

Abstract
A novel access to phosphonic acids via Pd-catalyzed tandem carbon-phosphorus bond formation – oxidation processes was developed. The procedures involve atom-economical and environmentally friendly functionalization reactions of hypophosphorous acid (H3PO2) and H-phosphinic acids [RP(O)(OH)(H)].
Keywords: phosphonic acid, H-phosphinic acid, hypophosphorous acid
Over the last few years, the quest for synthetic efficiency has gained remarkable importance, partly due to the need to reduce waste.1 Given the increasing biological and synthetic impact of organophosphorus compounds,2 it has become crucial to develop atom-economical methods that address the functionalization of P-H bonds from basic feedstocks. Homogeneous catalysis is an ever increasingly useful and versatile alternative for the construction of C-P bonds.3 Indeed, metal-catalyzed P-H bond activation has already shown some potential in the synthesis of various phosphines (free and protected),4 phosphonates,5 phosphine oxides,6 phosphinates,7 and more recently H-phosphinates.8 Our laboratory has demonstrated that in spite of being powerful reducing agents, hypophosphorous compounds (ROP(O)H2) can effectively participate in metal-catalyzed processes involving C-P bond formation, leading to H-phosphinic acids and their derivatives.9 Of particular relevance are the Pd-catalyzed addition of hypophosphorous acid (H3PO2) across unsaturated carbon linkages,8a–b and the Pd-catalyzed dehydrative allylic substitution of H3PO2 with allylic alcohols,10 which are both unique and highly atom-efficient transformations. H-phosphinic acids [RP(O)(OH)(H)] are characterized by the presence of a phosphinylidene [P(O)(H)] moiety that works as a bridge between the P(V) and P(III) forms via a tautomeric equilibrium,11 and provides them with a unique versatility for further functionalization.8c,12
Guided by the prospect of exploiting the flexibility of H-phosphinates as synthons for the preparation of other organophosphorus compounds, we became interested in developing new reactions that involve formal P-H bond activation processes, particularly those which can fulfill “green chemistry” requirements.13 In this regard, we discovered that H-phosphinates were sensitive towards oxidation into phosphonic acids, particulary under an air atmosphere and in the presence of catalytic amounts of transition metals. Since phosphonic acids have been extensively studied as phosphate analogs in catalytic enzymatic pathways, and their biological, biochemical, and synthetic importance are diverse,14 a convenient direct synthesis of these important compounds is highly desirable.
The literature syntheses of phosphonic acids generally involve the preparation of the phosphonate alkyl esters, which requires an additional deprotection step by treatment with trimethylsilyl bromide,15 or acidic hydrolysis at high temperatures.16 However, surprisingly few methods for their direct preparation via C-P bond formation have been described, such as the low-yielding free-radical mediated addition of phosphorous acid (H3PO3) to olefins,17 where polymerization is difficult to avoid.17,18 Another viable approach to phosphonic acids uses tris(trimethylsilyl) phosphite [P(OTMS)3] in Arbuzov reactions with electrophiles (i.e. iodides, bromides, triflates, aziridine and oxazolidine ring-opening, etc),19 as well as in Ni-catalyzed cross-couplings with aryl iodides and bromides.20 This approach is not convenient because it requires strict anhydrous conditions and wasteful silylation. Furthermore, the synthesis of pure P(OTMS)3 is cumbersome.19a On the other hand, phosphonic acids have also been previously prepared from H-phosphinic acids, however the strong oxidizing agents and/or harsh conditions that are usually required are major drawbacks,12a,21 along with the fact that, until recently, few effective synthetic methodologies for H-phosphinates were available.22
Initially, we studied a simultaneous P-H bond functionalization starting from H3PO2. This consisted of a one pot, one step hydrophosphinylation8a – oxidation process (Scheme 1), avoiding in this way the isolation of the intermediate H-phosphinic acid 1.
Scheme 1.
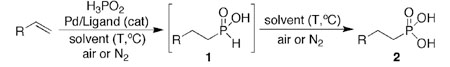
A series of experiments was performed in order to investigate the influence of the catalyst, solvent, temperature, air and water in the outcome of the reaction (Table 1). In a typical procedure, a DMF solution of freshly concentrated H3PO2 was heated with 1-octene in the presence of Pd2dba3 (1 mol%) and xantphos (2 mol%) under an air atmosphere. At 110°C, we observed a smooth conversion of the in situ formed octyl-H-phosphinic acid 3 into the corresponding phosphonic acid 4 (entry 1), along with minor amounts of H3PO3 and H3PO4. The use of undried, reagent grade DMF (110°C) proved to be optimum, and a decrease in the temperature and/or replacement of DMF by CH3CN significantly slowed down the oxidation step (entries 2 and 3). When the reaction was performed with anhydrous DMF under a nitrogen atmosphere, H-phosphinic acid 3 was the main product (entry 4). However, the presence of oxygen completely reverses this behavior and leads mainly to the oxidized product 4 (entry 6). As expected, the C-P bond-forming step proceeds very inefficiently with H3PO2 in the absence of Pd catalyst (entry 7). Other experiments probed the influence of stoichiometry and catalyst loading (entries 8 and 9). It was found that 1.5 – 2 equiv of H3PO2 and 2 mol% of Pd provide excellent yields of the phosphonic acid 4 (entry 8), while lowering the amount of catalyst markedly decreases the yield of 4, and H-phosphinic acid 3 becomes the exclusive product (entry 9). This, along with control experiments performed on isolated H-phosphinic acids,23 supports an active role of the Pd catalyst in the oxidation step. Remarkably, the reaction still proceeds in excellent yield when conducted with the commercially available aqueous solution of H3PO2 (50 wt. %) (entry 10). It should be noted that the use of concentrated versus aqueous H3PO2 did not show a significant impact on the formation of 4 (entry 4 vs. 5), contrary to the presence of nitrogen versus air in the reaction atmosphere (entries 4 vs. 6 and 5 vs. 10). Additionally, the concentration of the reactants also plays a key role, and 0.5 to 1 M appears to be optimum.
Table 1.
Optimization of the tandem hydrophosphinylation – oxidation of 1-octenea

