

Abstract
The reaction of carboxylic acids with carbodiimides is reviewed, and an “introverted” carboxylic acid is proposed as a means of trapping reactive intermediates along the reaction pathway. The introverted acid is a cavitand with the carboxylic function directed toward the floor of the cavity. Its reaction with diisopropyl carboodiimide gives a covalent adduct that is either the elusive O-acylisourea or the commonly encountered N-acylurea.
Keywords: Introverted Acid, Carbodiimide, N-acylurea, O-acylisourea, Reaction Mechanism
Introduction
Typical carboxylic acids react with dicyclohexylcarbodiimide (DCC) and amines to give amides, (Scheme 1) in a process so broadly used in peptide synthesis, particularly on the solid phase, that its importance would be hard to overstate. The terminal acylating agent (that which reacts with the amine) likely varies with the concentrations and the components in the specific system,1,2,3 but the initial acylating agent is undoubtedly the O-acylisourea.4,5 Support for this intermediate comes from kinetic methods and stereochemical probes,6 rather than direct observation. A report involving direct NMR observation7 was subsequently shown to be a misassignment,8 but an old claim of TLC separation of an O-acylisourea persists,9 and a more recent publication describes the crystallization of a series of O-acylisoureas from coumarin-3-carboxylic acid derivatives and DCC. 10
Scheme 1.
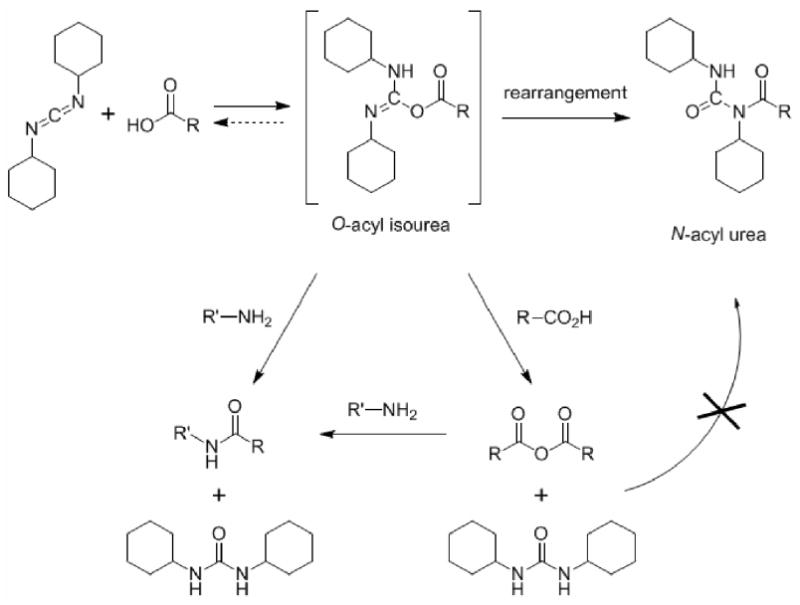
Typical reactions of carboxylic acids and amines with dicyclohexylcarbodiimide.
A number of factors make the O-acylisourea elusive: its formation may be reversible, limiting its steady state concentration to a vanishingly small one; its rearrangement to the N-acyl urea4 – a pesky side product frequently encountered in peptide synthesis – provides another pathway for its disappearance. This stable product arises from conventional carboxylic acids and DCC by what appears to be an intramolecular O-to-N acyl rearrangement. But even if the rearrangement to the N-acylurea is suppressed, the indiscriminate acylation of any available nucleophiles by the O-acylisourea insures its rapid disappearance. In the absence of added nucleophiles, the carboxylic acid itself is, of course, present and the symmetrical anhydride is formed. DeTar and Silverstein4 showed that the N-acylurea is readily formed at higher temperatures and established that it does not arise from the acylation of the urea with the symmetrical anhydride.
In other studies Benoiton11 obtained high yields of N-acylureas from the reaction of symmetrical anhydrides of BOC-protected amino acids with carbodiimides (Scheme 2).
Scheme 2.
