

Abstract
We describe the properties of a series of oligomeric polyfluorophores assembled on the DNA backbone. The eleven oligomers (oligodeoxyfluorosides, ODFs), 4 to 7 monomers in length, were composed of only two fluorescent monomers and a spacer in va ried sequences, and were designed to test how fluorescent nucleobases can interact electronically to yield complexity in fluorescence emission. The monomer fluorophores were deoxyribosides of pyrene and perylene, which emit light in violet and blue wavele ngths respectively. The experiments show that simple variation of sequence and spacing can dramatically change fluorescence, yielding emission maxima ranging from 380 to 557 nm and visible colors from violet to orange-red. Fluorescence lifetimes data, excitation spectra, and absorption data point to a number of multi-fluorophore electronic interactions, including pyrene-pyrene and perylene-perylene excimers, pyrene-perylene exciplexes, as well as monomer dye emissions, contributing to the final spectral outcomes. Thus, two simple fluorophores can be readily combined to give emissions over much of the visible spectrum, all requiring only a single excitation. The results demonstrate that fluorescent nucleobases in oligomeric form can act cooperatively as electronic units, and that fluorophore sequence in such oligomers is as important as fluorophore composition in determining fluorescence properties.
Keywords: pyrene, perylene, excimer, exciplex, Stokes shift
Introduction
Fluorescent nucleobase analogs are becoming increasingly employed in biotechnological applications involving nucleic acids, and as biophysical probes of interactions involving DNA and RNA. These compounds are useful alternatives to classical labeling of nucleic acids, offering a more well-defined structural localization and closer interaction with the center of the double helix. As such they have been employed recently as reporters of DNA dynamics1, charge transfer2, DNA polymerases3 and other enzymes.4
The number of fluorescent nucleobase analogs has increased greatly in recent years,5 however, they are typically employed one at a time in biological assays. Although this gives simplicity in yielding a predictable emission outcome, the use of one fluorescent nucleoside overlooks one of the most important properties of DNA: namely, that adjacent bases are stacked on one another and interact electronically. We have hypothesized that if this stacked interaction were exploited intentionally with multiple fluorescent nucleosides, it could lead to useful new properties involving cooperative electronic interactions. Among the properties that might arise include tunable excitation and emission, large Stokes shifts, high emission intensity, and sensing capabilities.
As part of this strategy, we recently introduced the concept of polyfluorophores assembled on a DNA backbone.6 These molecules resemble DNA (except that their “bases” are all hydrocarbon or heterocyclic fluorophores) (Fig. 1), and are readily prepared in programmed sequences on a DNA synthesizer. Because they retain the polyanionic backbone, polyfluors are inherently water soluble and are readily appended to natural oligonucleotides for DNA detection schemes using automated synthesis.7 Polyfluors have been demonstrated with large Stokes shifts (200+ nm), a property that aids in avoiding interference from the excitation light.7b Furthermore, single wavelength excitation combined with variable Stokes shifts can yield a range of tunable emissions, removing the need for expensive filter sets and multiple excitation sources. It has been our hypothesis that varied polyfluor sequences would yield electronic complexity from variable stacked neighbors, just as DNA properties depend on nearest neighbor interactions. To survey the general complexity of outcomes available, we have initially focused on combinatorial approaches to fluorescent oligomers. Previous libraries of polyfluors have employed combinatorial methods and up to 10 different individual “fluorosides” to produce as many as 14,000+ tetrameric fluorophores.7c Although a wide variability of molecular properties is indeed apparent from the screening approach, it leaves unanswered the questions of how complexity arises: What are the mechanisms by which simple fluorophores interact as neighbors in a DNA scaffold, and how do simple changes in composition and sequence affect the properties? In this study we test these issues explicitly, and we have found that the combination of just two fluorescent nucleosides in designed sequences can yield surprisingly varied emissions ranging from violet (375 nm) to orange (600 nm).
Figure 1.
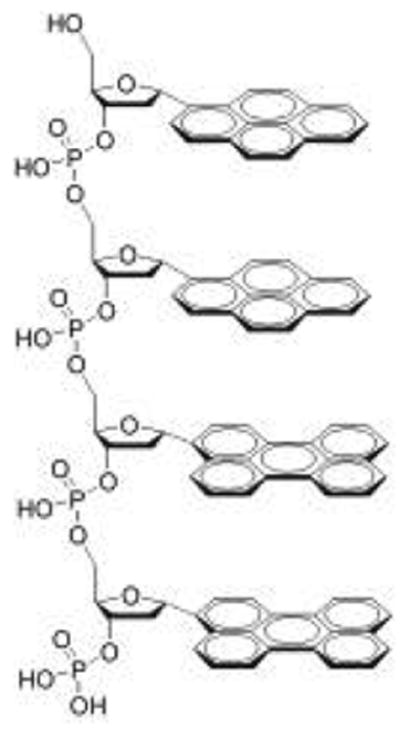
Structure of a representative oligodeoxyfluoroside showing the possible stacking of pyrene and perylene aromatic “bases” analogous to DNA.