| entry | H3PO2b aq/concd (equiv) | Pd/L (mol %) | air/N2 | solvent | T (°C) |
31P NMR and/or [isolated]c yields (%)
|
|
|---|---|---|---|---|---|---|---|
| 3 | 4 | ||||||
| 1 | concd (1) | 2 | air | DMF | 110 | 0 | 74
[72] |
| 2 | concd (1) | 2 | air | DMF | 85 | 34 | 53 |
| 3 | concd (1) | 2 | air | CH3CN | 82 | 46 | 34 |
| 4 | concd (1) | 2 | N2 | DMF (anh)d | 110 | 82 | 10 |
| 5 | aq (1) | 2 | N2 | DMF | 110 | 45 | 31 |
| 6 | concd (1) | 2 | air | DMF (anh)d,e | 110 | 13 | 77 |
| 7 | concd (1) | 0 | air | DMF | 110 | 6 | 0 |
| 8 | Concd (1.5–2) | 2 | air | DMF | 110 | 0 | 100
[100] |
| 9 | concd (2) | 0.05 | air | DMF | 110 | 100 | 0 |
| 10 | aq (2) | 2 | air | DMF | 110 | 0 | 100
[100] |
Unless otherwise noted, reactions were conducted in a one-step mode, using reagent grade, undried solvents [0.5 M]. Reaction times: 20–24 h.
Concentrated H3PO2 was obtained by rotary evaporation of the commercial aqueous solution (50 wt. %). See Supplementary data for details.
Isolated yield after extractive workup.
Freshly vacuum-distilled DMF (from CaH2).
Drierite trap was used to avoid moisture from air.
Pursuant to the design of a more general tandem C-P bond formation – oxidation process using H3PO2 as precursor, other Pd-catalyzed reactions that lead to H-phosphinic acids were conducted under the optimized conditions for the hydrophosphinylation-oxidation of 1-octene (Method A). Various unsaturated substrates, allylic alcohols and aryl halides were examined in hydrophosphinylation,8a allylation10 and cross-coupling reactions,24 respectively. However, we discovered that the presence of oxygen during the C-P bond-forming step was not always optimum in all cases. Therefore a stepwise process (Method B) where the H-phosphinic acid is preformed under our published conditions (under N2, with concentrated H3PO2) followed by in situ Pd-catalyzed oxidation (under air) provided overall better yields of products than the simultaneous version (Table 2). In accordance with the literature, Pd2dba3/xantphos (2 mol% Pd) worked efficiently as a catalytic system for hydrophosphinylation and allylation reactions (entries 1–8), while Pd(OAc)2/dppp (2 mol%) with Et3N as base (3 eq) proved successful in cross-coupling (entries 9–11). The products were generally isolated in moderate to good yields by an aqueous extractive workup and, if required, recrystallization was performed (entries 9–11). However, N-containing phosphonic acids (entries 4–5) are soluble in water and ion-exchange chromatography was required for their isolation.
Table 2.
Synthesis of phosphonic acids via tandem C-P bond formation – oxidation reactions