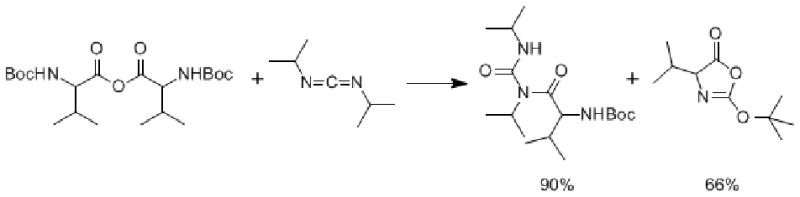
N-acylureas from the reaction of symmetrical anhydrides of BOC-protected amino acids with carbodiimides
We have been intrigued with the behavior of the reactive intermediates along this pathway for some time and report here our efforts to trap them with the aid of an unconventional carboxylic acid. Carboxylic acids are especially difficult to place in a sterically demanding environment; their oxygens are exposed and convoluted molecular architectures must be created to bring intramolecular elements near them in space. Cleft-like structures with convergent functional groups are used in molecular recognition studies and offer models for naturally-occurring receptors such as enzyme active sites. 12,13,14,15,16,17 The introverted acid 18,19,20 1 (Fig. 1) is a promising example, and its structure hints at its unique properties. The oxygens are in a well-defined cavity surrounded by four aromatic walls, held in place by intramolecular hydrogen bonds. The carboxyl O-H bond is directed toward the floor of the cavity, and is trained on small molecules held inside. The inversion/rotation rates of amines inside the cavity are profoundly slowed, and the isolation of the acid and bases in a hydrophobic environment results in interactions that resemble those in protein interiors. The cavity as a molecular host has a volume of ~280 A3 in which a single guest enjoys a uniquely high (~6 Molar) concentration. Considerable energetic barriers including the rupture of hydrogen bonds must be overcome to allow passage to and from the cavity. Its isolation has been used to control the steric environment around the carboxylic acid – a derivative of Kemp’s triacid.21 This positioning slows some of its reactions,22 or precludes them altogether: a symmetrical anhydride of the acid, for example, is hard even to imagine. These properties – probably not found elsewhere in the world of carboxylic acids – tempted us to examine the acid’s reaction with a carbodiimide; to trap the O-acylisourea intermediate.
Figure 1.
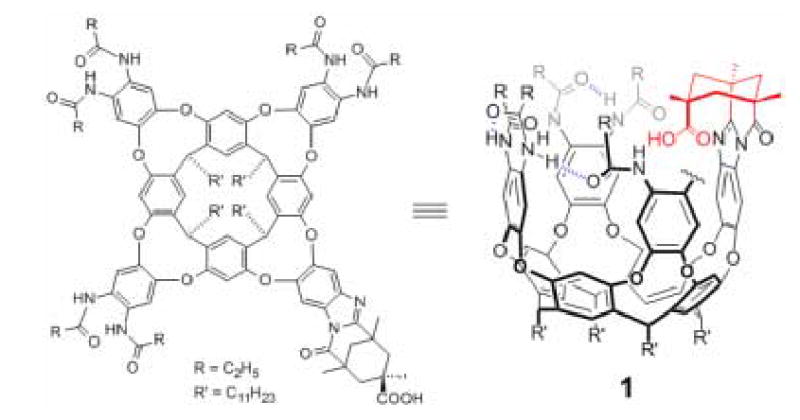
The introverted acid 1.
Results and Discussion
In the experiment, the reaction of acid 1 with diisopropylcarbodiimide at millimolar concentrations was very slow at room temperature (Scheme 3). The overnight reaction at 80 °C in C6D6 or C9D12 (deuterated mesitylene) barely proceeded to give the addition product. The stable product was isolated by evaporation of volatiles and washing with hexane. The electrospray ionization (ESI) mass spectrum provided clear evidence for the covalent adduct O-(or N) acylurea (m/z 2188, MH+; m/z 1095, [MH2]2+) and four methyl groups were seen in the upfield region of the 1H NMR spectrum (Fig. 2). The two doublet methyls were situated at δ -2.35 (d, J = 6.0 Hz, 3H) and -2.87 (d, J = 6.0 Hz, 3H), the other two broad-singlet methyls were situated at δ -2.29 (broad, 3H) and -2.63 (broad, 3H). Since 1 is chiral, the two methyls of each isopropyl group are diastereotopic, and the aromatic walls that line the cavity create an anisotropy that causes NMR signals of the guest species to shift upfield (note that they appear upfield of tetramethylsilane). The IR spectrum showed no trace of the N=C=N stretch but was otherwise uninformative; many carbonyl stretching absorptions were evident in the 1800–1500 cm−1 range but this part of the spectrum of the adduct was indistinguishable from that of the starting acid. Treatment of the product with excess isobutyl amine, aqueous HCl (15% w/w) or methanol for two days showed no evidence of reaction.
Scheme 3.
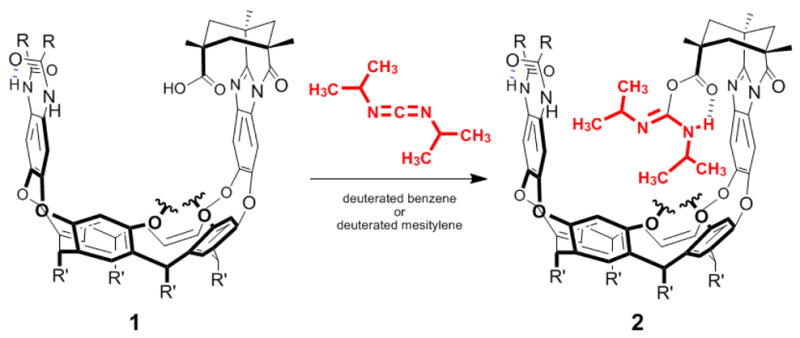
Trapping the reactive intermediate in the reaction of 1 with diisopropylcarbodiimide
Figure 2.