We selected the polycyclic aromatic hydrocarbon fluorophores pyrene and perylene as well characterized fluorophores having high quantum yields and different emission wavelengths. They are known to form both excimers and exciplexes upon photoexcitation of concentrated solutions, crystalline samples, or systems where two fluorophores are bound in close proximity.14,15,16 In addition, the absorption spectrum of perylene overlaps well with the pyrene emission spectrum offering the possibility of Förster resonance energy transfer (FRET) as another means of electronic interaction. To examine how sequence and composition might variably affect interactions, we constructed a series of rationally designed oligodeoxyfluorosides containing only pyrene, perylene and spacer nucleosides (Fig. 2).
Figure 2.
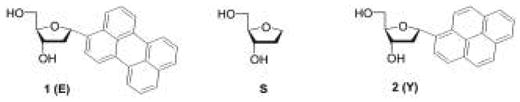
Deoxyriboside monomers used in this study. At left, α-perylene deoxyriboside, (1) also abbreviated as E in sequences. At right, α-pyrene nucleoside, (2), abbreviated Y. At center, an abasic dideoxyribose (S), used as a spacer.
Results
Preparation of ODF monomers and oligomers
A number of sequences were designed to test the effects of nearest neighbors, sequence, numbers of fluorophores, and spacing on fluorescence properties (Table 1). Compounds 3, 4 and 5 contained one, two and three perylene fluorophores respectively and were designed to evaluate perylene-perylene electronic interactions in the context of the DNA backbone (4, 5). Compounds 6–8 were similarly constructed using pyrene nucleosides. Compounds 7–9 contain one pyrene and one perylene moiety each and were designed to explore the spacer length dependence of any electronic interactions between the two (for example, exciplex or FRET). Finally, compounds 12 and 13 were designed with two pyrenes and two perylenes each, but the order of the monomers was different (3′-EYEY vs 3′-EEYY) to evaluate the effect of sequence when composition was the same.
Table 1.
Sequences and photophysical data for the compounds in this study. Measurements were taken in phosphate buffered saline (pH = 7.4) unless otherwise noted.
| Cpd | Sequence | λmax, abs (nm) | ε, cm−1 M−1 | λmax, em (nm) | Φem | τ (ns) |
|---|---|---|---|---|---|---|
| 1 | Ea | 440, 415, 393 | 39,000 | 443, 472, 503 | 0.88 | 4.3 ± 0.1 |
| 2 | Ya | 342, 326, 312 | 47,000 | 375, 394 | 0.12 | 51.0 ± 0.6 |
| 3 | ESSS | 440, 415 | 39,000 | 450
477 |
0.95 | 5.4 ± .1 |
| 4 | EESSS | 443, 420 | 43,000 | 450, 478, 563 | 0.06 | 7.0 ± 0.5 |
| 5 | EEESSS | 447, 422 | 62,000 | 450, 478, 557 | 0.03 | 6.2 ± 0.9 |
| 6 | YSSS | 344, 327 | 47,000 | 377, 396 | 0.35 | 198.0 ± 0. 3 |
| 7 | YYSSS | 343, 329 | 44,000 | 377, 396, 484 | 0.22 | 85.9 ± 1.4 |
| 8 | YYYSSS | 343, 329 | 67,000 | 492 | 0.12 | 116.5 ± 0.3 |
| 9 | EYSSS | 448, 421, 348, 327 | 39,000
42,000 |
490
490 |
0.39 (330 nm)
0.66 (420 nm) |
28.5 ± 0.7 (350 nm)
18.2 ± 0.2 (450 nm) |
| 10 | ESYSS | 448, 421, 348, 327 | 39,000
42,000 |
490
490 |
0.35 (330 nm)
0.61 (420 nm) |
27.6 ± 1.0 (350 nm)
16.5 ± 0.1 (450 nm) |
| 11 | ESSSY | 448, 421, 348, 327 | 39,000
42,000 |
465, 490
465, 490 |
0.40 (330 nm)
0.67 (420 nm) |
24.9 ± 0.5 (350 nm)
15.8 ± 0.1 (450 nm) |
| 12 | EYEYSSS | 451, 423, 346, 332 | 43,000
60,000 |
377, 396, 494; 481 | 0.02 (330 nm)
0.03 (450 nm) |
18.2 ± 0.1 (350 nm)
8.5 ± 0.1 (450 nm) |
| 13 | EEYYSSS | 451, 423, 347, 332 | 43,000
60,000 |
457, 485
454, 482 |
0.02 (330 nm)
0.03 (420 nm) |
14.9 ± 0.1 (350 nm)
27.1 ± 0.1 (450 nm) |
Free deoxyriboside. Data obtained in MeOH.
