

Abstract
A reinvestigation of the thiazole constituents from Cacospongia mycofijiensis, collected in Vanuatu, yielded known mycothiazole (3) plus a new derivative, mycothiazole-4,19-diol (6). The E stereochemistry at Δ14,15 of 3 has been revised to Z and the structural features of 6 are elucidated. These compounds, which presumably arise by the action of a polyketide–nonribosomal peptide synthetase (PKS/NRPS) hybrid, possess cytotoxic properties that need further exploration.
We continue to be intrigued by the parallel structural patterns present in compounds arising by the action of polyketide–nonribosomal peptide synthetase (PKS/NRPS) hybrids.1 Of particular interest are sponge metabolites possessing a central thiazole core because some of these frameworks are closely related to those isolated from terrestrial myxobacteria. A striking example is provided by the metabolites of Cacospongia mycofijiensis2 (previously known as Spongia mycofijiensis) versus those produced by various strains of myxobacteria. Such relationships are illustrated in Figure 1 by the structure of latrunculin A (1),3 a sponge-derived actin inhibitor,4 shown side-by-side with that of epothilone B (2),5 a tubulin inhibitor6 produced by the myxobacteria Sorangium cellulosum. Another illustrative set possessing a thiazole core with 1,3-substitution consists of mycothiazole (3),7 of sponge origin, which we reported several years ago, as compared to myxobacteria-derived substances such as melithiazol C (4),8 from Melittangium lichenicola, and myxothiazol A (5),9 found in as many as five myxobacterial genera.
Figure 1.
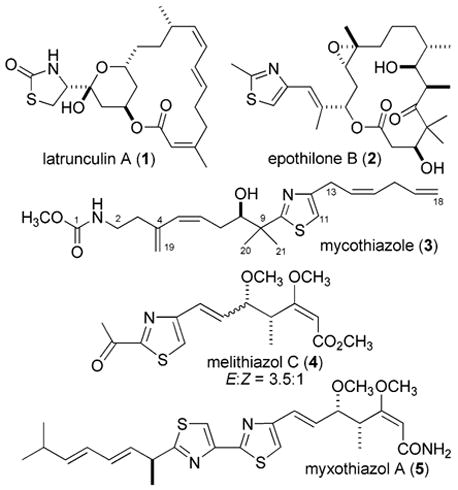
Compounds of PKS/NRPS origin having a disubstituted thiazole isolated from marine sponges and terrestrial myxobacteria.
While C. mycofijiensis contains 1 and 3, it is also a source of the fijianolides (syn. laulimalides),10 which are devoid of a thiazole functionality and of considerable current interest as microtubulin inhibitors.11 These three classes of compounds do not always co-occur, as we have discovered from ongoing study of C. mycofijiensis collected from Fiji, Vanuatu, Papua New Guinea, and Indonesia. One continuing goal is to use this variation to probe questions pertaining to their biosynthetic origin. Another goal has been to further exploit the biological properties of these compounds. We made a startling discovery during the course of scale-up isolation of fractions containing 3, which has a unique cytotoxicity profile12 and is reliably present in Vanuatu collections. After reexamining the NMR data of 3 it appeared that the E stereochemistry originally assigned at the C-14/C-15 double bond must be revised to Z. As discussed below, this finding explains discrepancies noted in comparing our natural to synthetic samples. A second discovery was that diol analogues of 3 were present as minor components in some extract fractions. Discussed below are the bases for the minor revision in structure of 3 plus the characterization of one of these new compounds, 6.