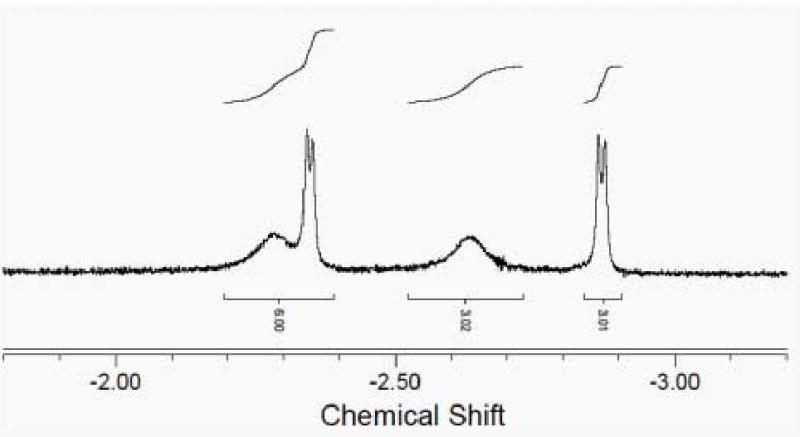
Upfield region of 1H NMR spectrum (600 MHz, 300 K, benzene-d6) in the reaction of 1 with diisopropylcarbodiimide.
Is the adduct the O-acyl or N-acylurea? The unreactivity is inconsistent with the former but a pathway to the latter is hard to defend. Assuming an intramolecular hydrogen bond and extended delocalization, the lowest energy conformation of the O-acylisourea has coplanar heteroatoms. (Fig. 3) Any external nucleophilic attack on the carbonyl carbon along the preferred angles23 is roughly perpendicular to this plane. One face of the function is already shielded by the benzimidazole wall, so the nucleophile must approach from the other face. Such an approach forces the oxygens into the π system of the cavitand’s odd wall as indicated in Figure 3a. The intramolecular O-to-N acyl shift requires the nitrogen nucleophile to assume a stereoelectronically improbable cyclization trajectory (Fig 3b) and a highly contorted intermediate for an already sterically challenged Kemp’s triacid derivative.
Figure 3.
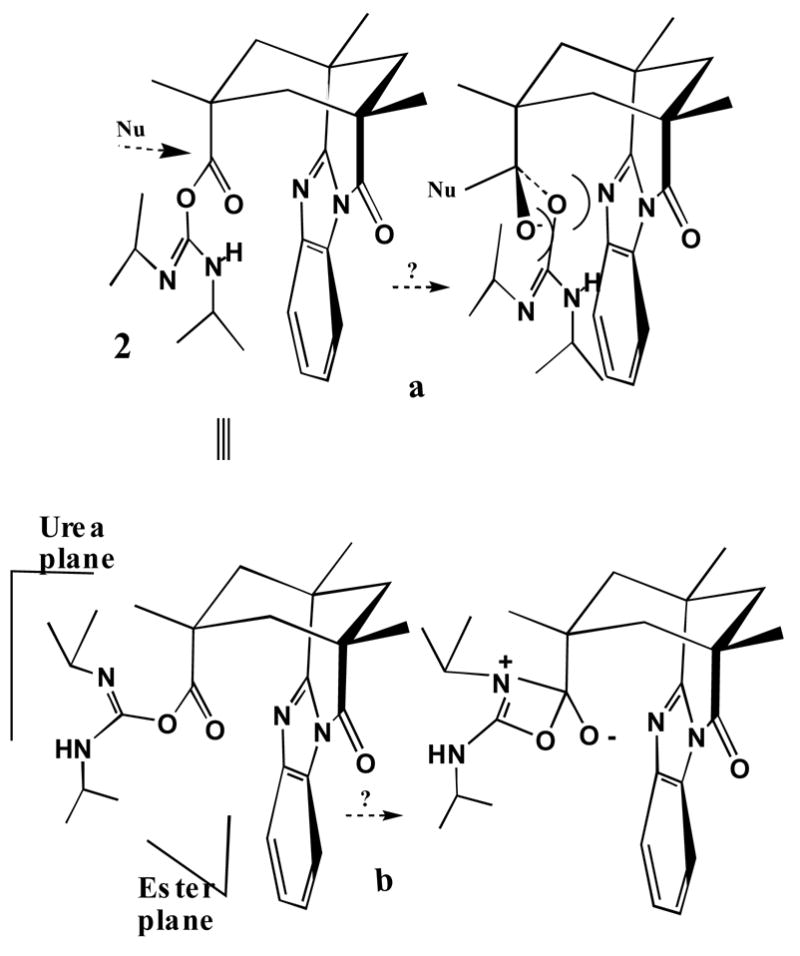
a) External nucleophiles and the O-acylisourea 2 and b) intramolecular rearrangement to a tetrahedral intermediate.
