

Abstract
Cystic Fibrosis (CF) lung disease, which is characterized by airway obstruction, chronic bacterial infection, and an excessive inflammatory response, is responsible for most of the morbidity and mortality. Early in life, CF patients become infected with a limited spectrum of bacteria, especially P. aeruginosa. New data now indicate that decreased depth of periciliary fluid and abnormal hydration of mucus, which impede mucociliary clearance, contribute to initial infection. Diminished production of the antibacterial molecule nitric oxide, increased bacterial binding sites (e.g., asialo GM-1) on CF airway epithelial cells, and adaptations made by the bacteria to the airway microenvironment, including the production of virulence factors and the ability to organize into a biofilm, contribute to susceptibility to initial bacterial infection. Once the patient is infected, an overzealous inflammatory response in the CF lung likely contributes to the host's inability to eradicate infection. In response to increased IL-8 and leukotriene B4 production, neutrophils infiltrate the lung where they release mediators, such as elastase, that further inhibit host defenses, cripple opsonophagocytosis, impair mucociliary clearance, and damage airway wall architecture. The combination of these events favors the persistence of bacteria in the airway. Until a cure is discovered, further investigations into therapies that relieve obstruction, control infection, and attenuate inflammation offer the best hope of limiting damage to host tissues and prolonging survival.
Keywords: cystic fibrosis, cystic fibrosis transmembrane conductance regulator, inflammation, lung, Pseudomonas aeruginosa
Introduction
Cystic fibrosis (CF) is an autosomal recessive disease caused by lack of function of a cAMP-regulated chloride channel, called CFTR (for the cystic fibrosis transmembrane conductance regulator), which normally resides at the apical surface of many epithelial cell types. Epithelial cells in the sweat glands, salivary glands, airways, nasal epithelium, vas deferens in males, bile ducts, pancreas, intestinal epithelium, as well as many other sites normally express CFTR. The function of CFTR is important in many of these organs, for its absence causes disease. However, the most important site of disease, which accounts for much of the morbidity and mortality in CF, is the lung. Early in life, patients become infected with bacteria, and eventually Pseudomonas aeruginosa becomes the predominant organism. Chronic infection leads to bronchiectasis, respiratory failure, and death [1]. The mechanism by which a defect in chloride transport leads to suppurative disease in the lung, but not elsewhere, is only now being elucidated.
Vulnerability to infection in CF occurs only in the airways, and not at other sites such as skin or urinary tract, so there is no systemic immune defect in CF. However, excess inflammation occurs at other sites: the prevalence of inflammatory bowel disease and pancreatitis is markedly increased [2,3]. Nevertheless, there is unquestionably something special about the lung, which is intended to be sterile, yet is continuously challenged by inhaled pathogens. Bacteria, when inhaled in small quantities, are ordinarily cleared without provoking significant inflammation. The lungs of patients with CF do not deal with this challenge appropriately. In this review, we ask two questions: Why do the lungs of patients with CF become infected? And why do they not clear these infections?
Why do CF patients become infected?
Mechanical factors
In the lung, the CFTR channel is found in surface airway epithelial cells and the cells of the submucosal glands [4]. Recent functional data indicate that there may be CFTR expression in the alveolar epithelium [5], and some of the migratory cells in the lung as well, including lymphocytes [6]. However, the most obvious defects in the lungs of CF patients appear to arise from defective salt transport across the airway epithelium and failure to properly hydrate airway secretions. CFTR is a cAMP-regulated chloride channel, so in CF, chloride secretion through CFTR (and any chloride channel whose activity depends on active CFTR, such as the outwardly rectifying chloride channel) is reduced, as is the amount of water which follows the salt. Although the calcium regulated chloride channel is upregulated in CF, this channel, at least in the murine airway, appears not to contribute to surface fluid depth. In the basal state, the depth of airway surface fluid in CF mice is reduced compared to normal mice [7]. Since calcium-regulated chloride channels induce secretion when stimulated in both normal and CF murine airways, reduced basal state fluid depth in CF patients indicates the lack of participation of such channels in the maintenance of basal state fluid balance. In addition, CFTR lives up to its name as a "conductance regulator" and affects the function of many other channels in the epithelium [8]. Notable among them is the amiloride-sensitive epithelial sodium channel (ENaC), which accounts for the bulk of salt and water transport in the airways [9]. ENaC is expressed in airway and alveolar epithelium, and is responsible for the reabsorption of sodium (with water following) from airway surface liquid. Such resorption is necessary to maintain the relatively constant depth of airway surface fluid in spite of marked reduction of cross sectional area of the airway surface from the alveoli to the trachea. ENaC is downregulated by functional CFTR: in the absence of CFTR function, ENaC activity increases [10-13]. This increase in activity increases salt and water reabsorption across the epithelium. The combination of increased resorption and decreased secretion results in too little fluid in the airways of patients with CF, although the ionic composition of the fluid remains normal [14-18].
Although it is difficult to measure salt and water content of airway surface directly in uninfected lungs of patients with CF, it is possible to make such measurements in mice engineered with defects in CFTR. The first such mouse to be developed was the S489X mouse, a "knockout" mouse in which a stop codon has been inserted at position 489. Since this first mouse was engineered, several other knockout mice and mice with the ΔF508 mutation and other amino acid substitutions have been produced. These mice, for the most part, lack function of the CFTR chloride channel. However, their clinical manifestations differ from those of humans. The CF mice reliably have intestinal obstruction, which is usually the dominant and fatal manifestation. On the other hand, these mice do not spontaneously develop lung infection, though they are more vulnerable to direct inoculation with various CF pathogens. This feature allows pristine, uninfected lungs with the CF ion transport defect to be studied. Direct measurements of sodium, chloride, potassium, and calcium concentrations, as well as osmolarity, in the airway surface liquid in the trachea of living CF and non-CF mice, and measurements in well-differentiated cultured airway epithelial cells grown at the air-liquid interface support this "isotonic, low-volume" hypothesis for the result of the ion transport abnormalities in the airways of patients with CF [7,14-18]. Measured ion composition and osmolarity of the airway surface liquid is comparable in CF and non-CF mice. However, the depth of the airway surface fluid is less in CF mice, and fluid volume is reduced atop well-differentiated CF airway epithelial cell cultures compared to non-CF. The "low volume" hypothesis predicts that reduced airway surface liquid volume interferes with proper ciliary function, reducing mucociliary clearance. In CF mice, mucociliary clearance also is reduced. However, reductions in mucociliary clearance have been difficult to demonstrate unequivocally in CF patients. The measurements themselves are quite variable, which may be part of the difficulty, although the same techniques demonstrate changes in mucociliary clearance with drug interventions, as well as the markedly reduced mucociliary clearance that occurs in patients with primary ciliary dyskinesia. Interpretation of results in CF patients is complicated by the secondary effects of disease, which are unevenly distributed throughout the lung. Nevertheless, since failure of mucociliary clearance is an important link in the logical chain connecting CFTR dysfunction with infection in the "low volume" model, it seems important to evaluate this mechanism further in patients.
Besides abnormal periciliary fluid depth, the CF defect probably leads to abnormal mucus hydration as well. Mucus is packaged into granules before secretion and unfolds during the secretory process. The water content of CF mucus is reduced, even at sites that are not infected (such as the uterine cervix) [19]. This reduced water content may contribute to abnormal properties that make mucus difficult to clear in CF. However, in experimental model systems, clearance is affected only in minor ways over a wide range of viscosity and hydration of mucus [20,21]. Therefore, it is likely that other factors combine with the properties of the mucus itself to produce the putative reduction in mucociliary clearance in CF.
During the later stages of disease, impaction of mucus and failure of mucociliary clearance are unequivocally present. In patients with bronchiectasis, stagnant pools of secretions collect in the saccular dilations of the bronchi. Histological evaluation of CF airways from patients who have died, or undergone lung transplantation or resection, show that dense plaques of mucus become adherent to the epithelial surface, at least during the later stages of disease in these patients. Bacteria in this mucous layer cannot be cleared normally. It is not certain that such dense mucus plaques plastered to the airway wall occur in the earlier stages of disease. However, as the disease progresses, stagnation of secretions and failure to clear bacteria clearly contributes to the maintenance of pulmonary infection.
