

Abstract
Four amphidinolide E stereoisomers, amphidinolide E (1), 2-epi-amphidinolide E (2), 19-epi-amphidinolide E (3), and 2-epi-19-epi-amphidinolide E (4), have been synthesized via the judicious union of aldehyde 5, allylsilanes 7 or 8, acids 9 or 10, and vinylstannane 6. The C19 stereocenters of the C19 epimeric allylsilanes 7 and 8 were introduced via crotylboration reactions early in the synthesis. [3+2]-Annulation reactions of aldehyde 5 with allylsilanes 7 and 8 were employed to set the core tetrahydrofuran units of 1–4. Finally, the C2 stereocenter was installed by esterification using acid 9, without incident, or with acid 10, in which case an unexpected and completely stereoselective inversion of C2 occurs.
Keywords: amphidinolide E stereoisomers, [3+2] annulation reaction, esterification of Fe(CO)3-complexed dienoic acid
1. Introduction
The amphidinolides are a family of biologically active macrolides isolated from the dinoflagellate Amphidinium sp.1 Many of the amphidinolides possess striking cytotoxic properties. Furthermore, this family of natural products exhibits a high degree of structural diversity despite being isolated from a common source. As a consequence, the amphidinolides have attracted considerable interest as targets for synthesis and biological evaluation. Total syntheses of amphidinolides A,2 J,3 K,4 P,5 T,6 W,7 X8 and Y9 have been reported.
Amphidinolide E10 (1) is a 19-membered biologically active1c macrolactone featuring an embedded 2,5-cis-tetrahydrofuran (Figure 1). This structural motif is common within the amphidinolide family. However, the C(1)-C(6) α-chiral, β,γ,δ,ε-dienoate moiety is unique to amphidinolide E. Lee has recently reported the total synthesis of amphidinolide E, 11 while Gurjar12 and Marshall13 have published studies toward the synthesis of this interesting natural product.
Figure 1. Retrosynthetic analysis of amphidinolide E.
As part of a program directed towards the synthesis of tetrahydrofuran-containing natural products14 using a [3+2] annulation strategy, 15,16 we developed and reported a convergent and stereoselective total synthesis of amphidinolide E.17 In the course of these studies, we encountered an unexpected and highly selective C2 inversion during an esterification reaction (25 + 10 → 39) that ultimately led to the inadvertent synthesis of 2-epi-amphidinolide E (2).18 Initially, we were unaware of this C2 inversion and were faced with the conundrum as to why 2-epi-amphidinolide E (2), which at the time we thought was structure 1, did not have spectroscopic properties that matched the data reported in the literature for amphidinolide E (1).10 We describe herein the various structural correlation experiments undertaken to unravel this problem, which ultimately led to the syntheses of amphidinolide E (1), 2-epi-amphidinolide E (2), 19-epi-amphidinolide E (3), and 2-epi-19-epi-amphidinolide E (4).
2. Results and discussion
2.1 Synthesis of 2-epi-amphidinolide E
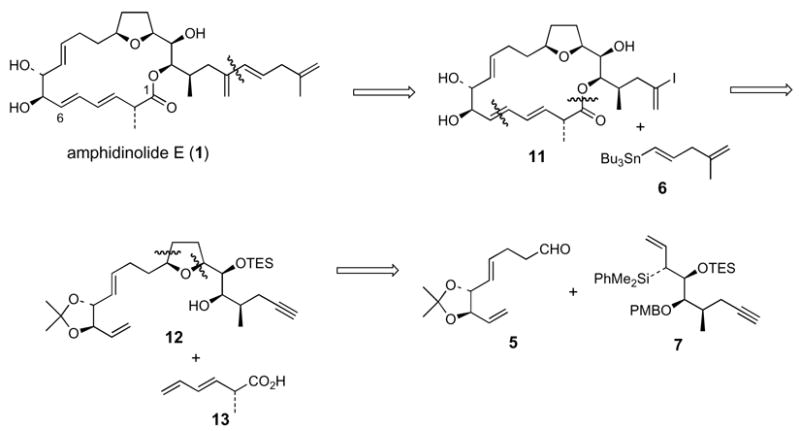
We envisioned that amphidinolide E could be obtained by the Stille19 cross coupling of vinyl iodide 11 with vinylstannane 6 (Figure 1). Macrocycle 11 would be accessed in two steps via esterification of 12 with dienoic acid 13, followed by ring closing metathesis. Finally, we anticipated that the tetrahydrofuran fragment 12 would arise from the product of the [3+2] annulation of aldehyde 5 and allylsilane 7.14a,15
The synthesis of aldehyde 5 began with the Swern oxidation of alcohol 14,20 which is available in five steps from commercially available isopropylidene dimethyl D-tartrate (Scheme 1). Treatment of the aldehyde with vinyl magnesium bromide followed by a Johnson orthoester Claisen rearrangement21 of the allylic alcohol intermediate afforded methyl ester 15 in 60% overall yield. Reduction of 15 with DIBAL (−78 °C) yielded the targeted aldehyde 5.
Scheme 1. Synthesis of aldehyde 5.
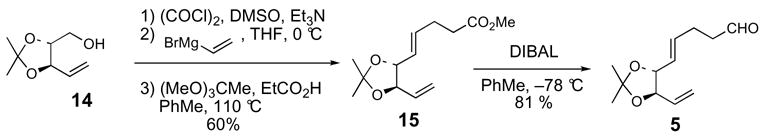
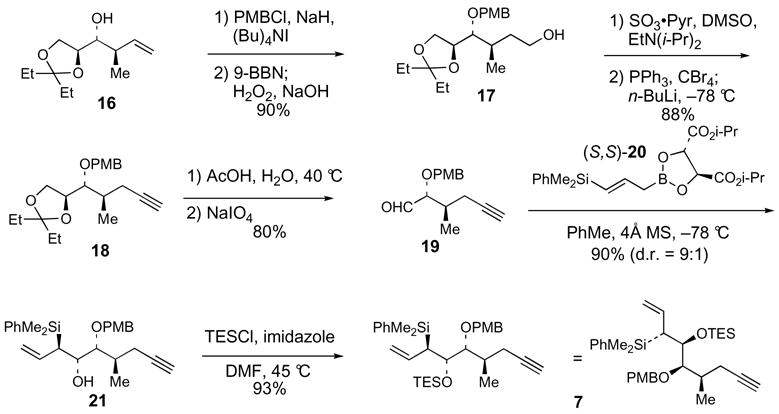
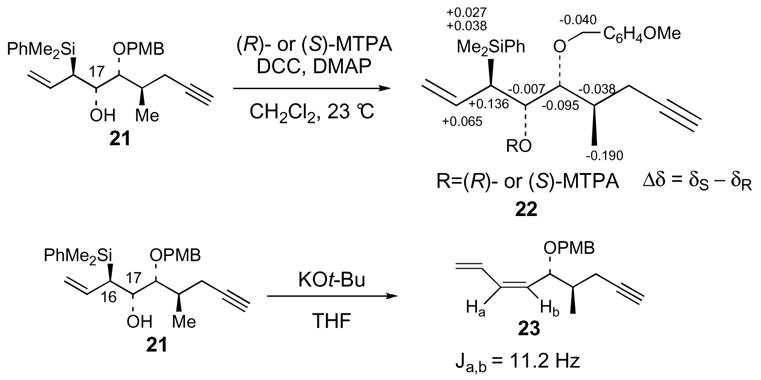
Allylsilane 7 was synthesized starting from homoallylic alcohol 16,22 which is available with high diastereoselectivity from the asymmetric (E)-crotylboration23 of L-glyceraldehyde pentylidene ketal24 (Scheme 2). Protection of 16 as the p-methoxybenzyl ether followed by hydroboration-oxidation of the vinyl group provided primary alcohol 17 (90% yield). Oxidation of 17, by using SO3·pyridine and DMSO,25 and subsequent Corey-Fuchs26 homologation of the aldehyde furnished alkyne 18 (88%). Acidic hydrolysis of the pentylidene ketal protecting group and oxidative cleavage of the resulting diol afforded aldehyde 19. anti-Silylallylboration of 19 was accomplished with 9:1 selectivity (90% yield) by using (E)-γ-silylallylboronate (S,S)-20.27 Protection of the β-hydroxy allylsilane 21 as the triethylsilyl ether provided allylsilane coupling partner 7. Mosher ester analysis of alcohol 21 confirmed the C17(S) hydroxyl stereocenter (Scheme 3).28 In addition, the C16-C17 relative stereochemistry was confirmed via basic Peterson elimination29 of 21, which afforded the Z diene 23.
Scheme 2. Synthesis of allylsilane 7.
Scheme 3. Assignment of the C17 absolute and C16-C17 relative stereochemistry of allylsilane 21.
Initial [3+2] annulations using excess aldehyde 5, with respect to allylsilane 7, and substiochiometric amounts of BF3·Et2O afforded low yields of product 24 (entry 1, Scheme 4). On the other hand, use of stoichiometric or excess amounts of allylsilane 7 and stoichiometric amounts of Lewis acid led to improved yields of 24 (entries 3–5). The optimum reaction stiochiometry, 2.5 equiv of 7 and 1 equiv of 5, led to 24 in 48% yield and d.r. >20:1. Use of SnCl4 as the Lewis acid led to trace amounts of 24 and significant decomposition of 7 (entry 2).30 Excess allylsilane 7 was recovered in excellent yield for all reactions using BF3·Et2O. The modest yield of 24 is due to the propensity of 5 to cyclotrimerize under the reaction conditions to give 26. Slow syringe pump addition of 5 into a −78 °C solution of allylsilane 7 and BF3·Et2O failed to improve the yield. In addition, conducting the reaction at temperatures higher than −78 °C resulted in significant Peterson elimination of 7.
Scheme 4. [3+2] annulation of 5 and 7.
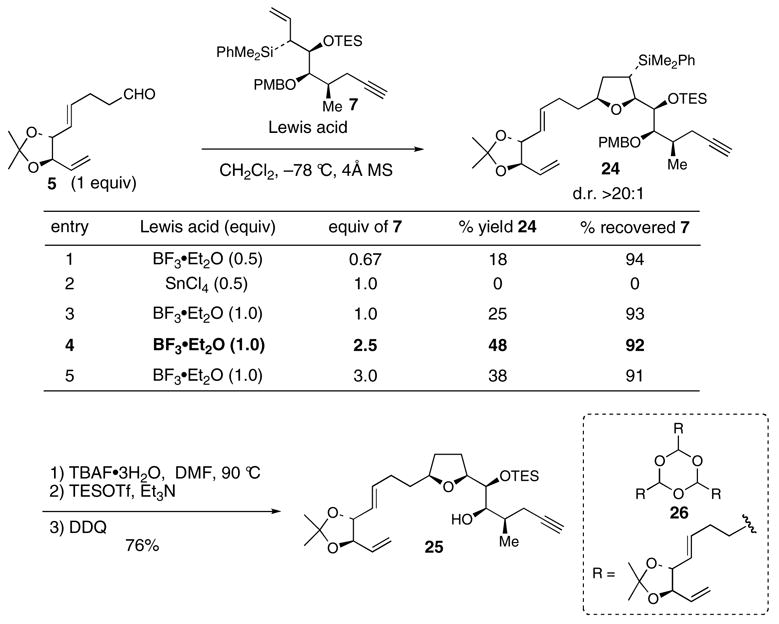
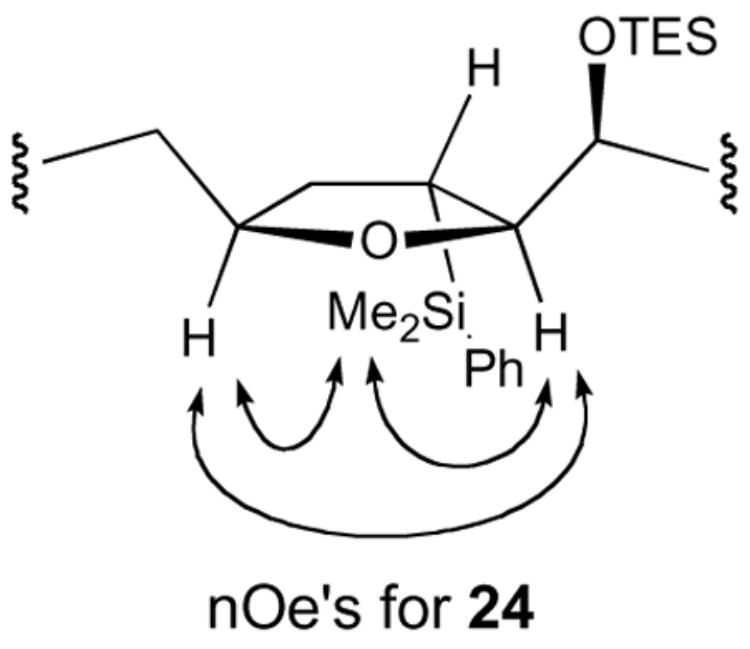
Treatment of [3+2] adduct 24 with solid TBAF·3H2O in DMF at 90 °C effected smooth sp3 C–Si bond scission with concomitant removal of the triethylsilyl ether (Scheme 4).31 Reintroduction of the TES ether, and subsequent oxidative removal of the p-methoxybenzyl group32 gave alcohol 25. The cis-THF stereochemistry of 24 was confirmed via nOe’s shown Figure 2.
Figure 2. nOe analysis of 24.
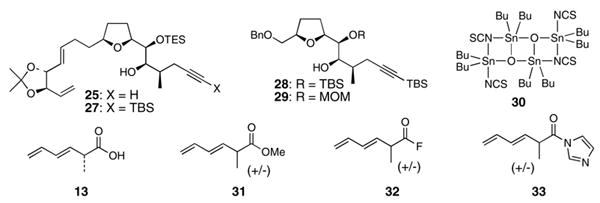
Esterification of the C18 hydroxyl group of 25 (or related intermediates 27, 28, and 29) with dienoic acid 1333 (or various derivatives of 13) proved to be extremely challenging (Table 1). Use of excess amounts (10–20 equiv.) of 13 and various coupling reagents invariably failed. The list of unsuccessful esterification reactions included attempts to use the modified Yamaguchi conditions34 (entry 1), use of mild peptide coupling conditions35 (entries 2, 3 and 6), use of Otera’s transesterification catalyst 3036 (entry 5), attempted coupling of the tributyltin ether of 29 with the acylfluoride 32 (entry 7), and generation of the lithium alkoxide of 29 followed by treatment with acylfluoride 32 (entry 8). Kita37 has developed a two step esterification protocol involving initial formation of a 1-ethoxyvinyl ester derivative of the acid coupling partner using 1-ethoxyacetylene and {RuCl2(p-cymene)}2. This 1-ethoxyvinyl ester derivative is then treated with the alcohol coupling partner and a catalytic amount of a Bronsted acid to achieve the esterification. Lee and co-workers11 have successfully employed this methodology in a macrolactonization fashion for amphidinolide E. Unfortunately, the Kita conditions proved to be unsuccessful in our intermolecular reaction (entry 9). Whereas in most cases the alcohol was recovered unscathed from these unsuccessful experiments, the acid component was recovered as the fully conjugated, diene migrated species 34. No more than trace quantities of ester products corresponding to acids 13 or 34 could be isolated from these experiments.
Table 1. Attempted esterification reactions.
| entry | reaction | conditions | Results | |
|---|---|---|---|---|
| 1 | 27 + 13 | 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, THF, rt to reflux | decomposition of acid
↓ |
|
| 2 | 27 + 13 | EDCI•MeI, DMAP, CH2Cl2, 0 to 23 °C | ||
| 3 | 28 + 13 | PyBrOP, i-Pr2NEt, CH2Cl2, 0 to 23 °C | ||
| 4 | 28 + 33 | Tol., 23 to 100 °C | ||
| 5 | 28 + 31 | 30 (10 mol %), Tol., 50 °C | ||
| 6 | 29 + 13 | DCC, HOBt, THF, 23 to 50 °C | ||
| 7 | 29 + 32 | 29, (Bu3Sn)2O, PhH, reflux; then add 32, CH2Cl2 | ||
| 8 | 29 + 32 | 29, LDA, −78 °C, THF; then 32 or 33, THF | ||
| 9 | 25 + 13 | 13, HCCOEt, {RuCl2(p-cymene)}2, PhMe, 0 to 23°C; then add 25, CSA (10 mol %), PhMe, 50 °C |
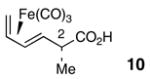
We reasoned that use of a “diene protected” acid 10 might be effective to avoid the problems encountered in attempted esterifications reactions of acid 13. The synthesis of acid 10 began with the Evans methylation38 of oxazolidinone 3539 to afford product 36 in 79% yield (Scheme 5). Treatment of 36 with Fe2(CO)9 in benzene at reflux gave a separable 1:1 mixture of 37 and 38. Hydrolysis of the acyloxazolidinone units of 37 and 38 furnished acids 10 and 940 in 62% and 58% yield, respectively.
Scheme 5. Synthesis of acids 9 and 10.

