

Abstract
Fluorescence anisotropy has been used to monitor the effect of ligands on a mobile loop over the active site of tyrosine hydroxylase. Phe184 in the center of the loop was mutated to tryptophan, and the three native tryptophan residues were mutated to phenylalanine to form an enzyme with a single tryptophan residue in the mobile loop. The addition of 6-methyl-5-deazatetrahydropterin to the enzyme resulted in a significant increase in the fluorescence anisotropy. The addition of phenylalanine did not result in a significant change in the anisotropy in the presence or absence of the deazapterin. The Kd value for the deazapterin was unaffected by the presence of phenylalanine. Qualitatively similar results were obtained with apoenzyme, except that the addition of phenylalanine led to a slight decrease in anisotropy. Frequency-domain lifetime measurements showed that the distribution of lifetimes was unaffected by both the amino acid and deazapterin. Frequency-domain anisotropy analyses were consistent with a decrease in the motion of the sole tryptophan in the presence of the deazapterin. This could be modeled as a decrease in the cone angle for the indole ring of about 12°. The data are consistent with a model in which binding of a tetrahydropterin results in a change in the conformation of the surface loop required for proper formation of the amino acid binding site.
The aromatic amino acid hydroxylases make up a small family of non-heme iron monooxygenases that catalyze physiologically essential reactions (1, 2). Phenylalanine hydroxylase (PheH)1 in the liver catalyzes the first step in the catabolism of excess phenylalanine in the diet to tyrosine. Tyrosine hydroxylase (TyrH), present in the brain and adrenal gland, catalyzes the rate-limiting step in the biosynthesis of the catecholamine neurotransmitters, the hydroxylation of tyrosine to form dihydroxyphenylalanine (DOPA). Tryptophan hydroxylase in the brain and the gastric system (3) catalyzes the rate-limiting step in the formation of the neurotransmitter serotonin, the hydroxylation of tryptophan to 5-hydroxytryptophan. All three enzymes use tetrahydrobiopterin as the physiological source of the electrons necessary for the monooxygenation reaction. A variety of structural studies have established that each eukaryotic enzyme is composed of diverse N-terminal regulatory domains and homologous catalytic and C-terminal tetramerization domains (4-9). The catalytic domains contain all of the residues required for catalysis and substrate specificity (10, 11). In light of the homology of the catalytic domains and the similarities of the reactions catalyzed by the three enzymes, it is generally assumed that the catalytic mechanisms are the same. This has allowed for mechanistic studies with all three enzymes to be used to develop a common catalytic mechanism (2). While the order of substrate addition is not definitively established for all three, a tetrahydropterin and an amino acid must be bound before any reaction with oxygen occurs in each case (12). The subsequent chemical reactions occur in two stages: the formation of a highly reactive hydroxylating intermediate, proposed to be an FeIVO species (2), and subsequent hydroxylation of the side chain of the amino acid.
Only in the case of rat TyrH has the steady-state mechanism of a eukaryotic member of this family been described (13). In this case, the tetrahydropterin binds first, followed by oxygen and then tyrosine. The lack of a reaction between oxygen and the tetrahydropterin in the absence of an amino acid substrate has led to the proposal that a conformational change occurs upon binding of an amino acid substrate to the enzyme–tetrahydropterin complex (14). Such a conformational change would prevent the formation of the highly reactive FeIVO intermediate in the absence of an appropriate substrate. Recent structures of the catalytic domain of PheH containing the iron in the catalytically active ferrous form have provided evidence for such a structural change upon binding of the amino acid. No significant difference can be observed in the overall fold of the protein in the presence and absence of tetrahydrobiopterin, although there are some changes in the degree of disorder of two water molecules and the glutamate that act as ligands to the iron (15). In contrast, the structure of PheH with both tetrahydrobiopterin and an amino acid bound to the FeII enzyme exhibits significant differences from the structure with only pterin bound (16). The most dramatic is the movement of a surface loop containing residues 132–148. The hydroxyl group of Tyr138 moves 21 Å, from a surface position to the active site, where it packs against residues that make up the amino acid binding site. Structural studies of TyrH and TrpH have not provided direct evidence for a similar structural change in these enzymes. Structures are available for the dihydropterin complexes of the ferric forms of the catalytic domains of both enzymes, but none is available of a structure with an amino acid substrate bound. In the case of TyrH, no significant structural differences are detectable between the enzyme with no substrate bound (17) and that with dihydrobiopterin alone (18). However, the residues in the center of the mobile loop seen in PheH are missing from these structures. In the structure of TrpH with dihydropterin bound, the position of the loop is closer to that seen in the presence of the amino acid in PheH than in its absence (19).
The available structures of PheH imply that this surface loop moves upon binding the amino acid substrate and, thus, may act as the critical conformational change gating oxygen activation. While the mechanistic studies suggest that all three aromatic amino acid hydroxylases have similar mechanisms, the limited structural information for TyrH and TrpH does not address the effects of substrate binding on the protein conformation. Residues 178–193 of TyrH correspond to PheH residues 132–148. Alanine-scanning mutagenesis of these residues in TyrH has established that they are indeed important for catalysis; mutagenesis of residues 180–189 perturbs both the kcat value and the Km value for tyrosine, while having little effect on the Km value for the tetrahydropterin (20). We describe here steady-state and time-resolved fluorescence anisotropy experiments designed to probe more directly the effects of substrates on the conformation of these residues in TyrH.
EXPERIMENTAL PROCEDURES
Materials
Custom oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA). Restriction endonucleases were from New England Biolabs, Inc. (Beverly, MA). Pfu DNA polymerase was obtained from Stratagene USA (La Jolla, CA). 6-Methyltetrahydropterin (6MePH4) was purchased from B. Schircks Laboratories (Jona, Switzerland). Leupeptin and pepstatin were obtained from Peptides International (Louisville, KY). Catalase was obtained from Roche (Gaithersburg, MD). Distilled glycerol was from Invitrogen (Carlsbad, CA). All other reagents were of the highest purity commercially available.