Failure to kill bacteria properly
Prior to birth, the lungs are bathed in amniotic fluid, and appear to be normal in CF. At the time of birth, however, marked changes in fluid flux across the airway occur, the air-liquid interface at the epithelial surface is established, and the pattern of ion transport is altered. On histopathologic examination of the lungs of uninfected neonates with CF who died of meconium ileus, there was little histopathology and no inflammation. However, there is widening of the orifices of the submucosal glands, as if already, in the prenatal period, plugging of the ducts has occurred [22]. However, airway tissue retrieved from CF fetuses and implanted into the backs of immunosuppressed mice shows submucosal collection of neutrophils, which do not reach the lumen unless some stimulus is applied [23]. These xenografts also appear to produce an excess of interleukin (IL)-8, and so may be primed to respond vigorously to inflammatory stimuli [24]. After birth, the infant airway is challenged repeatedly with small doses of bacteria. Nonspecific airway defenses such as defensins, lysozyme, and nitric oxide (NO) ordinarily dispatch these small inocula. However, in the airways of patients with CF, the non-specific defenses may be compromised. Although there has been considerable attention paid to the possibility of defective function of defensins in the airways of patients with CF, it now appears that these molecules are intact and function normally. However, the major isoform of nitric oxide synthase (NOS) in airway epithelial cells, NOS-2, is reduced in CF airway epithelium [25]. Reduction in local NO production probably compromises the host's ability to handle small bacterial inocula.
The lack of NOS-2 expression in CF airway epithelial cells appears to be related directly to CFTR function. In CF mice, NOS-2 expression in epithelia is markedly reduced [25]. In CF mice in which the basic defect is partially corrected by transgenic expression of human CFTR driven by the FABP promoter in the gut only, NOS-2 expression is restored only in the epithelium of the gut, not in the airway. In mice that have received human CFTR by gene transfer; the cells that show the immunohistochemical signal of hCFTR also have NOS-2 activity restored. Similar results are obtained in CF models in cell culture. Cells with a CF phenotype have markedly reduced NOS-2 expression at the mRNA and protein level compared to their matched non-CF controls. These results are also confirmed by immunohistochemical staining for NOS-2 in human postmortem tracheal samples. At first glance, reduction in NOS-2 expression in the airways of patients with CF is puzzling because NOS-2 is transcribed in response to nuclear factor-kappaB (NF-κB) activation, which is known to be increased in the CF epithelium. However, NOS-2 transcription also requires phosphorylated signal transducer and activator of transcription (Stat)-1. Further investigation demonstrated that Stat-1 is present in abundance in the CF epithelial cells and is normally phosphorylated. However, the protein inhibitor of activated Stat 1 is also markedly upregulated [26]. This protein binds to phosphorylated Stat-1 and prevents its transcriptional activity. This mechanism predicts that other Stat-1 regulated proteins might also be downregulated in CF. This prediction is confirmed for interferon regulatory factor-1 and regulated on activation normal T cell expressed and secreted. Thus, dysregulation of Stat-1 activity may well have broad functional consequences in CF for the defense of the airway.
One hypothesis for the specific defense of the airway against inhaled P. aeruginosa is that CFTR itself constitutes a specific receptor for this organism [27]. If CFTR is lacking at the airway epithelial surface, then this receptor is absent. Once bound to CFTR, P. aeruginosa are internalized, the epithelial cell undergoes apoptosis, and is sloughed, thereby clearing the internalized organisms. However, this hypothesis does not explain how patients with activation or channel mutants of CFTR, that reach the cell surface (e.g., G551D), are just as vulnerable to P. aeruginosa infection as are patients that lack CFTR at the cell surface.
If the nonspecific defenses of the airway are compromised, this, in combination with reduced clearance of bacteria, may supply a stimulus sufficient to recruit an inflammatory response in the CF airway, whereas a comparable bacterial challenge to a normal infant would simply result in killing by nonspecific host defense mechanisms and efficient clearance. Once the bacteria initiate an inflammatory response, many other systems are called into play, some of which are deleterious to the airway.
Abnormal retention of specific bacteria in the airway
The post-translational modification of cell surface and secreted molecules may be altered to facilitate binding of P. aeruginosa and other infecting agents. Several mechanisms for retention of bacteria in the airways of patients with CF have been proposed. Asialo-GM1, a ligand for P. aeruginosa, S. aureus, and H. influenzae, is increased on CF airway epithelial cells [28], sufficient to explain a two-fold increase in bacterial binding [29], a modest change, but one which could become important over time. Some investigators have questioned the importance of adherence, since histopathologic examination reveals relatively few bacteria in apposition to the epithelial surface, suggesting that the dense mucus layer may prevent access [30,31]. However, these studies were performed on samples taken at autopsy or transplantation, and thus represent end-stage lung disease. At this point, bacteria have adapted to their environment by a downregulation of the genes for pilin and flagelin, two important epithelial adhesins. Even at this late stage in the disease process, however, other bacteria, notably Burkholderia cepacia, can penetrate the mucus layer and even the epithelial barrier [32], suggesting that the dense mucus is not an absolute barrier to epithelial access. Earlier in the disease process, however, when pilin and flagelin are expressed by P. aeruginosa, the classical infectious disease paradigm of adherence followed by infection may still be valid. In addition, even if surface binding of bacteria is not quantitatively important in the establishment of infection, it may be important for the initiation of an exuberant inflammatory response [33]. The binding of pilin to asialo-GM1 activates the pro-inflammatory transcription factor NF-κB and induces production of the neutrophil chemoattractant IL-8 [34,35]. Flagelin, like pilin, extends from the bacterial cell surface and promotes P. aeruginosa retention in the airways of patients with CF by binding to mucin oligosaccharides via flagellar cap protein FliD [36,37] and to epithelial cell oligosaccharides where it stimulates the production of pro-inflammatory cytokines [38]. Specific attachment of bacteria, combined with the putative compromise of mucociliary clearance as a direct result of the salt transport defect, promotes retention of bacteria in the mucus of the airways of patients with CF and allows them to multiply there. The relationship between mutant CFTR and these pathophysiologic processes is shown in Figure 1.
Figure 1.
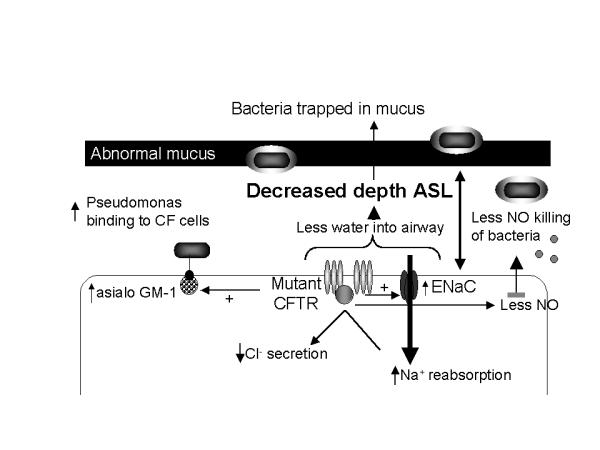
Impact of mutant cystic fibrosis transmembrane conductance regulator (CFTR) on cellular physiology. Mutant CFTR promotes initial bacterial infection by upregulating epithelial cell adhesion molecules for bacteria such as asialo-GM1 and by decreasing production of innate host defense molecules such as nitric oxide (NO). Defects in CFTR also lead to increased sodium absorption through the epithelial sodium channel (ENaC) and decreased chloride secretion. Water follows its concentration gradient and results in decreased depth of airway surface liquid. Bacterial persistence is promoted by alterations in airway wall architecture, impaired host defense mechanisms, an excessive inflammatory response, and adaptations made by the bacteria to the microenvironment of the cystic fibrosis airway.