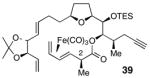
Gratifyingly, use of (CO)3Fe-complexed dienoic acid 10 (1.6 equiv) resulted in an efficient esterification with alcohol 25 under the modified Yamaguchi conditions (Scheme 6). However, the ester product 39 is the unexpected, C2 inverted isomer. Since 39 was formed as a single diastereomer, we had no reason to suspect inversion at C2 and therefore proceeded forward with the synthesis under the assumption that the 2S stereochemistry of 10 had been preserved after the esterification reaction. It was not until much later (see section 2.4) that we became aware of the C2 inversion in this reaction.
Scheme 6. Completion of 2-epi-amphidinolide E synthesis.
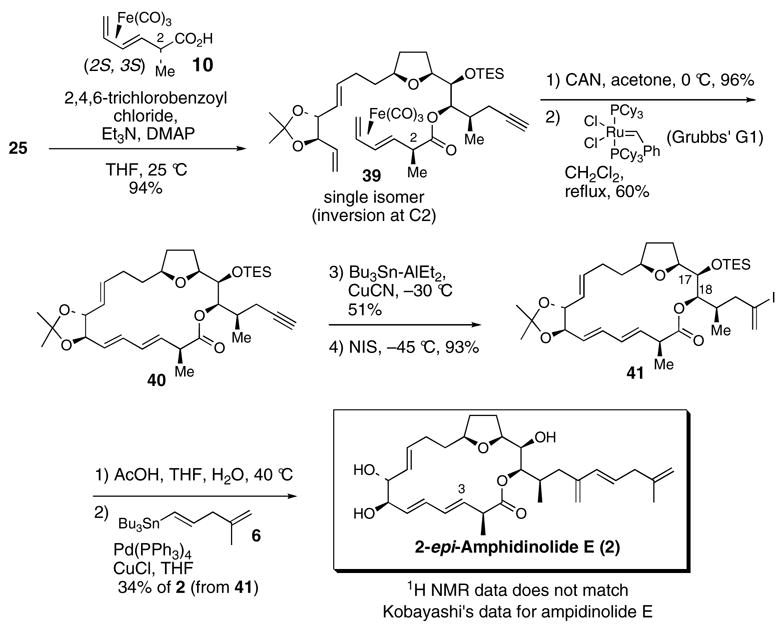
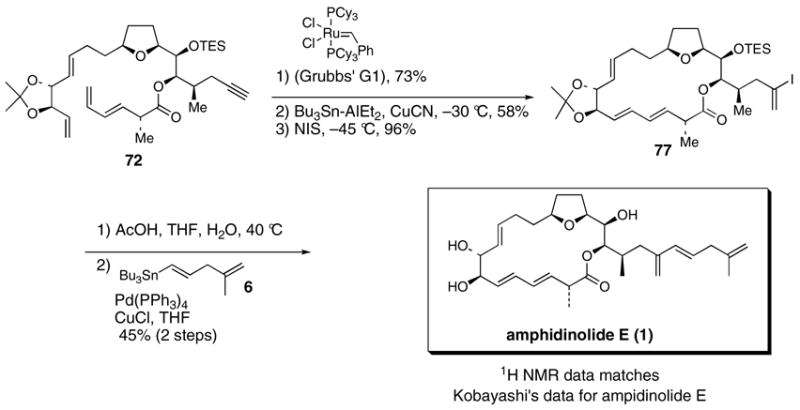
Oxidative decomplexation of the (CO)3Fe-unit of 39 (96% yield) followed by ring closing metathesis41,42 (60% yield) afforded the 19-membered macrocycle 40. Furthermore, an inseparable mixture of products thought to arrive by enyne metathesis was also isolated (15% yield). Use of the more active Grubbs’ second generation or Grubbs-Hoveyda catalysts resulted in significant decomposition of the polyene substrate. Diene and triene forming ring closing metathesis macrocyclizations can sometimes be plague with products containing rings smaller than desired.42a,b However, none of the smaller macrocycles (16-membered ring and smaller) were observed for the ring closure our polyene substrate. In addition, ruthenium catalyzed ring closing metathesis reactions of substrates containing internal alkynes,43 unprotected terminal alkynes,44 and protected terminal alkynes (silylated45 or dicobalt complexed46) are rare.
Stannylalumination-protonolysis47 of the alkyne unit of 40 followed by iododestannylation of the resultant vinylstannane gave vinyl iodide 41. Acidic hydrolysis of both the triethylsilyl and acetonide protecting groups afforded a 10:1 inseparable mixture of the C18 and C17 lactones. Stille cross coupling of the mixture of lactonic iodides with vinylstannane 612 followed by HPLC purification afforded 2-epi-amphidinolide E (2), spectroscopic data for which did not match Kobayashi’s spectroscopic data for amphidinolide E (1). The most egregious spectroscopic disagreement between 2 and natural amphidinolide E (1) was the chemical shift for the H3 proton (6.00 ppm for 2 vs. 5.59 ppm for natural 1).
2.2 Structural correlations of our intermediates with Kobayashi’s amphidinolide E degradation products
In an effort to determine the structural discrepancies between natural amphidinolide E (1) and what we believed was our “synthetic amphidinolide E” (2), we proceeded to repeat Kobayashi’s stereochemical assignments10b for 1 using our synthetic intermediates as correlation compounds.
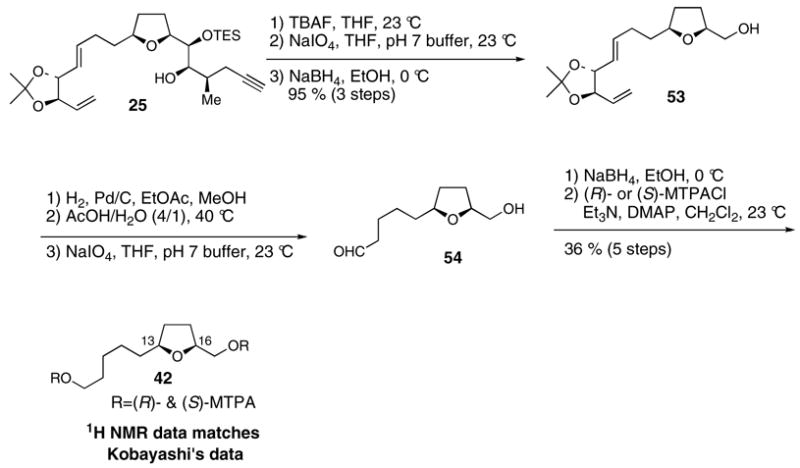
Kobayashi and co-workers transformed natural amphidinolide E (1) into two degradation products, the C8-C17 tetrahydrofuran containing fragment 42 and the C1-C7 fragment 43 (Scheme 7).10 The enantiomer of 42, namely compound 44, was independently synthesized by Kobayashi, thereby leading to the assignment of 13S and 16S stereochemistry in natural amphidinolide E. The 2R stereochemistry of natural amphidinolide E was assigned by analogy to data for a small set of structures containing primary Mosher esters with adjacent methyl-branched stereocenters.48 This precedent indicated that the difference in chemical shifts for the diastereotopic C1 methylene protons were typically smaller for (S)-MTPA esters when the adjacent methyl-branched stereocenter has R stereochemistry. We were concerned about the reliability of this method for absolute stereochemical assignment, given the small number of literature examples, and therefore resolved to make an unequivocal stereochemical assignment for C2 by independently synthesizing the C1-C7 fragment 43 from a chiral pool starting material, aldehyde 4549 (Scheme 8). Olefination of aldehyde 45 afforded product 46.50 Hydrogenation of 46 in EtOAc-MeOH over Pd/C occurred with concomitant hydrolysis of the primary TBS ether. Oxidative removal of the PMB ether of 47 followed by Mosher ester formation yielded the C1-C7 fragment 43. The 1H NMR data for our synthetic 43 matched Kobayashi’s data exactly. Therefore C2 of 43, and hence also of amphidinolide E, is R.
Scheme 7. Kobayashi’s absolute stereochemical assignment of 13S, 16S, and 2R.
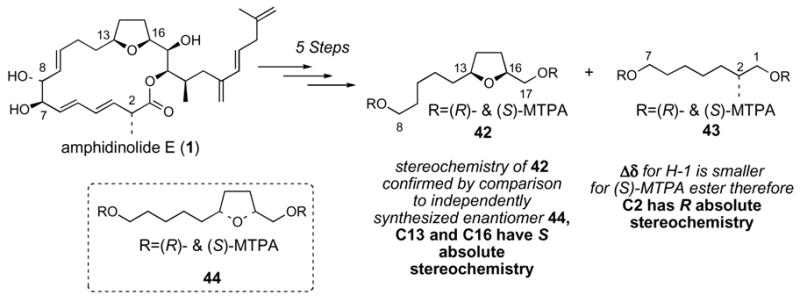
Scheme 8. Synthetic confirmation of 2R stereochemistry of 43.
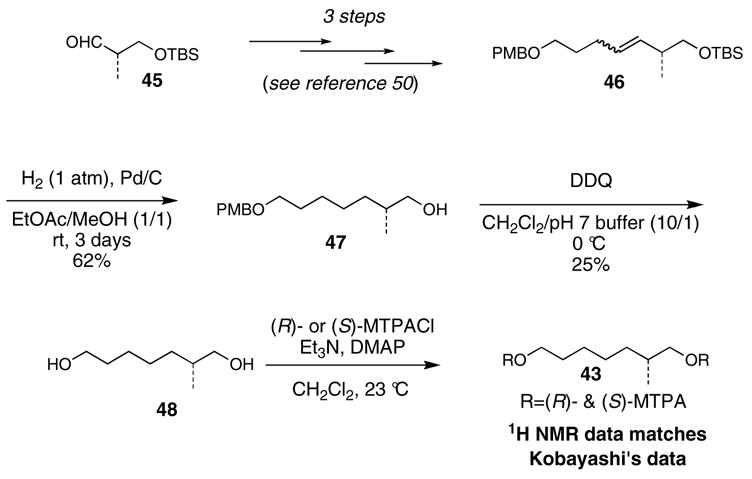
Kobayashi and co-workers treated natural amphidinolide E with 2,2-dimethoxypropane and p-toluenesulfonic acid to generate the C7-C8 acetonide derivative 49 (Scheme 9).10 The magnitude of the H7-H8 coupling constant and the indicated NOESY correlation peaks of 49 formed the basis for assignment of threo C7-C8 relative stereochemistry. The C7 and C8 absolute stereochemistry was assigned by application of the exciton chirality method51 for the 7,8-bis-cinnamoyl ester derivative 50. The CD spectrum of 50 showed a negative first Cotton effect (λext 324 nm, Δε -14.3) and positive second Cotton effect (λext 289 nm, Δε +18.9), indicating 7R and 8R absolute stereochemistry.52
Scheme 9. Kobayashi’s relative and absolute stereochemical assignment of C7 and C8.
Kobayashi’s NMR analysis of the 7,8,17-tris-MTPA ester derivative 51 confirmed the 7R and 8R assignments and determined the 17R stereocenter of natural amphidinolide E (Scheme 10a).10 Deprotection of our ring closing metathesis product 40, followed by Mosher ester formation afforded the synthetic correlation Mosher triester 52 (Scheme 10b). Mosher triester 52 lacks the complete side chain of Kobayashi’s intermediate 51. Therefore, the magnitudes of the chemical shift differences for the (S) versus (R)-MTPA ester derivatives were not expected to be identical. However, if the stereocenters in 51 and 52 were the same, we expected the directionalities of the chemical shift differences (Δδ = δS − δR) to correlate. This held true at every position except for C17 and C3. We thought this discrepancy was an indication that we had incorrectly assigned the C16 stereochemistry of 52, and ultimately 40 and 25, as 16S and that perhaps the correct stereochemistry is 16R. Therefore, our intermediate 25 was transformed into Kobayashi’s C8-C17 fragment 42 in eight standard steps (Scheme 11). The 1H NMR data for our synthetic 42 matched Kobayashi’s data, thereby confirming that our original 16S stereochemical assignment was correct.
Scheme 10.
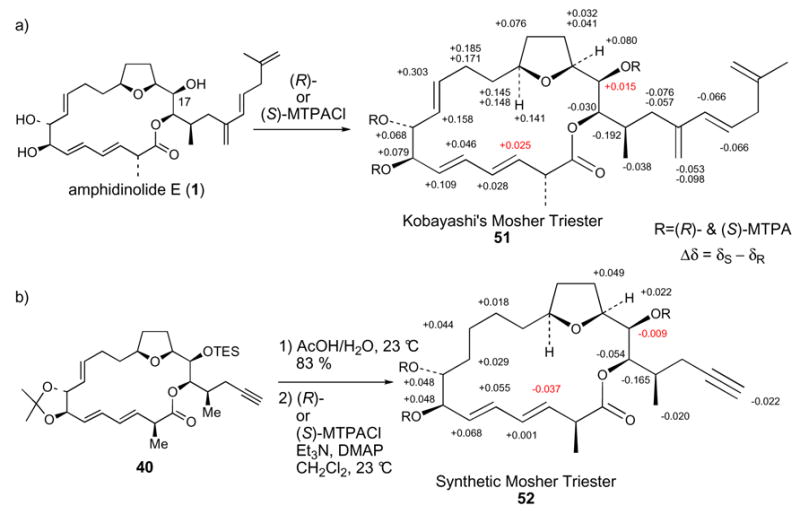
a) Kobayashi’s absolute stereochemical assignment of C17 (also confirming C7 and C8). b) Comparison of Mosher ester data for synthetic 52 and natural product derivative 51.
Scheme 11. Confirmation of 13S and 16S stereochemistry of 25.
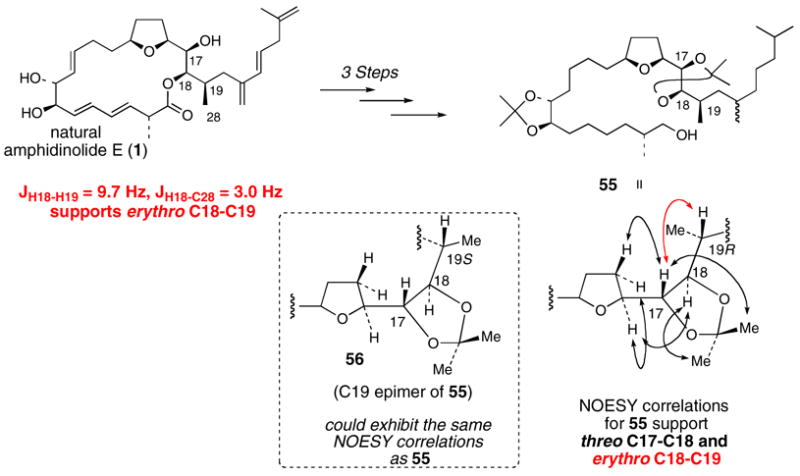
Kobayashi and co-workers assigned the C18 and C19 stereocenters by synthesizing the bis-acetonide intermediate 55 from natural amphidinolide E in three steps (Scheme 12).10 The NOESY correlation peaks of 55 established a threo C17-C18 relationship. In addition, the H18-H19 and H18-C28 coupling constants of 1 supported a erythro C18-C19 relationship. The red NOESY peak in 55 was also used to support the C18-C19 relative assignment. We felt that the NOESY peaks shown for 55 were not unique for the 19R (erythro C18-C19) stereochemical assignment of 55. It is possible that the C19-epimer of 55, compound 56 (19S instead of 19R), could exhibit the same NOESY peaks and coupling constant data. Therefore, we considered the possibility that C19 might have been originally misassigned.
Scheme 12. Kobayashi’s C17-C18 and C18-C19 relative stereochemical assignment.
Based upon the correlation experiments described above, we thought that we had confirmed the C7, C8, C13, C16, C17 and the C2 stereochemistry of both natural amphidinolide E as well as our synthetic intermediates. In addition, we felt that Kobayashi’s C18 stereochemical assignment was irrefutable. Therefore, we concluded at this stage that the structure of natural amphidinolide E most likely was 57, with 19S instead of 19R stereochemistry (Figure 3).
Figure 3. Postulated revised structure of amphidinolide E.
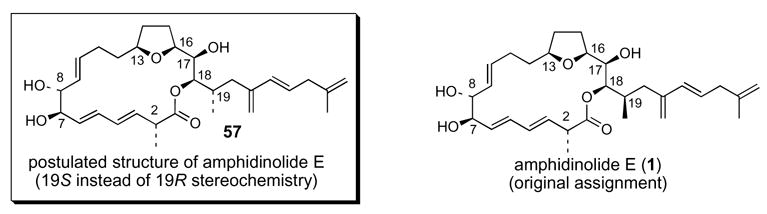
2.3 Synthesis of 2-epi-19-epi-amphidinolide E
The 19S stereochemistry in the possible alternative structure of amphidinolide E (57) was incorporated into our synthetic route by using Z-(S,S)-crotylboronate 5923 for the asymmetric crotylboration of L-glyceraldehyde pentylidene ketal 58.24 This experiment provided homoallylic alcohol 60 in good diastereoselectivity (Scheme 13). Elaboration of 60 into allylsilane 8 was accomplished using steps analogous to our original route shown previously in Scheme 2.