| entry | subsrate | methoda | catalyst | base (equiv) | product | 1st step time (h) | 2nd step time (h) | isolated yield,b [NMR yield] (%) |
|---|---|---|---|---|---|---|---|---|
| 1a |
|
A | Pd2dba3/xantphos | - |

|
20 | - | 100 |
| 1b | B | 12 | 24 | 100 | ||||
| 2a |

|
A | Pd2dba3/xantphos | - |

|
50 | - | 81 |
| 2b | B | 12 | 64 | 97 | ||||
| 3 |
|
A | Pd2dba3/xantphos | - |

|
50 | - | 95 |
| 4 |
|
B | Pd2dba3/xantphos | - |
|
12 | 50 | 86c |
| 5 |
|
B | Pd2dba3/xantphos | - |

|
12 | 20 | [81] |
| 6 |
|
B | Pd2dba3/xantphos | - |

|
12 | 50 | 91 |
| 7 |
|
B | Pd2dba3/xantphos | - |

|
12 | 22 | 89 |
| 8 |

|
B | Pd2dba3/xantphos | - |

|
15 | 50 | [92] |
| 9 |

|
B | Pd(OAc)2/dppp | Et3N (3) |

|
15 | 22 | 82d |
| 10 |
|
B | Pd(OAc)2/dppp | Et3N (3) |
|
15 | 22 | 55d |
| 11 |
|
B | Pd(OAc)2/dppp | Et3N (3) |
|
15 | 42 | 52d |
See Supplementary data for details of the procedures. Method A: one step, 2 equiv 50% aqueous H3PO2, DMF, 110°C, air. Method B: two steps, 2 equiv concentrated H3PO2; 1st step: P-C bond formation: DMF, 85°C, N2; 2nd step: oxidation: DMF, 110°C, air.
Unless otherwise noted, products were isolated in good purity (>95%) by a simple extractive workup.
Purified by ion-exchange chromatography.
Purified by recrystallization.
Even though the mechanism of this transformation is not completely clear at this time, it could proceed either, via a direct P-H bond activation where the palladium inserts into the P-H bond of the H-phosphinic acid3b–c,3f or, most probably, through an indirect process where the metal complex first activates molecular oxygen present in the solution forming a highly reactive species (as a radical), that in turn reacts with the H-phosphinic acid.25 Further work will be required to delineate the mechanism of the reaction. In summary, an efficient step- and atom-economical method to access phosphonic acids was developed.
Supplementary Material
Supplementary data
Representative experimental procedures and spectroscopic data. The supplementary data is available online with the paper in Science Direct.
Acknowledgments
We gratefully acknowledge the National Institute of General Medical Sciences/NIH (1R01 GM067610), and the Chemistry Department at Texas Christian University for the financial support of this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.For reviews, see: Trost BM. Science. 1991;254:1471–1477. doi: 10.1126/science.1962206.Trost BM. Acc Chem Res. 2002;35:695–705. doi: 10.1021/ar010068z.
- 2.For general references, see: Quin LD. A Guide to Organophosphorus Chemistry. Wiley; New York: 2000. Hartley FR, editor. The Chemistry of Organophosphorus Compounds. Vol. 4 Wiley; New York: 1996. Corbridge DE. Chemistry, Biochemistry and Technology. Elsevier; Amsterdam: 2000. Phosphorus 2000.Engel R, Cohen JI. Synthesis of Carbon-Phosphorus Bonds. 2. CRC Press; Boca Raton: 2003. Engel R, editor. Handbook of Organophosphorus Chemistry. Marcel Dekker; New York: 1992.