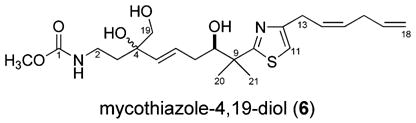
Subsequent investigations by others have scrutinized the original structure of mycothiazole (3). Isolation work on a Vanuatu sponge characterized as Dactylospongia sp. provided 3 as a major component, but its properties were not discussed.7b The first total synthesis of 3 defined the 8R absolute stereochemistry while also pointing out discrepancies among the optical rotation values and the 13C NMR chemical shifts between the synthetic and natural samples.13 A formal enantioselective synthesis14 and three additional partial syntheses15 of 3 were silent on any further comparisons. The key differences in 13C NMR shifts between the natural and synthetic samples occur at two allylic sites (see Table S1 for the full comparative data sets) and are C-13 (δ 34.7), C-16 (δ 36.6) for the synthetic sample13,14 versus C-13 (δ 29.4), C-16 (δ 31.5) for the natural product.7a These differences are consistent with an upfield shift expected at an allylic position for a disubstituted double bond in an E versus Z configuration.16 Thus, from this comparison it was clear that our original assignment of an E configuration at C-14/C-15 in 3 was incorrect. An NOE measurement on 3 provided definitive evidence for the revision to Z as shown here, and the results of Table 1 (spectra shown in Figures S4 and S5) substantiate that the synthetic samples and the natural products are indeed diastereomers. In the first NOE experiment (Table 1 “A”) irradiation of H-13 (δ 3.33) resulted in an enhancement at H-16 (δ 2.69) that was stronger than that to H-11 and H-14. The complementary experiment (Table 1 “B”) involving irradiation at H-16 gave parallel results. Finally, reevaluating the 3J14,15, originally estimated as 18 Hz but now remeasured as 10.7 Hz, clarified the source of the original assignment error. The 1H NMR spectrum of 3 measured at 300 MHz (benzene-d6, Figure S1) had H-15 (δ 5.62) as a complex multiplet. Alternatively, the enhanced resolution at 600 MHz (benzene-d6) allowed deciphering the J value by spectral simulation, and the couplings to the H-15 multiplet (centered at δ 5.50) are 10.7, 7.5, and 1.5 Hz.
Table 1.
| expt | atom irrad | NOE | rel enhanc inten |
|---|---|---|---|
| A | 13 | 11 | 1.3 |
| 14 | 1.0 | ||
| 16 | 2.4 | ||
| B | 16 | 11 | 1.0 |
| 13 | 6.6 | ||
| 15 | 3.1 | ||
| 17 | 1.5 | ||
| 18/18′ | 1.5 |
Relative enhanced intensity data based on setting the height of the weakest difference NOE peak as “1”.
Actual spectrum is shown in Figure S4.
Actual spectrum is shown in Figure S5.
The LCMS data collected during the reisolation of 3 was used to identify the other accompanying metabolites (see Figure S9) consisting of 1,3 fijianolides A and B,10a and polar fractions with m/z values 34 amu greater than 3. There were three such substances observed, and it was concluded that these were dihydrodiols of 3. Only one such compound, mycothiazole-4,19-diol (6), was purified in sufficient amounts for further characterization. Its molecular formula of C22H34N2O5S was established from the HRESIMS m/z 439.2271 [M + H]+ (Δ −0.84 mmu of calcd). The NMR properties of 6 versus 3 provided the reference point to focus on sites within this molecular structure that had been modified. Comparing the NMR data shown in Table 2 versus those of the literature7 indicated the following differences: (a) no 13C peaks were present in 6 for the C-4/C-19 double bond of 3, (b) a 4 ppm shift difference was observed for C-7, (c) a 6 ppm shift difference was seen at C-5, and (d) in 6 there were three resonances for the hydroxyl protons. Further confirmation of the three OH groups came from the δC’s 73.7 (C-4), 77.3 (C-8), and 68.8 (C-19), and their placement on the carbon skeleton was guided by 2D NMR data. These included a gHMBC correlation from H2-19 to C-5, along with gHMBC correlations shown in Figure 2 from OH-4 to C-3, C-4, C-5, and C-19, from H3-20/H3-21 to C-8, and from H-7 to C-8. The double-bond stereochemistry was deduced on the basis of both 1H J’s and 13C shifts. The resonances for vinyl protons H-5, H-6, H-14, and H-15 were clearly resolved (Figure S7), and the measured couplings 3J5,6) 16 Hz and 3J14,15) 10.7 Hz were diagnostic for the E and Z stereochemistry assigned, respectively. Two other observations were consistent with this conclusion and included (a) the 4 ppm downfield shift noted above at C-7 for 6 versus 3 and (b) the nearly identical shifts at C-13/C-14 for both compounds. The 8R configuration shown here for 6 is based on a biosynthetic analogy to 3, because additional experimental data could not be obtained due to compound instability.