6-Methyl-5-deazatetrahydropterin (6M5DPH4) was synthesized essentially as described by Moad et al. (21) but was purified by ion-exchange chromatography to eliminate a fluorescent contaminant present in the trifluoroacetate salt. A solution of 4.7 mmol (1.0 g) of 6-methyl-5-deazapterin in 12.0 mL of trifluoroacetic acid was hydrogenated in the presence of 110 mg of platinum dioxide under 45 psi of hydrogen. After 3.5 h, the catalyst was removed by filtration. The filtrate was then neutralized with saturated NaHCO3 and washed with ethyl acetate. The aqueous solution was loaded onto a Dowex 50W column and eluted with 0.01 M HCl. 6M5DPH4 was finally recrystallized using 50% methanol–water. 1H NMR [dimethyl sulfoxide (DMSO)] δ: 6.24 (d, 1H, J = 2.1 Hz), δ 2.38 (m, 1H, J = 2.3 Hz), δ 2.73–3.09 (dd, 2H, J = 2.8 Hz), δ 1.79 (d, 2H, J = 2.7 Hz), δ 0.94 (m, 3H). m/e = 181.0 (M+).
Cloning and Purification of Mutant Enzymes
Site-directed mutagenesis was done using the QuikChange site-directed mutagenesis method (Stratagene). Plasmids were purified using kits from Qiagen, Inc. (Valencia, CA). DNA sequencing of the mutated plasmids was performed using the BigDye kit of ABI (Foster City, CA). Mutant proteins were expressed in Escherichia coli BL21 Star (DE3) cells as previously described for wild-type rat TyrH (11); a 5 mL Hitrap–Heparin column was used in place of the Heparin–Sepharose column. The protein samples were dialyzed against 50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) at pH 7.0, 10% glycerol, and 75 μM diethylenetriaminepentaacetic acid prior to being loaded onto the Hitrap–Heparin column; this was eluted with a linear gradient of 0–0.8 M NaCl in 75 μM diethylenetriaminepentaacetic acid, 10% glycerol, and 50 mM HEPES at pH 7.0. Fractions containing TyrH were pooled and precipitated by making the solution 65% saturated in ammonium sulfate. The pellet was resuspended in 100 mM KCl, 10% glycerol, 1 μM pepstatin, 1 μM leupeptin, and 50 mM HEPES at pH 7.0 to give a concentration of ∼300 μM TyrH. To obtain an enzyme containing one iron atom per subunit, ferrous ammonium sulfate was added to the purified enzyme as previously described (22). To obtain apoenzyme, the purified protein was treated with ethylenediaminetetraacetic acid (EDTA) as previously described (23) The final stoichiometry of iron/enzyme was determined by atomic absorption spectroscopy (24). All of the enzymes used in this study contained 0.9–1.2 equiv of iron for the holoenzyme or less than 0.2 equiv for the apoenzyme. The purified enzymes were stored at −80 °C.
Enzyme Assays
The concentration of TyrH was determined using a value for A2800.1% of 1.04 and a molecular weight of 56 000 (25). A colorimetric end point assay was used to measure DOPA formation (13); standard conditions were ∼0.5 μM enzyme, 400 μM 6MePH4, 100 μM tyrosine, 100 μg/mL catalase, 1 mM dithiothreitol (DTT), 10 μM ferrous ammonium sulfate, and 50 mM HEPES at pH 7.0 and 25 °C. For determination of the Km value for tyrosine, assays contained 400 μM 6MePH4 and 10–360 μM tyrosine. For determination of the Km value for 6MePH4, assays contained 100 μM tyrosine and 10–360 μM 6MePH4. Rates of formation of tyrosine from phenylalanine were measured by monitoring the increase in absorbance at 275 nm (26); the conditions were ∼0.4 μM enzyme, 200 μM 6MePH4, 60 μg/mL catalase, 10 μM ferrous ammonium sulfate, 5 mM DTT, and 25 mM EPPS at pH 7.0, with concentrations of phenylalanine from 0.5 to 12.6 mM. To obtain steady-state kinetic parameters, initial rate data were fit to the Michaelis–Menten equation using KaleidaGraph (Synergy Software). Inhibition data for 6M5DPH4 were fit to the equation for the competitive inhibitor using Igor Pro (WaveMetrics, Inc.).
Steady-State Fluorescence Measurements
Steady-state fluorescence intensity and anisotropy were measured on an ISS Koala Model fluorometer equipped with a xenon arc lamp (27). Light of 300 nm wavelength was passed through a Glan–Thompson vertical polarizer, and the fluorescence between 310 and 500 nm was analyzed. For anisotropy measurements, emission intensities were collected through a 2 mm thick Schott WG-345 cut-on filter and a Glan–Thompson polarizer. The inner-filter effect because of 6M5DPH4 was corrected with a factor obtained by titrating 6M5DPH4 against N-acetyl-l-tryptophanamide (NATA).2 All fluorescence studies were performed in 25 mM EPPS at pH 7.0 with a buffer blank correction. A protein concentration of ∼3 μM was used for all steady-state fluorescence measurements. Changes in the steady-state anisotropy as a function of the ligand concentration were fit to eq 1
| (1) |
where r is the anisotropy at a given ligand concentration, rmax is the anisotropy at saturating ligand concentrations, L is the ligand concentration, and Kd is the dissociation constant.
Frequency-Domain Fluorescence Measurements
Frequency-domain measurements were performed on an ISS K2 multi-frequency fluorometer equipped with digital acquisition electronics and fast Fourier transform data processing capability (27). The vertically polarized 300 nm line from a Spectra Physics model 2045 argon ion laser was selected by passing through a Melles Griot-FIU004 filter, which removes the 275 nm line produced by a deep-UV mode. The exciting light was modulated with a pockell cell, and the modulated emission was passed through a Glan–Thompson polarizer. For lifetime measurements, the emission was detected with the polarizer oriented at 54.7° to the vertical axis to avoid polarization artifacts (28). NATA, with a lifetime of 2.85 ns, in 0.1 M phosphate buffer at pH 7 was used as a reference sample. Phase shift and modulation data for each sample relative to NATA were obtained at each excitation frequency. For frequency-domain anisotropy measurements, the emission was observed through the polarizer, which was rotated between vertical (parallel) and horizontal (perpendicular) orientations. The differential phase shift was obtained from the difference between the perpendicular and parallel components of the emission, and the modulation ratio was obtained from the ratio of the parallel to the perpendicular components of the modulated emission as a function of the frequency. Samples contained ∼12 μM TyrH in 25 mM EPPS at pH 7.0. Data analysis was performed using Globals Unlimited software from the Laboratory for Fluorescence Dynamics, University of Illinois.