Scheme 13. Synthesis of 19-epi-allylsilane 8.
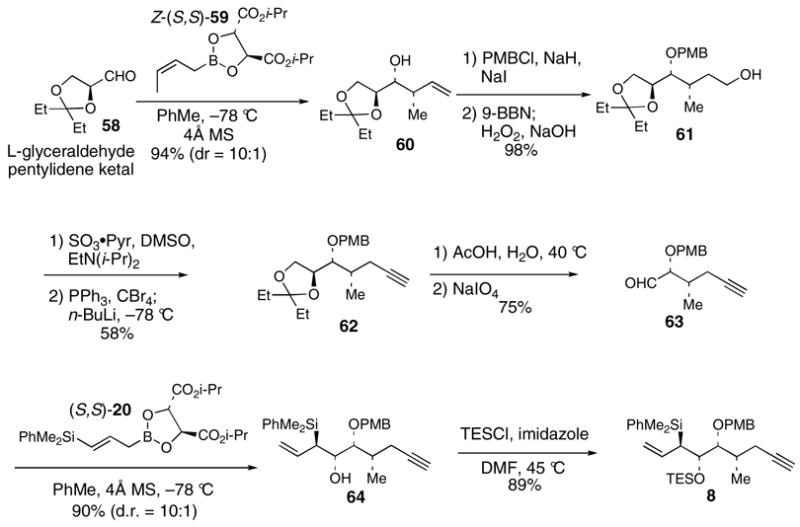
The [3+2] annulation reaction between allylsilane 8 (3 equiv) and aldehyde 5 provide 65 with >20:1 selectivity in 61% yield (Scheme 14). The excess 8 can be recovered with excellent efficiency (92%). The [3+2] adduct 65 was transformed into alcohol 67 using the same sequence as described for the synthesis of 25 (Scheme 4).
Scheme 14. [3+2] Annulation of 5 and 8.
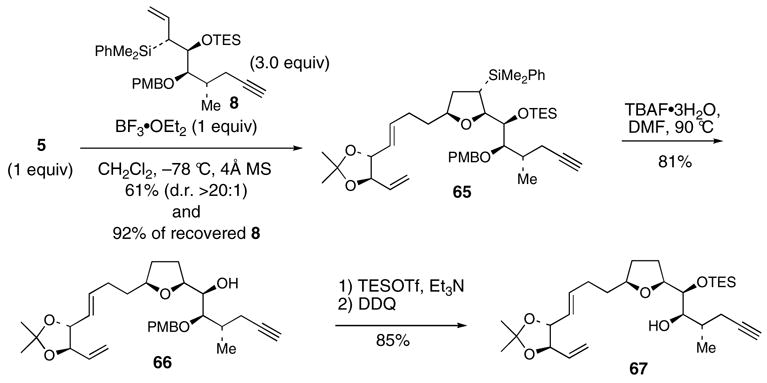
The C16-C17 relative stereochemistry of 8 was confirmed via basic Peterson elimination to afford the Z diene 68 (Scheme 15). Assessment of the C17 absolute stereochemistry was attempted by direct esterification of the hydroxyl group in 8 with (R) and (S)-MTPA-Cl. However, the hydroxyl group of 8 is quite hindered and no reaction was observed. Therefore, confirmation of the C17 absolute stereochemistry was accomplished by the Mosher ester analysis of alcohol 66.
Scheme 15. Confirmation of the C16-C17 relative and C17 absolute stereochemistry.
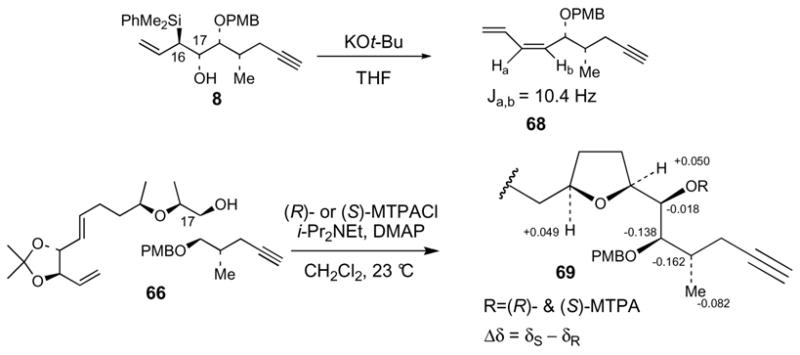
Esterification of alcohol 67 with acid 10 afforded 70 as single diastereomer with complete inversion of the C2 stereochemistry (Scheme 16). Again, we had no reason to suspect inversion at C2 due to the high diastereoselectivity and the very clean “spot to spot” nature of the reaction. Elaboration of 70 into 2-epi-19-epi-amphidinolide E (4) was accomplished using the same chemistry as described for the synthesis of 2-epi-amphidinolide E (2) (Scheme 6). However, it should be mentioned that a small amount of the presumed enyne side products was once again observed during the ring closing metathesis reaction (10%). In addition, the acidic deprotection of 71 afforded a 2:1 inseparable mixture of the regioisomeric C18 (desired) and C17 lactones. Stille coupling of this mixture with vinylstannane 6 followed by HPLC separation of the isomers afforded pure 4. To our dismay, spectroscopic properties of 2-epi-19-epi-amphidinolide E (4), which at this stage we thought was structure 57, once again did not match Kobayashi’s data for natural amphidinolide E.
Scheme 16. Completion of 2-epi-19-epi-amphidinolide E synthesis.
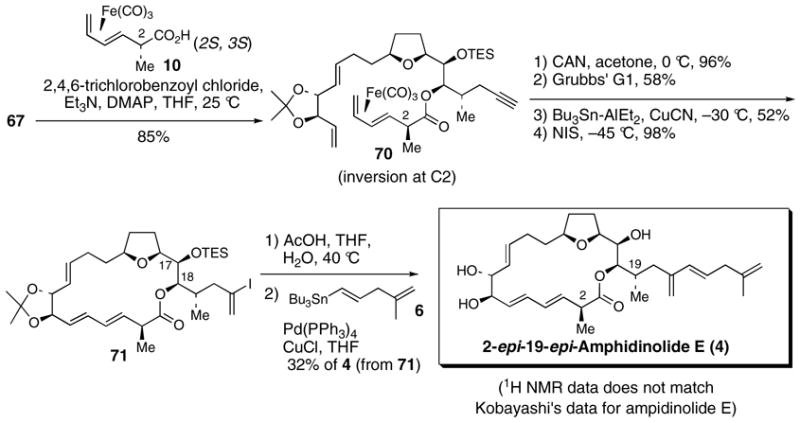
2.4 Discovery of the C2 inversion problem and synthesis of amphidinolide E (1) and 19-epi-amphidinolide E (3)
After synthesizing what we thought were structures 1 and 57 (i.e. syntheses of 2-epi-amphidinolide E (2) and 2-epi-19-epi-amphidinolide E (4)) only to arrive at material that did not match Kobayashi’s natural amphidinolide E, we decided to revisit the original 19R series of compounds in order to obtain more insight into the true structure of amphidinolide E (reaction pathway B, Scheme 17). Critically, we had consumed all of our supply of acid diastereomer 10 and decided to use the large supply of diastereomer 9 that had accumulated in our laboratory (reaction pathway A, Scheme 17). Use of either acid diastereomer 9 or 10 should lead to polyene 72 after oxidative decomplexation of the (CO)3Fe unit if C2 inversion were not occurring. To our surprise, we discovered that esterification-decomplexation reactions using 9 and 10 did not converge to one compound. Instead, they each separately yielded different polyenes 72 and 73 as single diastereomers. Before this discovery, we had always thought that the esterification-decomplexation sequence involving 10 was yielding polyene 72. Instead, it became absolutely clear based on the following examples that use of acid 10 in this sequence afforded polyene 73, proceeding through 39, with clean inversion at C2.
Scheme 17. Divergent behavior of acids 9 and 10 in the modified Yamaguchi esterification reaction of 25.
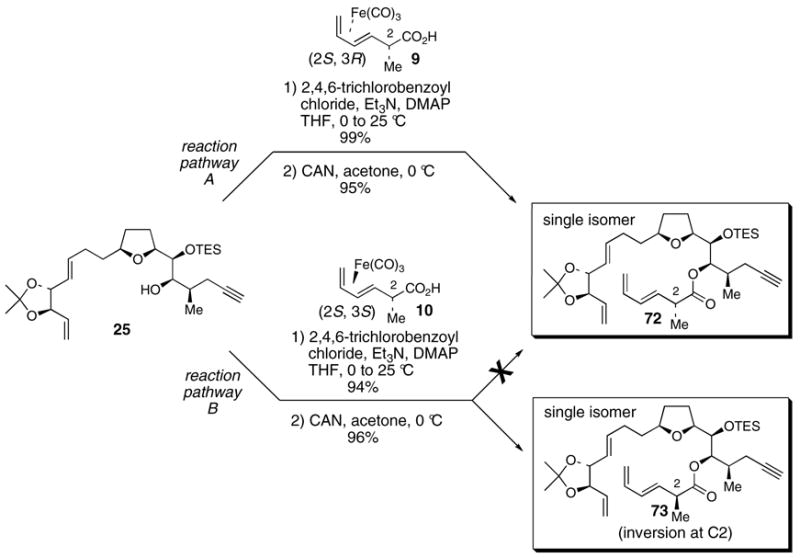
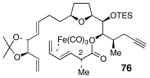
The discovery of separate, divergent reaction pathways for acids 9 and 10 prompted the evaluation of the C2 stereocenter in intermediates both prior to and after the esterification reaction (Table 2). In addition, the starting material used in the synthesis of both 9 and 10, oxazolidinone 36, was also evaluated. Table 2 summarizes these correlation experiments. Compounds 36, 9, 10, 76 and 39 were transformed into diene 75. The optical rotations of 75 from each reduction sequence were then compared with material independently synthesized from aldehyde 45,49 using the method of Keck.53 The 2R stereochemistry of 36 was verified prior to complexation of the (CO)3Fe unit and hydrolysis of the acyloxazolidinone (entry 1, Table 2). The 2S stereochemistry of acids 9 and 10 was also verified prior to being subjected to the esterification reactions (entries 2 & 3, Table 2). Furthermore, the C2 stereochemistry of ester 76, the product of esterification of 25 with acid 9, was confirmed as 2S (entry 4, Table 2). On the other hand, the data in entry 5 indisputably affirms that inversion at C2 occurs when acid 10 is used in the esterification reaction of 25.
Table 2. Assessment of C2 stereochemistry of 36, 9, 10, 76, and 39.
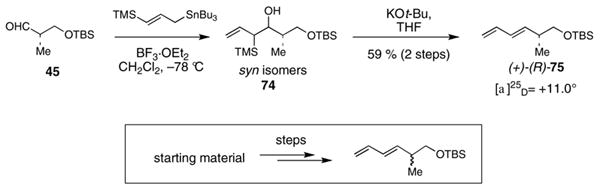
| entry | starting material | steps | overall yield | product [(+)- or (−)-75] |
|---|---|---|---|---|
|
1) LAH | |||
| 1 | 2) TBSCl, Imidazole | 76% | (+)-(R)-75 | |
|
1) BH3·SMe2, THF | |||
| 2 | 2) TBSCl, Imidazole | 84% | (+)-(R)-75 | |
| 3) CAN | ||||
|
1) BH3·SMe2, THF | |||
| 3 | 2) TBSCl, Imidazole | 68% | (+)-(R)-75 | |
| 3) CAN | ||||
|
1) DIBAL, −78 °C | |||
| 4 | 2) TBSCl, Imidazole | 56% | (+)-(R)-75 | |
| 3) CAN | ||||
|
1) DIBAL, −78 °C | |||
| 5 | 2) TBSCl, Imidazole | 61% | (−)-(S)-75 | |
| 3) CAN |
We were pleased to find that subjection of polyene 72 to the same sequence of reactions used to synthesized 2-epi-amphidinolide E (2) and 2-epi-19-epi-amphidinolide E (4) afforded synthetic amphidinolide E (1), which had spectroscopic properties that matched natural amphidinolide E (Scheme 18). Interestingly, it should be noted that, unlike the 2-epi-series, the acidic deprotection of 77 only afforded the desired C18 lactone and none of the undesired C17 regioisomeric lactone. As observed before, a mixture of products thought to arrive by enyne metatesis was also isolated in 10 % yield.
Scheme 18. Completion of the synthesis of amphidinolide E.
Furthermore, we also synthesized the final C2 and C19 stereochemical permutation, 19-epi-amphinolide E (3) (Scheme 19). Only the C18 lactone was observed in the deprotection of 79. Furthermore, only a 5% yield of presumbed enyne products was observed during the RING CLOSING METATHESIS reaction. During the course of the synthesis of 3, we treated 78 with K2CO3 (0.9 equiv) in methanol at 50 °C for 3 h and obtained a 2.6:1 mixture of C2 diastereomers favoring 78. This experiment establishes that the epimerizations observed in the esterifications of 25 (Scheme 6) and 67 (Scheme 16) with the diastereomeric Fe(CO)3-complexed dienoic acid (2S,3S)-10 are contrathermodynamic, and therefore also kinetically controlled.18
Scheme 19. Completion of the synthesis of 19-epi-amphidinolide E.
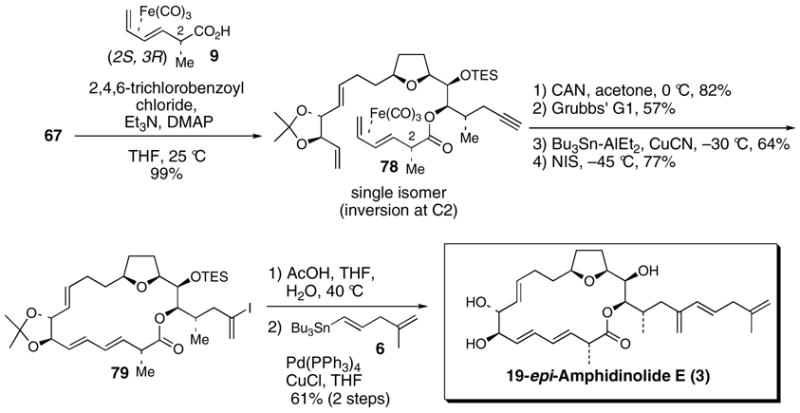
We hypothesize that ketene intermediates may be involved in the esterification reactions with (CO)3Fe-complexed acids 9 and 10 (Scheme 20). Ketene intermediates have previously been implicated by Fürstner and co-workers in studies of the Yamaguchi macrolactonization directed towards iejimalide B.54 Activation of acid 10 followed by elimination could afford ketene intermediate 81. Addition of the alcohol coupling partner (HO-R) to 81 and subsequent reformation of the C2 stereocenter via diastereoselective protonation of enol 82 anti to the (CO)3Fe unit would yield the observed C2 invert product 80. Alternatively, DMAP could add to the ketene intermediate 81.55 Diastereoselective protonation of enolate 83 followed by alkoxide addtion to acyl pyridinium 84 could also afford product 80.
Scheme 20. Proposed esterification pathway.
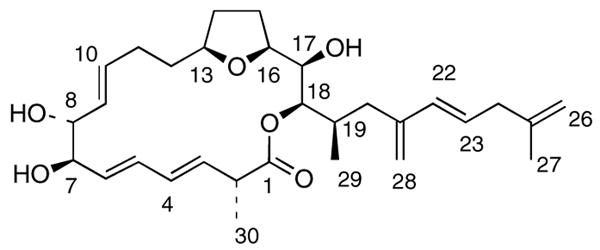
Matching spectroscopic details for natural and synthetic amphidinolide E are summarized in Table 3, confirming the validity of the originally assigned structure. Characteristic 1H NMR data for all four amphidinolide stereoisomers (1–4) are presented in Table 4. It is noteworthy, that the H3 resonance is substantially shifted up field relative to amphidinolide E for both C2 epimers (2 and 4). Furthermore, the C30-Me resonance is also shifted up field for 2 and 4. On the other hand, the C29-Me is shifted downfield in the 19-epi-series (3 and 4). Only the data for synthetic 1 matches that of the natural product.