Table 2.
NMR Dataa of Mycothiazole-4,19-diol (6) in DMSO-d6
| position | δC | δH mult, J (Hz) | gHMBCb |
|---|---|---|---|
| 1 | 156.6 | ||
| 2 | 36.1 | 2.98 m | |
| 3 | 36.7 | 1.55 m | |
| 4 | 73.7 | ||
| 5 | 135.0 | 5.35 bd, 16 | 4,7 |
| 6 | 129.8 | 5.62 m | 4 |
| 7 | 35.2 | 1.98 m | 5, 6, 8 |
| 1.85 m | |||
| 8 | 77.3 | 3.57 m | |
| 9 | 45.3 | ||
| 10 | 177.7 | ||
| 11 | 112.8 | 7.07 bs | 12, 13 |
| 12 | 154.0 | ||
| 13 | 29.2 | 3.45 d, 7.5 | 11, 12, 14, 15 |
| 14 | 127.6c | 5.69 dtt, 10.7, 7.3, 1.5 | |
| 15 | 127.9c | 5.51 dtt, 10.6, 7.3, 1.7 | |
| 16 | 31.3 | 2.87 dddt, 7.5, 6.0, 1.5, 2.0 | 14, 15, 17, 18 |
| 17 | 136.7 | 5.82 ddt, 16.8, 10.3, 6.2 | |
| 18 | 115.1 | 5.06 ddt, 16.9, 2.0, 1.8 | 16 |
| 4.98 ddt, 10.3, 2.0, 1.8 | |||
| 19 | 68.8 | 3.16 bt, 5 | 5 |
| 20 | 25.9d | 1.32 s | 8, 9, 10, 21 |
| 21 | 23.6d | 1.29 s | 8, 9, 10, 20 |
| O-Me | 51.1 | 3.48 s | 1 |
| NH | 6.87 bt, 5.5 | ||
| OH-4 | 4.27 d, 3.7 | 3, 4, 5, 19 | |
| OH-8 | 4.52 m | ||
| OH-19 | 4.81 bt, 6.3 |
500/125 MHz 1H/13C.
δH to δC.
Interchangeable assignments.
Figure 2.
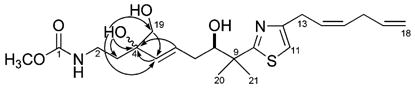
Selected gHMBC correlations for mycothiazole-4,19-diol (6).
From a biosynthetic perspective the structures of mycothiazole (3) and diol 6 are unique given their PKS/NRPS signature that includes a bisalkylated thiazole core. The only other example of this chemotype from sponge natural products chemistry is pateamine,17 which has a 1,3-substituted thiazole embedded in a macro-cyclic ring and is highly cytotoxic to P388 cancer cells. Efforts are continuing in our lab to further explore the biological properties of mycothiazole and its analogues with the goal of strengthening a case for their therapeutic potential. The NCI mean graph data12 are encouraging and indicate 3 is selective against several tumor lines such as DMS 114 (small cell lung cancer) and NCI-H23 (non-small cell lung cancer). In addition, the closest COMPARE analysis match in the NCI database is methotrexate (NSC 740 formerly amethopterin), an antimetabolite used clinically to treat certain cancers, severe psoriasis, and adult rheumatoid arthritis. The biological data obtained from a disk diffusion assay18 show potent in vitro murine solid tumor selectivity, because murine colon cells (C-38) are 3 times more sensitive to 3 versus murine leukemia cells (L1210). The next steps to clarify the therapeutic potential of mycothiazole (3) are underway and will be reported in due course.