The synthesis of the phosphoramidites of 1 and 2 was carried out as previously described.7 The alpha anomers were used for synthetic expedience by the chlorosugar route;8 this is not expected to hinder stacking since alpha nucleosides can also stack and form helices in a DNA backbone. The oligodeoxyfluorosides were prepared using 3′-phosphate CPG supports employing standard phosphoramidite chemistry. Each sequence contained three spacers to insure aqueous solubility of the hydrophobic aromatics. Yields of the oligomers ranged from 58% to 98% overall based on trityl monitoring. They were purified by HPLC, and masses of the isolated compounds were confirmed by MALDI-TOF mass spectrometry.
Optical properties of fluorophore monomers
The absorption and emission of fluorosides 1 and 2 have been described previously,7 but were measured here again as a reference (Fig. 3). Pyrene absorbs with a maximum at 342 nm with well defined vibronic progressions at 326 and 312 nm. Emission maxima are 376 and 396 nm, giving a moderate Stokes shift of 34 nm. Perylene deoxyriboside, 1, absorbs at slightly longer wavelength of 440 nm with additional bands at 415 and 394 nm. Emission of 1 occurs at 450 nm, a Stokes shift of only 10 nm, with an additional prominent transition at 480 nm. Quantum yields in air-saturated methanol were 0.88 and 0.12 for 1 and 2 respectively. We measured emission lifetimes under the same conditions; they were 4.3 ± 0.1 ns for 1 and 51.0 ± 0.6 ns for 2, similar to the reported values for the parent hydrocarbons.12
Figure 3.
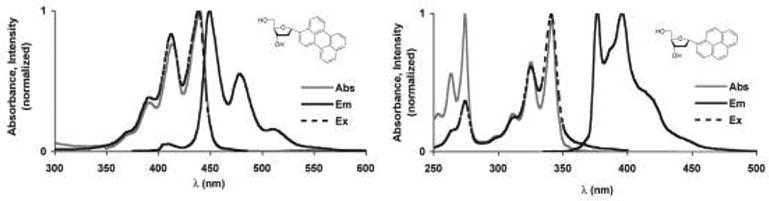
Absorption, emission and excitation spectra of nucleosides 1 and 2 in methanol.
Optical properties of pyrene oligomers 6–8
We first examined the absorption spectra of the pyrene series of oligonucleotides, 6–8. Absorption of 6, which contains a single pyrene, was virtually identical to the parent nucleoside, 2, showing clear vibronic progressions beyond the 342 nm transition (Fig. 4). However, the oligonucleotides containing more than one pyrene fluorophore, 7 and 8, exhibited substantial peak broadening as well as a shift in the relative peak heights. A prominent shoulder exists at wavelengths longer than the 342 nm transition and yet there is an apparent increase in the relative intensity of the shorter wavelength peak at 326 nm. Aggregates of dyes are known to interact electronically and exhibit changes in their absorption spectra as a result of transition dipole coupling.10,11,17 Adjacent pyrenes on a single ODF strand most likely exist in a staggered or oblique geometry, which gives rise to band-splitting and could explain the spectral broadening observed in the absorbance spectra of 7 and 8.
Figure 4.
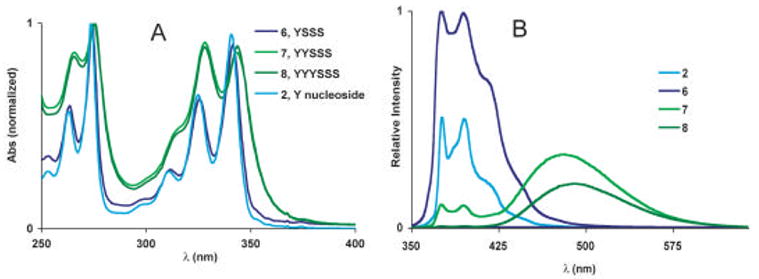
Absorption spectra (A), and emission spectra (B) of the pyrene containing series of oligomers (6–8). Data were measured in phosphate buffered saline (pH = 7.4) with ODFs at 10 μM.
We found that intramolecular aggregation of the pyrenes could be reversed thermally, by addition of a surfactant to aqueous solutions or by switching to an organic solvent (data not shown). We found that over the range of 25 to 95 °C, there is only a very small effect (1–2% change). An isosbestic point was observed at 331 nm as the relative peak intensities shifted slightly to the longer wavelength transition. This behavior was reversible and reproducible at different optical densities. The effect was more pronounced with the addition of surfactants such as Tween, Triton or sodium dodecyl sulfate (SDS) which may disrupt the hydrophobic association of neighboring pyrenes. Taken together, the electronic properties and denaturation behavior strongly suggest that in aqueous solution at room temperature, the hydrophobic pyrene moieties are in a stacked geometry.