For reasons mentioned above, the intermediacy of the symmetrical anhydride is out of the question. In addition to the conformation shown for 1, a “kite-shaped” conformation with the four walls flipped outward, has been characterized for related molecule.24 It is likely that this conformation is an intermediate that appears during guest uptake and release by 1.25 Recent work also establishes a conformation with only 2 walls flipped outward. 26 An alternate, dissociative mechanism leading to the N-acylurea 3 via an acylium ion has little appeal under the mild conditions here. Even so, the N-acylurea structure for the adduct cannot be excluded.
Conclusion
Related molecule-within-molecule assemblies are providing physical organic chemistry with a new tool for the study of reactive intermediates, either as the covalently-bonded structures, carcerands27,28,29 and cryptophanes,30 or reversibly formed capsules held together with hydrogen bonds,31,32,33 and with metal-ligand interactions.34,35,36 The Kemp’s triacid derivative of the present case is, admittedly, unique; extrapolation of these results to other acids is not without peril. If, indeed, the adduct is the elusive O-acylisourea, its isolation is due to the cavitand’s ability shut down some reaction pathways to reveal intermediates along others. Only crystallography can resolve the structure of the adduct as neither NMR nor IR is up to the task. Further progress in this admittedly narrow investigation must now await the growth of suitable crystals for x-ray analysis. In the meantime, we note that confinement of reactants in synthetic capsules and other limited spaces has already given rise to exclusive regioselectivities37 and even unthinkable regioselectivity 38 in some cycloaddition reactions. The stabilization of unseen reaction intermediates39 and ground states unknown in free solution40 has also been observed. Even carbodiimides have been encapsulated, where their reactivities are much modified.41,42,43 Accordingly, encapsulation is already unlocking secrets of reaction pathways and provides realistic models for reactions confined to enzyme interiors.
Experimental
General
All chemicals were purchased from Aldrich Chemical Company and were used without purification unless otherwise specified. The cavitand 1 was prepared from corresponding resorcinarene 4 as follows (Scheme 4) in an improvement on the published procedure (18). Proton (1H) and Carbon (13C) NMR spectra were recorded on a Bruker DRX-600 (600 MHz) spectrometer. Proton (1H) chemical shifts, reported in parts per million (ppm), were indirectly referenced to external tetramethylsilane employing resonances due to trace protiated solvent as an internal references. Abbreviations are as follows: s, singlet; d, doublet; t, triplet; m, multiplet; brs, broad singlet. Deuterated NMR solvents were obtained from Cambridge Isotope Laboratories, Inc., and used without further purification. Electrospray ionization time-of-flight reflection experiments were performed on an Agilent Electrospray ionization time-of-flight mass spectrometer.
Scheme 4.
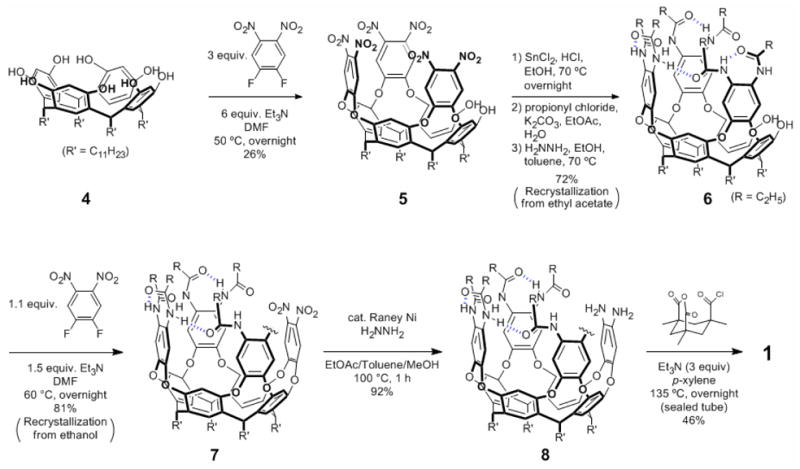
An improved procedure for synthesis of the introverted acid 1
Synthesis of hexa-amide cavitand (6)