Table 3. Spectroscopic comparison of natural and synthetic amphidinolide E (1) 1H NMR Data.
| Proton | Natural (Kobayashi, 600 MHz) | Synthetic (Roush, 400 MHz) |
|---|---|---|
| 2 | 3.26 (1H, dq, J = 10.0, 6.8 Hz) | 3.26 (1H, m) |
| 3 | 5.59 (1H, dd, J = 14.0, 10.0) | 5.59 (1H, m) H3, H10 & H23 overlap |
| 4 | 6.20 (1H, dd, J = 14.0, 10.6) | 6.20 (1H, m) H4 and H5 overlap |
| 5 | 6.16 (1H, dd, J = 14.5, 10.6) | 6.16 (1H, m) H4 and H5 overlap |
| 6 | 5.53 (1H, dd, J = 14.5, 8.5) | 5.53 (1H, dd, J = 14.8, 8.8) |
| 7 | 3.88 (1H, t, J = 8.5) | 3.89 (1H, t, J = 8.8) |
| 8 | 3.95 (1H, t, J = 8.5) | 3.95 (1H, t, J = 8.4) |
| 9 | 5.27 (1H, dd, J = 15.6, 8.5) | 5.27 (1H, dd, J = 14.4, 7.6) |
| 10 | 5.64 (1H, m) | 5.64 (1H, m) H3, H10 & H23 overlap |
| 11a | 2.23 (1H, m) | 2.23 (1H, m) |
| 11b | 1.82 (1H, m) | 1.82 (1H, m) |
| 12a | 1.76 (1H, m) | 1.76 (1H, m) |
| 12b | 1.48 (1H, m) | 1.48 (1H, m) |
| 13 | 3.41 (1H, m) | 3.40 (1H, m) |
| 14a | 1.40 (1H, m) | 1.40 (1H, m) |
| 14b | 1.25 (1H, m) | 1.25 (1H, m) |
| 15a | 1.58 (1H, m) | 1.58 (1H, m) overlapping w/water |
| 15b | 1.33 (1H, m) | 1.33 (1H, m) |
| 16 | 3.56 (1H, dt, J = 7.5, 7.1) | 3.56 (1H, m) |
| 17 | 3.72 (1H, dt, J = 7.5, 4.5) | 3.72 (1H, m) |
| 18 | 4.66 (1H, d, J = 8.3) | 4.66 (1H, d, J = 9.2) |
| 19 | 2.25 (1H, m) | 2.25 (1H, m) |
| 20a | 2.40 (1H, d, J = 13.4) | 2.40 (1H, d, J = 14.0) |
| 20b | 1.79 (1H, m) | 1.79 (1H, m) |
| 22 | 6.05 (1H, d, J = 15.9) | 6.05 (1H, d, J = 15.2) |
| 23 | 5.71 (1H, dt, J = 15.9, 6.8) | 5.71 (1H, m) H3, H10 & H23 overlap |
| 24 | 2.78 (2H, br d, J = 6.8) | 2.78 (2H, m) |
| 26a | 4.75 (1H, s) | 4.75 (1H, s) |
| 26b | 4.71 (1H, s) | 4.71 (1H, s) |
| 27 | 1.72 (3H, s) | 1.72 (3H, s) |
| 28a | 4.98 (1H, s) | 4.98 (1H, s) |
| 28b | 4.87 (1H, s) | 4.87 (1H, s) |
| 29 | 0.92 (3H, d, J = 6.6) | 0.92 (3H, d, J = 6.8) |
| 30 | 1.25 (3H, d, J = 6.8) | 1.25 (3H, d, J = 6.8) |
Table 4. Partial spectroscopic details of the four amphidinolide E stereoisomers 1–4.
| C(H) | Synthetic Amphidinolide E (1) | 2-epi-Amphidinolide E (2) | 19-epi-Amphidinolide E (3) | 2-epi-19-epi-Amphidinolide E (4) |
|---|---|---|---|---|
| 2 | 3.26 (1H, m) | 3.35 (1H, q, J = 5.2) | 3.26 (1H, m) | 3.34 (1H, m) |
| 3 | 5.67 (3H, m) 3, 10, & 23 overlap | 6.00 (1H, m) | 5.60 (3H, m) 3, 6, & 10 overlap | 5.98 (1H, m) |
| 4 | 6.19 (2H, m) 4 & 5 overlap | 6.20 (2, m) 4 & 5 overlap | 6.19 (2, m) 4 & 5 overlap | 6.20 (2, m) 4 & 5 overlap |
| 5 | 6.19 (2H, m) | 6.20 (2, m) | 6.19 (2, m) | 6.20 (2, m) |
| 4 & 5 overlap | 4 & 5 overlap | 4 & 5 overlap | 4 & 5 overlap | |
| 6 | 5.53 (1H, dd, J = 14.4, 8.8) | 6.00 (3H, m) 6, 10, & 23 overlap | 5.60 (3H, m) 3, 6, & 10 overlap | 5.63 (2H, m) 6 & 10 overlap |
| 7 | 3.89 (1H, t, J = 8.8) | 3.95 (2H, app dt, J = 8.4, 19.2) 7 & 8 overlap | 3.88 (1H, t, J = 8.8) | 3.96 (2H, m) 7 & 8 overlap |
| 8 | 3.95 (1H, t, J = 8.4) | 3.95 (2H, app dt, J = 8.4, 19.2) 7 & 8 overlap | 3.95 (1H, t, J = 8.4) | 3.96 (2H, m) 7 & 8 overlap |
| 9 | 5.27 (1H, dd, J = 14.4, 7.6) | 5.27 (1H, dd, J = 7.6, 14.4) | 5.27 (1H, dd, J = 8.0, 15.2) | 5.30 (1H, dd, J = 7.6, 15.2) |
| 10 | 5.67 (3H, m) 3, 10, & 23 overlap | 6.00 (3H, m) 6, 10, & 23 overlap | 5.60 (3H, m) 3, 6, & 10 overlap | 5.63 (2H, m) 6 & 10 overlap |
| 13 | 3.40 (1H, m) | 3.43 (1H, m) | 3.40 (1H, m) | 3.42 (1H, m) |
| 16 | 3.56 (1H, m) | 3.50 (1H, m) | 3.56 (1H, app q, J = 8.8) | 3.50 (1H, m) |
| 17 | 3.72 (1H, m) | 3.69 (1H, app t, J = 5.2) | 3.78 (1H, d, J = 7.2) | 3.74 (1H, m) |
| 18 | 4.66 (1H, d, J = 9.2) | 4.66 (1H, d, J = 9.6) | 4.68 (1H, d, J = 10.4) | 4.69 (1H, d, J = 10.4) |
| 22 | 6.05 (1H, d, J = 15.2) | 6.04 (1H, d, J = 16.0) | 6.05 (1H, d, J = 15.6) | 6.05 (1H, d, J = 16.0) |
| 23 | 5.67 (3H, m) 3, 10, & 23 overlap | 6.00 (3H, m) 6, 10, & 23 overlap | 5.87 (1H, dt, J = 6.8, 16.0) | 5.85 (1H, dt, J = 7.2, 15.6) |
| 26a | 4.75 (1H, s) | 4.74 (1H, s) | 4.73 (1H, s) | 4.74 (1H, s) |
| 26b | 4.71 (1H, s) | 4.70 (1H, s) | 4.70 (1H, s) | 4.70 (1H, s) |
| 27 | 1.72 (3H, s) | 1.72 (3H, s) | 1.71 (3H, s) | 1.72 (3H, s) |
| 28a | 4.98 (1H, s) | 4.98 (1H, s) | 4.98 (1H, s) | 4.98 (1H, s) |
| 28b | 4.87 (1H, s) | 4.86 (1H, s) | 4.88 (1H, s) | 4.88 (1H, s) |
| 29 | 0.92 (3H, d, J = 6.8) | 0.91 (3H, d, J = 6.8) | 0.81 (3H, d, J = 6.4) | 0.82 (3H, d, J = 6.8) |
| 30 | 1.25 (3H, d, J = 6.8) | 1.34 (3H, d, J = 7.2) | 1.25 (3H, d, J = 6.8) | 1.34 (3H, d, J = 6.8) |
3. Conclusion
In conclusion, we have synthesized four stereoisomers of amphidinolide E, namely amphidinolide E (1), 2-epi-amphidinolide E (2), 19-epi-amphidinolide E (3), and 2-epi-19-epi-amphidinolide E (4). This constitutes a rigorous verification of the stereochemistry of amphidinolide E.56 In the course of the studies toward 1, we discovered an unexpected and highly selective C2 inversion in the esterification reaction of (CO)3Fe-complexed dienoic acid 10. Insight into the possible mechanism of this epimerization, the context of which depends on the steric environment of the alcohol, has been published elsewhere.18 Results of the biological evaluation of 2, 3, and 4 will be reported in due course.
4. Experimental
General Experimental Details
All reaction solvents were purified before use. Tetrahydrofuran, dichloromethane, diethyl ether, and toluene were purified by passing through a solvent column composed of activated A-1 alumina. Unless indicated otherwise, all reactions were conducted under an atmosphere of argon using flame-dried or oven-dried (170 °C) glassware. Four Å molecular sieves were activated under high vacuum with heat (180 °C) for 12 h and re-activated by thorough flame-drying immediately prior to use.
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on commercial instruments at 400 or 500 MHz. Carbon-13 nuclear magnetic resonance (13C NMR) spectra were recorded at 100 and 125 MHz, respectively. The proton signal for residual non-deuterated solvent (δ 7.26 for CHCl3) was used as an internal reference for 1H NMR spectra. For 13C NMR spectra, chemical shifts are reported relative to the δ 77.2 resonance of CHCl3. Coupling constants are reported in Hz. Infrared (IR) spectra were recorded as films on a FTIR instrument. Opitcal rotations were measured on a polarimeter using a quartz cell with 1 mL capacity and a 10 cm path length. Mass spectra were recorded on a commercial spectrometer.
Analytical thin layer chromatography (TLC) was performed on Kieselgel 60 F254 glass plates pre-coated with a 0.25 mm thickness of silica gel. The TLC plates were visualized with UV light and/or by staining with Hanessian solution (ceric sulfate and ammonium molybdate in aqueous sulfuric acid). Column chromatography was generally performed using Kieselgel 60 (230–400 mesh) silica gel, typically using a 50–100:1 weight ratio of silica gel to crude product.
HPLC purifications were performed by using a HPLC system composed of two Varian Prostar pumps (model 210) connected to normal phase columns. Samples were loaded into the system with a 2 mL Rheodyne 7125 injector and were detected using a Varian Prostar UV and a Varian RI detector.
(E)-5-((4R,5R)-2,2-Dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-pent-4-enoic acid methyl ester (15)
To a −78 °C solution of (COCl)2 (3.45 mL, 39.4 mmol) in CH2Cl2 (80 mL) was added DMSO (3.50 mL, 49.2 mmol) in CH2Cl2 (10 mL). The reaction was stirred at −78 °C for 15 min, then alcohol 1420 (3.12 g, 19.7 mmol) in CH2Cl2 (10 mL) was added. The reaction was stirred for 20 min at −78 °C followed by the addition of triethylamine (16.4 mL, 118 mmol). The mixture was allowed to warm to 0 °C. After 30 min, the reaction was diluted with Et2O (300 mL), upon which a white precipitate forms (triethylamine hydrochloride). The slurry was filtered through a 1 inch pad of Celite and concentrated to afford the aldehyde, a yellow oil, which was immediately used in the next reaction.
To a 0 °C solution of the crude aldehyde in THF (60 mL) was added vinylmagnesium bromide (60 mL of a 1.0M THF solution, 60 mmol). The reaction was stirred for 2.5 h, and then quenched with saturated aqueous NaHCO3 (50 mL) and extract with Et2O (20 mL × 3). The organic phase was washed with brine (50 mL), dried over anhydrous MgSO4, filtered, and concentrated to afford a mixture of diastereomeric allylic alcohols as a yellow oil. This oil was used immediately in the next reaction.
To the mixture of diastereomeric allylic alcohols, from the preceding step, in toluene (66 mL) was added trimethyl orthoacetate (12.5 mL, 98.5 mmol) and propionoic acid (0.3 mL, 3.94 mmol). The reaction was fitted with a condenser and placed in a 110 °C oil bath for 18 h. The solution was then quenched with 3 mL of triethylamine and concentrated. The crude product was purified by flash column chromatography to yield methyl ester 15 (2.83 g, 60% over 3 steps) as a colorless oil: [α]25D = −132° (c 0.99, CHCl3); 1H NMR (400MHz, CDCl3) δ 5.74-5.83 (m, 2H), 5.48 (dd, J = 6.4, 15.2 Hz, 1H), 5.33 (d, J = 17.2 Hz, 1H), 5.24 (d, J = 10.4 Hz, 1H), 4.04 (app q, J = 6.8 Hz, 2H), 3.67 (s, 3H), 2.36-2.44 (m, 4H), 1.44 (s, 3H), 1.43 (s, 3H); 13C NMR (100MHz, CDCl3) δ 172. 8, 134.0, 133.7, 126.9, 118.3, 108.7, 82.0, 81.6, 51.3, 33.1, 27.3, 26.8, 26.7; IR (neat) 2987, 2874, 1740, 1437, 1371 cm−1; HRMS (ES+) m/z for C12H18O3Na [M+Na]+ calcd 263.1259, found 263.1255.
(E)-5-((4R,5R)-2,2-Dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-pent-4-enal (5)
To a −78 °C solution of methyl ester 15 (2.25 g, 9.36 mmol) in toluene (31 mL) was added DIBAL (9.36 mL of a 1.0M hexane solution, 9.36 mmol) dropwise such that the internal temperature was below −70 °C. After being stirred for 30 min, the reaction was quenched with saturated aqueous sodium potassium tartrate (Rochelle’s salt) (40 mL) and diluted with Et2O (20 mL). The mixure was stirred at room temperature for 3 h and extracted with Et2O (20 mL × 3). The organic phase was washed with brine (50 mL), dried over anhydrous MgSO4, filtered, and concentrated. The crude product was purified by flash column chromatography to afford aldehyde 5 (1.59 g, 81%) as a colorless oil: [α]25D = −28.7° (c 1.41, CHCl3); 1H NMR (400MHz, CDCl3) δ 9.73 (bs, 1H), 5.70-5.85 (m, 2H), 5.49 (bdd, J = 6.0, 15.6 Hz, 1H), 5.34 (d, J = 17.2 Hz, 1H), 5.24 (d, J = 10.4 Hz, 1H), 4.00-4.10 (m, 2H), 2.52-2.60 (m, 2H), 2.35-2.45 (m, 2H), 1.44 (s, 3H), 1.43 (s, 3H); 13C NMR (100MHz, CDCl3) δ 201.3, 134.1, 133.8, 127.2, 118.8, 109.0, 82.2, 81.8, 42.8, 27.0, 27.0, 24.7; IR (neat) 3085, 2987, 2875, 1726, 1379, 1371, 1239 cm−1; HRMS (ES+) m/z for C13H20O3Na [M+Na]+ calcd 233.1154, found 233.1245.
(3R,4R)-4-((S)-2,2-Diethyl-[1,3]dioxolan-4-yl)-4-(4-methoxy-benzyloxy)-3-methyl-butan-1-ol (17)
To a 0 °C slurry of NaH (1.69 g, 70.6 mmol) and Bu4NI (1.7 g, 4.7 mmol) in THF (157 mL) was added homoallylic alcohol 1622 (10.1 g, 47.0 mmol) followed by p-methoxybenzyl chloride (6.38 mL, 47. 0 mmol). The reaction was fitted with a condenser and refluxed for 16 h. The reaction was quenched with sat. aq. NH4Cl (50 mL) and water (50 mL) and extracted with EtOAc (25 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afforded the p-methoxybenzyl ether (15.15 g, 96%) as a colorless oil: [α]25D = −41° (c 1.53, CHCl3); 1H NMR (400MHz, CDCl3) δ 7.25 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 5.86 (ddd, J = 8.0, 10.4, 17.2 Hz, 1H), 5.04 (app t, J = 17.6 Hz, 2H), 4.60 (AB, J = 10.8 Hz, 1H), 4.55 (AB, J = 11.2 Hz, 2H), 4.05-4.10 (m, 1H), 4.00 (dd, J = 6.0, 7.6 Hz, 1H), 3.81 (s, 3H), 3.77 (d, J = 7.6 Hz, 1H), 3.52 (dd, J = 3.6, 6.0 Hz, 1H), 2.50-2.54 (m, 1H), 1.57-1.70 (m, 4H), 1.09 (d, J = 6.8 Hz, 3H), 0.89 (dt, J = 9.6, 7.6 Hz, 6H); 13C NMR (100MHz, CDCl3) δ 159.1, 140.1, 130.1, 129.3, 115.0, 113.7, 112.1, 83.1, 77.3, 74.0, 66.9, 55.2, 40.8, 29.7, 29.0, 17.0, 8.2, 8.1; IR (neat) 3073, 2972, 1613, 1514, 1249 cm−1; HRMS (ES+) m/z for C20H30O4Na [M+Na]+ calcd 357.2042, found 357.2044.