Experimental Section
General Experimental Procedures
LCMS analysis was performed using an analytical 5 μm C18 ODS column with a photodiode array (PDA) detector along with an evaporative light scattering (ELS) detector for compound detection with an electrospray ionization time-of-flight (ESITOF) mass spectrometer for mass detection. Preparative HPLC was performed using a 6 μm C18 ODS column using an ELS detector. Semipreparative HPLC was performed with a 5 μm C18 ODS column using a single wavelength (λ = 254 or 230 nm) for compound detection. High-resolution MS was obtained using an ESITOF mass spectrometer.
Biological Material, Collection, and Identification
Specimens of Cacospongia mycofijiensis2 (coll. no. 02600) (3.7 kg wet weight) were collected using scuba in 2002 from Mele Bay, Vanuatu, at depths of 15–20 m. Taxonomic identification was based on comparison of the biological features to other voucher samples in our repository. The secondary metabolite chemistry is also consistent with this identification. Voucher specimens and underwater photos are available.
Extraction and Isolation
Samples were preserved in the field according to our standard laboratory procedures.19 The sponge was extracted 3× with methanol, and then the resultant oil was partitioned using a modified solvent partition scheme as described previously.20
Pure compounds were obtained as follows: (Figure S9) a portion of the 02600 FD was fractionated using preparatory reversed-phase HPLC to give 11 fractions. Preparatory fraction 9 (P9, 112 mg) was then separated using semipreparative reversed-phase HPLC to give eight fractions (P9, H1–H8). P9H8 (10.4 mg) contained pure mycothiazole (3). P9H4 (4.6 mg) was further fractionated to yield four fractions, one of which (P9H4H2, 2.6 mg) contained pure 6.
Mycothiazole (3): whitish powder; −13.7 (c 0.6, MeOH), −3.5 (c 0.6, CHCl3); 1H NMR (benzene-d6, 600 MHz) δ 6.31 (1H, s, H-11), 5.78 (1H, ddd, J ) 11.4, 1.8, 0.6 Hz, H-5), 5.72 (1H, ddt, J ) 16.9, 10.6, 6.4 Hz, H-17), 5.69 (1H, s, OH), 5.67 (1H, ddd, J ) 11.4, 9.0, 3.6 Hz, H-6), 5.62 (1H, dtt, J ) 10.8, 7.8, 1.2 Hz, H-14), 5.50 (1H, dtt, J ) 10.7, 7.5, 1.5 Hz, H-15), 5.36 (1H, m, NH), 5.02 (1H, dq, J ) 17.4, 1.8 Hz, H-18), 4.97 (1H, dq, J ) 10.2, 1.8 Hz, H-18′), 4.95 (1H, bs, H-19), 4.89 (1H, bs, H-19′), 3.79 (1H, dd, J ) 10.5, 2.1 Hz, H-8), 3.48 (1H, s, OCH3), 3.33 (2H, d, J ) 7.2 Hz, H-13), 3.28 (1H, dddd, J ) 13.8, 7.2, 6.6, 6.0 Hz, H-2), 3.11 (1H, dddd, J ) 13.8, 8.4, 6.0, 4.8 Hz, H-2′), 2.69 (2H, ddd, J ) 7.8, 6.6, 1.2 Hz, H-16), 2.56 (1H, dddd, J ) 14.4, 10.2, 9.0, 0.6 Hz, H-7), 2.30 (1H, dddd, J ) 13.8, 8.4, 6.0, 0.6 Hz, H-3), 2.20 (1H, dddd J ) 14.4, 3.6, 2.4, 1.8 Hz, H-7′), 2.02 (1H, dddd, J ) 13.8, 6.6, 4.8, 1.2 Hz, H-3′), 1.34 (3H, s, Me-20), 1.28 (3H, s, Me-21). Additional NMR data are in Figure S3 (CDCl3, 500 MHz) and in Figure S6.