- 3.For recent reviews on metal-catalyzed C-P bond formation, see: Schwan A. Chem Soc Rev. 2004;33:218–224. doi: 10.1039/b307538a.Tanaka M. Top Curr Chem. 2004;232:25–54.Alonso F, Beletskaya IP, Yus M. Chem Rev. 2004;104:3079–3159. doi: 10.1021/cr0201068.Baillie C, Xiao J. Curr Org Chem. 2003;7:477–514.Beletskaya IP, Kazankova MA. Russ J Org Chem. 2002;38:1391–1430.Wicht DK, Glueck DS. In: In Catalytic Heterofunctionalization. Togni A, Grützmacher H, editors. Wiley-VCH; Weinheim: 2001. pp. 143–170. and references therein.
- 4.For a review on hydrophosphination that includes catalytic methods for P-H bond activation: Delacroix O, Gaumont AC. Curr Org Chem. 2005;9:1851–1882. For examples of metal-catalyzed hydrophosphination: Komeyama K, Kobayashi D, Yamamoto Y, Takehira K, Takaki K. Tetrahedron. 2006;62:2511–2519.Join B, Mimeau D, Delacroix O, Gaumont AC. Chem Commun. 2006:3249–3251. doi: 10.1039/b607434k.Shulyupin MO, Trostyanskaya IG, Kazanova MA, Beletskaya IP. Russ J Org Chem. 2006;42:17–22.Ohmiya H, Yorimitsu H, Oshima K. Angew Chem Int Ed. 2005;44:2368–2370. doi: 10.1002/anie.200500255.Kawaoka AM, Douglass MR, Marks TJ. Organometallics. 2003;22:4630–4632.Jérôme F, Monnier F, Lawicka H, Dérien S, Dixneuf PH. Chem Commun. 2003:696–697. doi: 10.1039/b212408d.Wicht DK, Kourkine IV, Kovacik I, Glueck DS, Concolino TE, Yap GPA, Incarvito CD, Rheingold AL. Organometallics. 1999;18:5381–5394.Costa E, Pringle PG, Worboys K. Chem Commun. 1998:49–50.
- 5.For examples of metal-catalyzed hydrophosphorylation, see: Han LB, Tanaka M. J Am Chem Soc. 1996;118:1571–1572.Han LB, Mirzaei F, Zhao CQ, Tanaka M. J Am Chem Soc. 2000;122:5407–5408.Zhao CQ, Han LB, Goto M, Tanaka M. Angew Chem Int Ed. 2001;40:1929–1932.Zhao CQ, Han LB, Tanaka M. Organometallics. 2000;19:4196–4198.Reichwein JF, Patel MC, Pagenkopf BL. Org Lett. 2001;3:4303–4306. doi: 10.1021/ol016989r.Reichwein JF, Pagenkopf BL. J Org Chem. 2003;68:1459–1463. doi: 10.1021/jo026834c.Levine AM, Stockland RA, Jr, Clark R, Guzei I. Organometallics. 2002;21:3278–3284.Stockland RA, Jr, Levine AM, Giovine MT, Guzei IA, Cannistra JC. Organometallics. 2004;23:647–656.Shulyupin MO, Francio G, Beletskaya IP, Leitner W. Adv Synth Catal. 2005;347:667–672.
- 6.For examples of metal-catalyzed synthesis of phosphine oxides via hydrophosphinylation, see: Han LB, Choi N, Tanaka M. Organometallics. 1996;15:3259–3261.Han LB, Hua R, Tanaka M. Angew Chem, Int Ed. 1998;37:94–96.Han LB, Zhao CQ, Tanaka M. J Org Chem. 2001;66:5929–5932. doi: 10.1021/jo010337z.Allen A, Jr, Ma L, Lin W. Tetrahedron Lett. 2002;43:3707–3710.Van Rooy S, Cao C, Patrick BO, Lam A, Love JA. Inorg Chim Acta. 2006;359:2918–2923.Niu M, Fu H, Jian Y, Zhao Y. Chem Commun. 2007:272–274. doi: 10.1039/b613416e.