To a solution of the p-methoxybenzyl ether (15.1 g, 45.3 mmol) in THF (181 mL) was added 9-BBN (272 mL of a 0.5 M THF solution, 136 mmol). The reaction was fitted with a condenser, refluxed for 3 h, cooled to 0 °C and quenched with water (25 mL). The mixure was then treated with 2N NaOH aq. (227 mL) followed by 30% (w/w) H2O2 (46.3 mL) and the biphasic mixture was stirred at room temperature for 17 h. The aqueous phase was extracted with EtOAc (50 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford 17 (15.1 g, 94%) as a colorless oil: [α]25D = −27° (c 0.63, CHCl3); 1H NMR (500MHz, CDCl3) δ 7.24 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 4.56 (s, 2H), 4.16 (app q, J = 6.5 Hz, 1H), 4.07 (dd, J = 6.0, 8.0 Hz, 1H), 3.80 (s, 3H), 3.75 (app t, J = 8.0 Hz, 1H), 3.70-3.76 (m, 1H), 3.60-3.64 (m, 1H), 3.46 (dd, J = 4.5, 6.0 Hz, 1H), 2.02-2.07 (m, 1H), 1.95 (dd, J = 4.5, 6.0 Hz, 1H), 1.73-1.79 (m, 1H), 1.58-1.67 (m, 4H), 1.03 (d, J = 7.0 Hz, 3H), 0.89 (dt, J = 7.0, 5.5 Hz, 6H); 13C NMR (125MHz, CDCl3) δ 159.2, 130.3, 129.3, 113.7, 112.6, 83.3, 77.3, 76.3, 67.7, 60.5, 55.2, 34.9, 32.1, 29.7, 29.0, 16.3, 8.2; IR (neat) 3436, 2971, 2881, 1613, 1514, 1249 cm−1; HRMS (ES+) m/z for C20H32O5Na [M+Na]+ calcd 375.2147, found 375.2141.
(S)-2,2-Diethyl-4-[(1R,2R)-1-(4-methoxy-benzyloxy)-2-methyl-pent-4-ynyl]-[1,3]dioxolane (18)
To a 0 °C solution of alcohol 17 (15.0 g, 42.6 mmol) in CH2Cl2 (142 mL) was added DMSO (9.1 mL, 128 mmol), i-Pr2NEt (22.2 mL, 128 mmol) and SO3·Pyr (20.3 g, 128 mmol). The reaction was stirred at 0 °C for 30 min, then quenched with sat. aq. Na2S2O3 (100 mL) and extracted with CH2Cl2 (30 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford the aldehyde (13.39 g, 89%) as a colorless oil: [α]25D = −30° (c 2.2, CHCl3); 1H NMR (500MHz, CDCl3) δ 9.73 (app t, J = 2.0 Hz, 1H), 7.23 (d, J = 9.0 Hz, 2H), 6.87 (d, J = 9.0 Hz, 2H), 4.56 (AB, J = 11.0 Hz, 1H), 4.53 (AB, J = 11.0 Hz, 1H), 4.12 (dd, J = 6.5, 13.0 Hz, 1H), 4.06 (dd, J = 6.5, 8.0 Hz, 1H), 3.80 (s, 3H), 3.73 (t, J = 8.0 Hz, 1H), 3.40 (dd, J = 4.5, 6.0 Hz, 1H), 2.65 (ddd, J = 2.0, 6.0, 7.5 Hz, 1H), 2.43-2.48 (m, 1H), 2.37 (ddd, J = 2.0, 7.5, 9.5 Hz, 1H), 1.57-1.67 (m, 4H), 1.06 (d, J = 7.5, Hz, 3H), 0.88 (t, J = 7.0 Hz, 6H); 13C NMR (125MHz, CDCl3) δ 202.2, 159.2, 130.2, 129.4, 113.7, 113.0, 82.2, 76.1, 73.3, 67.6, 55.2, 47.1, 30.3, 29.7, 28.9, 16.5, 8.2, 8.2; IR (neat) 2971, 2934, 2724, 2721, 1724, 1514, 1249 cm−1; HRMS (ES+) m/z for C20H30O5Na [M+Na]+ calcd 373.1991, found 373.1984.
To a 0 °C solution of PPh3 (24.9 g, 94.87 mmol) in CH2Cl2 (182 mL) was added CBr4 (15.7 g, 47.4 mmol). The reaction was warmed to room temperature for 30 min and then cooled back to 0 °C. To this mixture was added the aldehyde from the preceding step (12.8 g, 36.5 mmol) in CH2Cl2 (5 mL). The reaction was stirred for 30 min and then diluted with hexane (400 mL), upon which a white precipitate formed (Ph3P=O). The slurry was filtered through Celite and concentrated. The residue was dissolve in hexane (300 mL) to precipitate more Ph3P=O. The slurry was filtered through Celite and again concentrated. The residual oil was dissolved in THF (100 mL), cooled to −78 °C and treated with n-BuLi (32.4 mL of 2.29M hexane solution, 74.3 mmol). The reaction was stirred for 1h and then quenched with sat. aq. NH4Cl (100 mL) and extracted with EtOAc (50 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. Purification of the crude product by flash column chromatography afforded 18 (11.0 g, 98%) as a colorless oil: [α]25D = −7.6° (c 0.89, CHCl3); 1H NMR (400MHz, CDCl3) δ 7.25 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 8.8 Hz, 2H), 4.62 (AB, J = 10.8 Hz, 1H), 4.54 (AB, J = 11.2 Hz, 1H), 4.17 (dt, J = 6.0, 8.0 Hz, 1H), 4.03 (dd, J = 6.0, 8.0 Hz, 1H), 3.80 (s, 3H), 3.77 (t, J = 8.0 Hz, 1H), 3.57 (t, J = 6.0 Hz, 1H), 2.27-2.39 (m, 2H), 1.98 (app t, J = 3.2 Hz, 1H), 1.91-1.98 (m, 1H), 1.56-1.71 (m, 4H), 1.10 (d, J = 7.2 Hz, 3H), 0.90 (app q, J = 7.6 Hz, 6H); 13C NMR (100MHz, CDCl3) δ 159.2, 130.5, 129.4, 113.7, 112.8, 83.2, 81.2, 76.5, 73.7, 69.4, 67.0, 55.2, 34.9, 29.7, 29.0, 22.1, 15.7, 8.2, 8.1; IR (neat) 3295, 2971, 1613, 1514 cm−1; HRMS (ES+) m/z for C21H30O4Na [M+Na]+ calcd 369.2042, found 369.2037.
(2R,3R)-2-(4-Methoxy-benzyloxy)-3-methyl-hex-5-ynal (19)
To alkyne 18 (4.84 g, 14.0 mmol) was added a 4:1 mixture of AcOH and water (47 mL). The reaction mixture was heated to 40 °C for 6 h and then was diluted with 50 mL of EtOAc. Solid NaHCO3 (20 g) was slowly added portionwise and then the mixture was extracted with EtOAc (25 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford the diol (3.39 g, 87%) as a colorless oil: [α]25D = +13.6° (c 0.59, CHCl3); 1H NMR (400MHz, CDCl3) δ 7.27 (d, J = 8.8 Hz, 2H), 6.89 (d, J = 8.8 Hz, 2H), 4.64 (AB, J = 11.2 Hz, 1H), 4.61 (AB, J = 11.2 Hz, 1H), 3.80 (s, 3H), 3.69-3.84 (m, 3H), 3.58 (dd, J = 4.4, 7.2 Hz, 1H), 2.33-2.46 (m, 3H), 2.18 (dd, J = 4.0, 8.0 Hz, 1H), 2.03 (t, J = 2.4 Hz, 1H), 1.96-2.02 (m, 1H), 7.27 (d, J = 8.8 Hz, 2H), 1.08 (d, J = 6.8 Hz, 3H); 13C NMR (100MHz, CDCl3) δ 159.4, 130.3, 129.6, 113.9, 83.8, 83.0, 74.8, 71.5, 69.9, 63.3, 55.3, 34.4, 21.9, 16.3; IR (neat) 3413, 3306, 2936, 1612, 1515, 1249 cm−1; HRMS (ES+) m/z for C16H22O4Na [M+Na]+ calcd 301.1416, found 301.1416.
To a 0 °C solution of the diol (3.39 g, 12.2 mmol) in THF (20 mL) and pH 7 buffer (20 mL) was added NaIO4 (3.13 g, 14.6 mmol). The reaction was stirred for 4 h, quenched with sat. aq. Na2S2O3 (25 mL) and extracted with EtOAc (25 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated to afford pure 19 (2.76 g, 92%) as a colorless oil: [α]25D = +80° (c 2.26, CHCl3); 1H NMR (500MHz, CDCl3) δ 9.65 (app d, J = 3.0 Hz, 1H), 7.27 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 4.59 (d, J = 11.5 Hz, 1H), 4.47 (d, J = 11.5 Hz, 1H), 3.81 (s, 3H), 3.60 (dd, J = 3.0, 10.0 Hz, 1H), 2.34-2.36 (m, 2H), 2.11-2.17 (m, 1H), 1.98 (app t, J = 2.5 Hz, 1H), 1.04 (d, J = 7.0 Hz, 3H); 13C NMR (125MHz, CDCl3) δ 203.5, 159.5, 129.8, 129.2, 113.8, 85.6, 81.9, 72.8, 70.4, 55.2, 34.0, 21.3, 15.3; IR (neat) 3292, 2967, 2837, 1731, 1515, 1249 cm−1; HRMS (ES+) m/z for C15H18O3Na [M+Na]+ calcd 269.1154, found 269.1147.
(3R,4S,5R,6R)-3-(Dimethylphenylsilanyl)-5-(4-methoxy-benzyloxy)-6-methyl-non-1-en-8-yn-4-ol (21)
To a −78 °C slurry of aldehyde 19 (5.95 g, 24.2 mmol) and 4Å mol. sieves (4.8 g) in toluene (20 mL) was added (S,S)-2027 (61 mL of a 1.0M solution in toluene, 60.4 mmol). The reaction was stirred at −78 °C for 18 h and then quenched with 2N NaOH aq. (100 mL). The biphasic mixture was filtered through Celite and extracted with EtOAc (30 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford 21 (9.19 g, 90%) as a colorless oil: [α]25D = −6° (c 2.48, CHCl3); 1H NMR (400MHz, CDCl3) δ 7.55-7.57 (m, 2H), 7.34-7.36 (m, 3H), 7.25 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 5.98 (dt, J = 10.4, 21.5 Hz, 1H), 5.03 (d, J = 10.4 Hz, 1H), 4.85 (d, J = 21.5 Hz, 1H), 4.58 (d, J = 13.0 Hz, 1H), 4.49 (d, J = 13.5 Hz, 1H), 3.81 (s, 3H), 3.73-3.77 (m, 1H), 3.31 (dd, J = 3.2, 6.8 Hz, 1H), 2.43 (d, J = 4.0 Hz, 1H), 2.08-2.18 (m, 1H), 1.91-1.98 (m, 3H), 1.08 (d, J = 7.2 Hz, 3H), 0.39 (s, 3H), 0.34 (s, 3H); 13C NMR (100MHz, CDCl3) δ 159.3, 137.9, 134.8, 134.1, 130.4, 129.4, 129.0, 127.6, 114.5, 113.9, 85.5, 83.6, 74.9, 71.1, 69.3, 55.3, 39.2, 34.1, 20.3, 17.9, -3.8, -4.2; IR (neat) 3560, 3304, 2961, 1613, 1514 cm−1; HRMS (ES+) m/z for C26H34O3SiNa [M+Na]+ calcd 445.2175, found 445.2176.
1-[(1R,2S,3R)-3-(Dimethyl-phenyl-silanyl)-1-((R)-1-methyl-but-3-ynyl)-2-triethylsilanyloxy-pent-4-enyloxymethyl]-4-methoxy-benzene (7)
To a solution of 21 (1.01 g, 2.39 mmol) in DMF (2.5 mL) was added imidazole (0.50 g, 7.4 mmol) and triethylsilylchloride (1.21 mL, 7.17 mmol). The reaction was heated to 45 °C for 17 h and then quenched with water (15 mL) and extracted with Et2O (25 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford 7 (1.19 g, 93%) as a colorless oil: [α]25D = +31° (c 1.30, CHCl3); 1H NMR (400MHz, CDCl3) δ 7.49-7.52 (m, 2H), 7.29-7.35 (m, 3H), 7.18 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 6.12 (dt, J = 10.8, 17.2 Hz, 1H), 4.91 (dd, J = 10.4, 2.0 Hz, 1H), 4.79 (dd, J = 17.2, 1.6 Hz, 1H), 4.30 (AB, J = 18.8 Hz, 1H), 4.27 (d, J = 12.4 Hz, 1H), 4.50-4.70 (m, 1H), 3.81 (s, 3H), 3.22 (dd, J = 3.6, 8.4 Hz, 1H), 2.31-2.36 (m, 2H), 2.19 (dt, J = 3.2, 16.8 Hz, 1H), 2.09-2.13 (m, 1H), 1.94 (app t, J = 2.4 Hz, 1H), 1.02 (d, J = 6.4 Hz, 3H), 0.91 (t, J = 8.4 Hz, 9H), 0.50-0.57 (m, 6H), 0.34 (s, 3H), 0.27 (s, 3H); 13C NMR (100MHz, CDCl3) δ 159.1, 138.0, 136.5, 134.3, 130.8, 129.3, 128.9, 127.6, 113.6, 113.2, 85.3, 83.4, 72.6, 71.5, 69.5, 55.2, 37.0, 32.9, 22.8, 17.0, 7.1, 5.6, -3.2, -4.2; IR (neat) 3309, 2956, 2877, 1613, 1514, 1248 cm−1; HRMS (ES+) m/z for C32H48O3Si2Na [M+Na]+ calcd 559.3040, found 559.3044.
(4R,5R)-4-((E)-4-{(2S,4S,5R)-4-(Dimethyl-phenyl-silanyl)-5-[(1S,2R,3R)-2-(4-methoxy-benzyloxy)-3-methyl-1-triethylsilanyloxy-hex-5-ynyl]-tetrahydro-furan-2-yl}-but-1-enyl)-2,2-dimethyl-5-vinyl-[1,3]dioxolane (24)
A 25-mL round bottom flask was charged with aldehyde 5 (1.06 g, 5.04 mmol), allylsilane 7 (8.12 g, 15.1 mmol), activated 4 Å molecular sieves (2.0 g) and dichloromethane (10 mL). The slurry was stirred at room temperature for 10 min and then cooled to −78 °C. The cooled reaction was then treated with BF3·OEt2 (0.64 mL, 5.04 mmol, freshly distilled from calcium hydride). The reaction mixture was stirred at −78 °C for 21 h and then quenched with triethylamine (1 mL). The mixture was diluted with sat. aq. NaHCO3 (60 mL) and Et2O (50 mL) and filtered through Celite. The aqueous phase was extracted with Et2O (30 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. Purification of the crude product by flash column chromatography afforded 24 (1.19 g, 48%; (6.27 g of allylsilane 7 was recovered)) as a colorless oil with >20:1 diastereoselectivity: [α]25D = +23° (c 0.76, CHCl3); 1H NMR (400MHz, CDCl3) δ 7.47 (app dd, J = 1.6, 7.2 Hz, 2H), 7.29-7.38 (m, 3H), 7.24 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 5.74-5.82 (m, 2H), 5.41 (b dd, J = 7.2, 15.2 Hz, 1H), 5.32 (d, J = 17.6 Hz, 1H), 5.22 (d, J = 10.4 Hz, 1H), 4.51 (d, J = 10.8 Hz, 1H), 4.39 (d, J = 10.8 Hz, 1H), 4.05 (app dd, J = 6.8, 12.4 Hz, 3H), 3.81 (s, 3H), 3.71 (m, 1H), 3.58 (d, J = 5.6 Hz, 1H), 3.32 (app t, J = 6.8 Hz, 1H), 2.03-2.34 (m, 5H), 1.94 (t, J = 2.4 Hz, 1H), 1.79-1.83 (m, 1H), 1.58-1.69 (m, 3H), 1.44 (s, 3H), 1.43 (s, 3H), 1.10 (d, J = 6.8 Hz, 3H), 0.95 (t, J = 8.0 Hz, 9H), 0.51-0.61 (m, 6H), 0.32 (s, 3H), 0.32 (s, 3H); 13C NMR (100MHz, CDCl3) δ 158.9, 137.6, 136.4, 134.3, 133.8, 130.9, 129.1, 128.9, 127.8, 125.5, 118.2, 113.2, 108.6, 83.8, 83.2, 82.1, 80.2, 78.5, 77.2, 73.6, 73.1, 69.2, 55.1, 35.2, 34.5. 34.3, 29.3, 27.0, 26.9, 26.2, 22.1, 17.1, 7.1, 5.2, -4.1; IR (neat) 3309, 2955, 2250, 2115, 1614, 1514 cm−1; HRMS (ES+) m/z for C44H66O6Si2Na [M+Na]+ calcd 769.4296, found 769.4307.