Mycothiazole-4,19-diol (6): whitish powder; −2.5 (c 0.04, MeOH); HRESITOFMS m/z 439.2271 [M + H]+ (Δ −0.84 mmu of calcd for C22H34N2O5S); 1H NMR (DMSO-d6, 500 MHz), see Table 2 and Figure S7; 13C NMR (DMSO-d6,125 MHz), see Table 2 and Figure S8.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (R01 CA 47135), an equipment grant from NSF CHE-0342912 and NIH RR19918 (NMR), and a supplement to NIH CA 52955 (ESITOFMS).
Footnotes
Supporting Information Available: 1H and 13C NMR spectra of 3 and 6, simulated versus experimental 1H NMR spectra of 3 for H-15, selected 1H-1H NOE NMR irradiations of 3, the isolation scheme, and 13C comparison data table for synthetic 3 versus natural 3 and 6. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Cane DE. Chem Rev. 1997;97:2463–2464. doi: 10.1021/cr970097g. [DOI] [PubMed] [Google Scholar]; (b) Edwards DJ, Marquez BL, Nogle LM, McPhail K, Goeger DE, Roberts MA, Gerwick WH. Chem Biol. 2004;11:817–833. doi: 10.1016/j.chembiol.2004.03.030. [DOI] [PubMed] [Google Scholar]; (c) Sieber SA, Marahiel MA. Chem Rev. 2005;105:715–738. doi: 10.1021/cr0301191. [DOI] [PubMed] [Google Scholar]
- 2.Sanders ML, Van Soest RWM. Biologie. 1996;88:117–122. [Google Scholar]
- 3.Kashman Y, Groweiss A, Shmueli U. Tetrahedron Lett. 1980;21:3629–3632. [Google Scholar]; (b) Kakou Y, Crews P, Bakus GJ. J Nat Prod. 1987;50:482–484. [Google Scholar]
- 4.Spector I, Shochet NR, Kashman Y, Groweiss A. Science. 1983;219:493–495. doi: 10.1126/science.6681676. [DOI] [PubMed] [Google Scholar]; (b) Spector I, Shochet NR, Blasberger D, Kashman Y. Cell Motility Cytoskel. 1989;13:127–144. doi: 10.1002/cm.970130302. [DOI] [PubMed] [Google Scholar]; (c) Ayscough KR, Stryker J, Pokala N, Sanders M, Crews P, Drubin DG. J Cell Biol. 1997;137:399–416. doi: 10.1083/jcb.137.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Vilozny B, Amagata T, Mooberry SL, Crews P. J Nat Prod. 2004;67:1055–1057. doi: 10.1021/np0340753. [DOI] [PubMed] [Google Scholar]
- 5.Höfle G, Bedorf N, Steinmetz H, Schomburg D, Gerth K, Reichenbach H. Angew Chem, Int Ed Engl. 1996;35:1567–1569. [Google Scholar]
- 6.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 7.Crews P, Kakou Y, Quinoa E. J Am Chem Soc. 1988;110:4365–4368. [Google Scholar]; (b) Cutignano A, Bruno I, Bifulco G, Casapullo A, Debitus C, Gomez-Paloma L, Riccio R. Eur J Org Chem. 2001:775–778. doi: 10.1021/np010053+. [DOI] [PubMed] [Google Scholar]