Under a nitrogen atmosphere, to the mixture of resorcinarene 4 (26.4 g, 24 mmol) and 1,2-difluoro-4,5-dinitrobenzene (14.6 g, 72 mmol) in DMF (700 mL) at 50 °C was added Et3N (20.0 mL, 144 mmol) dropwise over 10 min. After overnight stirring, the reaction mixture was allowed to cool to ambient temperature, and the mixture was poured into 1N HCl aq. (960 mL). The solid materials were filtered off, washed with water, and then completely dissolved into CH2Cl2 (1500 mL). The solution was transferred into a separatory funnel, and the separated organic layer was dried over Na2SO4, and filtered, and concentrated in vacuo to give the crude as dark brown solid materials. The crude was purified by column chromatography (CH2Cl2/EtOAc = 100/0 ~ 100/1 ~ 100/2) to afford the hexa-nitro cavitand 5 as yellow solid materials in 26% yield (10.0 g). To the cavitand 5 (7.2 g, 4.5 mmol) and SnCl2·H2O (27 g. 120 mmol) was added a mixture of EtOH (320 mL)/conc. HCl aq. (80 mL). After overnight stirring at 70 °C, the volatiles were evaporated. To the residue was added EtOAc (1000 mL), followed by the slow addition of aqueous K2CO3 (85 g, 500 mL). Then propionyl chloride (25 mL) was added dropwise over 20 min. After vigorous stirring for 30 min, the mixture was transferred into a separatory funnel, and the aqueous phase was extracted with EtOAc (200 mL×4). The combined organic phases were dried over Na2SO4, filtered, and concentrated in vacuo. To the resultant materials was added toluene (100 mL)/ethanol (100 mL), followed by hydrazine (1M THF solution, 60 mL). The mixture was stirred at 70 °C for 1 h, and allowed to cool to ambient temperature, and all the volatiles were removed in vacuo. The residue was purified by silica gel column chromatography (EtOAc/CH2Cl2 = 70/30) to give yellow materials, which were recrystallized from EtOAc (13 mL/g) to afford snow-white crystals of 6 (5.6 g, 72%). HRMS (ESI, MH+) Calcd For C108H149N6O14: 1754.1126. Found: 1754.1198 (error < 1ppm). 1H NMR (600 MHz, acetone-d6) δ 9.53 (s, 2H), 9.41 (s, 2H), 9.12 (s, 2H), 8.95 (s, 2H), 7.89 (s, 2H), 7.74 – 7.67 (m, 8H), 7.50 (s, 2H), 6.75 (s, 2H), 5.87 (t, J = 7.8 Hz, 1H), 5.75 (t, J = 7.8 Hz, 2H), 4.37 (t, J = 7.8 Hz, 1H), 2.56 – 2.27 (m, 20H), 1.44 – 1.21 (m, 80H), 0.89 – 0.87 (m, 12H).
Synthesis of hexa-amide di-nitro cavitand (7)
To the mixture of hexa-amide cavitand 6 (1.80g, 1.03 mmol) and 1,2-difluoro-4,5-dinitrobenzene (230 mg, 1.13 mmol) in DMF (30 mL) at 60 °C was added Et3N (0.22 mL, 1.55 mmol) dropwise over 2 min. After overnight stirring, the reaction mixture was allowed to cool to ambient temperature, and the volatiles were removed in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2/EtOAc = 4/1), followed by recrystallization from ethanol (15 mL) to afford the target compound 7 as a pale orange powder (1.61 g, 81%). HRMS (ESI, MH+) Calcd For C114H149N8O18: 1918.0984. Found: 1918.0990 (error < 0.3 ppm). 1H NMR (600 MHz, CDCl3) δ 9.45 (s, 2H), 9.30 (s, 2H), 8.21 – 8.19 (brs, 4H), 7.68 (brs, 2H), 7.55 (s, 4H), 7.36 – 7.35 (m, 2H), 7.31 – 7.30 (m, 6H), 5.83 (t, J = 8.4 Hz, 2H), 5.80 (t, J = 8.4 Hz, 1H), 5.61 (t, J = 8.4 Hz, 1H), 2.52 – 2.21 (m, 20H), 1.49 – 1.25 (m, 80H), 0.94 – 0.91 (m, 12H).
Synthesis of hexa-amide di-amino cavitand (8)
The di-nitro starting compound 7 (200 mg, 0.104 mmol) was dissolved in a mixture of EtOAc (6 mL) and toluene (2 mL). To this solution was added a catalytic amount of Raney Nickel (pre-washed with methanol, 2 mL × 5) suspended in MeOH (6 mL), followed by the addition of anhydrous hydrazine (0.3 mL), and the flask was filled with N2. The flask was fitted with a N2 balloon then immersed in a pre-heated oil-bath (100 °C). After vigorous stirring for 1 h, the (mostly colorless) reaction mixture was allowed to cool to ambient temperature, filtered through a pad of celite, and concentrated in vacuo to give 8 as pale brown solid (178 mg, 92%). ESI-MS m/z: 1858 (MH+), 1880 (MNa+). 1H NMR (600 MHz, CDCl3) δ 9.49 (s, 2H), 9.34 (s, 2H), 8.73 (s, 2H), 7.55 (s, 2H), 7.50 (s, 2H), 7.42 (s, 2H), 7.25 – 7.23 (m, 4H), 7.21 – 7.20 (m, 4H), 6.97 (s, 2H), 5.76 (t, J = 7.8 Hz, 1H), 5.71 (t, J = 8.4 Hz, 2H), 5.66(t, J = 7.8 Hz, 1H), 5.47 (brs, 2H), 5.37 (brs, 2H), 2.49 – 2.16 (m, 20H), 1.43 – 1.15 (m, 80H), 0.89 – 0.87 (m, 12H).