RESULTS
Cloning and Characterization of Tryptophan Mutants
Tyr138 is in the middle of a mobile loop in PheH that shows a large displacement when both a tetrahydropterin and an amino acid substrate are bound (16). The residue corresponding to Tyr138 in TyrH, Phe184, was mutated to tryptophan to generate a fluorescent probe whose anisotropy could be monitored. TyrH contains tryptophan residues at positions 166, 233, and 372. To avoid interference from the intrinsic fluorescence of these residues, they were sequentially mutated to phenylalanine. The resulting protein, W166F/F184W/W233F/W372F TyrH (F3W TyrH), has a single tryptophan residue at position 184 in the mobile loop. All of the mutant proteins expressed well in E. coli and could be purified using the protocol developed for the wild-type enzyme. The steady-state kinetic parameters with tyrosine and 6MePH4 as substrates were measured for all of the mutant enzymes and are summarized in Table 1. While some of the mutant enzymes that do not contain all four mutations exhibit perturbations in both Km and kcat values, in the case of the F3W enzyme, the Km value for tyrosine and the kcat value are similar to the wild-type values, while the Km value for the tetrahydropterin increases only about 3-fold. This suggests that catalysis and binding are not significantly perturbed in this mutant protein.
Table 1.
Characterization of TyrH Mutant Enzymesa
| enzyme |
Ktyrb (μM) |
K6MePH4c (μM) |
kcat (min−1) |
|---|---|---|---|
| wild-type TyrH | 80 ± 18 | 45 ± 4 | 182 ± 26 |
| W372F | 8 ± 1 | 45 ± 5 | 44 ± 3 |
| W166F/W372F | 90 ± 4 | 180 ± 18 | 243 ± 5 |
| W166F/W233F/W372F | 90 ± 6 | 162 ± 15 | 276 ± 7 |
| F184W/W372F | 17 ± 5 | 31 ± 4 | 64 ± 6 |
| W166F/F184W/W372F | 65 ± 5 | 129 ± 4 | 184 ± 5 |
| F3W | 64 ± 6 | 116 ± 11 | 165 ± 5 |
Conditions: 0.2 μM enzyme, 100 μg/mL catalase, 50 mM HEPES at pH 7.0, 10 μM ferrous ammonium sulfate, and 1 mM DTT at 25 °C.
6MePH4 (380 μM) with varied concentrations of tyrosine.
Tyrosine (100 μM) with varied concentrations of 6MePH4.
It was not possible to carry out the fluorescence anisotropy experiments with the normal substrates for TyrH, tyrosine and tetrahydropterin. Tyrosine itself is fluorescent, precluding its use. However, phenylalanine is a relatively good substrate for wild-type TyrH, with a V/K value about 15% that for tyrosine (29). Phenylalanine is also a reasonably good substrate for F3W TyrH, with a Km value of 0.90 ± 0.16 mM and a kcat value of 55 ± 3 min−1 (results not shown), and, consequently, was used to analyze the effects of amino acid binding. An alternative to tetrahydropterin was also required, both to prevent interference with the fluorescence of the tryptophan residue and to prevent turnover. However, dihydropterins, while used as pterin analogues in several of the structure determinations because they are not reactive, are also fluorescent. Instead, 6-methyl-5-deazatetrahydropterin was used as a ligand. This compound has previously been described as a tetrahydropterin analogue; the replacement of N(5) with carbon renders the deazatetrahydropterin airstable (21). Moreover, 6M5DPH4 has been shown to act as an inhibitor of TyrH (30). In the case of F3W TyrH, 6M5DPH4 has a Ki value of 140 ± 19 μM versus 6MPH4 as the varied substrate (results not shown), comparable to the value for the wild-type enzyme.
Steady-State Fluorescence Experiments
Steady-state fluorescence anisotropy provides a method for monitoring the movement of an intrinsic fluorophore attached to a larger protein. The mobility of a fluorophore buried in the active site of a protein is expected to be much lower than that of one on a mobile surface loop. This decrease in mobility should result in greater anisotropy. The intrinsic tryptophan fluorescence emission of F3W TyrH was determined in the presence and absence of ligands. The fluorescence intensity of the enzyme in the presence of either phenylalanine or 6M5DPH4 is not significantly different from that in the absence of these ligands (results not shown). The steady-state fluorescence anisotropy of the single tryptophan in the F3W TyrH was measured next. The average anisotropy of the enzyme in the absence of ligands is 0.177 ± 0.001. A small increase in the anisotropy is seen in the presence of 10 mM phenylalanine (Figure 1);3 while the precision of this change is low in a single experiment, it is reproducible. A much larger increase in anisotropy is observed with 6M5DPH4 alone than with phenylalanine alone (Figure 1).4 When both phenylalanine and 6M5DPH4 are added, the increase in anisotropy is comparable to that seen with 6M5DPH4 alone. The increase in anisotropy seen in the presence of 6M5DPH4 is concentration-dependent (Figure 2), yielding a Kd value of 218 ± 85 μM for 6M5DPH4 in the absence of phenylalanine and 239 ± 60 μM in its presence. These Kd values agree reasonably well with the Ki value determined from steady-state kinetics.
Figure 1.