We next examined the emission properties of 6–8. The emission spectrum of 6 is similar to 2, with identical maxima, and a moderate amount of broadening. Solutions of 6 appeared violet-blue when irradiated with a hand-held UV lamp. The quantum yield of 2 in methanol has been previously reported as 0.12, so it was somewhat surprising that Φem for 6 was 0.35 even in the more polar buffer solution. An increase in the emission lifetime was also observed (τ = 198.0 ± 0.3 ns). It is possible that the addition of the three solubilizing abasic sites function essentially as a surfactant creating a hydrophobic environment around the pyrene fluorophore leading to the increased quantum yield.12a We confirmed this by obtaining the quantum yield of 2 in buffer solution with 2.0 % w/v SDS added. In this case, the quantum yield increased by a factor of 4.5 (Φem = 0.54). Molecular modeling (Fig. 4) suggests that the YSSS sequence can adopt a curled structure shielding one face of the fluorophore from the polar aqueous environment. The use of aliphatic spacers may be a general strategy that can be employed with the polyfluors to achieve greater quantum yields in aqueous solutions or other polar environments.
The fluorescence spectra of sequences containing two or three pyrenes (7 and 8, respectively) show a broad, featureless peak near 490 nm. Similar emission behavior was recently reported for the same pyrenes conjugated to DNA.7b,8 Two small peaks corresponding to pyrene monomer emission can still be seen in the dimer spectrum of 7, but are nearly absent in the trimer 8. Pyrene is well known to form excimers when two pyrenes are in close proximity and one is promoted to an excited state.14,15 The disappearance of the peaks corresponding to pyrene monomer emission in the spectra of 7 and 8 and concomitant appearance of the featureless, low energy peak at 490 nm can be assigned to excimer formation. Solutions of 7 and 8 appear bright green to the eye as the fluorescence is dominated by the pyrene excimer. Quantum yields decrease to 0.22 for 7 and 0.12 for 8 and the emission lifetimes are attenuated as well (Table 1).
Optical properties of perylene -containing oligomers 3–5
We next turned to the perylene series, 3–5. The data show that many of the same trends observed in the pyrene series are found in the perylene series as well. Sequence 3 containing one perylene had an absorbance maximum at 442 nm and was virtually identical to solutions of the perylene nucleoside in MeOH. Spectral broadening was found in the case of the sequences containing more than one perylene. As seen in the pyrene series, for 4 and 5 there exist both a red shoulder as well as an increase in the relative intensities of the higher energy transitions at 415 and 393 nm, suggestive of band-splitting resulting from oblique or staggered aggregates. Switching to different organic solvents such as DMF, or addition of surfactants to aqueous solutions of 4 and 5 reduced the apparent aggregation, and the spectra became more defined (data not shown).
The emission spectrum of 3, which contains only one perylene unit, was identical to the parent nucleoside and appeared bright blue in aqueous solutions (Φem = 0.95). With the addition of a second perylene fluorophore as in 4, there appeared a second peak centered at 563 nm. This broad, featureless peak of lower energy than the monomer emission can be explained by the formation of excimers as seen with the pyrene oligomers above. Solutions of 4 appeared orange when irradiated with a hand-held UV lamp (λmax = 365 nm) although they were not as intense (Φem = 0.06) as solutions of 3. With three perylenes (5), the excimer peak became even more pronounced (relative to monomer). The intensity of the excimer emission in 4 and 5 varied with concentration suggesting some degree of intermolecular aggregation may present in solutions above 0.25 μM. This switching between emission profiles may indicate different conformations in different solvent environments. We are currently investigating the conformational and photophysical behavior of these perylene oligomers in more detail.
Optical properties of mixed fluorophore oligomers 9–11
Sequences 9–11 contain one pyrene and one perylene with zero, one and three spacers separating them. The absorption spectra of oligomers 9–11 at first glance appeared simply to be the combination of pyrene and perylene absorption spectra (Fig. 7). However, closer inspection revealed that the absorbance maxima due to the perylene subunit are red shifted by 7 nm (448 nm vs 441 nm) with respect to the monomer. For the peaks corresponding to the pyrene subunit, the shift is 3 nm. This would indicate that there is some degree of coupling between the pyrene and perylene chromophores in these sequences.
Figure 7.