(1S,2R,3R)-1-{(2S,5S)-5-[(E)-4-((4R,5R)-2,2-Dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-but-3-enyl]-tetrahydro-furan-2-yl}-3-methyl-1-triethylsilanyloxy-hex-5-yn-2-ol (25)
To a solution of [3+2] adduct 24 (1.81 g, 2.42 mmol) in DMF (2.5 mL) was added TBAF•3H2O (3.82 g, 12.1 mmol). The reaction was fitted with a condenser and placed in a 90 °C oil bath for 72 h. Additional TBAF•3H2O (2.0 g, 6.34 mmol) was added to the reaction three times during the 72 h period; at hour 8, hour 32 and hour 56. After 72 h, the reaction was diluted with pH 7 buffer (50 mL) and Et2O (30 mL). The aqueous phase was extracted with Et2O (30 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford the alcohol product (1.03 g, 85%) as a colorless oil: [α]25D = +6.4° (c 0.39, CHCl3); 1H NMR (400MHz, CDCl3) δ 7.28 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 5.73-5.81 (m, 2H), 5.41 (app bddd, J = 1.6, 6.0, 15.6 Hz, 1H), 5.32 (d, J = 16.4 Hz, 1H), 5.21 (dd, J = 1.2, 10.4 Hz, 1H), 4.58 (app q, J = 10.8 Hz, 2H), 4.03 (app q, J = 6.8 Hz, 2H), 3.93 (app q, J = 7.2 Hz, 1H), 3.85 (quint., J = 6.4 Hz, 1H), 3.78 (s, 3H), 3.45-3.50 (m, 1H), 3.35 (dd, J = 2.0, 8.0 Hz, 1H), 5.23 (bd, J = 6.8 Hz, 1H), 2.34 (ddq, J = 2.4, 6.8, 16.8 Hz, 2H), 2.05-2.24 (m, 3H), 1.99 (t, J = 2.4 Hz, 1H), 1.89-1.95 (m, 1H), 1.78-1.86 (m, 1H), 1.64-1.75 (m, 2H), 1.46-1.60 (m, 2H), 1.43 (s, 3H), 1.42 (s, 3H), 1.08 (d, J = 6.8 Hz, 3H); 13C NMR (100MHz, CDCl3) δ 159.2, 135.9, 134.2, 130.4, 129.5, 125.9, 118.4, 113.6, 108.7, 83.1, 82.1, 82.0, 81.6, 80.4, 79.2, 73.5, 73.1, 69.9, 55.1, 35.1, 34.2, 31.0, 29.0, 27.0, 27.0, 21.8, 16.2; IR (neat) 3536, 3296, 2984, 2934, 1613, 1514, 1248 cm−1; HRMS (ES+) m/z for C30H42O6Na [M+Na]+ calcd 521.2879, found 521.2879.
To a 0 °C solution of the alcohol from the preceding step (1.2 g, 2.41 mmol) and triethylamine (0.67 mL, 4.82 mmol) in dichloromethane (8 mL) was added triethylsilyl trifluoromethanesulfonate (0.65 mL, 2.89 mmol). After 5 min the reaction was quenched with sat. aq. NaHCO3 (30 mL) and Et2O (30 mL). The aqueous phase was extracted with Et2O (30 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford the triethylsilyl ether (1.39 g, 94%) as a colorless oil: [α]25D = +11° (c 0.36, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 8.8 Hz, 2H), 5.75-5.85 (m, 2H), 5.44 (bdd, J = 7.2, 15.2 Hz, 1H), 5.34 (d, J = 17.2 Hz, 1H), 5.23 (dd, J = 0.8, 10.4 Hz, 1H), 4.55 (s, 2H), 4.05 (app q, J = 6.4 Hz, 2H), 3.93 (app q, J = 6.8 Hz, 1H), 3.80 (s, 3H), 3.75-3.79 (m, 1H), 3.74 (dd, J = 3.2, 6.8 Hz, 1H), 3.29 (dd, J = 3.2, 8.8 Hz, 1H), 2.28-2.40 (m, 2H), 2.09-2.24 (m, 3H), 1.97 (t, J = 2.8 Hz, 1H), 1.79-1.94 (m, 2H), 1.61-1.70 (m, 2H), 1.51-1.59 (m, 2H), 1.45 (s, 3H), 1.44 (s, 3H), 1.10 (d, J = 6.8 Hz, 3H), 0.96 (t, J = 8.0 Hz, 9H), 0.55-0.72 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 159.0, 136.3, 134.3, 131.0, 129.0, 125.7, 118.4, 113.6, 108.8, 83.2, 83.1, 82.2, 82.2, 80.4, 78.2, 75.8, 71.9, 69.6, 55.2, 35.3, 33.2, 31.1, 29.2, 27.8, 27.0, 26.9, 22.5, 16.5, 7.0, 5.2; IR (neat) 3308, 2954, 2875, 1612, 1514, 1247, 1057 cm−1; HRMS (ES+) m/z for C36H56O6SiNa [M+Na]+ calcd 635.3744, found 635.3754.
To a 0 °C solution of the triethylsilyl ether (0.621 g, 1.01 mmol) in dichloromethane (10 mL) and pH 7 buffer (1 mL) was added DDQ (0.46 g, 2.02 mmol). The reaction was stirred for 1 h, and then quenched with sat. aq. NaHCO3 (40 mL) and Et2O (30 mL). The aqueous phase was extracted with Et2O (30 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. Purification of the crude product by flash column chromatography afforded 25 (0.49 g, 99%) as a colorless oil: [α]25D = +13° (c 0.18, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.74-5.83 (m, 2H), 5.43 (app bddd, J = 1.2, 6.0, 15.2 Hz, 1H), 5.33 (d, J = 17.2 Hz, 1H), 5.23 (dd, J = 0.8, 10.4 Hz, 1H), 4.05 (app q, J = 6.4 Hz, 2H), 3.76 (m, 2H), 3.67 (d, J = 7.2 Hz, 1H), 3.18 (t, J = 9.6 Hz, 1H), 2.50 (d, J = 9.6 Hz, 1H), 2.48 (dt, J = 3.6, 16.4 Hz, 1H), 2.08-2.20 (m, 2H), 1.95 (t, J = 2.4 Hz, 1H), 1.84-1.97 (m, 2H), 1.74-1.80 (m, 1H), 1.61-1.66 (m, 1H), 1.59-1.46 (m, 3H), 1.44 (s, 3H), 1.43 (s, 3H), 0.99 (d, J = 6.8 Hz, 3H), 0.95 (t, J = 8.0 Hz, 9H), 0.66 (app dsept., J = 7.6, 19.2 6H); 13C NMR (100MHz, CDCl3) δ 136.2, 134.3, 125.9, 118.5, 108.8, 83.1, 82.2, 82.2, 81.3, 78.7, 74.5, 74.4, 69.3, 35.7, 35.2, 30.8, 29.2, 27.4, 27.1, 27.0, 22.1, 15.7, 7.0, 5.3; IR (neat) 3524, 3310, 2954, 2875, 1379, 1238, 1054 cm−1; HRMS (ES+) m/z for C28H48O5SiNa [M+Na]+ calcd 515.3169, found 515.3171.
(4R,5S)-4-Methyl-3-((E)-(R)-2-methyl-hexa-3,5-dienoyl)-5-phenyl-oxazolidin-2-one (36)
To a −78 °C solution of oxazolidinone 3539 (8.75 g, 32.2 mmol) in THF (90 mL) was added NaHMDS (8.28 g, 45.1 mmol) in THF (10 mL). The reaction was stirred at −78 °C for 1 h and then treated with methyl trifluoromethanesulfonate (5.47 mL, 48.4 mmol). The reaction was quenched after 3 h with sat. aq. NH4Cl (100 mL) and Et2O (50 mL). The aqueous phase was extracted with Et2O (50 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered, and concentrated. Analysis of the crude product by 1H NMR indicated a 5:1 mixture of diastereomers in favor of 36. The crude product was purified by flash column chromatography in 20% Et2O/hexane (minor isomer, Rf = 0.21; major isomer, Rf = 0.36) to afford 36 (7.30 g, 79%) as a colorless oil: [α]25D = −27° (c 0.55, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.35-7.44 (m, 3H), 7.30-7.32 (m, 2H), 6.20-6.37 (m, 2H), 5.83 (dd, J = 8.4, 15.2 Hz, 1H), 5.66 (d, J = 7.2 Hz, 1H), 5.21 (app d, J = 17.6 Hz, 1H), 5.09 (app d, J = 10.8 Hz, 1H), 4.53 (quint., J = 7.6 Hz, 1H), 4.74 (quint., J = 6.8 Hz, 1H), 1.33 (d, J = 7.2 Hz, 3H), 0.90 (d, J = 6.4 Hz, 3H); 13C NMR (100MHz, CDCl3) δ 174.4, 152.6, 136.5, 133.2, 132.7, 132.6, 128.7, 128.7, 125.6, 117.3, 78.8, 55.1, 40.8, 17.3, 14.5; IR (neat) 2981, 2934, 1782, 1699, 1356, 1197 cm−1; HRMS (ES+) m/z for C17H21NO4Na [M+Na]+ calcd 308.1263, found 308.1262.
Tricarbonyl[(4R,5S)-4-Methyl-3-((E)-(2S,3R)-2-methyl-hexa-3,5-dienoyl)-5-phenyl-oxazolidin-2-one]iron (38) and Tricarbonyl[(4R,5S)-4-Methyl-3-((E)-(2S,3S)-2-methyl-hexa-3,5-dienoyl)-5-phenyl-oxazolidin-2-one]iron (37)
To a solution of oxazolidinone 36 ( 2.0 g, 7.0 mmol) in benzene (23 mL) was added diiron(nonacarbonyl) (3.8 g, 10.5 mmol). The reaction was fitted with a condenser and refluxed for a total of 24 h. Additional diiron(nonacarbonyl) (1.5 g, 4.12 mmol) and benzene (10 mL) was added to the reaction at hour 6 and hour 20. After 24 hours, the reaction was cooled to room temperature, filtered through Celite with an Et2O (25 mL) wash and concentrated to afford a 1:1 mixture of 38 and 37. The crude product mixture was separated by flash column chromatography (10% Et2O/hexanes to 40% Et2O/hexanes with 37 (0.78 g) eluting before 38 (0.71 g), 38 and 37 combined yield of 50%).
Data for 38 (yellow solid): Rf = 0.33 (30% Et2O/hexane); [α]25D = +8° (c 0.1, CHCl3); 1H NMR (400MHz, CDCl3) δ 7.36-7.45 (m, 3H), 7.36 (app d, J = 6.8 Hz, 2H), 5.71 (d, J = 7.2 Hz, 1H), 5.46 (dd, J = 4.8, 8.4 Hz, 1H), 5.22-5.27 (m, 1H), 4.76 (quint., J = 6.8 Hz, 1H), 3.82-3.40 (m, 1H), 1.78 (app dd, J = 1.6, 6.8 Hz, 1H), 1.37 (d, J = 6.8 Hz, 3H), 1.15 (app t, J = 8.8 Hz, 1H), 0.88 (d, J = 6.4 Hz, 3H), 0.39 (app dd, J = 2.0, 9.6 Hz, 1H); 13C NMR (100MHz, CDCl3) δ 210.8, 175.2, 152.9, 133.2, 128.9, 128.8, 125.7, 86.4, 81.9, 78.9, 64.7, 55.1, 41.0, 40.4, 19.9, 14.5; IR (neat) 2984, 2047, 1970, 1779, 1697, 1355 cm−1; HRMS (ES+) m/z for C20H19FeNO6Na [M+Na]+ calcd 448.0459, found 448.0464.
Data for 37 (yellow solid): Rf = 0.50 (30% Et2O/hexane); [α]25D = −83° (c 0.1, CHCl3); 1H NMR (400MHz, CDCl3) δ 7.30-7.45 (m, 5H), 5.71 (d, J = 7.2 Hz, 1H), 5.28-5.33 (m, 2H), 4.84 (quint., J = 6.8 Hz, 1H), 3.65-3.78 (m, 1H), 1.40 (d, J = 6.8 Hz, 3H), 1.36 (app dd, J = 7.6, 10.0 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H), 0.45 (app dd, J = 2.8, 8.8 Hz, 1H); 13C NMR (100MHz, CDCl3) δ 211.4, 174.6, 152.6, 133.2, 128.7, 125.6, 87.1, 82.7, 79.2, 62.9, 55.4, 43.4, 40.4, 26.3, 22.3, 14.3; IR (neat) 2977, 2046, 1978, 1782, 1700, 1342 cm−1; HRMS (ES+) m/z for C20H19FeNO6Na [M+Na]+ calcd 448.0459, found 448.0462.
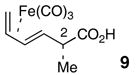
Tricarbonyl[(E)-(2S,3R)-2-Methyl-hexa-3,5-dienoic acid]iron (9)
To a 0 °C solution of oxazolidinone 38 (1.15 g, 2.70 mmol) in THF (21 mL) and water (7 mL) was added LiOH (0.194 g, 8.11 mmol). The reaction was stirred for 1.5 h and then quenched with 1M HCl (25 mL) and Et2O (25 mL). The aqueous phase was extracted with Et2O (25 mL × 3). The organic phase was dried over anhydrous MgSO4, filtered and concentrated. The crude carboxylic acid was purified by flash column chromatography to afford 9 (0.423 g, 58%) as a yellow solid: [α]25D = +11° (c 0.1, CHCl3); 1H NMR (400MHz, CDCl3) δ 10.40 (bs, 1H), 5.38-5.44 (m, 1H), 5.25-5.30 (m, 1H), 2.32 (bs, 1H), 1.81 (d, J = 6.4 Hz, 1H), 1.35 (d, J = 6.4 Hz, 3H), 0.94 (app t, J = 9.2 Hz, 1H), 0.38 (d, J = 7.6 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 211.1, 181.1, 87.1, 82.3, 63.1, 44.0, 40.5, 19.0; IR (neat) 2983, 2049, 1971, 1705 cm−1; HRMS (EI+) m/z for C9H10FeO4 [M-CO]+ calcd 237.9928, found 237.9918. The spectroscopic data obtained for 9 were fully consistent with data for racemic 9 previously published by Donaldson.40
Tricarbonyl[(E)-(2S,3S)-2-Methyl-hexa-3,5-dienoic acid]iron (10)
To a room temperature solution of oxazolidinone 37 (1.08 g, 2.54 mmol) in THF (18 mL) and water (6 mL) was added LiOH (0.304 g, 12.7 mmol). The reaction was stirred for 6.5 h and then quenched with 1M HCl (25 mL) and Et2O (25 mL). The aqueous phase was extracted with Et2O (25 mL × 3). The organic phase was dried over anhydrous MgSO4, filtered and concentrated. The crude carboxylic acid was purified by flash column chromatography to afford 10 (0.421 g, 62%) as a yellow solid: [α]25D = −140° (c 0.39, CHCl3); 1H NMR (400MHz, CDCl3) δ 11.26 (bs, 1H), 5.22-5.30 (m, 2H), 2.37-2.45 (m, 1H), 1.74-1.78 (m, 1H), 1.38 (d, J = 6.8 Hz, 3H), 1.05 (app dd, J = 8.0, 10.0 Hz, 1H), 0.36 (app dd, J = 4.4, 8.8 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 211.0, 181.0, 86.4, 82.2, 62.1, 45.2, 39.8, 21.4; IR (neat) 2979, 2934, 2049, 1974,1708 cm−1; HRMS (EI+) m/z for C9H10FeO4 [M-CO]+ calcd 237.9928, found 237.9924.
(E)-(R)-2-Methyl-hexa-3,5-dienoic acid (13)
To a 0 °C solution of oxazolidinone 36 (0.536 g, 1.88 mmol) and 30 % (w/w) H2O2 (2.3 mL, 22.6 mmol) in THF (6.3 mL) was added LiOH (0.24 g, 5.6 mmol). The reaction was stirred for 1.5 h and then quenched with 1M HCl (20 mL) and Et2O (20 mL). The aqueous phase was extracted with Et2O (20 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford 13 (0.151 g, 64 %): [α]25D = −47° (c 1.05, CHCl3); 1H NMR (500 MHz, CDCl3) δ 11.07 (bs, 1H), 6.34 (dt, J = 10.5, 17.0 Hz, 1H), 6.17 (dd, J = 10.5, 15.0 Hz, 1H), 5.77 (dd, J = 8.0, 15.5 Hz, 1H), 5.20 (d, J = 18.0 Hz, 1H), 5.09 (d, J = 10.0 Hz, 1H), 3.22 (quint., J = 7.5 Hz, 1H), 1.32 (d, J = 7.0 Hz, 3H),; 13C NMR (100MHz, CDCl3) δ 181.1, 136.4, 132.6, 132.0, 118.1, 117.4, 42.6, 17.0; IR (neat) 3088, 2980, 1709, 1414, 1212, 1003 cm−1; HRMS (ESI-TOF) m/z for C7H10NO2Na [M-H] − calcd 125.0608, found 125.0607.