- 7.For examples of metal-catalyzed synthesis of phosphinates via hydrophosphinylation, see: Han L-B, Zhao C-Q, Onozawa S-y, Goto M, Tanaka M. J Am Chem Soc. 2002;124:3842–3843. doi: 10.1021/ja025816+.Han LB, Zhang C, Yazawa H, Shimada S. J Am Chem Soc. 2004;126:5080–5081. doi: 10.1021/ja0494297.Han LB, Ono Y, Yazawa H. Org Lett. 2005;7:2909–2911. doi: 10.1021/ol0508431.
- 8.For examples of metal-catalyzed synthesis of H-phosphinates via hydrophosphinylation, see: Deprèle S, Montchamp JL. J Am Chem Soc. 2002;124:9386–9387. doi: 10.1021/ja0261978.Deprèle S, Montchamp JL. Org Lett. 2004;6:3805–3808. doi: 10.1021/ol0484198.Ribière P, Bravo-Altamirano K, Antczak M, Hawkins J, Montchamp JL. J Org Chem. 2005;70:4064–4072. doi: 10.1021/jo050096l.
- 9.For a review, see: Montchamp JL. J Organomet Chem. 2005;690:2388–2406.
- 10.Bravo-Altamirano K, Montchamp JL. Org Lett. 2006;8:4169–4171. doi: 10.1021/ol061828e. [DOI] [PubMed] [Google Scholar]
- 11.Stawinski J, Kraszewski A. Acc Chem Res. 2002;35:952–960. doi: 10.1021/ar010049p. [DOI] [PubMed] [Google Scholar]
- 12.For examples of functional group interconversion related to H-phosphinic acids, see: Frank AW. Chem Rev. 1961;60:389–424.Nifant’ev EE. Russ Chem Rev. 1978;47:835.Yudelevich VI, Sokolov LB, Ionin BI. Russ Chem Rev. 1980;49:46.Frank AW. In: Organic Phosphorus Compounds. Chapter 10 Kosolapoff GM, Maier L, editors. Vol. 4. Wiley; New York: 1972. Soulier E, Clement J-C, Yaouanc J-J, Des Abbayes H. Tetrahedron Lett. 1998;39:4291–4294.Anderson NG, Ciaramella BM, Feldman AF, Lust DA, Moniot JL, Moran L, Polomski RE, Wang SSY. Org Process Res Dev. 1997;1:211–216.
- 13.Van Aken K, Strekowski L, Patiny L. Beilstein J Org Chem. 2006;2:3. doi: 10.1186/1860-5397-2-3. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selected references: Savignac P, Iorga B. Modern Phosphonate Chemistry. CRC Press; Boca Raton: 2003. Engel R. Chem Rev. 1977;77:349–367.Palacios F, Alonso C, de los Santos JM. Chem Rev. 2005;105:899–931. doi: 10.1021/cr040672y.Ma JA. Chem Soc Rev. 2006;35:630–636. doi: 10.1039/b517100h.Kafarski P, Lejczak B. Phosphorus, Sulfur Silicon Related Elem. 1991;63:193–215.Dembitsky VM, Al Quntar AAA, Haj-Yehiaa A, Srebnik M. Mini-Rev Org Chem. 2005;2:91–109.Romanenko VD, Kukhar VP. Chem Rev. 2006;106:3868–3935. doi: 10.1021/cr051000q.Moonen K, Laureyn I, Stevens CV. Chem Rev. 2004;104:6177–6215. doi: 10.1021/cr030451c.Blackburn MG. Chem Ind (London) 1981:134–144.Krise JP, Stella VJ. Adv Drug Delivery Rev. 1996;19:287–310.Flett DS. J Organomet Chem. 2005;690:2426–2438.Fields SC. Tetrahedron. 1999;55:12237–12273.
- 15.McKenna CE, Higa MT, Cheung NH, McKenna MC. J Tetrahedron Lett. 1977:155–158. [Google Scholar]