Adaptation of bacteria to live in the CF airway
It is impossible to consider the host's response to infection without considering the character of the infecting organisms. In CF, chronic endobronchial bacterial infections display a limited spectrum of organisms including H. influenzae, S. aureus, P. aeruginosa, B. cepacia, Stenotrophomonas maltophilia, and Alcaligenes xylosoxidans [39,40]. While H. influenzae and S. aureus may predominate early in life, over 30% of patients three years of age and 80% of young adults are chronically infected with P. aeruginosa [39-41], a ubiquitous and highly adaptable, aerobic, Gram-negative bacillus that is motile by means of a single polar flagellum. It is non-pathogenic in normal hosts, but becomes a pathogen in individuals with weakened defenses [42]. P. aeruginosa, like a few other CF pathogens, has the ability to develop resistance to multiple antibiotics. The mechanism of resistance is in large part due to the presence of efflux pumps, which, in addition to antibiotics, export detergents, dyes, and homoserine lactones [43]. Also, the CF airway confers special selective advantages to P. aeruginosa. Some strains of P. aeruginosa have the ability to mutate rapidly in the lungs of patients with CF. These hypermutable strains demonstrate an increased ability to resist antibiotics [44]. In explanted lung tissue obtained from end-stage CF patients, some investigators demonstrated that P. aeruginosa resides primarily within the intraluminal mucus [45], thus suggesting that the mucociliary escalator is an important host defense mechanism for those bacteria trapped in mucus (Fig. 2). In the airways of patients with CF, P. aeruginosa initially continues its usual non-mucoid phenotype, but ultimately produces mucoid exopolysaccharide (MEP) or alginate, which gives its colonies their typical appearance. A steep oxygen concentration gradient exists between the airway lumen and the interior of the mucus [45]. P. aeruginosa responds to the hypoxic environment of mucus by producing even more MEP [45,46], which contributes to the persistence of P. aeruginosa in the CF airway by interfering with host defenses and delivery of antibiotics to the bacterial cell. Although MEP is highly antigenic, antibodies directed against it are not effective opsonins, but they can participate in formation of immune complexes that intensify local tissue damage [47-49]. P. aeruginosa also produces an alginate lyase enzyme that cleaves MEP and may allow spread of the organism to contiguous sites [50]. Although the conversion to mucoidy typically is associated with decreased virulence [51], the decline in lung function that occurs in CF is actually accelerated after P. aeruginosa assumes the mucoid phenotype [52]. This implies that the progressive deterioration in lung function experienced by a CF patient is due more to the long-term deleterious effects of host inflammatory responses rather than direct damage from the bacteria itself. Residence in the CF lung also seems to alter the properties of P. aeruginosa lipopolysaccharide (LPS): LPS isolated from a large percentage of CF patients has little or no O-side chain, conferring a rough appearance to colonies when grown on agar plates. Changes at this site in the molecule may have clinical implications because complement fixation occurs on the O-side chain. Furthermore, P. aeruginosa may synthesize specific structures in the lipid-A moiety of its endotoxin, which provoke increased host inflammatory responses and resistance to antimicrobial peptides [53]. In addition to the advantages conferred upon it by the appropriate environmental conditions, P. aeruginosa itself possesses special characteristics that allow it to persist in the lungs of patients with CF, including the production of virulence factors and the ability to organize into a biofilm.
Figure 2.
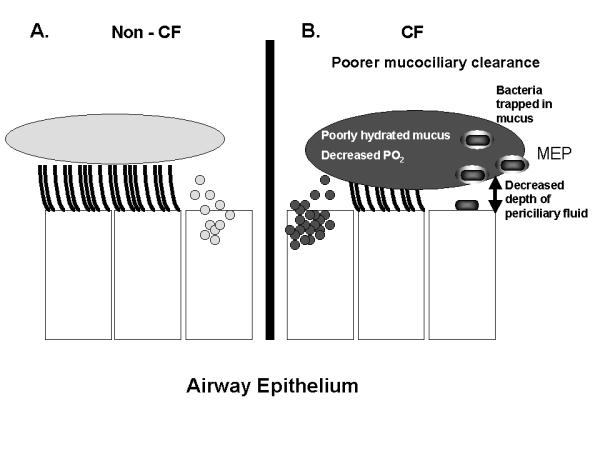
Schematic Representation of the mucociliary escalator in the non-cystic fibrosis and cystic fibrosis (CF) airways. In the non-CF airway (Fig. 2A), where the depth of the periciliary fluid is normal, islands of mucus float on top and are propelled upward toward the mouth by the coordinated beating of cilia. In the CF airway (Fig 2B), the mucus is poorly hydrated and hypoxic. Because of the decreased depth of the periciliary fluid, the abnormal mucus is plastered down upon the cilia, thus inhibiting normal ciliary beating. Eventually the bacteria present in the airway become trapped in the mucus and adapt to the local environment. In the case of P. aeruginosa, this includes production of mucoid exopolysaccharide (MEP) and organization into a biofilm.
Numerous P. aeruginosa virulence factors contribute to its pathogenicity in CF by altering the host's defenses. Pseudomonas elastase and alkaline protease are proteolytic enzymes that may damage host tissues, disrupt tight junctions, and impair opsonophagocytosis [54]. Pseudomonas elastase degrades immunoglobulins, coagulation factors, complement components, cytokines, and alpha proteinase inhibitor [55] and stimulates mucin release from goblet cells [56], likely enhancing the already increased production of mucin that occurs in the CF airway. Pseudomonas elastase is more potent than neutrophil elastase, on a per mg basis, with respect to elastin degradation, and thus may contribute to CF lung pathology, even though the predominant elastase in CF sputum is from neutrophils [57]. Exotoxin A promotes tissue necrosis by inhibiting protein synthesis in eukaryotic cells by a similar mechanism to that described for diphtheria toxin. Exotoxin A catalyzes the transfer of the ADP-ribosyl moiety of nicotinamide adenine dinucleotide onto elongation factor 2, which then is inactive in protein synthesis. Exotoxin A also attracts neutrophils into the lungs of mice [58]. Exoenzyme S is an ADP-ribosyltransferase that disrupts eukaryotic cell signal transduction, stimulates actin reorganization, inhibits tissue regeneration, serves as a potent T lymphocyte mitogen, maintains the site of infection by promoting P. aeruginosa adhesion, and is cytotoxic, especially to epithelial cells, [59-64]. Phospholipase C hydrolyzes lecithin, decreases the neutrophil's respiratory burst, and stimulates IL-8 release by monocytes in vitro. It also induces local production of tumor necrosis factor-alpha (TNF-α), IL-1β, interferon-gamma, macrophage inflammatory protein-1α, and macrophage inflammatory protein-2 in addition to stimulating neutrophil infiltration, thereby likely contributing to the vigorous inflammatory response seen in the CF airway [58]. Pigments such as pyocyanin bind iron, inhibit the growth of other bacteria, and inhibit ciliary beat frequency [65,66]. Since P. aeruginosa virulence factors increase with acute pulmonary exacerbations and decrease after the administration of systemic antibiotics [67,68], virulence factors may contribute, at least in part, to acute deteriorations in lung function.
Recently, the ability of P. aeruginosa to organize into a biofilm has garnered much attention. Donlan and Costerton define a biofilm as "a microbially derived sessile community characterized by cells that are irreversibly attached to a substratum or interface or to each other, are embedded in a matrix of extracellular polymeric substances that they have produced, and exhibit an altered phenotype with respect to growth rate and gene transcription" [69]. Biofilm formation protects bacteria from changes in environmental conditions, antibiotics, and host defenses, and thus may consolidate the ability of the bacterium to persist in the airways of patients with CF. Bacteria within a biofilm communicate with one other via a mechanism known as quorum sensing, which also downregulates virulence factors, allowing the bacteria to live in symbiosis with the host [70]. The altered phenotype of bacteria in a biofilm may have clinical importance. Growth characteristics differ significantly for bacteria in a biofilm than for those in the free-living, planktonic state. Antibiotic sensitivity testing performed on bacteria in the planktonic state, as occurs in the clinical microbiology laboratory, may not accurately reflect the true sensitivities of bacteria in a biofilm [71]. This difference may account for the clinical efficacy of macrolide antibiotics that has been described in CF [72,73]. Quorum sensing signals provide a promising potential therapeutic target in CF.