Synthesis of the introverted acid (1)
To the sealed tube charged with 8 (449 mg, 0.24 mmol) and Kemp’s triacid chloride anhydride (62 mg, 0.24 mmol) was added a mixture of p-xylene (6 mL) and Et3N (0.10 mL, 0.72 mmol), then flushed with argon and sealed. The mixture was stirred for 30 min at room temperature, then the tube was immersed in an oil bath pre-heated at 135 °C (the dark gray cloudy mixture becomes a yellow solution). After overnight stirring, the tube was allowed to cool to ambient temperature, and the reaction mixture was filtered through a pad of cotton, and concentrated in vacuo to give brown solid materials. The residue was dissolved in CH2Cl2 (40 mL) and to this solution was added 1N HCl aq. (10 mL). The mixture was transferred into a separatory funnel, and the organic phase was washed with 1N HCl aq. (10 mL), and dried over Na2SO4, filtered, and concentrated in vacuo to give brown crude solid (466 mg). Purification by column chromatography (47 g silica gel, CHCl3/EtOAc = 1/0 ~ 4/1 ~ 1/1) afforded 1 230 mg (46%) as a white powder. HRMS (ESI, MH+) Calcd For C126H165N8O17: 2062.2287. Found: 2062.2242 (error < 2.2 ppm). 1H NMR (600 MHz, CDCl3) δ 9.92 (s, 1H), 9.47 (s, 1H), 9.06 (s, 1H), 8.57 (s, 1H), 8.24 (s, 1H), 8.10 (s, 1H), 8.05 (s, 1H), 7.79 (s, 1H), 7.77 (s, 1H), 7.66 (s, 1H), 7.48 (s, 1H), 7.29 – 7.27 (m, 3H), 7.25 – 7.23 (m, 6H), 7.21 – 7.20 (m, 2H), 5.84 (t, J = 8.4 Hz, 1H), 5.79 (t, J = 8.4 Hz, 1H), 5.73 (t, J = 8.4 Hz, 1H), 5.66 (t, J = 8.4 Hz, 1H), 2.75 – 2.14 (m, 24H), 1.59 – 1.16 (m, 98H), 0.92 – 0.88 (m, 15H).
13C NMR (150 MHz, CDCl3): δ 175.9, 175.3, 174.8, 173.9, 173.5, 173.4 (2 coincident resonances), 172.7, 159.5, 156.2, 155.5, 155.4, 155.2, 155.0, 154.9, 154.8, 154.6, 151.8, 151.6, 151.3, 150.9, 150.8, 150.1, 150.0, 149.6, 141.0, 136.3, 136.2, 136.0, 135.9, 135.8, 135.7, 135.6, 135.4, 130.5, 128.8, 128.5, 128.4, 128.3, 127.9, 126.5, 124.2, 124.1, 124.0, 123.7, 123.0, 122.6, 122.5, 121.8, 121.2, 119.5, 116.8, 116.5, 116.4, 116.1, 115.6, 111.8, 47.5, 46.9, 44.6, 42.5, 41.9, 35.4, 34.0, 33.9, 33.8, 33.7, 33.6, 33.3, 32.4, 33.0, 32.2, 31.8, 31.6, 30.9, 30.5, 30.3(1), 30.2(8), 30.2(6), 30.2(5), 30.1, 30.0(9), 30.0(3), 29.8, 29.3, 28.6, 28.5, 28.4(8), 28.4(2), 23.1, 14.6, 11.1, 10.9, 10.6, 10.5, 9.5; numbers in parentheses indicate resonances that differ by hundredths of a ppm (i.e. 30.09 and 30.03).