- 8.Bohlendorf B, Herrmann M, Hecht HJ, Sasse F, Forche E, Kunze B, Reichenbach H, Hofle G. Eur J Org Chem. 1999:2601–2608. doi: 10.7164/antibiotics.52.721. [DOI] [PubMed] [Google Scholar]; (b) Weinig S, Hecht HJ, Mahmud T, Müller R. Chem Biol. 2003;10:939–952. doi: 10.1016/j.chembiol.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 9.Trowitzsch W, Höfle G, Sheldrick WS. Tetrahedron Lett. 1981;22:3829–3832. [Google Scholar]; (b) Silakowski B, Schairer HU, Ehret H, Kunze B, Weinig S, Nordsiek G, Brandt P, Blöcker H, Höfle G, Beyer S, Müller R. J Biol Chem. 1999;274:37391–37399. doi: 10.1074/jbc.274.52.37391. [DOI] [PubMed] [Google Scholar]; (c) Trowitzsch W, Höfle G, Sheldrick WS. Tetrahedron Lett. 2000;56:1681–1684. [Google Scholar]
- 10.Quinoa E, Kakou Y, Crews P. J Org Chem. 1988;53:3642–3644. [Google Scholar]; (b) Corley DG, Herb R, Moore RE, Scheuer PJ, Paul VJ. J Org Chem. 1988;53:3644–3646. [Google Scholar]
- 11.Mooberry SL, Randall-Hlubek DA, Leal RM, Hegde SG, Hubbard RD, Zhang L, Wender PA. Proc Natl Acad Sci USA. 2004;101:8803–8808. doi: 10.1073/pnas.0402759101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gapud EJ, Bai R, Ghosh AK, Hamel E. Mol Pharmacol. 2004;66:133–121. doi: 10.1124/mol.66.1.113. [DOI] [PubMed] [Google Scholar]; (c) Gallagher BM, Jr, Zhao H, Pesant M, Fang FG. Tetrahedron Lett. 2005;46:923–926. [Google Scholar]
- 12.The NIH NSC number of 3 is 647640, and the NCI data set is shown in Figure S10.
- 13.Sugiyama H, Yokokawa F, Shioiri T. Org Lett. 2000;2:2149–2152. doi: 10.1021/ol000128l. [DOI] [PubMed] [Google Scholar]; (b) Sugiyama H, Yokokawa F, Shioiri T. Tetrahedron. 2003;59:6579–6593. [Google Scholar]
- 14.Flohic A, Meyer C, Cossy J. Org Lett. 2005;7:339–342. doi: 10.1021/ol047603q. [DOI] [PubMed] [Google Scholar]
- 15.Serra G, Mahler G, Manta E. Heterocycles. 1998;48:2035–2048. [Google Scholar]; (b) Rodriguez-Conesa S, Paloma C, Jiménez C, Rodríguez J. Tetrahedron Lett. 2001;42:6699–6702. [Google Scholar]; (c) Mahler G, Serra G, Manta E. Synth Commun. 2005;35:1481–1492. [Google Scholar]
- 16.Crews P, Rodríguez J, Jaspars MJ. Organic Structure Analysis. Oxford University Press; New York: 1998. [Google Scholar]
- 17.Northcote PT, Blunt JW, Munro MHG. Tetrahedron Lett. 1991;32:6411–6414. [Google Scholar]
- 18.Bioassay procedures are described in: Valeriote F, Grieshaber CK, Media J, Pietraszkewicz H, Hoffmann J, Pan M, McLaughlin S. J Exp Ther Oncol. 2002;2:228–236. doi: 10.1046/j.1359-4117.2002.01038.x.
- 19.Sperry S, Valeriote FA, Corbett TH, Crews P. J Nat Prod. 1998;61:241–247. doi: 10.1021/np970467w. [DOI] [PubMed] [Google Scholar]
- 20.Thale Z, Johnson T, Tenney K, Wenzel PJ, Lobkovsky E, Clardy J, Media J, Pietraszkiewicz H, Valeriote FA, Crews P. J Org Chem. 2002;67:9384–9391. doi: 10.1021/jo026459o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.