No article on bacterial infections in CF would be complete without at least mentioning B. cepacia. What was once thought to be a single organism, "B cepacia" actually includes several related organisms now known as B. cepacia complex [74,75]. Although infrequent pathogens in CF, organisms of the B. cepacia complex often have major clinical impact. The clinical course after acquisition of B. cepacia complex organisms spans the spectrum of no discernable clinical change to severe and rapidly progressive respiratory failure, often associated with bacteremia and death ("cepacia syndrome") [75]. Organisms of the B. cepacia complex, especially the organisms implicated in cepacia syndrome, have been proposed to provoke a more robust host inflammatory response than P. aeruginosa with respect to production of TNF-α by monocyte cell lines in vitro, neutrophil recruitment, and priming of the neutrophil respiratory burst [76,77]. However, this increased inflammatory response has not been documented in CF patients [78].
Chronic endobronchial bacterial infection with one or more typical organisms is the hallmark of CF lung disease. The host inflammatory response in CF to the bacterial infection dictates the clinical manifestations of the lung disease. In general, CF patients experience a progressive decline in pulmonary function that is punctuated by intermittent exacerbations, which are characterized by increased cough, sputum production, anorexia, and malaise. Antimicrobial therapy for CF bacterial infections, especially for P. aeruginosa, frequently requires the administration of a combination of two or more antibiotics due to the bacteria's ability to become resistant to a single agent. Moreover, CF patients typically require greater than normal antibiotic doses to penetrate the large endobronchial mucus sink and to counter the altered pharmacokinetics that occur in CF patients due to increased volume of distribution from malnutrition and, for some drugs, increased renal clearance [42]. Most CF patients return to pre-exacerbation pulmonary function values after completing a course of parenteral antibiotics, but this is not always the case. It is possible that the cumulative effects of multiple pulmonary exacerbations contribute to the decline in lung function and that those patients with more frequent and/or severe exacerbations have shorter life spans.
Why do patients with CF fail to clear bacterial infection?
Excess inflammation provides an environment favorable to bacterial growth
CF infants develop bacterial infection early, and respond to it with a vigorous inflammatory response. Epithelial cells respond to bacteria and their products by increasing production of cytokines such as IL-6, IL-8, granulocyte macrophage colony stimulating factor (GM-CSF), expression of intercellular adhesion molecule-1, and production of mucins. IL-8, a potent chemokine, attracts neutrophils to the inflammatory site, where their transepithelial passage is facilitated by intercellular adhesion molecule-1 and their survival prolonged by GM-CSF. When lung macrophages encounter bacteria, they respond not only by producing their own IL-8, but also by producing TNF-α and IL-1β, which in turn can drive epithelial cell production of pro-inflammatory molecules by a signaling pathway different from that accessed by the bacterial products. Quickly, the airway recruits large numbers of neutrophils, which early in the course of the infection, are often able to contain the bacteria. Initial infections are frequently cleared, and colonization that is only intermittent is common in the first few years of life.
The inflammatory process appears to go awry in the lungs of patients with CF, even in infancy. Clinical studies indicate that, for a given lung bacterial burden, the neutrophil and IL-8 responses of CF infants, measured in bronchoalveolar lavage fluid, are excessive compared to those of normal infants. This is true whether all organisms recovered from the lung are considered, or whether analysis is restricted only to infants whose cultures reveal only H. influenzae [79,80]. Inflammation is in excess in CF even if the neutrophil and IL-8 responses are adjusted for the amount of endotoxin in the bronchoalveolar lavage. Initially, this response seems to contain the bacteria. Indeed, in other, cross-sectional studies of inflammatory responses in CF bronchoalveolar lavage fluid, many CF infants have no detectable bacteria, but even some of these infants have a modest inflammatory response, which exceeds that observed in other, uninfected, non-CF infants undergoing bronchoalveolar lavage [81]. However, other studies show that at least some CF infants, particularly those who have never had lung infection, have no detectable inflammatory response [82]. The picture emerges of a lung which, although initially pristine and uninflamed, mounts an excessive inflammatory response to bacterial stimulation, which continues to reverberate even after the infection is controlled. Eventually, all the factors, which serve to retain bacteria in the CF lung, overwhelm the defenses of the lung, even the phagocytic defenses, and the bacterial signals for inflammation persist. At this point, the excessive inflammatory response becomes deleterious and even promotes continuing infection.
One striking feature of CF airways disease is the progressive accumulation of neutrophils over a period of years. This "acute inflammation" never converts to a more "chronic" pattern. Since neutrophils do not survive long after exiting the circulation, there must be a persistent stimulus to attract these neutrophils. There is certainly an excess of chemoattractants such as IL-8 and leukotriene B4 recovered in bronchoalveolar lavage fluid [83,84]. Bacteria provide additional chemoattractants. The neutrophils may survive longer in the airways of patients with CF because of the production of excess GM-CSF and the relative lack of IL-10 [83,85,86], which, when present, promotes neutrophil apoptosis. When present in excess, neutrophils and their products actually impair the host's ability to clear bacterial infection. Neutrophil elastase, in particular, interacts with airway epithelial cells to promote the transcription of IL-8 and macromolecular secretion, further fueling airway inflammation and obstruction [87-91]. Elastase cleaves IgG at the hinge region [92,93]. Since macrophages use antibodies to ingest P. aeruginosa, opsonophagocytosis is reduced in the presence of excess elastase. On the other hand, neutrophils employ complement for opsonophagocytosis of P. aeruginosa. This system consists of two receptors, CR-1 and CR-3 and two complement opsonins, C3b (ligand for CR-1) and C3bi (ligand for CR-3). Elastase cleaves the CR1 receptor and the C3bi ligand, so that neither of the opsonin-receptor pairs is left intact [94,95]. Thus, all the usual mechanisms of ingestion of P. aeruginosa are crippled in the presence of free elastase activity (Fig. 3). In one study of CF patients, all patients above the age of one year, and many of those less than one year of age, had excess neutrophil elastase activity in their bronchoalveolar lavage fluid [96]. Most patients over 1 year of age have concentrations in excess of 1 μM. Since the opsonins and receptors are cleaved at concentrations of free elastase of 10-8 M, 1 μM is more than sufficient to turn the vicious cycle of inflammation and infection, and to destroy the fabric of the lung.
Figure 3.
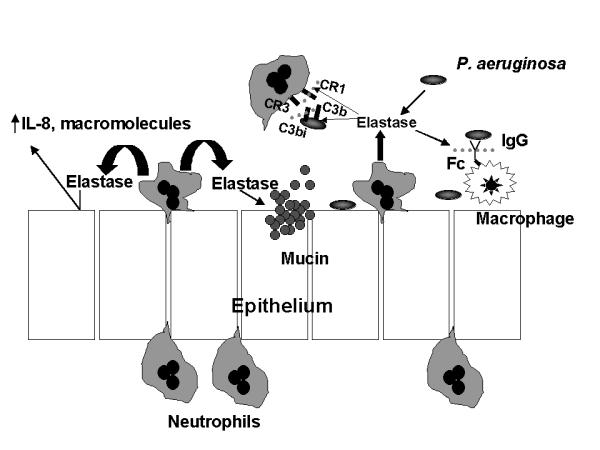
Adverse effects of elastase on host defense mechanisms and inflammation. In the cystic fibrosis airway, the concentration of elastase exceeds the concentration of inhibitors of elastase by several hundred to several thousand fold. While the vast majority of elastase is produced by the neutrophil, a small but significant amount is derived from bacteria. In addition to causing structural damage directly, elastase stimulates the production of pro-inflammatory mediators such as IL-8, which further induces neutrophil influx. Elastase also impairs mucociliary clearance by direct effects on ciliary function and by stimulating increased mucus production. Elastase inhibits opsonophagocytosis by cleaving the Fc portion of immunoglobulin G and complement receptors on both the neutrophil (CR1) and P. aeruginosa (C3bi), resulting in an opsonin-receptor mismatch.