The adduct of the acid 1 with diisopropylcarbodiimide
To a flask charged with acid 1 (16 mg, 7.76 ×10−3 mmol) in 1.5 mL of mesitylene-d12 (or 1.5 mL of benzene-d6) was added a 1.5 mL mesitylene-d12 (or 1.5 mL of benzene-d6) solution of diisopropylcarbodiimide (3.9 mg, 31.0 ×10−3 mmol). The tube was dipped into a preheated oil bath at 80 °C. After overnight heating, the volatiles were evaporated under vacuum. The resultant residue was thoroughly washed with hexane (10 mL), and dried overnight in vacuo to give white solid materials (10 mg, 60%). 1H NMR (600 MHz, 300 K, mesitylene-d12) 9.88 – 9.78 (m, 2H), 9.23 – 9.11 (m, 2H), 8.64 – 8.56 (m, 2H), 8.48 (s, 1H), 8.15 – 8.06 (m, 2H), 7.91 (s, 1H), 7.75 – 7.72 (m, 1H), 7.64 – 7.54 (m, 6H), 7.32 – 6.96 (m, 6H), 6.24 – 6.04(m, 4H), 2.71 – 2.29 (m, 17H), 1.60 – 0.47 (m, 128H), −2.48 (d, J = 6.0 Hz, 3H), −2.80 (d, J = 6.0 Hz, 2H), −3.01 (d, J = 6.0 Hz, 1H). HRMS (ESI, MH+) Calcd For C133H179N10O17; 2188.3443. Found; 2188.3391. ESI-MS (m/z): 2210 (MNa+), 2188 (MH+), 1095 ([MH2]2+).
Figure 4.
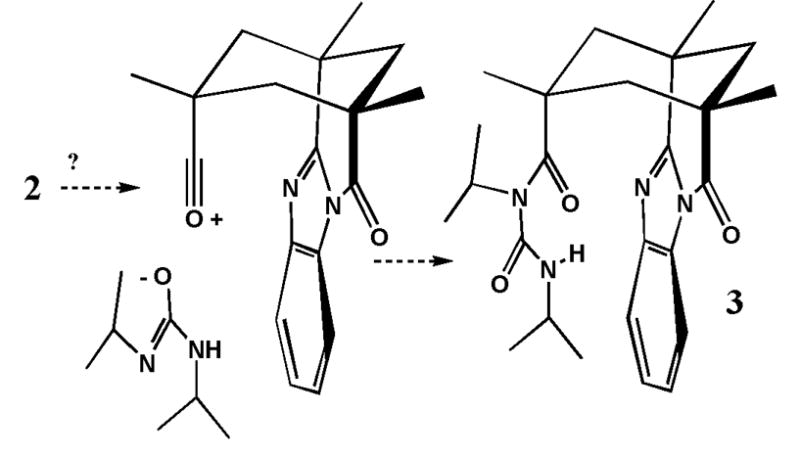
Possible rearrangement to the N-acylurea via an acylium ion.
Acknowledgments
We are grateful for financial support from the Skaggs Institute for Research and the National Institutes of Health (GM 50714). We are pleased to thank Dr. John Thomas for assistance with mass spectrometry. Dr. T. Iwasawa is a Skaggs Postdoctoral Fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Rebek J, Jr, Feitler D. J Am Chem Soc. 1973;95:4052. [Google Scholar]
- 2.Rebek J, Jr, Feitler D. J Am Chem Soc. 1974;96:1606. doi: 10.1021/ja00812a061. [DOI] [PubMed] [Google Scholar]
- 3.Rebek J., Jr Tetrahedron. 1979;35:723. [Google Scholar]
- 4.De Tar DF, Silverstein R. J Am Chem Soc. 1966;88:1013. [Google Scholar]
- 5.De Tar DF, Silverstein R. J Am Chem Soc. 1966;88:1020. [Google Scholar]
- 6.Rebek J, Jr, Zimmerman S, Brown D. J Am Chem Soc. 1975;97:4407. [Google Scholar]
- 7.Bates SH, Jones JH, Witty MJ. J Chem Soc Chem Comm. 1980:773. [Google Scholar]
- 8.Benoiton NL, Chen FMF. J Chem Soc Chem Comm. 1981;543:5127. [Google Scholar]
- 9.Arendt A, Kolodziejczyk A. Tetrahedron Lett. 1978:3867. [Google Scholar]
- 10.Bonsignore L, Cottiglia F, Maccioni AM, Secci D, Lavagna SM. J Heterocyclic Chem. 1995;32:573. [Google Scholar]
- 11.Benoiton NL, Chen FMF. J Chem Soc Chem Comm. 1981:1225. [Google Scholar]
- 12.Rebek J, Jr, Marshall L, Wolak R, Parris K, Killoran M, Askew B, Nemeth D, Islam N. J Am Chem Soc. 1985;107:7476. [Google Scholar]
- 13.Wilcox CS, Greer LM, Lynch V. J Am Chem Soc. 1987;109:1865. [Google Scholar]
- 14.Rebek J., Jr Science. 1987;235:1478. doi: 10.1126/science.3823899. [DOI] [PubMed] [Google Scholar]
- 15.Rebek J., Jr Angew Chem Int Ed Eng. 1990;29:245. [Google Scholar]
- 16.Nowick JS, Ballester P, Ebmeyer F, Rebek J., Jr J Am Chem Soc. 1990;112:8902. [Google Scholar]
- 17.Park TK, Schroeder J, Rebek J., Jr J Am Chem Soc. 1991;113:5125. [Google Scholar]
- 18.Renslo AR, Rebek J., Jr Angew Chem, Int Ed, Engl. 2000;39:3281. doi: 10.1002/1521-3773(20000915)39:18<3281::aid-anie3281>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 19.Wash PL, Renslo AR, Rebek J., Jr Angew Chem, Int Ed Engl. 2000;40:1221. [PubMed] [Google Scholar]