2-epi-Amphidinolide E (2) series
Tricarbonyl[(E)-(2R,3S)-2-Methyl-hexa-3,5-dienoic acid (1R,2R)-1-((R)-{(2S,5S)-5-[(E)-4-((4R,5R)-2,2-dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-but-3-enyl]-tetrahydro-furan-2-yl}-triethylsilanyloxy-methyl)-2-methyl-pent-4-ynyl ester]iron (39)
To 0 °C solution of 25 (0.435 g, 0.883 mmol), 10 (0.332 g, 1.24 mmol), triethylamine (0.37 mL, 2.65 mmol), and DMAP (0.108 g, 0.883 mmol) in THF (1.8 mL) was added 2,4,6-trichlorobenzoyl chloride (0.19 mL, 1.24 mmol). The reddish brown solution was stirred at 0 °C for 1 h and allowed to warm to room temperature over another 1 h. After complete consumption of 25 was observed by TLC analysis, the reaction was quenched with sat. aq. NaHCO3 (30 mL) and Et2O (30 mL). The aqueous phase was extracted with Et2O (25 mL × 3). The organic phase was washed with sat. aq. NH4Cl, brine, dried over anhydrous MgSO4, filtered and concentrated. Purification of the crude product by flash column chromatography afforded 39 (0.614 g, 94%) as a colorless oil: [α]25D = −3.8° (c 0.87, CHCl3); 1H NMR (400MHz, CDCl3) δ 5.75-5.85 (m, 2H), 5.40-5.48 (m, 1H), 5.35-5.39 (m, 1H), 5.33 (d, J = 16.4 Hz, 1H), 5.22-5.29 (m, 1H), 5.24 (d, J = 10.4 Hz, 1H), 4.72 (d, J = 8.8 Hz, 1H), 4.03-4.08 (m, 2H), 3.67-3.78 (m, 1H), 3.69 (dd, J = 2.0, 7.2 Hz, 1H), 3.58 (app q, J = 6.8 Hz, 1H), 2.00-2.34 (m, 7H), 1.96 (t, J = 2.4 Hz, 1H), 1.77-1.94 (m, 3H), 1.59-1.69 (m, 1H), 1.50-1.59 (m, 1H), 1.40-1.50 (m, 1H), 1.44 (s, 3H), 1.44 (s, 3H), 1.34 (d, J = 6.8 Hz, 3H), 1.09 (d, J = 6.8 Hz, 3H), 0.92-1.02 (m, 1H), 0.96 (t, J = 7.6 Hz, 9H), 0.57-0.73 (m, 6H), 0.35 (b d, J = 9.2 Hz, 1H); 13C NMR (100MHz, CDCl3) δ 211.1, 173.5, 135.9, 134.2, 125.9, 118.2, 108.7, 87.0, 82.2, 82.1, 82.1, 80.4, 78.4, 77.3, 75.1, 69.7, 63.8, 44.5, 40.3, 35.1, 33.3, 30.8, 29.1, 27.6, 27.0, 27.0, 22.0, 19.3, 16.1, 6.9, 5.3; IR (neat) 3311, 2954, 2050, 1980, 1732, 1237 cm−1; HRMS (ES+) m/z for C38H56FeO9SiNa [M+Na]+ calcd 763.2941, found 763.2955.
(E)-(S)-2-Methyl-hexa-3,5-dienoic acid (1R,2R)-1-((R)-{(2S,5S)-5-[(E)-4-((4R,5R)-2,2-dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-but-3-enyl]-tetrahydro-furan-2-yl}-triethylsilanyloxy-methyl)-2-methyl-pent-4-ynyl ester (73)
To a 0 °C solution of 39 (0.574 g, 0.775 mmol) in acetone (8 mL) was added cerium ammonium nitrate (CAN) (0.935 g, 1.70 mmol). The reaction was stirred for 1.5 h, then quenched with triethylamine (2 mL) and diluted with sat. aq. NaHCO3 (50 mL) and Et2O (50 mL). The aqueous phase was extracted with Et2O (30 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford 73 (0.447 g, 96%) as a colorless oil: [α]25D = +22° (c 0.98, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.31 (dt, J = 10.0, 17.2, 1H), 6.15 (dd, J = 10.4, 15.2 Hz, 1H), 5.72-5.85 (m, 3H), 5.40-5.49 (m, 1H), 5.33 (d, J = 16.4 Hz, 1H), 5.23 (d, J = 10.0 Hz, 1H), 5.18 (d, J = 16.2, 1H), 5.07 (d, J = 11.2 Hz, 1H), 4.73 (app dd, J = 2.4, 8.8 Hz, 1H), 4.02-4.08 (m, 2H), 3.57-3.77 (m, 3H), 3.21 (quint., J = 7.2 Hz, 1H), 1.96-2.26 (m, 5H), 1.94 (t, J = 2.4 Hz, 1H), 1.77-1.98 (m, 2H), 1.58-1.68 (m, 1H), 1.37-1.58 (m, 3H), 1.44 (s, 3H), 1.43 (s, 3H), 1.30 (d, J = 7.2 Hz, 3H), 1.08 (d, J = 6.8 Hz, 3H), 0.96 (t, J = 8.0 Hz, 9H), 0.57-0.73 (m, 6H); 13C NMR (100MHz, CDCl3) δ 173.4, 136.2, 135.8, 134.2, 132.3, 132.1, 125.8, 118.1, 116.8, 108.6, 82.1, 82.1, 82.0, 80.3, 78.3, 77.2, 75.1, 69.6, 42.9, 35.0, 33.2, 30.8, 29.0, 27.5, 26.9, 26.8, 21.8, 17.0, 16.0, 6.8, 5.2; IR (neat) 3311, 2953, 2876, 2050, 1983, 1738, 1732, 1240 cm−1; HRMS (ES+) m/z for C35H56O6SiNa [M+Na]+ calcd 623.3744, found 623.3737.
(4E,11E,13E)-(1S,6R,10R,15S,18R,19R,20S)-8,8,15-Trimethyl-18-((R)-1-methyl-but-3-ynyl)-19-triethylsilanyloxy-7,9,17,23-tetraoxa-tricyclo[18.2.1.06,10]tricosa-4,11,13-trien-16-one (40)
To a solution of polyene 73 (50 mg, 0.083 mmol) in dichloromethane (83 mL) was added Grubbs’ first generation catalyst (14 mg, 0.017 mmol) in dichloromethane (2 mL). The reaction was fitted with a condenser, refluxed for 18 h and condensed. The crude product was purified by flash column chromatography to afford 40 (29 mg, 60%) as a colorless oil. In addition, an inseparable mixture of products thought to arrive by enyne metathesis (7 mg, 15%) was also isolated. Spectroscopic data for 40: [α]25D = −57° (c 0.57, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.15-6.26 (m, 2H), 5.93 (dd, J = 4.8, 14.8 Hz, 1H), 5.71 (ddd, J = 3.6, 9.6, 15.2 Hz, 1H), 5.56 (dd, J = 8.4, 14.0 Hz, 1H), 5.34 (dd, J = 8.0, 14.8 Hz, 1H), 4.55 (d, J = 9.2 Hz, 1H), 4.03 (dt, J = 8.8, 21.2 Hz, 2H), 3.68 (d, J = 8.4 Hz, 1H), 3.28-3.37 (m, 1H), 3.18-3.28 (m, 2H), 2.19-2.36 (m, 3H), 1.82-2.03 (m, 3H), 1.62-1.73 (m, 1H), 1.40-1.55 (m, 2H), 1.43 (s, 6H), 1.31 (d, J = 6.8 Hz, 3H), 1.13-1.28 (m, 3H), 1.06 (d, J = 6.8 Hz, 3H), 0.95 (t, J = 8.0 Hz, 9H), 0.53-0.70 (m, 6H); 13C NMR (100MHz, CDCl3) δ 172.5, 138.1, 135.9, 134.6, 128.9, 127.5, 125.9, 109.0, 83.1, 82.8, 82.2, 79.9, 78.5, 77.2, 75.0, 69.4, 43.3, 33.1, 32.1, 29.6, 28.5, 27.2, 27.1, 27.1, 22.3, 15.5, 12.0, 7.1, 5.6; IR (neat) 3310, 2935, 2874, 1726, 1238, 1052 cm−1; HRMS (ES+) m/z for C33H52O6SiNa [M+Na]+ calcd 595.3431, found 595.3438.
(4E,11E,13E)-(1S,6R,10R,15S,18R,19R,20S)-18-((R)-3-Iodo-1-methyl-but-3-enyl)-8,8,15-trimethyl-19-triethylsilanyloxy-7,9,17,23-tetraoxa-tricyclo[18.2.1.06,10]tricosa-4,11,13-trien-16-one (41)
To a 0 °C solution of i-Pr2NH (0.45 mL, 3.2 mmol) in THF (3 mL) was added n-BuLi (1.24 mL of a 2.41M solution in hexanes, 3.0 mmol). The reaction was allowed to stir for 30 min and then cooled to −30 °C. To this mixture was added Bu3SnH (0.80 mL, 3.0 mmol). After 1h, Et2AlCl (1.7 mL of a 1.8M solution in toluene, 3.0 mmol) was added. The reaction was stirred at −30 °C for another 1.5 h and then used immediately in the stannylalumination-protonolysis of 40.
To a −30 °C solution of 40 (23 mg, 0.40 mmol) in THF (1 mL) was added Bu3Sn-AlEt2 (0.57 mL of the 0.42M solution from above, 0.24 mmol), followed by CuCN (1 mg, 0.012 mmol). The bright orange solution was stirred for 1 h at −30 °C, then quenched with sat. aq. NH4Cl (20 mL) and Et2O (20 mL). This mixture was stirred vigorously at room temp for 15 min. The aqueous phase was extracted with Et2O (10 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. Purification of the crude product by flash column chromatography afforded the vinylstannane product (18 mg, 51%) as a colorless oil: [α]25D = −78° (c 0.12, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.15-6.28 (m, 2H), 5.96 (dd, J = 4.8, 14.4 Hz, 1H), 5.72 (ddd, J = 3.2, 9.2, 15.2 Hz, 1H), 5.64 (app t, 3JSn-H = 69.2 Hz, 1H), 5.57 (dd, J = 8.4, 14.0 Hz, 1H), 5.34 (dd, J = 8.4, 15.2 Hz, 1H), 5.16 (app t, 3JSn-H = 31.6 Hz, 1H), 4.49 (d, J = 10.0 Hz, 1H), 4.04 (app dt, J = 8.8, 20.4 Hz, 1H), 3.74 (d, J = 8.4 Hz, 1H), 3.28-3.36 (m, 1H), 3.18-3.28 (m, 2H), 2.28-2.40 (m, 3H), 1.82-2.13 (m, 4H), 1.62-1.74 (m, 1H), 1.40-1.55 (qm, 7H), 1.44 (s, 6H), 1.13-1.38 (m, 11H), 0.85-1.00 (m, 24H), 0.79 (d, J = 6.4 Hz, 3H), 0.55-0.70 (m, 6H); 13C NMR (100MHz, CDCl3) δ 172.6, 153.9, 138.2, 136.0, 134.9, 131.1, 128.7, 127.3, 127.0, 125.8, 109.0, 83.1, 82.3, 80.1, 79.9, 74.9, 45.3, 43.3, 32.7, 32.1, 29.6, 29.2, 29.1, 29.0, 28.5, 27.4, 27.2, 27.1, 14.8, 13.7, 12.0, 9.5, 7.1, 5.6; IR (neat) 2955, 1727, 1378, 1239, 1052 cm−1; HRMS (ES+) m/z for C45H80O6SiSnNa [M+Na]+ calcd 887.4644, found 887.4666.
To a −45 °C solution of the vinylstannane from the preceding step (20 mg, 0.023 mmol) in dichloromethane (1 mL) was added NIS (6 mg, 0.03 mmol). The reaction was stirred at −45 °C for 1 h and then quenched with sat. aq. Na2S2O3 (30 mL) and extracted with Et2O (20 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford 41 (15 mg, 93%) as a colorless oil: [α]25D = −72° (c 0.43, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.17-6.27 (m, 2H), 6.02 (s, 1H), 5.93 (dd, J = 5.2, 14.4 Hz, 1H), 5.73 (ddd, J = 3.6, 9.6, 15.2 Hz, 1H), 5.72 (s, 1H), 5.58 (dd, J = 8.8, 14.0 Hz, 1H), 5.34 (dd, J = 8.4, 15.2 Hz, 1H), 4.55 (d, J = 9.6 Hz, 1H), 4.03 (app dt, J = 8.8, 23.6 Hz, 2H), 3.72 (d, J = 8.4 Hz, 1H), 3.28-3.35 (m, 1H), 3.19-3.28 (m, 2H), 2.29-2.50 (m, 3H), 1.82-2.05 (m, 3H), 1.65-1.73 (m, 1H), 1.45-1.57 (m, 1H), 1.43 (s, 6H), 1.33 (d, J = 6.8 Hz, 3H), 1.12-1.27 (m, 3H), 0.98 (t, J = 8.0 Hz, 9H), 0.86 (d, J = 6.4 Hz, 3H), 0.56-0.72 (m, 6H); 13C NMR (100MHz, CDCl3) δ 172.8, 138.1, 136.0, 134.5, 129.0, 127.5, 127.0, 125.8, 111.5, 109.0, 83.1, 82.2, 80.0, 78.6, 74.9, 48.3, 43.3, 32.8, 32.0, 29.6, 28.5, 27.2, 27.1, 27.1, 14.6, 12.0, 7.2, 5.7; IR (neat) 2933, 2873, 1723, 1239, 1052 cm−1; HRMS (ES+) m/z for C33H53IO6SiNa [M+Na]+ calcd 723.2554, found 723.25563
2-epi-amphidinolide E (2)
A solution of vinyl iodide 41 (44 mg, 0.063 mmol) in a mixture of AcOH, THF and water (4/1/1 ratio) (1.5 mL) was heated to 40 °C for 7h. The mixture was then carefully poured into a separatory funnel containing Et2O (40 mL) and sat. aq. NaHCO3 (60 mL). The pH of the aqueous layer was adjusted to ~7 using solid NaHCO3. The aqueous phase was extracted with Et2O (20 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. Analysis of the crude product by 1H NMR indicated a 10:1 mixture of the desired C(18) and undesired C(17) lactones.
To the crude mixure of vinyl iodide-containing lactones (from above) and CuCl (23 mg, 0.23 mmol) in THF (0.5 mL) was added vinylstannane 612 (78 mg, 0.21 mmol), followed by Pd(PPh3)4 (10 mg, 0.0084 mmol) in THF (0.5 mL). The reaction was stirred at room temperature for 26 h, and then diluted with Et2O (30 mL), filtered through Celite and concentrated. The crude product was purified by flash column chromatography using 10% methanol/chloroform to afford material that was still contaminated with an organotin impurity. The stannane impurity was removed by HPLC purification with 100% ethyl acetate eluent on a normal phase, Varian Dynamax Microsorb 60-8 Si, 250 × 21.4 mm column. The retention time for 2-epi-amphidinolide E was 8 min. The flow rate was 18 mL/min. 2-epi-amphidinolide E was detected using UV absorption (λ = 254 nm and 280 nm) and RI detection. Using the above conditions 11 mg (34% from 41) of pure 2-epi-amphidinolide E (2) was isolated: [α]25D = −80° (c 0.15, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.14-6.25 (m, 2H), 6.04 (d, J = 16.0 Hz, 1H), 5.95-6.04 (m, 1H), 5.54-5.74 (m, 3H), 5.30 (dd, J = 7.2, 15.2 Hz, 1H), 4.98 (s, 1H), 4.86 (s, 1H), 4.74 (s, 1H), 4.70 (s, 1H), 4.66 (d, J = 9.6 Hz, 1H), 3.95 (app dt, J = 8.4, 19.2 Hz, 2H), 3.69 (app t, J = 5.2 Hz, 1H), 3.46-3.53 (m, 1H), 3.40-3.46 (m, 1H), 3.35 (quint., J = 5.2 Hz, 1H), 2.72-2.84 (m, 2H), 2.22-2.48 (m, 5H), 1.60-1.95 (m, 5H), 1.72 (s, 3H), 1.25-1.52 (m, 4H), 1.34 (d, J = 7.2 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H); 13C NMR (100MHz, CDCl3) δ 172.9, 144.7, 144.1, 134.8, 134.2, 134.1, 133.4, 131.2, 129.7, 129.6, 128.2, 116.1, 110.9, 79.9, 78.7, 77.9, 77.3, 76.6, 73.2, 43.1, 41.5, 36.3, 32.8, 32.3, 30.2, 29.1, 27.5, 22.7, 15.5, 13.4; IR (neat) 3413, 2933, 1723, 1454, 1238, 992 cm−1; HRMS (ES+) m/z for C30H44O6Na [M+Na]+ calcd 523.3036, found 523.3016.
Synthesis of 19-epi-series Precursors
(1R,2S)-1-((S)-2,2-Diethyl-[1,3]dioxolan-4-yl)-2-methyl-but-3-en-1-ol (60)
To a −78 °C slurry of L-glyceraldehyde pentylidene ketal 5824 (12.1 g, 76.5 mmol) and 4Å mol. sieves (9 g) in toluene (100 mL) was added Z-(S,S)-crotylboronate 5923 (153 mL of a 1.0M solution in toluene, 153 mmol). The reaction was stirred at −78 °C for 18 h and then quenched with 2N NaOH aq. (300 mL). The biphasic mixture was filtered through Celite and extracted with EtOAc (75 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The crude product was purified by flash column chromatography to afford 60 (15.4 g, 94%) as a colorless oil: [α]25D = −45° (c 4.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.71 (ddd, J = 8.0, 10.0, 17.2 Hz, 1H), 5.04 (d, J = 10.0 Hz, 1H), 5.00 (s, 1H), 4.00-4.08 (m, 1H), 3.94 (app t, J = 7.6 Hz, 1H), 3.81 (app t, J = 7.6 Hz, 1H), 3.60-3.66 (m, 1H), 2.18-2.28 (m, 2H), 1.52-1.68 (m, 4H), 1.06 (d, J = 6.4 Hz, 3H), 0.80-0.91 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 140.2, 115.4, 112.4, 76.8, 73.8, 65.1, 40.7, 29.1, 15.3, 8.2, 7.9; IR (neat) 3481, 2974, 1641, 1463 cm−1; HRMS (ES+) m/z for C12H22O3Na [M+Na]+ calcd 237.1467, found 237.1460.