Structural damage to the lung allows for mechanical retention of secretions and retention of bacteria
We have argued here that inflammation in the CF lung occurs in excess compared to the response mounted by non-CF individuals, and that it is ultimately ineffective against the bacteria. The result of all this is that CF infants develop bacterial infections very early in life. In the beginning, colonization may be intermittent, but eventually, it becomes chronic. The special binding properties of P. aeruginosa, combined with its ubiquitous presence in our environment and therefore regular exposure, and its particular ability to adjust quickly and perfectly to conditions in the CF lung likely account for its predilection for the CF lung. Eventually most patients with CF acquire this organism, develop a vigorous and persistent neutrophilic inflammatory response, and settle into a vicious cycle of airway obstruction, infection, and excess inflammation that results in lung destruction, further damage to the clearance processes, and additional vulnerability to infection or phenotypic transformation of the P. aeruginosa into a biofilm, which is impossible to eradicate despite the most vigorous antibiotic therapy.
The persistent bacterial stimulation of an overzealous inflammatory response results in excess neutrophils and neutrophil products in the airways. Many of the proteases secreted by the neutrophil are capable of digesting the structural proteins of the CF lung, including collagen and elastin. Small breaks in the epithelial barrier expose these structural proteins, and the normal antiprotease defenses are overwhelmed by the massive quantities of enzymes released by the enormous neutrophil infiltration. Reactive oxygen species are also potent agents of tissue damage. The antioxidant defenses of the lung are markedly reduced in CF, although the reasons for this are not entirely clear. It has been speculated that CFTR transports glutathione as well as chloride ion, and in the absence of functional CFTR, less glutathione reaches the airway to defend against oxidant damage. In any event, reduced oxidant defenses can be demonstrated even in the uninfected airways of CF mice [97]. Another class of proteases is also elevated in the bronchoalveolar lavage fluid of patients with CF. Matrix metalloproteinases, implicated in remodeling of inflamed areas of the lung, are found in excess in the lungs of patients with CF. These proteinases can be produced by epithelial cells, and their transcription is activated by NF-κB. All of these damaging proteases and oxidants combine to destroy the supporting structures of the airway and ultimately lead to bronchiectasis. Once the fabric of the airway wall is compromised, outpouching of the airway wall (saccular bronchiectasis) occurs. In these damaged areas, pooling of secretions and failure of clearance is inevitable. It is rare that infection can be cleared once such structural damage has occurred. In the later stages of the disease, all of the complications of bronchiectasis of any cause emerge in patients with CF – engorgement of the bronchial blood vessels with risk for massive hemoptysis, persistent secretions and cough, and persistent bacterial infection that is impossible to clear. However, all of the features noted above that allow bacteria to be retained in the CF lung in the first place are still present, and all of the abnormalities in signaling that make for increased inflammatory responses are also in play. Therefore, the progression of bronchiectasis in the lungs of patients with CF tends to be more rapid than it is in patients with bronchiectasis of other causes, such as post-infectious bronchiectasis or bronchiectasis associated with primary ciliary dyskinesia. Many patients with bronchiectasis of non-CF etiology survive well into adulthood or even old age, whereas such survival is rare in patients with CF.
Summary and conclusions
The lungs of patients with CF are vulnerable to bacterial infection, and once the infection becomes established, it is not eradicated despite prolonged and vigorous antibiotic and airway clearance therapy. This aspect of the disease has long provided an inviting therapeutic target, though it is essentially a rear guard action which delays but does not prevent the progression of the lung disease. Successful strategies to prevent the initial colonization, assist in the clearance of initial infections, prevent the adaptation of P. aeruginosa to the CF lung environment, or even to limit the excess inflammatory response (although not the response required to kill the bacteria), would have great therapeutic benefit to patients with CF. Indeed, a number of strategies have been proposed to interfere at each of these steps: aerosolized dextrans to prevent pseudomonas adherence, intravenous IgG to assist in clearance, and many proposed anti-inflammatory treatments to limit the excessive inflammation already have reached clinical trial. Once infection has been established, lung damage might be slowed by inhibiting the excess of oxidants in the CF airway or by inhibiting the proteolytic damage to the structural proteins of the airways with antiproteases (or, at a more fundamental level, limiting the access of the neutrophils to the airway). Drugs aimed at these steps are also in development. Strategies directed at the basic defect, if applied sufficiently early in the course of the disease, might abort the entire process and provide the best therapeutic result of all. Once structural damage has occurred, however, bronchiectasis may take on a life of its own, and even complete correction of the underlying genetic defect may not completely halt disease progression. For these patients, further development of the means to control the inflammatory response and its consequences likely will be necessary.
Abbreviations
CF cystic fibrosis
CFTR cystic fibrosis transmembrane conductance regulator
ENaC epithelial sodium channel
GM-CSF granulocyte macrophage colony stimulating factor
IL interleukin
LPS lipopolysaccharide
MEP mucoid exopolysaccharide
NF-κB nuclear factor-kappaB
NO nitric oxide
NOS nitric oxide synthase
Stat signal transducer and activator of transcription
TNF-α tumor necrosis factor-alpha
Contributor Information
James F Chmiel, Email: jxc34@po.cwru.edu.
Pamela B Davis, Email: pbd@po.cwru.edu.
References
- Davis PB, Drumm ML, Konstan MW. State of the Art: Cystic Fibrosis. Am J Resp Crit Care Med. 1996;154:1229–1256. doi: 10.1164/ajrccm.154.5.8912731. [DOI] [PubMed] [Google Scholar]
- Lloyd-Still JD. Crohn's disease and cystic fibrosis. Dig Dis Sci. 1994;39:880–885. doi: 10.1007/BF02087437. [DOI] [PubMed] [Google Scholar]
- Taylor CJ, Aswani N. The pancreas in cystic fibrosis. Paediatr Respir Rev. 2002;3:77–81. doi: 10.1053/prrv.2002.0183. [DOI] [PubMed] [Google Scholar]
- Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiol Rev. 1999;79(1 Suppl):S215–S255. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- Fang X, Fukuda N, Barbry P, Sartori C, Verkman AS, Matthay MA. Novel role for CFTR in fluid absorption from the distal airspaces of the lung. J Gen Physiol. 2002;119:199–207. doi: 10.1085/jgp.119.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubien JK. CFTR may play a role in regulated secretion by lymphocytes: a new hypothesis for the pathophysiology of cystic fibrosis. Pflugers Arch. 2001;443(Suppl 1):S36–S39. doi: 10.1007/s004240100641. [DOI] [PubMed] [Google Scholar]