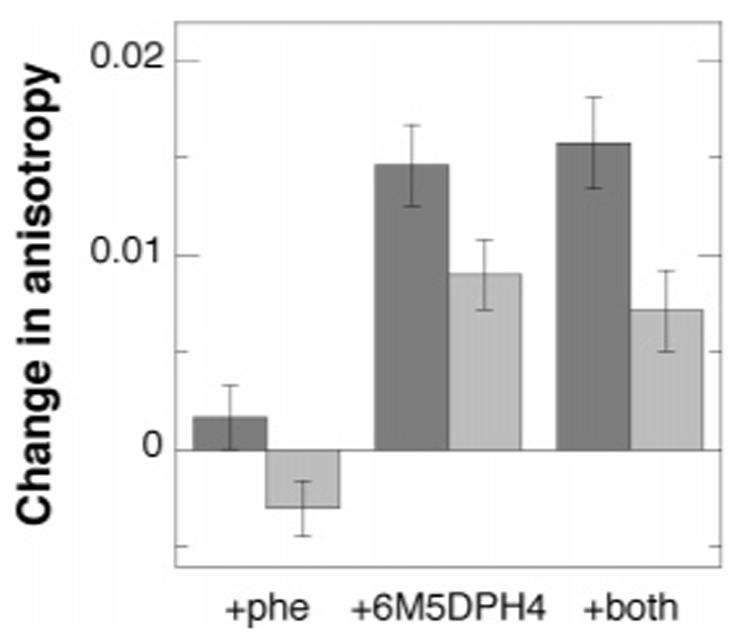
Effects of ligands and iron on the steady-state fluorescence anisotropy of F3W TyrH: dark gray, holoenzyme; light gray, apoenzyme. The samples contained 10 mM phenylalanine and/or 500 μM 6M5DPH4, as indicated, in 25 mM EPPS buffer at pH 7.
Figure 2.
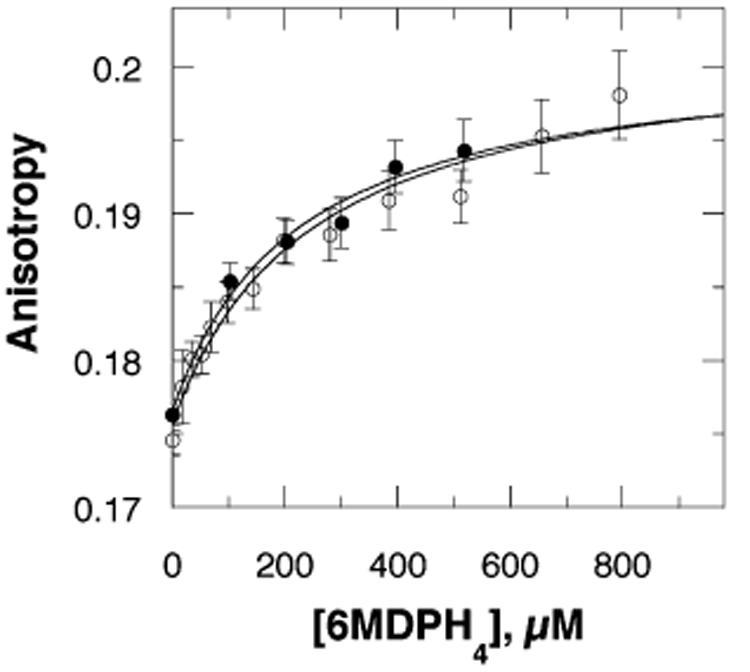
Concentration dependence of the effect of 6M5DPH4 on the steady-state fluorescence anisotropy of F3W TyrH in the absence (●) or presence (○) of 10 mM phenylalanine in 25 mM EPPS buffer at pH 7 and 25 °C. The lines are from fits of the data to eq 1.
The magnitude of the anisotropy change also depends upon whether the enzyme contains iron (Figure 1). In case of the apoenzyme, the average anisotropy is consistently less than that of the holoenzyme, 0.157 ± 0.001. The addition of phenylalanine to the apoenzyme results in a small decrease in the anisotropy. In contrast, the addition of 6M5DPH4 increases the anisotropy, while the presence of both ligands increases it somewhat less. In both cases, the magnitudes of the increases are less than with the holoenzyme.
Frequency-Domain Lifetime Measurements
As shown in eq 2
| (2) |
a change in anisotropy (r) from the initial value (r0) could occur because of a change in either the fluorescence lifetime (τ) or the rotational correlation time (θ) of the fluorophore (31). The frequency-domain method (32) was used to determine the lifetime of the single tryptophan residue in F3W TyrH, measuring the changes in the phase and modulation of the fluorescence emission as a function of the frequency of the modulated excitation. The phase and modulation of the samples at each frequency of excitation were determined in the presence and absence of both ligands (Figure 3). The lifetimes of tryptophan residues are frequently analyzed as sums of exponential decays. However, as discussed by Alcala et al. (33, 34), an often more useful alternative is to analyze the decay behavior as a continuous distribution of lifetimes, characterized by a central lifetime C and a width W. Consequently, the present data were analyzed as a Gaussian distribution of lifetimes using eq 3 (35)
| (3) |
The data fit well to the model, yielding the numerical results in Table 2. The central lifetime, around 4.1–4.4 ns, changes very little upon binding the ligands. Therefore, the change in steady-state anisotropy upon 6M5DPH4 binding can be attributed to a change in the rotational characteristics of the sole tryptophan rather than its lifetime.
Figure 3.
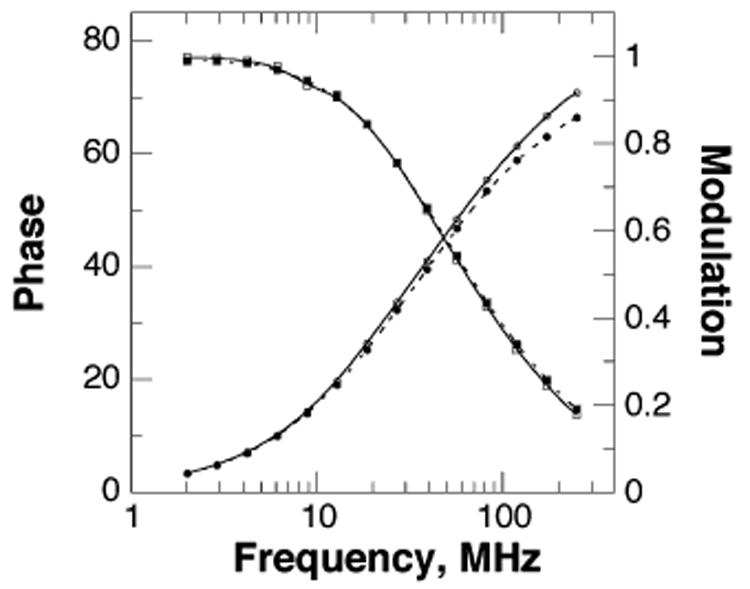
Variation of the phase (○ and ●) and modulation (□ and ■) with frequency for F3W TyrH alone (○ and □) or in the presence of 10 mM phenylalanine and 500 μM 6M5DPH4 (● and ■). The lines represent the best fit of the data to a Gaussian distribution of lifetimes.
