubMed] [Google Scholar]
- Saier, M.H., Jr. 2000. Families of transmembrane sugar transport proteins. Mol. Microbiol. 35:699–710. [DOI] [PubMed] [Google Scholar]
- Saier, M.H., Jr., J.T. Beatty, A. Goffeau, K.T. Harley, W.H. Heijne, S.C. Huang, D.L. Jack, P.S. Jahn, K. Lew, J. Liu, et al. 1999. The major facilitator superfamily. J. Mol. Microbiol. Biotechnol. 1:257–279. [PubMed] [Google Scholar]
- Salas-Burgos, A., P. Iserovich, F. Zuniga, J.C. Vera, and J. Fischbarg. 2004. Predicting the three-dimensional structure of the human facilitative glucose transporter glut1 by a novel evolutionary homology strategy: insights on the molecular mechanism of substrate migration, and binding sites for glucose and inhibitory molecules. Biophys. J. 87:2990–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty, M., J.N. Loeb, K. Vikstrom, and B.F. Ismail. 1993. Rapid activation of GLUT-1 glucose transporter following inhibition of oxidative phosphorylation in clone 9 cells. J. Biol. Chem. 268:17225–17232. [PubMed] [Google Scholar]
- Simpson, I., A. Carruthers, and S. Vanucci. 2007. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J. Cereb. Blood Flow Metab. 10.1038/sj.jcbfm.9600521 [DOI] [PMC free article] [PubMed]
- Simpson, I.A., and S.W. Cushman. 1986. Hormonal regulation of mammalian glucose transport. Annu. Rev. Biochem. 55:1059–1089. [DOI] [PubMed] [Google Scholar]
- Smith, R.M., M.J. Charron, N. Shah, H.F. Lodish, and L. Jarett. 1991. Immunoelectron microscopic demonstration of insulin-stimulated translocation of glucose transporters to the plasma membrane of isolated rat adipocytes and masking of the carboxyl-terminal epitope of intracellular GLUT4. Proc. Natl. Acad. Sci. USA. 88:6893–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultzman, L.A., and A. Carruthers. 1999. Stop-flow analysis of cooperative interactions between GLUT1 sugar import and export sites. Biochemistry. 38:6640–6650. [DOI] [PubMed] [Google Scholar]
- Takakura, Y., S.L. Kuentzel, T.J. Raub, A. Davies, S.A. Baldwin, and R.T. Borchardt. 1991. Hexose uptake in primary cultures of bovine brain microvessel endothelial cells. I. Basic characteristics and effects of D-glucose and insulin. Biochim. Biophys. Acta. 1070:1–10. [DOI] [PubMed] [Google Scholar]
- Takata, K., T. Kasahara, M. Kasahara, O. Ezaki, and H. Hirano. 1990. Erythrocyte/HepG2-type glucose transporter is concentrated in cells of blood-tissue barriers. Biochem. Biophys. Res. Commun. 173:67–73. [DOI] [PubMed] [Google Scholar]
- Takata, K., H. Hirano, and M. Kasahara. 1997. Transport of glucose across the blood-tissue barriers. Int. Rev. Cytol. 172:1–53. [DOI] [PubMed] [Google Scholar]
- Taverna, R.D., and R.G. Langdon. 1973. Glucose transport in white erythrocyte ghosts and membrane derived vesicles. Biochim. Biophys. Acta. 298:422–428. [DOI] [PubMed] [Google Scholar]
- Wang, W., P.A. Hansen, B.A. Marshall, J.O. Holloszy, and M. Mueckler. 1996. Insulin unmasks a COOH-terminal Glut4 epitope and increases glucose transport across T-tubules in skeletal muscle. J. Cell Biol. 135:415–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinglass, A.B., and H.R. Kaback. 2000. The central cytoplasmic loop of the major facilitator superfamily of transport proteins governs efficient membrane insertion. Proc. Natl. Acad. Sci. USA. 97:8938–8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser, M.B., M. Razin, and W.D. Stein. 1983. Kinetic tests of models for sugar transport in human erythrocytes and a comparison of fresh and cold–stored cells. Biochim. Biophys. Acta. 727:379–388. [DOI] [PubMed] [Google Scholar]
- Xu, W., K. Tanaka, A.Q. Sun, and G. You. 2006. Functional role of the C terminus of human organic anion transporter hOAT1. J. Biol. Chem. 281:31178–31183. [DOI] [PubMed] [Google Scholar]
- Zeidel, M.L., A. Albalak, E. Grossman, and A. Carruthers. 1992. Role of glucose carrier in human erythrocyte water permeability. Biochemistry. 31:589–596. [DOI] [PubMed] [Google Scholar]
- Zottola, R.J., E.K. Cloherty, P.E. Coderre, A. Hansen, D.N. Hebert, and A. Carruthers. 1995. Glucose transporter function is controlled by transporter oligomeric structure. A single, intramolecular disulfide promotes GLUT1 tetramerization. Biochemistry. 34:9734–9747. [DOI] [PubMed] [Google Scholar]