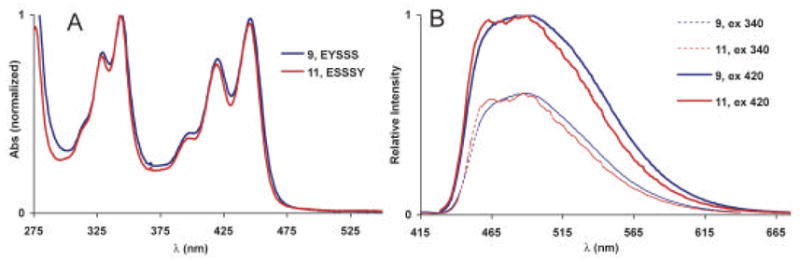
Absorption (A) and emission spectra (B) of 9 and 11. The spectra of 10 are virtually identical to 9 and are not shown. Data were measured in phosphate buffered saline (pH = 7.4) with ODFs at 10 μM.
The emission spectra of 9–11 were nearly identical and varied only slightly with excitation wavelength. A broad peak centered at 491 nm is indicative of an excited state interaction between pyrene and perylene, i.e. exciplex, as the emitting state. A small (~5 nm) shoulder was observed when 9 was excited at 420 nm, while the red shoulder of 11 was diminished to a small degree when excited at 330 nm. A small degree of definition was visible in the emission spectrum of 11 at 465 nm and may correspond to perylene monomer emission resulting from FRET when pyrene is excited at 330 nm. This would be expected as there is considerable spacing between pyrene and perylene in this sequence with three S monomers between them. Yet, the dominant emission of 11 still appears to result from exciplex formation. This is further supported by the emission lifetime decays (Table 1), which were virtually the same indicating that the same mechanism is responsible for the emission in all the sequences. This was true regardless of whether the excitation wavelength was 350 nm (favoring excitation of pyrene) or 450 nm (favoring excitation of perylene): in both cases an exciplex resulted even when the two fluorophores were separated by three spacers. It is possible that the two hydrophobic aromatic species are closely associated in solution, which may be conformationally allowed by the flexibility of the backbone.
Optical properties of oligonucleotides 12–13
The final two oligomers (12 and 13) contained the same four fluorescent monomers but in different sequences. The absorbance spectra were found to display significantly broadened peaks. They were similar to the sum of the corresponding difluorophore sequences (4 and 7) but with an additional 4 nm bathochromic shift. The spectral broadening may be explained by exciton coupling between pyrenes as in 7 and between perylenes as in 4. The small red shift can be explained by electronic interactions between pyrene and perylene as seen in the absorbance spectra of 9–11.
The emission spectrum of 12 was observed to switch between two states depending on the excitation wavelength. When excited at 330 nm, both pyrene monomer emission as well as the pyrene-perylene exciplex emission were visible. Thus, aqueous solutions of 12 appeared very bright and almost white, under UV excitation, as most of the visible spectrum is covered. The prominence of the pyrene monomer fluorescence in the emission spectrum implies that excited state interactions between pyrene and perylene are not as efficient as in 9–11, nor is there efficient energy transfer to perylene by a FRET mechanism. Switching to 420 nm excitation completely eliminated the pyrene emission and only the pyrene-perylene exciplex emission was visible. This is not unexpected, as pyrene does not absorb appreciably beyond 375 nm.
In contrast to 12, the emission spectrum of 13 did not vary significantly with excitation wavelength, displaying only emission above 440 nm and stretching beyond 650 nm. We assign this broad peak to be a conglomerate of three modes of emission including perylene monomer, exciplex and excimer emission. The two defined peaks at 454 and 484 nm can be assigned to perylene monomer type emission as found in the spectra of 1 or 3. The broad shoulder found between 550 and 650 nm can be assigned as emission from the excimer of perylene as seen in the spectra of 4 and 5. When 13 is excited at 330 nm, the strong emission band between 475 and 550 overlaps well with the emission of the EY containing sequence, 9, and therefore may be ascribed to exciplex emission. Solutions containing 13 appeared yellow, although they were not exceptionally bright due to the relatively low quantum yield (Φem = 0.03).
Discussion
An overview of the oligomers in this series shows remarkable variation in emission using simple combinations of only two component fluorophores. By varying the sequences and numbers of pyrene and perylene nucleosides, a wide range of optical properties can be achieved, as clearly visible in Figure 9. Violet and blue emission is achieved by using sequences containing only one pyrene (6) or one perylene (3) fluorescent nucleoside. A range of green emitting sequences can be achieved either by excimers of pyrene (7 and 8) or by formation of the pyrene -perylene exciplex (9, 10, 11). By incorporating more than one perylene, orange emission is produced (4) although a sequence of three perylenes (5) produces only a weakly emissive orange-red polyfluor in strongly polar media. Sequences with four fluorophores exhibit quite complex emission profiles consisting of more than one emitting species. Their photoluminescence can vary greatly in the EYEY sequence, 12, appearing a bright blue -green with excitation < 400 nm due to pyrene monomer and pyrene-perylene exciplex emission, while 13 (EEYY) appears yellow due to contributions of pyrene-perylene exciplex and perylene excimer emission.
Figure 9.