Table 2.
Effects of Ligands on the Fluorescence Lifetimes of the Tryptophan in F3W TyrH
| additions | C (ns)a | W (ns)a |
|---|---|---|
| none | 4.47 ± 0.04 | 2.53 ± 0.06 |
| phenylalanine (10 mM) | 4.24 ± 0.04 | 2.81 ± 0.06 |
| 6-methyl-5-deaza-tetrahydropterin (500 μM) | 4.38 ± 0.04 | 2.90 ± 0.06 |
| 6-methyl-5-deaza-tetrahydropterin (500 μM) plus phenylalanine (10 mM) | 4.13 ± 0.05 | 3.01 ± 0.07 |
From a fit of the data to eq 3.
Frequency-Domain Anisotropy Measurements
The different modes of rotation associated with a fluorescent moiety can be identified and quantified by dynamic anisotropy; this is not possible with steady-state anisotropy measurements. Vertically polarized, sinusoidally modulated light was used to obtain the phase difference between the perpendicular and parallel phase components of the emission and the ratio of the parallel to perpendicular modulation components of the emitted light for F3W TyrH in the absence and presence of ligands. As shown in Figure 4, the presence of the pterin again has a greater effect than the presence of the amino acid. The contribution of a rapid motion is evident from the phase-angle distributions at the higher frequencies. For a quantitative analysis, the data were fit to a model describing a fluorophore that experiences a hindered local motion plus a global rotation (eq 4), as would be expected for a single tryptophan attached to a much larger protein (27, 36, 37)
| (4) |
Here, r0 is the anisotropy at time 0 and is independent of the rotational motion, θ1 and θ2 are the rotational correlation times associated with the two modes of rotation with θ1 ≪ θ2, and r∞ is the anisotropy that persists for a long time relative to the short rotational correlation time, θ1. Thus, r∞ reflects the slow global motion of the protein, and the amplitude of the local motion of the tryptophan is reflected in r – r∞. When each data set was analyzed independently, similar values of θ1 and θ2 were obtained for each. Consequently, a global analysis was performed of all of the data sets in the presence and absence of ligands, using the same values of θ1, θ2, and r0 throughout. This yielded values of θ1, θ2, and r0 of 1.89 ± 0.25 ns, 115 ± 10 ns, and 0.22 ± 0.03, respectively; the values of r0 – r∞ are given in Table 3. This value for θ2 is that predicted for the rotational correlation time of a spherical protein with a molecular weight of about 280 000, in reasonable agreement with the molecular weight of TyrH of 224 000. The amplitude of the local motion when phenylalanine alone was present was close to that in the absence of ligands, but there was a decrease in r0 – r∞ in the presence of 6M5DPH4, irrespective of the presence of phenylalanine. To obtain a measure of the change in the local motion of the tryptophan in the presence of 6M5DPH4, the local motion was modeled as a rotation within a cone. The half-angle of rotation ϕ was calculated using eq 5 and can be viewed as indicative of the amplitude of the hindered motion (37-39)
| (5) |
This analysis showed that the cone angle of the tryptophan on the loop decreases about 12° in the presence of 6M5DPH4, irrespective of whether phenylalanine is present (Table 3).
Figure 4.
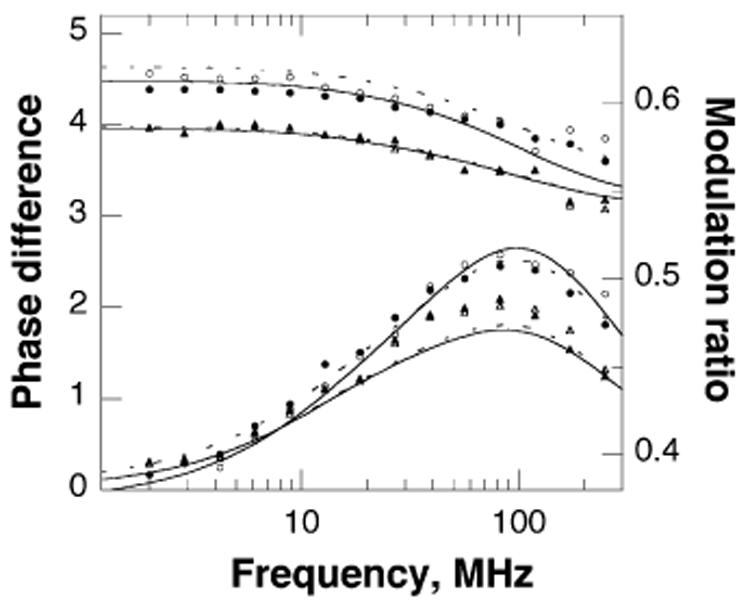
Frequency dependence of the modulation ratio (upper four lines) and phase difference (lower lines) of the tryptophan fluorescence of F3W TyrH for the enzyme alone (●) or in the presence of 10 mM phenylalanine (○), 500 μM 6M5DPH4 (▲), or both (△). The curves represent the best fit of the data to a model describing a hindered local motion plus a global rotation; the fits to data for samples in the presence of phenylalanine are given by dashed lines.
Table 3.
Effects of Ligands on the Pre-exponential Amplitude and Cone Angle of the Tryptophan in F3W TyrH
DISCUSSION
Fluorescence anisotropy provides a sensitive probe for changes in the mobility of a small chromophore such as a tryptophan residue attached to a much larger protein. Application of this method to analysis of the effects of ligands on the motion of the putative mobile loop in TyrH required mutagenesis of the three endogenous tryptophans to phenylalanine and introduction of a tryptophan in place of Phe184, the residue showing the greatest change in position on this loop in PheH. The steady-state kinetic parameters of the resulting enzyme, F3W TyrH, establish that these four mutations did not significantly perturb the catalytic properties of the enzyme. Thus, this mutant enzyme serves as a valid model for probing conformational changes in TyrH.