- 20.Butterfield SM, Rebek J., Jr J Am Chem Soc. 2006;128:15366. doi: 10.1021/ja0663374. [DOI] [PubMed] [Google Scholar]
- 21.Kemp DS, Petrakis KS. J Org Chem. 1981;46:5140. [Google Scholar]
- 22.Rebek J, Marshall L, McManis J, Wolak R. J Org Chem. 1986;51:1649. [Google Scholar]
- 23.Bürgi HB, Dunitz JD, Lehn JM, Wipff G. Tetrahedron. 1974;30:1563. [Google Scholar]
- 24.Moran JR, Ericson JL, Dalcanale E, Bryant JA, Knobler CB, Cram DJ. J Am Chem Soc. 1991;113:5707. [Google Scholar]
- 25.Rudkevich DM, Hilmersson G, Rebek J., Jr J Am Chem Soc. 1997;41:9911. [Google Scholar]
- 26.Gottschalk T, Jaun B, Diederich F. Angew Chem, Int Ed. 2007;46:260. doi: 10.1002/anie.200603366. [DOI] [PubMed] [Google Scholar]
- 27.Sherman JC, Knobler CC, Cram DJ. J Am Chem Soc. 1991;113:2194. [Google Scholar]
- 28.Cram DJ, Cram JM. Container Molecules and their Guests. Royal Society of Chemistry; Cambridge: 1994. [Google Scholar]
- 29.Warmuth R, Yoon J. Acc Chem Res. 2001;34:95. doi: 10.1021/ar980082k. [DOI] [PubMed] [Google Scholar]
- 30.Collet A, Dutasta JP, Lozach B, Canciell J. In: Topics in Current Chemistry, 165:Chemistry 1-Directed Synthesis and Molecular Recognition. Weber E, editor. Springer Verlag; Berlin: 1993. pp. 103–129. [Google Scholar]
- 31.Conn MM, Rebek J., Jr Chem Rev. 1997;97:1647. doi: 10.1021/cr9603800. [DOI] [PubMed] [Google Scholar]
- 32.Sherman JC. Tetrahedron. 1995;51:3395. [Google Scholar]
- 33.Shivanyuk A, Paulus EF, Böhmer V. Angew Chem, Int Ed. 1999;38:2906. doi: 10.1002/(sici)1521-3773(19991004)38:19<2906::aid-anie2906>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 34.Yoshizawa M, Kusakawa T, Fujita M, Yamaguchi K. J Am Chem Soc. 2000;122:6311. [Google Scholar]
- 35.Parac TN, Caulder DL, Raymond KN. J Am Chem Soc. 1998;120:8003. doi: 10.1021/ja0104507. [DOI] [PubMed] [Google Scholar]
- 36.Ziegler M, Brumaghim JL, Raymond KN. Angew Chemie, Intl Ed Engl. 2000:4119. doi: 10.1002/1521-3773(20001117)39:22<4119::aid-anie4119>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Rebek J., Jr Org Lett. 2002;4:327. doi: 10.1021/ol0168115. [DOI] [PubMed] [Google Scholar]
- 38.Yoshizawa MI, Tamura M, Fujita M. Science. 2006;312:251. doi: 10.1126/science.1124985. [DOI] [PubMed] [Google Scholar]
- 39.Dong VM, Fiedler D, Carl B, Bergman RG, Raymond K. J Am Chem Soc. 2006;128:14464. doi: 10.1021/ja0657915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iwasawa T, Mann E, Rebek J., Jr J Am Chem Soc. 2006;128:9308. doi: 10.1021/ja062768a. [DOI] [PubMed] [Google Scholar]
- 41.Heinz T, Rudkevich DM, Rebek J., Jr Angew Chem Int Ed Engl. 1999;38:1136. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1136::AID-ANIE1136>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Körner S, Craig SL, Rudkevich DM, Rebek J., Jr Nature. 2002;415:385. doi: 10.1038/415385b. [DOI] [PubMed] [Google Scholar]
- 43.Chen J, Körner S, Craig SL, Lin S, Rudkevich DM, Rebek J., Jr Proc Natl Acad Sci USA. 2002;99:2593. doi: 10.1073/pnas.052706499. [DOI] [PMC free article] [PubMed] [Google Scholar]