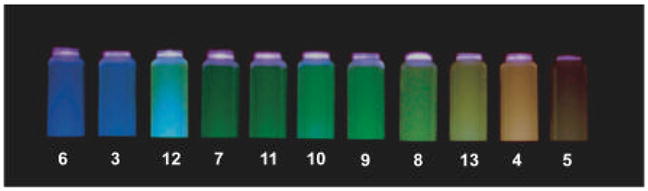
Photograph of ODFs 3–13 in PBS buffer using transilluminator (365 nm) for excitation. Blue emission is achieved with sequences containing only one pyrene (6) or perylene (3) residue. A range of greens results from excimers of pyrene (7, 8) or pyrene-perylene exciplexes (9–11). Excimers of perylene (4,5) appear orange.
Conclusion
Taken together, this set of simple polyfluors demonstrates that combinations of just two fluorescent nucleosides can produce a set fluorescent molecules that demonstrate cooperative electronic behavior, and yield complexity from simple components. Such compounds offer the useful property of having a single excitation but yielding multiple emission colors, much like quantum dots, but because they are discrete organic molecules they can be conjugated easily to nucleic acids and other biomolecules. It seems likely that such molecular strategies may find use in future biological assays and biophysical studies.
Experimental
General
Chemicals used for phosphoramidite synthesis were purchased from Aldrich or Acros. Reagents for oligonucleotide synthesis were purchased from Glen Research. Perylene phosphoramidite and pyrene phosphoramidite were prepared according to published procedures.7 Oligonucleotides were assembled on an Applied Biosystems 394 DNA/RNA Synthesizer. Purification was carried out utilizing a Shimadzu 10 Series HPLC with an Alltec C4 column with acetonitrile and H2O as eluents.
Oligomer synthesis and characterization
After initial HPLC purification, sequences were analyzed again using a standard concentration gradient of 30% → 80% AcCN in water over a 12 minute period to confirm their purity before subsequent photophysical characterizations. Single peaks were observed at the elution times listed in Table 2 and MALDI confirmed their composition.
Table 2.
Summary of synthesis and purification of ODFs.
| Cpd | Sequence | Yield | Elution Time (min) | Calc’d mass | MALDI (m/z) |
|---|---|---|---|---|---|
| 3 | ESSS | 98.3 | 4.19 | 988.16 | 987.6 |
| 4 | EESSS | 81.9 | 5.34 | 1419.26 | 1418.1 |
| 5 | EEESSS | 75.4 | 7.57 | 1848.36 | 1848.8 |
| 6 | YSSS | 95.9 | 3.96 | 938.15 | 939.5 |
| 7 | YYSSS | 87.5 | 4.77 | 1318.23 | 1319.8 |
| 8 | YYYSSS | 79.8 | 6.26 | 1698.31 | 1700.16 |
| 9 | EYSSS | 58.0 | 5.06 | 1368.25 | 1367.9 |
| 10 | ESYSS | 63.5 | 4.99 | 1368.25 | 1367.9 |
| 11 | ESSSY | 68.9 | 5.06 | 1368.25 | 1367.8 |
| 12 | EYEYSSS | 59.8 | 8.34 | 2178.42 | 2179.3 |
| 13 | EEYYSSS | 83.2 | 8.90 | 2178.42 | 2180.7 |
Optical measurements
Absorption measurements were performed on a Cary 100 Bio UV-vis spectrometer. Fluorescence studies were performed on a Jobin Yvon-Spex Fluorolog 3 spectrometer. Quantum yields were obtained on solutions with absorbances below 0.05 to minimize intermolecular effects and reabsorption processes. Fluorescein in 0.1 N NaOH was used as a reference for quantum yield measurements.18 Emission lifetime measurements were carried out on a PTI Easylife instrument using either 350 nm or 450 nm LED for excitation of pyrene or perylene respectively. Fitting and analysis was performed using Felix32 software.
Figure 5.
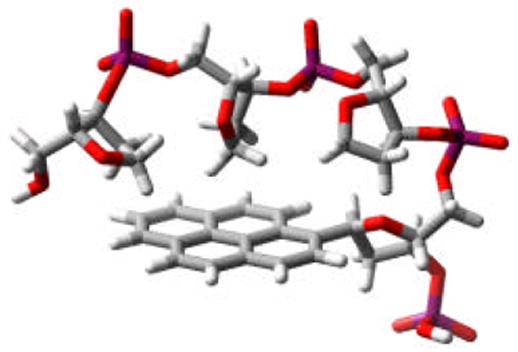
Modeled plausible conformation for 6 showing possible interaction of backbone with pyrene. (MMFFs force field, continuum water solvent, Macromodel).
Figure 6.