The naive expectation from the available structures of PheH was that the conformation and, consequently, the anisotropy of the single tryptophan residue in F3W TyrH would be unaffected by the binding of a pterin. Instead, the anisotropy would increase as the binding of an amino acid resulted in a protein conformation in which the single tryptophan was packed into the active site, restricting the motion of the fluorophore. In contrast, all of the analyses described here show that the binding of a pterin decreases the motion of the tryptophan residue significantly, while binding of an amino acid substrate has little effect. This rules out the simple model in which movement of Phe184 into the active site upon binding of an amino acid substrate is the key conformational change required before any reaction with oxygen can occur. While the results do not support the simple model based on structures of PheH, they are fully consistent with the steady-state kinetic mechanism of TyrH. The kinetic mechanism of the enzyme is ordered, with a tetrahydropterin being the first substrate to bind and the amino acid being the last (13). The observation of an ordered kinetic mechanism indicates that binding of one substrate is necessary to allow for a productive binding mode for a subsequent substrate. The present results establish that binding of a pterin results in a change in the mobility and, presumably, the conformation of a loop over the active site. If this conformational change results in the formation of the binding site for the amino acid, the present results provide a structural basis for the ordered kinetic mechanism. The present data are also consistent with the results of alanine-scanning mutagenesis of residues 178–193 (20). Removal of the side chains of residues in this loop by mutagenesis had little effect on the Km value for the tetrahydropterin but significantly altered that for tyrosine. To explain this result, it was proposed that binding the pterin resulted in a conformational change necessary for productive binding of the amino acid.
While the loop movement addressed here does not appear to be the critical conformational change required for catalysis, there are other possibilities for such a conformational change. Inspection of the available structures of ferrous PheH shows that several regions of the protein other than that analyzed here also move (16). However, the extent of movement is much less, and they are farther from the active site. In addition, the single glutamate ligand to the iron changes from monodentate to bidentate ligation. This would be expected to increase the electron density at the iron, lowering its redox potential and increasing its reactivity with oxygen. Support for this as a key change in determining the reactivity of the iron comes from the effect of mutating the metal ligand His331 in TyrH to glutamate. H331E TyrH is a tetrahydropterin oxidase, in that it catalyzes oxidation of tetrahydropterins in the absence of amino acid substrates (24). This suggests that the presence of a second negatively charged ligand mimics the effect of a bidentate carboxylate on the iron reactivity, bypassing the requirement for a change in the coordination of the single glutamate ligand in the wild-type enzyme.
The present data also address the effect of the change in the conformation of the loop on the local mobility of the indole ring tryptophan at its center. This motion can be described as a rotation within a cone of angle 2ϕ (37), such that a decrease in the range of motion is reflected by a decrease in the cone angle. Binding of the pterin results in a decrease of about 25% in the cone angle of the tryptophan in the loop of TyrH, reflecting a decrease in the mobility of the indole ring. The magnitude of the cone angle in the absence of bound pterin suggests that the indole ring does not have complete freedom of motion even in the absence of ligands.
The presence of iron affects the magnitude of the anisotropy, but the effects of ligands are qualitatively similar for the apoenzyme and holoenzyme. The lower anisotropy of the apoenzyme suggests that the apoenzyme is somewhat less structured, such that metal binding results in a more rigid protein. This would be consistent with the observation that iron stabilizes TyrH (40), although the apoenzyme is stable for days at 4 °C.5 X-ray crystallography does not show any difference in the structures of the catalytic domain of TyrH in the presence and absence of iron (17), but structures determined with crystals at low temperatures would not be expected to reveal small differences in protein stability. The effects of ligands on the anisotropy of the apoenzyme are consistent with a tightening up of the overall protein structure combined with the effects on loop mobility seen with the holoenzyme. While the effect of amino acid binding on the anisotropy of the holoenzyme is statistically too small to state conclusively that the amino acid binds, the effect on the anisotropy of the apoenzyme does support such a conclusion. An ability of TyrH to bind an amino acid substrate in the absence of pterin is also supported by steady-state kinetic studies; the substrate inhibition seen with tyrosine can be attributed to the binding of the amino acid to the free enzyme (13).
The conclusion from these studies that binding of the pterin rather than the amino acid has the greater effect on the motion of the 178–193 loop in TyrH contrasts with the most straightforward interpretation of the effects of substrates on the structure of PheH drawn from X-ray crystallography. Two explanations can be proposed for this discrepancy. One is that the two enzymes respond somewhat differently to the binding of ligands. PheH is an allosteric enzyme; phenylalanine activates the enzyme, and tetrahydrobiopterin antagonizes this effect (41). In contrast, TyrH is regulated by feedback inhibition by catecholamines, with no indication of allosteric effects (42). Thus, the PheH crystal structures may reflect binding modes different from those of TyrH. Alternatively, the energy barriers between the different conformations of the mobile loop examined here may be sufficiently low that the conformations in PheH crystals are determined by crystal-packing forces and do not accurately reflect the conformations in solution. Regardless of the explanation, the data here clearly establish that the conformation of this loop over the active site in TyrH is altered upon binding of a pterin rather than an amino acid substrate. This change in conformation appears to result in the formation of the amino acid binding site rather than serving to trigger oxygen activation.
Footnotes
Abbreviations: TyrH, tyrosine hydroxylase; F3W TyrH, W166F/F184W/W233F/W372F TyrH, an enzyme in which all of the intrinsic tryptophans have been replaced with phenylalanine and a single tryptophan has been introduced in place of Phe184; PheH, phenylalanine hydroxylase; DOPA, dihydroxyphenylalanine; 6MePH4, 6-methyltetrahydropterin; 6M5DPH4, 6-methyl-5-deazatetrahydropterin; NATA, N-acetyl-l-tryptophanamide.
This work was funded in part by NIH Grants GM47291 to P.F.F. and GM33216 to G.D.R. and Grants A1245 to P.F.F. and A1543 to G.D.R. from the Robert A. Welch Foundation.
A concentration of 500 μM 6M5DPH4 was routinely added to minimize any inner-filter effect, although this concentration is not saturating.
Preliminary experiments carried out with tyrosine as the amino acid ligand were qualitatively similar to those shown in Figure 1 for phenylalanine. However, the interference from the fluorescence of tyrosine and its limited solubility compared to phenylalanine precluded more thorough analyses.