- Tarran R, Loewen ME, Paradiso AM, Olsen JC, Gray MA, Argent BE, Boucher RC, Gabriel SE. Regulation of Murine Airway Surface Liquid Volume by CFTR and Ca(2+)-activated Cl(-) Conductances. J Gen Physiol. 2002;120:407–418. doi: 10.1085/jgp.20028599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger R, Mall M, Bleich M, Ecke D, Warth R, Riedemann N, Kunzelmann K. Regulation of epithelial ion channels by the cystic fibrosis transmembrane conductance regulator. J Mol Med. 1996;74:527–534. doi: 10.1007/BF00204979. [DOI] [PubMed] [Google Scholar]
- Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet. 1996;12:325–328. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Schreiber R, Nitschke R, Mall M. Control of epithelial Na+ conductance by the cystic fibrosis transmembrane conductance regulator. Pflugers Arch. 2000;440:193–201. doi: 10.1007/s004240000255. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Li J, Dubroff R, Ahn YJ, Foskett JK, Engelhardt J, Kleyman TR. Epithelial sodium channels regulate cystic fibrosis transmembrane conductance regulator chloride channels in Xenopus oocytes. J Biol Chem. 2000;275:13266–13274. doi: 10.1074/jbc.275.18.13266. [DOI] [PubMed] [Google Scholar]
- Briel M, Greger R, Kunzelmann K. Cl-transport by cystic fibrosis transmembrane conductance regulator (CFTR) contributes to the inhibition of epithelial Na+ channels (ENaCs) in Xenopus oocytes co-expressing CFTR and ENaC. J Physiol. 1998;508(Pt 3):825–836. doi: 10.1111/j.1469-7793.1998.825bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutts MJ, Rossier BC, Boucher RC. Cystic fibrosis transmembrane conductance regulator inverts protein kinase A-mediated regulation of epithelial sodium channel single channel kinetics. J Biol Chem. 1997;272:14037–14040. doi: 10.1074/jbc.272.22.14037. [DOI] [PubMed] [Google Scholar]
- Zahm JM, Baconnais S, Davidson DJ, Webb S, Dorin J, Bonnet N, Balossier G, Puchelle E. X-ray microanalysis of airway surface liquid collected in cystic fibrosis mice. Am J Physiol Lung Cell Mol Physiol. 2001;281:L309–L313. doi: 10.1152/ajplung.2001.281.2.L309. [DOI] [PubMed] [Google Scholar]
- Jayaraman S, Joo NS, Reitz B, Wine JJ, Verkman AS. Submucosal gland secretions in airways from cystic fibrosis patients have normal [Na(+)] and pH but elevated viscosity. Proc Natl Acad Sci U S A. 2001;98:8119–8123. doi: 10.1073/pnas.131087598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S, Song Y, Vetrivel L, Shankar L, Verkman AS. Noninvasive in vivo fluorescence measurement of airway-surface liquid depth, salt concentration, and pH. J Clin Invest. 2001;107:317–324. doi: 10.1172/JCI11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui H, Davis CW, Tarran R, Boucher RC. Osmotic water permeabilities of cultured, well-differentiated normal and cystic fibrosis airway epithelia. J Clin Invest. 2000;105:1419–1427. doi: 10.1172/JCI4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- Kopito LE, Kosasky HJ, Shwachman H. Water and electrolytes in cervical mucus from patients with cystic fibrosis. Fertil Steril. 1973;24:512–516. [PubMed] [Google Scholar]
- King M. Experimental models for studying mucociliary clearance. Eur Respir J. 1998;11:222–228. doi: 10.1183/09031936.98.11010222. [DOI] [PubMed] [Google Scholar]
- Atsuta S, Majima Y. Nasal mucociliary clearance of chronic sinusitis in relation to rheological properties of nasal mucus. Ann Otol Rhinol Laryngol. 1998;107:47–51. doi: 10.1177/000348949810700109. [DOI] [PubMed] [Google Scholar]
- Sturgess J, Imrie J. Quantitative evaluation of the development of tracheal submucosal glands in infants with cystic fibrosis and control infants. Am J Pathol. 1982;106:303–311. [PMC free article] [PubMed] [Google Scholar]
- Tirouvanziam R, de Bentzmann S, Hubeau C, Hinnrasky J, Jacquot J, Peault B, Puchelle E. Inflammation and infection in naive human cystic fibrosis airway grafts. Am J Respir Cell Mol Biol. 2000;23:121–127. doi: 10.1165/ajrcmb.23.2.4214. [DOI] [PubMed] [Google Scholar]
- Tabary O, Zahm JM, Hinnrasky J, Couetil JP, Cornillet P, Guenounou M, Gaillard D, Puchelle E, Jacquot J. Selective up-regulation of chemokine IL-8 expression in cystic fibrosis bronchial gland cells in vivo and in vitro. Am J Pathol. 1998;153:921–930. doi: 10.1016/S0002-9440(10)65633-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley TJ, Drumm ML. Inducible nitric oxide synthase expression is reduced in cystic fibrosis murine and human airway epithelial cells. J Clin Invest. 1998;102:1200–1207. doi: 10.1172/JCI2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley TJ, Elmer HL. In vivo alterations of IFN regulatory factor-1 and PIAS1 protein levels in cystic fibrosis epithelium. J Clin Invest. 2000;106:403–410. doi: 10.1172/JCI9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pier GB, Grout M, Zaidi TS, Olsen JC, Johnson LG, Yankaskas JR, Goldberg JB. Role of mutant CFTR in hypersusceptibility of cystic fibrosis patients to lung infections. Science. 1996;271:64–67. doi: 10.1126/science.271.5245.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiman L, Prince A. Pseudomonas aeruginosa pili bind to asialoGM1, which is increased on the surface of cystic fibrosis epithelial cells. J Clin Invest. 1993;92:1875–1880. doi: 10.1172/JCI116779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiman L, Cacalano G, Gruenert D, Prince A. Comparison of adherence of Pseudomonas aeruginosa to respiratory epithelial cells from cystic fibrosis patients and healthy subjects. Infect Immun. 1992;60:2808–2814. doi: 10.1128/iai.60.7.2808-2814.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltimore RS, Christie CD, Smith GJ. Immunohistopathologic localization of Pseudomonas aeruginosa in lungs from patients with cystic fibrosis. Implications for the pathogenesis of progressive lung deterioration. Am Rev Respir Dis. 1989;140:1650–1661. doi: 10.1164/ajrccm/140.6.1650. [DOI] [PubMed] [Google Scholar]
- Ulrich M, Herbert S, Berger J, Bellon G, Louis D, Munker G, Doring G. Localization of Staphylococcus aureus in infected airways of patients with cystic fibrosis and in a cell culture model of S. aureus adherence. Am J Respir Cell Mol Biol. 1998;19:83–91. doi: 10.1165/ajrcmb.19.1.3137. [DOI] [PubMed] [Google Scholar]
- Sajjan U, Corey M, Humar A, Tullis E, Cutz E, Ackerley C, Forstner J. Immunolocalisation of Burkholderia cepacia in the lungs of cystic fibrosis patients. J Med Microbiol. 2001;50:535–546. doi: 10.1099/0022-1317-50-6-535. [DOI] [PubMed] [Google Scholar]
- Scheid P, Kempster L, Griesenbach U, Davies JC, Dewar A, Weber PP, Colledge WH, Evans MJ, Geddes DM, Alton EW. Inflammation in cystic fibrosis airways: relationship to increased bacterial adherence. Eur Respir J. 2001;17:27–35. doi: 10.1183/09031936.01.17100270. [DOI] [PubMed] [Google Scholar]
- DiMango E, Ratner AJ, Bryan R, Tabibi S, Prince A. Activation of NF-kappaB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis respiratory epithelial cells. J Clin Invest. 1998;101:2598–2605. doi: 10.1172/JCI2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMango E, Zar HJ, Bryan R, Prince A. Diverse Pseudomonas aeruginosa gene products stimulate respiratory epithelial cells to produce interleukin-8. J Clin Invest. 1995;96:2204–2210. doi: 10.1172/JCI118275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora SK, Ritchings BW, Almira EC, Lory S, Ramphal R. The Pseudomonas aeruginosa flagellar cap protein, FliD, is responsible for mucin adhesion. Infect Immun. 1998;66:1000–1007. doi: 10.1128/iai.66.3.1000-1007.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramphal R, Arora SK, Ritchings BW. Recognition of mucin by the adhesin-flagellar system of Pseudomonas aeruginosa. Am J Respir Crit Care Med. 1996;154(4 Pt 2):S170–S174. doi: 10.1164/ajrccm/154.4_Pt_2.S170. [DOI] [PubMed] [Google Scholar]