- 16.Kosolapoff GM. J Am Chem Soc. 1945;67:1180–1182. doi: 10.1021/ja01228a066. [DOI] [PubMed] [Google Scholar]
- 17.Griffin CE, Wells HJ. J Org Chem. 1959;24:2049–2051. [Google Scholar]
- 18.Griffin CE. J Org Chem. 1960;25:665–666. [Google Scholar]
- 19.For examples, see: Sekine M, Okimoto K, Yamada K, Hata T. J Org Chem. 1981;46:2097–2107.Lazukina LA, Kukhar VP. Zh Obshch Khim. 1988;58:939–940.Nifant’ev EE, Popkova TN, Kukhareva TS, Bekker AR. Zh Obshch Khim. 1988;58:1550–1557.Vaghefi MM, Bernacki RJ, Dalley NK, Wilson BE, Robins RK. J Med Chem. 1987;30:1383–1391. doi: 10.1021/jm00391a020.Bhattacharya AK, Stolz F, Kurzeck J, Rüger W, Schmidt RR. Can J Chem. 2002;80:973–982. doi: 10.1016/s0968-0896(01)00371-6.Wedel M, Walter A, Montforts FP. Eur J Org Chem. 2001:1681–1687.
- 20.Demik NN, Kabachnik MM, Novikova ZS, Beletskaya IP. Izv Akad Nauk SSSR, Ser Khim. 1992;10:2432–2435. [Google Scholar]
- 21.H2O2: Andreev NA, Grishina ON, Enikeev KM. Zh Obshch Khim. 1982;52:1530–1537.H2O2/SO2: Barton DHR, Vonder Embse RA. Tetrahedron. 1998;54:12475–12496.I2/DMSO: Albouy D, Brun A, Munoz A, Etemad-Moghadam G. J Org Chem. 1998;63:7223–7230. doi: 10.1021/jo9805551.HgCl2 or Br2/H2O: Baylis E Keith, Campbell CD, Dingwall JG. J Chem Soc, Perkin Trans 1. 1984:2845–2853.Fastrez J, Jespers L, Lison D, Renard M, Sonveaux E. Tetrahedron Lett. 1989;30:6861–6684.HNO3: Guichard F. Chem Ber. 1899;32:1572–1581.I2/HI(AcOH) or HI: Hatt HH. J Chem Soc. 1933:776–786.Albouy D, Etemad-Moghadam G, Koenig M. Eur J Org Chem. 1999:861–868. doi: 10.1021/jo9622441.Zhukov YN, Khomutov AR, Osipova TI, Khomutov RM. Russ Chem Bul. 1999;48:1348–1351.CCl4/Et3N/H2O: Belakhov VV, Yudelevich VI, Ionin BI. Zh Obshch Khim. 1986;56:1749–1755.AcOH: Vysotskii VI, Levan’kov SV. Zh Obshch Khim. 1991;61:1315–1321.NaIO4: Karanewsky DS, Badia MC. Tetrahedron Lett. 1986;27:1751–1754.Thermolysis (250°C): Andreev NA, Grishina ON. Zh Obshch Khim. 1981;51:1743–1747.Kinetic and mechanistic studies of the oxidation of phenyl-H-phosphinic acid: Moondra A, Mathur A, Banerji KK. J Chem Soc, Dalton Trans. 1990:2697–2700.Sharma K, Mehrotra RN. Ind J Chem, Sect A. 2002;41A:270–278.Sharma K, Prakash A, Mehrotra RN. Bull Chem Soc Jpn. 1989;62:4009–4015.
- 22.Montchamp JL. Speciality Chemicals Magazine. 2006;26:44–46. [Google Scholar]
- 23.Pure H-phosphinic acids (i.e. alkyl- and allylic-) underwent complete oxidation into phosphonic acids when heated in DMF (under air) with catalytic amounts of palladium.
- 24.Montchamp JL, Dumond YR. J Am Chem Soc. 2001;123:510–511. [Google Scholar]
- 25.Shilov AE, Shul’pin GB. Chem Rev. 1997;97:2879–2932. doi: 10.1021/cr9411886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Representative experimental procedures and spectroscopic data. The supplementary data is available online with the paper in Science Direct.