A larger change in the anisotropy of F3W TyrH was seen in experiments designed to determine the phenylalanine concentration dependence of the anisotropy change, with a total change of about ⅓ of that seen with 6M5DPH4. However, these titrations required several hours to complete. In contrast, the anisotropy change seen immediately upon the addition of a saturating concentration of phenylalanine was quite small, as seen in Figure 1. In the case of 6Me5DPH4, the change in the anisotropy was independent of whether it was measured immediately after a high concentration was added directly or after several hours during a titration. This result suggested that the presence of phenylalanine can lead to a slow change in the conformation of TyrH. This conformational change is so much slower than catalysis that it is not likely to be catalytically relevant, in contrast to the much more rapid change seen upon pterin addition.
Frantom, P. A., and Fitzpatrick, P. F., unpublished observations.
REFERENCES
- 1.Fitzpatrick PF. The tetrahydropterin-dependent amino acid hydroxylases. Annu. Rev. Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 2.Fitzpatrick PF. Mechanism of aromatic amino acid hydroxylation. Biochemistry. 2003;42:14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walther DJ, Peter J, Bashammakh S, Hörtnagl H, Voits M, Fink H, Bader M. Synthesis of serotonin by a second tryptophan hydroxylase isoform. Science. 2003;299:76. doi: 10.1126/science.1078197. [DOI] [PubMed] [Google Scholar]
- 4.Ledley FD, DiLella AG, Kwok SCM, Woo SLC. Homology between phenylalanine and tyrosine hydroxylases reveals common structural and functional domains. Biochemistry. 1985;24:3389–3394. doi: 10.1021/bi00335a001. [DOI] [PubMed] [Google Scholar]
- 5.Grenett HE, Ledley FD, Reed LL, Woo SLC. Full-length cDNA for rabbit tryptophan hydroxylase: Functional domains and evolution of aromatic amino acid hydroxylases. Proc. Natl. Acad. Sci. U.S.A. 1987;84:5530–5534. doi: 10.1073/pnas.84.16.5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abate C, Smith JA, Joh TH. Characterization of the catalytic domain of bovine adrenal tyrosine hydroxylase. Biochem. Biophys. Res. Commun. 1988;151:1446–1453. doi: 10.1016/s0006-291x(88)80524-2. [DOI] [PubMed] [Google Scholar]
- 7.Daubner SC, Lohse DL, Fitzpatrick PF. Expression and characterization of catalytic and regulatory domains of rat tyrosine hydroxylase. Protein Sci. 1993;2:1452–1460. doi: 10.1002/pro.5560020909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Sa C, Arthur R, Jr., Kuhn DM. Expression and deletion mutagenesis of rat tryptophan hydroxylase fusion proteins: Delineation of the enzyme catalytic core. J. Neurochem. 1996;67:917–926. doi: 10.1046/j.1471-4159.1996.67030917.x. [DOI] [PubMed] [Google Scholar]
- 9.Kobe G, Jennings IG, House CM, Feil SC, Michell BJ, Tiganis T, Parker MW, Cotton RGH, Kemp BE. Regulation and crystallization of phosphorylated and dephosphorylated forms of truncated dimeric phenylalanine hydroxylase. Protein Sci. 1997;6:1352–1357. doi: 10.1002/pro.5560060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moran GR, Daubner SC, Fitzpatrick PF. Expression and characterization of the catalytic core of tryptophan hydroxylase. J. Biol. Chem. 1998;273:12259–12266. doi: 10.1074/jbc.273.20.12259. [DOI] [PubMed] [Google Scholar]
- 11.Daubner SC, Hillas PJ, Fitzpatrick PF. Characterization of chimeric pterin dependent hydroxylases: Contributions of the regulatory domains of tyrosine and phenylalanine hydroxylase to substrate specificity. Biochemistry. 1997;36:11574–11582. doi: 10.1021/bi9711137. [DOI] [PubMed] [Google Scholar]
- 12.Fitzpatrick PF. The aromatic amino acid hydroxylases. In: Purich DL, editor. Advances in Enzymology and Related Areas of Molecular Biology. John Wiley and Sons, Inc.; New York: 2000. pp. 235–294. [DOI] [PubMed] [Google Scholar]
- 13.Fitzpatrick PF. The steady-state kinetic mechanism of rat tyrosine hydroxylase. Biochemistry. 1991;30:3658–3662. doi: 10.1021/bi00229a010. [DOI] [PubMed] [Google Scholar]
- 14.Fitzpatrick PF. Studies of the rate-limiting step in the tyrosine hydroxylase reaction: Alternate substrates, solvent isotope effects, and transition state analogs. Biochemistry. 1991;30:6386–6391. doi: 10.1021/bi00240a006. [DOI] [PubMed] [Google Scholar]
- 15.Andersen OA, Flatmark T, Hough E. High resolution crystal structures of the catalytic domain of human phenylalanine hydroxylase in its catalytically active Fe(II) form and binary complex with tetrahydrobiopterin. J. Mol. Biol. 2001;314:279–291. doi: 10.1006/jmbi.2001.5061. [DOI] [PubMed] [Google Scholar]
- 16.Andersen OA, Stokka AJ, Flatmark T, Hough E. 2.0 Å Resolution crystal structures of the ternary complexes of human phenylalanine hydroxylase catalytic domain with tetrahydrobiopterin and 3-(2-thienyl)-l-alanine or l-norleucine: Substrate specificity and molecular motions related to substrate binding. J. Mol. Biol. 2003;333:747–757. doi: 10.1016/j.jmb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC. Crystal structure of tyrosine hydroxylase at 2.3 Å and its implications for inherited diseases. Nat. Struct. Biol. 1997;4:578–585. doi: 10.1038/nsb0797-578. [DOI] [PubMed] [Google Scholar]
- 18.Goodwill KE, Sabatier C, Stevens RC. Crystal structure of tyrosine hydroxylase with bound cofactor analogue and iron at 2.3 Å resolution: Self-hydroxylation of phe300 and the pterin-binding site. Biochemistry. 1998;37:13437–13445. doi: 10.1021/bi981462g. [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Erlandsen H, Haavik J, Knappskog PM, Stevens RC. Three-dimensional structure of human tryptophan hydroxylase and its implications for the biosynthesis of the neurotransmitters serotonin and melatonin. Biochemistry. 2002;41:12569–12574. doi: 10.1021/bi026561f. [DOI] [PubMed] [Google Scholar]