- Feldman M, Bryan R, Rajan S, Scheffler L, Brunnert S, Tang H, Prince A. Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect Immun. 1998;66:43–51. doi: 10.1128/iai.66.1.43-51.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind A, Bertele RM, Harms K, Horl G, Jungwirth R, Petermuller C, Przyklenk B, Weisslein-Pfister C. Qualitative and quantitative microbiological analysis of sputa of 102 patients with cystic fibrosis. Infection. 1987;15:270–277. doi: 10.1007/BF01644137. [DOI] [PubMed] [Google Scholar]
- Gilligan PH. Microbiology of airway disease in patients with cystic fibrosis. Clin Microbiol Rev. 1991;4:35–51. doi: 10.1128/cmr.4.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld M, Emerson J, Accurso F, Armstrong D, Castile R, Grimwood K, Hiatt P, McCoy K, McNamara S, Ramsey B, Wagener J. Diagnostic accuracy of oropharyngeal cultures in infants and young children with cystic fibrosis. Pediatr Pulmonol. 1999;28(2):321–328. doi: 10.1002/(sici)1099-0496(199911)28:5<321::aid-ppul3>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Pitt TL. Biology of Pseudomonas aeruginosa in relation to pulmonary infection in cystic fibrosis. J R Soc Med. 1986;79(Suppl 12):13–18. [PMC free article] [PubMed] [Google Scholar]
- Poole K. Multidrug efflux pumps and antimicrobial resistance in Pseudomonas aeruginosa and related organisms. J Mol Microbiol Biotechnol. 2001;3:255–264. [PubMed] [Google Scholar]
- Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High Frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, Birrer P, Bellon G, Berger J, Weiss T, Botzenhart K, Yankaskas JR, Randell S, Boucher RC, Doring G. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest. 2002;109:317–325. doi: 10.1172/JCI13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasil ML. Pseudomonas aeruginosa : biology, mechanisms of virulence, epidemiology. J Pediatr. 1986;108(5 Pt 2):800–805. doi: 10.1016/s0022-3476(86)80748-x. [DOI] [PubMed] [Google Scholar]
- Pier GB, Elcock ME. Nonspecific immunoglobulin synthesis and elevated IgG levels in rabbits immunized with mucoid exopolysaccharide from cystic fibrosis isolates of Pseudomonas aeruginosa. J Immunol. 1984;133:734–739. [PubMed] [Google Scholar]
- Pier GB, Takeda S, Grout M, Markham RB. Immune complexes from immunized mice and infected cystic fibrosis patients mediate murine and human T cell killing of hybridomas producing protective, opsonic antibody to Pseudomonas aeruginosa. J Clin Invest. 1993;91:1079–1087. doi: 10.1172/JCI116265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall BC, Carroll KC. Interaction between Pseudomonas aeruginosa and host defenses in cystic fibrosis. Semin Respir Infect. 1991;6:11–18. [PubMed] [Google Scholar]
- Boyd A, Chakrabarty AM. Role of alginate lyase in cell detachment of Pseudomonas aeruginosa. Appl Environ Microbiol. 1994;60:2355–2359. doi: 10.1128/aem.60.7.2355-2359.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke V, Robinson JO, Richardson CJ, Bundell CS. Longitudinal studies of virulence factors of Pseudomonas aeruginosa in cystic fibrosis. Pathology. 1991;23:145–148. doi: 10.3109/00313029109060814. [DOI] [PubMed] [Google Scholar]
- Demko CA, Byard PJ, Davis PB. Gender differences in cystic fibrosis: Pseudomonas aeruginosa infection. J Clin Epidemiol. 1995;48:1041–1049. doi: 10.1016/0895-4356(94)00230-n. [DOI] [PubMed] [Google Scholar]
- Ernst RK, Yi EC, Guo L, Lim KB, Burns JL, Hackett M, Miller SI. Specific lipopolysaccharide found in cystic fibrosis airway Pseudomonas aeruginosa. Science. 1999;286:1561–1565. doi: 10.1126/science.286.5444.1561. [DOI] [PubMed] [Google Scholar]
- Bainbridge T, Fick RB., Jr Functional importance of cystic fibrosis immunoglobulin G fragments generated by Pseudomonas aeruginosa elastase. J Lab Clin Med. 1989;114:728–733. [PubMed] [Google Scholar]
- Wretlind B, Pavlovskis OR. Pseudomonas aeruginosa elastase and its role in pseudomonas infections. Rev Infect Dis. 1983;5(Suppl):S998–S1004. doi: 10.1093/clinids/5.supplement_5.s998. [DOI] [PubMed] [Google Scholar]
- Klinger JD, Tandler B, Liedtke CM, Boat TF. Proteinases of Pseudomonas aeruginosa evoke mucin release by tracheal epithelium. J Clin Invest. 1984;74:1669–1678. doi: 10.1172/JCI111583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JC, Lucas BJ, Knee C, Renzetti M, Donahue J. Acute lung injury induced by Pseudomonas aeruginosa elastase in hamsters. Exp Lung Res. 1992;18:155–171. doi: 10.3109/01902149209020658. [DOI] [PubMed] [Google Scholar]
- Wieland CW, Siegmund B, Senaldi G, Vasil ML, Dinarello CA, Fantuzzi G. Pulmonary inflammation induced by Pseudomonas aeruginosa lipopolysaccharide, phospholipase C, and exotoxin A: role of interferon regulatory factor 1. Infect Immun. 2002;70:1352–1358. doi: 10.1128/IAI.70.3.1352-1358.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank DW. The exoenzyme S regulon of Pseudomonas aeruginosa. Mol Microbiol. 1997;26:621–629. doi: 10.1046/j.1365-2958.1997.6251991.x. [DOI] [PubMed] [Google Scholar]
- Bruno TF, Buser DE, Syme RM, Woods DE, Mody CH. Pseudomonas aeruginosa exoenzyme S is a mitogen but not a superantigen for human T lymphocytes. Infect Immun. 1998;66:3072–3079. doi: 10.1128/iai.66.7.3072-3079.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker NR, Minor V, Deal C, Shahrabadi MS, Simpson DA, Woods DE. Pseudomonas aeruginosa exoenzyme S is an adhesion. Infect Immun. 1991;59:2859–2863. doi: 10.1128/iai.59.9.2859-2863.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri JT. Pseudomonas aeruginosa exoenzyme S, a bifunctional type-III secreted cytotoxin. Int J Med Microbiol. 2000;290:381–387. doi: 10.1016/S1438-4221(00)80047-8. [DOI] [PubMed] [Google Scholar]
- Herard AL, Pierrot D, Hinnrasky J, Kaplan H, Sheppard D, Puchelle E, Zahm JM. Fibronectin and its alpha 5 beta 1-integrin receptor are involved in the wound-repair process of airway epithelium. Am J Physiol. 1996;271:L726–733. doi: 10.1152/ajplung.1996.271.5.L726. [DOI] [PubMed] [Google Scholar]
- Krall R, Sun J, Pederson KJ, Barbieri JT. In vivo rho GTPase-activating protein activity of Pseudomonas aeruginosa cytotoxin ExoS. Infect Immun. 2002;70:360–367. doi: 10.1128/IAI.70.1.360-367.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CD. Role of pyocyanin in the acquisition of iron from transferrin. Infect Immun. 1986;52:263–270. doi: 10.1128/iai.52.1.263-270.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson R, Pitt T, Taylor G, Watson D, MacDermot J, Sykes D, Roberts D, Cole P. Pyocyanin and 1-hydroxyphenazine produced by Pseudomonas aeruginosa inhibit the beating of human respiratory cilia in vitro. J Clin Invest. 1987;79:221–229. doi: 10.1172/JCI112787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwood K, Semple RA, Rabin HR, Sokol PA, Woods DE. Elevated exoenzyme expression by P. aeruginosa is correlated with exacerbations of lung disease in cystic fibrosis. Pediatr Pulmonol. 1993;15:135–139. doi: 10.1002/ppul.1950150302. [DOI] [PubMed] [Google Scholar]