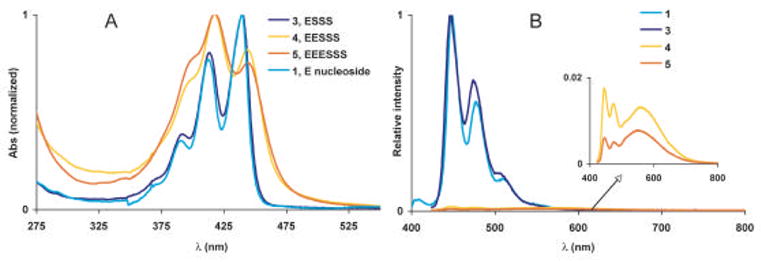
Absorption (A) and emission (B) of the perylene containing series of oligonucleotides (3–5). Data were measured in phosphate buffered saline (pH = 7.4) with ODFs at 10 μM.
Figure 8.
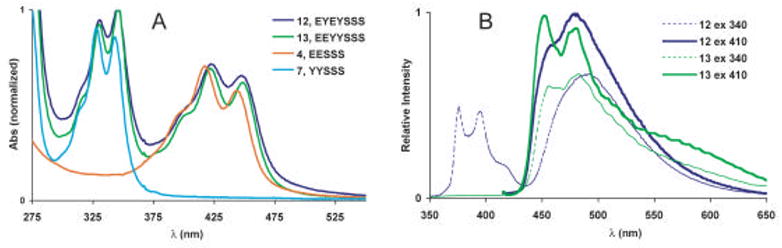
Absorption (A) and emission spectra (B) of 12 and 13. Data were measured in phosphate buffered saline (pH = 7.4) with ODFs at 10 μM
Acknowledgments
This work was supported by the U.S. National Institutes of Health (GM067201) and the U.S. Army Research Office. JNW acknowledges an NIH Postdoctoral Fellowship.
Footnotes
Contact Information: Eric T. Kool, Department of Chemistry, Stanford University, Stanford, CA 94305-5080, Phone: 650-724-4741, Fax: 650-725-0259, kool@stanford.edu
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Ward DC, Reich E, Stryer L. J Biol Chem. 1969;244:1228–1237. [PubMed] [Google Scholar]; (b) Coleman RS, Madaras ML. J Org Chem. 1998;63:5700–5703. [Google Scholar]; (c) Paris PL, Langenhan J, Kool ET. Nucleic Acids Res. 1998;26:3789–3793. doi: 10.1093/nar/26.16.3789. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pompizi I, Haberli A, Leumann CJ. Nucleic Acids Res. 2000;28:2702–2708. doi: 10.1093/nar/28.14.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Singleton SF, Shan F, Kanan MW, McIntosh CM, Stearman CJ, Helm JS, Webb KJ. Org Lett. 2001;3:3919–3922. doi: 10.1021/ol0167863. [DOI] [PubMed] [Google Scholar]; (f) Bradrick TD, Marino JP. RNA. 2004;10:1459–1468. doi: 10.1261/rna.7620304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kaul M, Barbieri CM, Pilch DS. J Am Chem Soc. 2004;126:3447–3453. doi: 10.1021/ja030568i. [DOI] [PubMed] [Google Scholar]; (h) Marti AA, Jockusch S, Li Z, Ju J, Turro NJ. Nucleic Acids Res. 2006;34:e50/1–e50/7. doi: 10.1093/nar/gkl134. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Kimura T, Kawai K, Majima T. Chem Commun. 2006:1542–1544. doi: 10.1039/b600026f. [DOI] [PubMed] [Google Scholar]; (j) Zhang L, Long H, Boldt GE, Janda KD, Schatz GC, Lewis FD. Org Biomol Chem. 2006;4:314–322. doi: 10.1039/b513694f. [DOI] [PubMed] [Google Scholar]
- 2.(a) Thornton NB, Wojtowicz H, Netzel T, Dixon DW. J Phys Chem B. 1998;102:2101–2110. [Google Scholar]; (b) Amann N, Huber R, Wagenknecht HA. Angew Chem Int Ed. 2004;43:1845–1847. doi: 10.1002/anie.200353153. [DOI] [PubMed] [Google Scholar]
- 3.(a) Liu C, Martin CT. J Mol Biol. 2001;308:465–475. doi: 10.1006/jmbi.2001.4601. [DOI] [PubMed] [Google Scholar]; (a) Kawai R, Kimoto M, Ikeda S, Mitsui T, Endo M, Yokoyama S, Hirao I. J Am Chem Soc. 2005;127:17286–17295. doi: 10.1021/ja0542946. [DOI] [PubMed] [Google Scholar]
- 4.Secrist JA, Barrio JR, Leonard NJ. Science. 1972;175:646–647. doi: 10.1126/science.175.4022.646. [DOI] [PubMed] [Google Scholar]