- 20.Daubner SC, McGinnis JT, Gardner M, Kroboth SL, Morris AR, Fitzpatrick PF. A flexible loop in tyrosine hydroxylase controls coupling of amino acid hydroxylation to tetrahydropterin oxidation. J. Mol. Biol. 2006;359:299–307. doi: 10.1016/j.jmb.2006.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moad G, Luthy CL, Benkovic PA, Benkovic SJ. Studies on 6-methyl-5-deazatetrahydropterin and its 4a adducts. J. Am. Chem. Soc. 1979;101:6068–6076. [Google Scholar]
- 22.Ramsey AJ, Fitzpatrick PF. Effects of phosphorylation on binding of catecholamines to tyrosine hydroxylase: Specificity and thermodynamics. Biochemistry. 2000;39:773–778. doi: 10.1021/bi991901r. [DOI] [PubMed] [Google Scholar]
- 23.Frantom PAS,J, Ragsdale SW, Fitzpatrick PF. Reduction and oxidation of the active site iron in tyrosine hydroxylase: Kinetics and specificity. Biochemistry. 2006;45:2372–2379. doi: 10.1021/bi052283j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fitzpatrick PF, Ralph EC, Ellis HR, Willmon OJ, Daubner SC. Characterization of metal ligand mutants of tyrosine hydroxylase: Insights into the plasticity of a 2-histi-dine-1-carboxylate triad. Biochemistry. 2003;42:2081–2088. doi: 10.1021/bi0271493. [DOI] [PubMed] [Google Scholar]
- 25.Andersson KK, Vassort C, Brennan BA, Que L, Jr., Haavik J, Flatmark T, Gros F, Thibault J. Purification and characterization of the blue-green rat phaeochromocytoma (PC12) tyrosine hydroxylase with a dopamine–Fe(III) complex reversal of the endogenous feedback inhibition by phosphorylation of serine-40. Biochem. J. 1992;284:687–695. doi: 10.1042/bj2840687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazarus RA, Dietrich RF, Wallick DE, Benkovic SJ. On the mechanism of action of phenylalanine hydroxylase. Biochemistry. 1981;20:6834–6841. doi: 10.1021/bi00527a015. [DOI] [PubMed] [Google Scholar]
- 27.Johnson JL, Reinhart GD. Influence of substrates and MgADP on the time-resolved intrinsic fluorescence of phosphofructokinase from Escherichia coli. Correlation of tryptophan dynamics to coupling entropy. Biochemistry. 1994;33:2644–2650. doi: 10.1021/bi00175a037. [DOI] [PubMed] [Google Scholar]
- 28.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2nd ed. Kluwer Academic/Plenum Publishers; New York: 1999. [Google Scholar]
- 29.Daubner SC, Melendez J, Fitzpatrick PF. Reversing the substrate specificities of phenylalanine and tyrosine hydroxylase: Aspartate 425 of tyrosine hydroxylase is essential for l-DOPA formation. Biochemistry. 2000;39:9652–9661. doi: 10.1021/bi000493k. [DOI] [PubMed] [Google Scholar]
- 30.Fitzpatrick PF. The pH dependency of binding of inhibitors to bovine adrenal tyrosine hydroxylase. J. Biol. Chem. 1988;263:16058–16062. [PubMed] [Google Scholar]
- 31.Perrin F. Polarization of light of fluorescence, average life of molecules in the excited state. J. Phys. Radium. 1926;7:390–401. [Google Scholar]
- 32.Gratton E, Jameson DM, Hall RD. Multifrequency phase and modulation fluorometry. Annu. Rev. Biophys. Bioeng. 1984;13:105–124. doi: 10.1146/annurev.bb.13.060184.000541. [DOI] [PubMed] [Google Scholar]
- 33.Alcala JR, Gratton E, Prendergast FG. Resolvability of fluorescence lifetime distributions using phase fluorometry. Biophys. J. 1987;51:587–596. doi: 10.1016/S0006-3495(87)83383-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alcala JR, Gratton E, Prendergast FG. Fluorescence lifetime distributions in proteins. Biophys. J. 1987;51:597–604. doi: 10.1016/S0006-3495(87)83384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alcala JR, Gratton E, Prendergast FG. Interpretation of fluorescence decays in proteins using continuous lifetime distributions. Biophys. J. 1987;51:925–936. doi: 10.1016/S0006-3495(87)83420-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lipari G, Szabo A. Effect of librational motion on fluorescence depolarization and nuclear magnetic resonance relaxation in macromolecules and membranes. Biophys. J. 1980;30:489–506. doi: 10.1016/S0006-3495(80)85109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munro I, Pecht I, Stryer L. Subnanosecond motions of tryptophan residues in proteins. Proc. Natl. Acad. Sci. U.S.A. 1979;76:56–60. doi: 10.1073/pnas.76.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lipari G, Szabo A. Effect of librational motion on fluorescence depolarization and nuclear magnetic resonance relaxation in macromolecules and membranes. Biophys. J. 1980;30:489–506. doi: 10.1016/S0006-3495(80)85109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gratton E, Alcala JR, Marriott G. Rotations of tryptophan residues in proteins. Biochem. Soc. Trans. 1986;14:835–838. doi: 10.1042/bst0140835. [DOI] [PubMed] [Google Scholar]
- 40.Gahn LG, Roskoski R., Jr. Thermal stability and CD analysis of rat tyrosine hydroxylase. Biochemistry. 1995;34:252–256. doi: 10.1021/bi00001a030. [DOI] [PubMed] [Google Scholar]
- 41.Xia T, Gray DW, Shiman R. Regulation of rat liver phenylalanine hydroxylase. III. Control of catalysis by (6R)-tetrahydrobiopterin and phenylalanine. J. Biol. Chem. 1994;269:24657–24665. [PubMed] [Google Scholar]
- 42.Ramsey AJ, Fitzpatrick PF. Effects of phosphorylation of serine 40 of tyrosine hydroxylase on binding of catecholamines: Evidence for a novel regulatory mechanism. Biochemistry. 1998;37:8980–8986. doi: 10.1021/bi980582l. [DOI] [PubMed] [Google Scholar]