- Jaffar-Bandjee MC, Lazdunski A, Bally M, Carrere J, Chazalette JP, Galabert C. Production of elastase, exotoxin A and alkaline protease in sputa during pulmonary exacerbation of cystic fibrosis in patients chronically infected by Pseudomonas aeruginosa. J Clin Microbiol. 1995;33:924–929. doi: 10.1128/jcm.33.4.924-929.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlan RM, Costerton JW. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev. 2002;15:167–193. doi: 10.1128/CMR.15.2.167-193.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature. 2000;407:762–764. doi: 10.1038/35037627. [DOI] [PubMed] [Google Scholar]
- Drenkard E, Ausubel FM. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature. 2002;416:740–743. doi: 10.1038/416740a. [DOI] [PubMed] [Google Scholar]
- Equi A, Balfour-Lynn I, Bush A, Rosenthal M. Long term azithromycin in children with cystic fibrosis: a randomised, placebo-controlled crossover trial. Lancet. 2002;360:978. doi: 10.1016/s0140-6736(02)11081-6. [DOI] [PubMed] [Google Scholar]
- Wolter J, Seeney S, Bell S, Bowler S, Masel P, McCormack J. Effect of long term treatment with azithromycin on disease parameters in cystic fibrosis: a randomised trial. Thorax. 2002;57:212–216. doi: 10.1136/thorax.57.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coenye T, Vandamme P, Govan JR, LiPuma JJ. Taxonomy and identification of the Burkholderia cepacia complex. J Clin Microbiol. 2001;39:3427–3436. doi: 10.1128/JCM.39.10.3427-3436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LiPuma JJ. Burkholderia cepacia. Management issues and new insights. Clin Chest Med. 1998;19:473–486. doi: 10.1016/s0272-5231(05)70094-0. [DOI] [PubMed] [Google Scholar]
- Zughaier SM, Ryley HC, Jackson SK. Lipopolysaccharide (LPS) from Burkholderia cepacia is more active than LPS from Pseudomonas aeruginosa and Stenotrophomonas maltophilia in stimulating tumor necrosis factor alpha from human monocytes. Infect Immun. 1999;67:1505–1507. doi: 10.1128/iai.67.3.1505-1507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JE, Stewart J, Barclay GR, Govan JR. Priming of neutrophil respiratory burst activity by lipopolysaccharide from Burkholderia cepacia. Infect Immun. 1997;65:4281–4287. doi: 10.1128/iai.65.10.4281-4287.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry J, Elborn JS, Nixon L, Shale DJ, Webb AK. Cystic fibrosis: Inflammatory response to infection with Burkholderia cepacia and Pseudomonas aeruginosa. Eur Respir J. 1999;14:435–438. doi: 10.1034/j.1399-3003.1999.14b32.x. [DOI] [PubMed] [Google Scholar]
- Noah TL, Black HR, Cheng PW, Wood RE, Leigh MW. Nasal and bronchoalveolar lavage fluid cytokines in early cystic fibrosis. J Infect Dis. 1997;175:638–647. doi: 10.1093/infdis/175.3.638. [DOI] [PubMed] [Google Scholar]
- Muhlebach MS, Stewart PW, Leigh MW, Noah TL. Quantitation of inflammatory response to bacteria in young cystic fibrosis and control patients. Am J Respir Crit Care Med. 1999;160:186–191. doi: 10.1164/ajrccm.160.1.9808096. [DOI] [PubMed] [Google Scholar]
- Khan TZ, Wagener JS, Bost T, Martinez J, Accurso FJ, Riches DWH. Early pulmonary inflammation in infants with cystic fibrosis. Am J Respir Crit Care Med. 1995;151:1075–1082. doi: 10.1164/ajrccm/151.4.1075. [DOI] [PubMed] [Google Scholar]
- Armstrong DS, Grimwood K, Carlin JB, Carzino R, Gutierrez JP, Hull J, Olinsky A, Phelan EM, Robertson CF, Phelan PD. Lower airway inflammation in infants and young children with cystic fibrosis. Am J Respir Crit Care Med. 1997;156:1197–1204. doi: 10.1164/ajrccm.156.4.96-11058. [DOI] [PubMed] [Google Scholar]
- Bonfield TL, Panuska JR, Konstan MW, Hillard KA, Hillard JB, Ghnaim H, Berger M. Inflammatory cytokines in cystic fibrosis lungs. Am J Respir Crit Care Med. 1995;152:2111–2118. doi: 10.1164/ajrccm.152.6.8520783. [DOI] [PubMed] [Google Scholar]
- Konstan MW, Walenga RW, Hilliard KA, Hilliard JB. Leukotriene B4 is markedly elevated in the epithelial lining fluid of patients with cystic fibrosis. Am Rev Respir Dis. 1993;148:896–901. doi: 10.1164/ajrccm/148.4_Pt_1.896. [DOI] [PubMed] [Google Scholar]
- Bonfield TL, Konstan MW, Burfeind P, Panuska JR, Hillard JB, Berger M. Normal bronchial epithelial cells constitutively produce the anti-inflammatory cytokine interleukin-10, which is downregulated in cystic fibrosis. Am J Respir Cell Mol Biol. 1995;13:257–261. doi: 10.1165/ajrcmb.13.3.7544594. [DOI] [PubMed] [Google Scholar]
- Bonfield TL, Konstan MW, Berger M. Altered respiratory epithelial cell cytokine production in cystic fibrosis. J Allergy Clin Immunol. 1999;104:72–78. doi: 10.1016/s0091-6749(99)70116-8. [DOI] [PubMed] [Google Scholar]
- McElvaney NG, Nakamura H, Birrer P, Hebert CA, Wong WL, Alphonso M, Baker JB, Catalano MA, Crystal RG. Modulation of airway inflammation in cystic fibrosis: In vivo suppression of interleukin-8 levels on the respiratory epithelial surface by aerosolization of recombinant secretory leukoprotease inhibitor. J Clin Invest. 1992;90:1296–1301. doi: 10.1172/JCI115994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura H, Yoshimura K, McElvaney NG, Crystal RG. Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in human bronchial epithelial cell line. J Clin Invest. 1992;89:1478–1484. doi: 10.1172/JCI115738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerhoff CP, Nadel JA, Basbaum CB, Caughey GH. Neutrophil elastase and cathepsin G stimulate secretion from cultured bovine airway gland serous cells. J Clin Invest. 1990;85:682–689. doi: 10.1172/JCI114492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster A, Ueki I, Nadel JA. Neutrophil elastase stimulates tracheal submucosa gland secretion that is inhibited by ICI 200,355. Am J Physiol. 1992;262:L86–L91. doi: 10.1152/ajplung.1992.262.1.L86. [DOI] [PubMed] [Google Scholar]
- Tegner H, Ohlsson K, Toremalm NG, von Mecklenbeurg C. Effect of human leukocyte enzymes on tracheal mucosa and mucociliary activity. Rhinology. 1979;17:199–206. [PubMed] [Google Scholar]
- Tosi MF, Berger M. Functional differences between the 40 kDa and 50 to 70 kDa IgG Fc receptors on human neutrophils revealed by elastase treatment and antireceptor antibodies. J Immunol. 1988;141:2097–2103. [PubMed] [Google Scholar]
- Tosi MF, Zakem H. Surface expression of Fc gamma receptor III (CD 16) on chemoattractant-stimulated neutrophils is determined by both surface shedding and translocation from intracellular storage compartments. J Clin Invest. 1992;90:462–470. doi: 10.1172/JCI115882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi MF, Zakem H, Berger M. Neutrophil elastase cleaves C3bi on opsonized pseudomonas as well as CR1 on neutrophils to create a functionally important opsonin receptor mismatch. J Clin Invest. 1990;86:300–308. doi: 10.1172/JCI114699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger M, Sorensen RU, Tosi MF, Dearborn DG, Doring G. Complement receptor expression on neutrophils at an inflammatory site, the pseudomonas-infected lung in cystic fibrosis. J Clin Invest. 1989;84:1302–1313. doi: 10.1172/JCI114298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrer P, McElvaney NG, Rudeberg A, Sommer CW, Liechti-Gallati S, Kraemer R, Hubbard R, Crystal RG. Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am J Respir Crit Care Med. 1994;150:207–213. doi: 10.1164/ajrccm.150.1.7912987. [DOI] [PubMed] [Google Scholar]
- Velsor LW, van Heeckeren A, Day BJ. Antioxidant imbalance in the lungs of cystic fibrosis transmembrane conductance regulator protein mutant mice. Am J Physiol Lung Cell Mol Physiol. 2001;281:L31–L38. doi: 10.1152/ajplung.2001.281.1.L31. [DOI] [PubMed] [Google Scholar]