- 5.(a) Seela F, Zulauf M, Sauer M, Deimel M. Helv Chim Acta. 2000;83:910–927. [Google Scholar]; (b) Lehbauer J, Pfleiderer W. Helv Chim Acta. 2001;84:2330–2342. [Google Scholar]; (c) Rao P, Benner SA. J Org Chem. 2001;66:5012–5015. doi: 10.1021/jo005743h. [DOI] [PubMed] [Google Scholar]; (d) Saito Y, Miyauchi Y, Okamoto A, Saito I. Chem Commun. 2004:1704–1705. doi: 10.1039/b405832a. [DOI] [PubMed] [Google Scholar]; (e) Okamoto A, Tanaka K, Fukuta T, Saito I. Chembiochem. 2004;5:958–963. doi: 10.1002/cbic.200400010. [DOI] [PubMed] [Google Scholar]; (f) Hwang GT, Seo YJ, Kim BH. J Am Chem Soc. 2004;126:6528–6529. doi: 10.1021/ja049795q. [DOI] [PubMed] [Google Scholar]; (g) Hawkins ME, Balis FM. Nucleic Acids Res. 2004;32:e62/1–e62/7. doi: 10.1093/nar/gnh060. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Sandin P, Wilhelmsson LM, Lincoln P, Powers VEC, Brown T, Albinsson B. Nucleic Acids Res. 2005;33:5019–5025. doi: 10.1093/nar/gki790. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Greco NJ, Tor Y. J Am Chem Soc. 2005;127:10784–10785. doi: 10.1021/ja052000a. [DOI] [PubMed] [Google Scholar]; (j) Ben GN, Glasser N, Ramalanjaona N, Beltz H, Wolff P, Marquet R, Burger A, Mely Y. Nucleic Acids Res. 2005;33:1031–1039. doi: 10.1093/nar/gki253. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Rao P, Benner SA. J Org Chem. 2001;66:5012–5015. doi: 10.1021/jo005743h. [DOI] [PubMed] [Google Scholar]
- 6.Gao J, Strässler C, Tahmassebi DC, Kool ET. J Am Chem Soc. 2002;124:11590–11591. doi: 10.1021/ja027197a. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ren RX-F, Chaudhuri NC, Paris PL, Rumney S, IV, Kool ET. J Am Chem Soc. 1996;118:7671–7678. doi: 10.1021/ja9612763. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cuppoletti A, Cho Y, Park JS, Strässler C, Kool ET. Bioconj Chem. 2005;16:528–534. doi: 10.1021/bc0497766. [DOI] [PubMed] [Google Scholar]; (c) Gao J, Watanabe S, Kool ET. J Am Chem Soc. 2004;126:12748–12749. doi: 10.1021/ja046910o. [DOI] [PubMed] [Google Scholar]
- 8.Chaudhuri NC, Ren Rex XF, Kool ET. Synlett. 1997;4:341–347. doi: 10.1055/s-1997-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cho Y, Kool ET. ChemBioChem. 2005;7:669–672. doi: 10.1002/cbic.200500515. [DOI] [PubMed] [Google Scholar]
- 10.Kasha M. Rev Mod Phys. 1959;31:162–169. and references within. [Google Scholar]
- 11.D’Ilario L, Martinelli A. Modelling Simul Mater Sci Eng. 2006;14:581–595. [Google Scholar]
- 12.(a) Geiger MW, Turro NJ. Photochem Photobiol. 1975;22:273–276. doi: 10.1111/j.1751-1097.1975.tb06749.x. [DOI] [PubMed] [Google Scholar]; (b) Johansson LB-Å, Molotkovsky JG, Bergelson LD. J Am Chem Soc. 1987;109:7374–7381. [Google Scholar]
- 13.Karpovich DS, Blanchard GJ. J Phys Chem. 1995;99:3951–3958. [Google Scholar]
- 14.Birks JB, Christophorou LG. Proc Roy Soc A. 1964;277:571–582. and references within. [Google Scholar]
- 15.(a) Tomkiewicz Y, Loewenthal E. Mol Cryst Liquid Cryst. 1969;6:211–225. [Google Scholar]; (b) Chu NYC, Kawaoka K, Kearns DR. J Chem Phys. 1971;55:3059–3067. [Google Scholar]
- 16.(a) Nishizawa S, Kato Y, Teramae N. J Am Chem Soc. 1999;121:9463–9464. [Google Scholar]; (b) Bodenant B, Fages F, Delville MH. J Am Chem Soc. 1998;120:7511–7519. [Google Scholar]
- 17.Herz AH. Adv Colloid Interface Sci. 1997;8:237–298. [Google Scholar]
- 18.Magde D, Wong R, Seybold PG. Photochem Photobiol. 2002;75:327–334. doi: 10.1562/0031-8655(2002)075<0327:fqyatr>2.0.co;2. [DOI] [PubMed] [Google Scholar]