(3S,4R)-4-((S)-2,2-Diethyl-[1,3]dioxolan-4-yl)-4-(4-methoxy-benzyloxy)-3-methyl-butan-1-ol (61)
Protection of alcohol 60 as a p-methoxybenzyl ether was accomplished using a procedure analogous to that outlined for the conversion of 16 to 17: NaH (3.45 g, 144 mmol), NaI (2.7 g, 18.0 mmol), THF (200 mL), alcohol 60 (15.4 g, 71.9 mmol) and p-methoxybenzyl chloride (6.38 mL, 47. 0 mmol) were used. The crude product was purified by flash column chromatography to afford the p-methoxybenzyl ether (23.8 g, 99%) as a colorless oil: [α]25D = −34° (c 2.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J = 9.2 Hz, 2H), 6.86 (d, J = 9.2 Hz, 2H), 5.88 (ddd, J = 7.2, 10.0, 17.2 Hz, 1H), 5.07 (d J = 17.2 Hz, 1H), 5.03 (d, J = 10.4 Hz, 1H), 4.61 (AB, J = 10.8 Hz, 1H), 4.55 (AB, J = 10.8 Hz, 1H), 4.12 (ddd, J = 6.4, 7.6, 11.6 Hz, 1H), 4.00 (dd, J = 6.0, 7.6 Hz, 1H), 3.78-3.83 (m, 1H), 3.80 (s, 3H), 3.51 (t, J = 5.2 Hz, 1H), 2.38-2.48 (m, 1H), 1.55-1.72 (m, 4H), 1.08 (d, J = 6.8 Hz, 3H), 0.87-0.93 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 159.1, 141.3, 130.7, 129.3, 114.4, 113.7, 112.5, 82.4, 76.9, 74.0, 66.5, 55.2, 40.2, 29.7, 29.0, 15.0, 8.2, 8.1; IR (neat) 2972, 1614, 1514, 1248 cm−1; HRMS (ES+) m/z for C20H30O4Na [M+Na]+ calcd 357.2042, found 357.2039.
The hydroboration-oxidation of the p-methoxybenzyl ether compound was accomplished using a procedure analogous to that outlined for the conversion of 16 to 17: the p-methoxybenzyl ether (23.8 g, 71.2 mmol), THF (50 mL) and 9-BBN (430 mL of a 0.5 M THF solution, 136 mmol) were used. The crude product was purified by flash column chromatography to afford 61 (24.8 g, 99%) as a colorless oil: [α]25D = −21° (c 2.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 4.55 (s, 2H), 4.14 (app q, J = 6.0 Hz, 1H), 4.07 (dd, J = 5.2, 7.6 Hz, 1H), 3.80 (s, 3H), 3.79 (app q, J = 7.6 Hz, 1H), 3.69-3.76 (m, 1H), 3.60-3.69 (m, 1H), 3.48 (dd, J = 2.8, 6.4 Hz, 1H), 1.98-2.06 (m, 1H), 1.72-1.82 (m, 1H), 1.59-1.70 (m, 4H), 1.50-1.59 (m, 1H), 0.99 (d, J = 6.8 Hz, 3H), 0.89 (q, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 159.2, 130.5, 129.3, 113.8, 112.5, 83.1, 76.7, 73.6, 67.7, 61.2, 55.3, 36.5, 32.5, 29.8, 29.0, 15.2, 8.2, 8.2; IR (neat) 3418, 2934, 2245, 1614 cm−1; HRMS (ES+) m/z for C20H32O5Na [M+Na]+ calcd 375.2147, found 375.2144.
(S)-2,2-Diethyl-4-[(1R,2S)-1-(4-methoxy-benzyloxy)-2-methyl-pent-4-ynyl]-[1,3]dioxolane (62)
The oxidation of 61 was accomplished using a procedure analogous to that outlined for the conversion of 17 to 18: alcohol 61 (24.8 g, 70.4 mmol), CH2Cl2 (240 mL), DMSO (15.3 mL, 216 mmol), i-Pr2NEt (39 mL, 216 mmol) and SO3·pyridine (34 g, 216 mmol) were used. The crude product was purified by flash column chromatography to afford the aldehyde (24.4 g, 99%) as a colorless oil: [α]25D = −18° (c 1.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 9.73 (s, 1H), 7.22 (d, J = 8.4 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 4.51 (AB, J = 10.8 Hz, 1H), 4.47 (AB, J = 11.2 Hz, 1H), 4.30-4.11 (m, 2H), 3.81 (s, 3H), 3.72-3.80 (m, 1H), 3.45 (bdd, J = 2.8, 6.8 Hz, 1H), 2.45-2.61 (m, 2H), 2.30-2.45 (m, 1H), 1.55-1.70 (m, 4H), 1.03 (d, J = 6.8 Hz, 3H), 0.85-0.93 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 202.0, 159.2, 130.2, 129.3, 113.7, 112.6, 82.2, 76.5, 73.2, 67.9, 55.2, 47.630.3, 29.7, 28.9, 15.0, 8.1, 8.1; IR (neat) 2972, 1724, 1514, 1249 cm−1; HRMS (ES+) m/z for C20H30O5Na [M+Na]+ calcd 373.1991, found 373.1985.
The Corey-Fuchs homologation of the aldehyde was accomplished using a procedure analogous to that outlined for the conversion of 17 to 18: the aldehyde (24.4 g, 69 mmol), PPh3 (30.9 g, 118 mmol), CH2Cl2 (235 mL), CBr4 (23.4 g, 70.6 mmol) and n-BuLi (46 mL of 2.48M hexane solution, 113 mmol) were used were used. Purification of the crude product by flash column chromatography afforded 62 (9.6 g, 59%) as a colorless oil: [α]25D = −8.5° (c 1.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.25 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 4.59 (AB, J = 10.8 Hz, 1H), 4.55 (AB, J = 10.8 Hz, 1H), 4.30-4.11 (m, 2H), 3.81 (s, 3H), 3.75-3.82 (m, 1H), 3.70 (bdd, J = 3.2, 6.4 Hz, 1H), 2.32 (ddd, J = 2.4, 8.0, 16.4 Hz, 1H), 2.20 (ddd, J = 2.4, 6.8, 16.4 Hz, 1H), 2.08 (dq, J = 4.0, 7.2 Hz, 1H), 2.02 (t, J = 2.4 Hz, 1H), 1.58-1.70 (m, 4H), 1.02 (d, J = 7.2 Hz, 3H), 0.90 (app dt, J = 7.6, 9.6 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 159.2, 130.6, 129.2, 113.7, 112.5, 83.2, 81.0, 76.8, 74.1, 69.6, 67.6, 55.1, 35.2, 29.0, 23.1, 14.2, 8.1; IR (neat) 3294, 2972, 1613, 1515, 1249 cm−1; HRMS (ES+) m/z for C21H30O4Na [M+Na]+ calcd 369.2042, found 369.2033.
(2R,3S)-2-(4-Methoxy-benzyloxy)-3-methyl-hex-5-ynal (63)
The deprotection of 62 was accomplished using a procedure analogous to that outlined for the conversion of 18 to 19: alkyne 62 (22.5 g, 64.9 mmol) and a 4:1 mixture of AcOH and water (215 mL). The crude product was purified by flash column chromatography to afford the diol product (16.9 g, 93%) as a colorless oil: [α]25D = +0.08° (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.28 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 4.61 (AB, J = 10.8 Hz, 1H), 4.55 (AB, J = 10.8 Hz, 1H), 3.81 (s, 3H), 3.70-3.82 (m, 3H), 3.65 (dd, J = 4.0, 6.0 Hz, 1H), 2.19-2.35 (m, 3H), 2.04-2.11 (m, 1H), 2.03 (t, J = 2.4 Hz, 1H), 1.94 (b t, J = 6.0 Hz, 1H), 1.08 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 159.2, 130.3, 129.4, 113.8, 83.1, 81.1, 74.3, 71.8, 69.9, 63.8, 55.2, 34.3, 23.3, 14.4; IR (neat) 3401, 3294, 2935, 1614, 1515, 1250, 1074, 1035 cm−1; HRMS (ES+) m/z for C16H22O4Na [M+Na]+ calcd 301.1416, found 301.1418.
Oxidative cleavage of the diol above was accomplished using a procedure analogous to that outlined for the conversion of 18 to 19: the diol product from the previous step (16.9 g, 60.7 mmol), THF (100 mL), pH 7 buffer (100 mL) and NaIO4 (15.6 g, 72.9 mmol) were used. The product was used without further purification. 63 (12.1 g, 81%) was a colorless oil: [α]25D = +69° (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 9.70 (d, J = 1.6 Hz, 1H), 7.29 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 8.4 Hz, 2H), 4.66 (d, J = 11.2 Hz, 1H), 4.50 (d, J = 11.2 Hz, 1H), 3.94 (bdd, J = 1.2, 3.2 Hz, 1H), 3.81 (s, 3H), 2.18-2.38 (m, 3H), 2.00 (t, J = 2.4 Hz, 1H), 1.00 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 204.2, 159.3, 129.6, 129.3, 113.7, 84.5, 82.1, 72.7, 70.2, 55.0, 34.7, 22.2, 14.0; IR (neat) 3291, 2936, 1731, 1514, 1250 cm−1; HRMS (ES+) m/z for C15H18O3Na [M+Na]+ calcd 269.1154, found 269.1146.
(3R,4S,5R,6S)-3-(Dimethylphenylsilanyl)-5-(4-methoxy-benzyloxy)-6-methyl-non-1-en-8-yn-4-ol (64)
Hydroxyallylsilane 64 was synthesized using a procedure analogous to that outlined for the conversion of 19 to 21: aldehyde 63 (11.5 g, 46.7 mmol), 4Å mol. sieves (8.6 g), toluene (80 mL) and (S,S)-20 (110 mL of a 1.0M solution in toluene, 110 mmol) were used. The crude product was purified by flash column chromatography to afford 64 (17.7 g, 90%) as a colorless oil: [α]25D = −12° (c 0.8, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.52-7.56 (m, 2H), 7.30-7.38 (m, 3H), 7.24 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 5.98 (dt, J = 10.4, 17.2 Hz, 1H), 5.02 (dd, J = 2.0, 10.4 Hz, 1H), 4.85 (dd, J = 2.0, 17.2 Hz, 1H), 4.59 (AB, J = 10.4 Hz, 1H), 4.52 (AB, J = 10.4 Hz, 1H), 3.80 (s, 3H), 3.65 (bdt, J = 2.4, 8.4 Hz, 1H), 3.52 (bdd, J = 2.8, 8.4 Hz, 1H), 2.28-2.31 (m, 1H), 2.20 (ddq, J = 2.4, 7.2, 16.8 Hz, 2H), 1.98 (t, J = 2.4 Hz, 1H), 1.85-1.92 (m, 1H), 1.82 (bd, J = 10.8 Hz, 1H), 0.82 (d, J = 7.2 Hz, 3H), 0.38 (s, 3H), 0.31 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.2, 137.8, 134.1, 134.0, 130.6, 129.3, 128.9, 127.6, 114.8, 113.9, 83.8, 83.3, 75.1, 71.6, 69.5, 55.2, 38.3, 34.1, 23.8, 13.4, -3.8, -4.3; IR (neat) 3564, 3303, 2963, 1613, 1514, 1248 cm−1; HRMS (ES+) m/z for C26H34O3SiNa [M+Na]+ calcd 445.2175, found 445.2173.
1-[(1R,2S,3R)-3-(Dimethyl-phenyl-silanyl)-1-((S)-1-methyl-but-3-ynyl)-2-triethylsilanyloxy-pent-4-enyloxymethyl]-4-methoxy-benzene (8)
Allylsilane 8 was protected as the triethylsilyl ether using a procedure analogous to that outlined for the conversion of 21 to 7: 64 (0.53 g, 1.3 mmol), DMF (45 mL), imidazole (0.26 g, 3.8 mmol) and triethylsilyl chloride (0.64 mL, 3.8 mmol) were used. The crude product was purified by flash column chromatography to afford 8 (0.60 g, 89%) as a colorless oil: [α]25D = +9.0° (c 1.4, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.48-7.54 (m, 2H), 7.29-7.38 (m, 3H), 7.18 (d, J = 8.8 Hz, 2H), 6.85 (d, J = 8.8 Hz, 2H), 6.05 (dt, J = 10.4, 17.2 Hz, 1H), 4.98 (dd, J = 2.0, 10.0 Hz, 1H), 4.82 (dd, J = 2.0, 17.6 Hz, 1H), 4.45 (d, J = 11.6 Hz, 1H), 4.28 (d, J = 11.2 Hz, 1H), 4.02 (dd, J = 2.0, 6.0 Hz, 1H), 3.81 (s, 3H), 3.20 (t, J = 5.2 Hz, 1H), 2.28 (dq, J = 2.4, 8.8 Hz, 1H), 2.18 (dd, J = 1.6, 10.4 Hz, 1H), 1.92-2.10 (m, 2H), 1.91 (t, J = 2.4 Hz, 1H), 0.96 (d, J = 6.0 Hz, 3H), 0.88 (t, J = 8.0 Hz, 9H), 0.46-0.56 (m, 6H), 0.35 (s, 3H), 0.30 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 158.9, 138.0, 135.7, 134.2, 131.1, 128.9, 128.8, 127.6, 114.3, 113.5, 84.5, 83.8, 73.1, 72.6, 69.1, 55.2, 38.2, 33.8, 23.8, 15.3, 7.1, 5.5, -3.1, -4.1; IR (neat) 3310, 2956, 1614, 1514, 1248, 1112 cm−1; HRMS (ES+) m/z for C32H48O3Si2Na [M+Na]+ calcd 559.3040, found 559.3030.
(4R,5R)-4-((E)-4-{(2S,4S,5R)-4-(Dimethyl-phenyl-silanyl)-5-[(1S,2R,3S)-2-(4-methoxy-benzyloxy)-3-methyl-1-triethylsilanyloxy-hex-5-ynyl]-tetrahydro-furan-2-yl}-but-1-enyl)-2,2-dimethyl-5-vinyl-[1,3]dioxolane (65)
Tetrahydrofuran 65 was synthesized using a procedure analogous to that outlined for 24: aldehyde 5 (0.079 g, 0.37 mmol), allylsilane 8 (0.60 g, 1.12 mmol), activated 4 Å molecular sieves (0.15 g), dichloromethane (0.75 mL) and BF3·OEt2 (47 μL, 0.37 mmol) were used. Purification of the crude product by flash column chromatography afforded 65 (0.171 g, 61%; 0.43 g of allylsilane 8 was recovered) as a colorless oil: [α]25D = +19° (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.45-7.50 (m, 2H), 7.36-7.35 (m, 3H), 7.24 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.8 Hz, 2H), 5.72-5.84 (m, 2H), 5.42 (bdd, J = 6.8, 15.2 Hz, 1H), 5.32 (d, J = 17.2 Hz, 1H), 5.21 (d, J = 10.4 Hz, 1H), 4.62 (d, J = 11.6 Hz, 1H), 4.39 (d, J = 11.6 Hz, 1H), 4.00-4.08 (m, 2H), 3.96 (d, J = 8.4 Hz, 1H), 3.78 (s, 3H), 3.66-3.75 (m, 1H), 3.64 (d, J = 6.8 Hz, 1H), 3.46 (bdd, J = 4.4, 6.4 Hz, 1H), 2.32-2.42 (m, 1H), 2.21-2.25 (m, 4H), 1.94 (s, 1H), 1.79-1.89 (m, 1H), 1.56-1.74 (m, 3H), 1.36-1.48 (m, 1H), 1.43 (s, 3H), 1.42 (s, 3H), 0.90-1.00 (m, 12H), 0.54-0.64 (m, 6H), 0.35 (s, 3H), 0.33 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 158.7, 137.7, 136.4, 134.3, 133.9, 131.3, 129.1, 128.6, 127.8, 125.6, 118.3, 113.5, 108.7, 84.2, 82.7, 82.2, 82.1, 81.2, 78.0, 74.0, 73.7, 69.1, 55.1, 35.2, 34.7, 34.5, 29.2, 27.0, 26.9, 25.4, 23.5, 14.9, 7.1, 5.3, -3.7, -4.4; IR (neat) 3308, 2955, 1614, 1514, 1248 cm−1; HRMS (ES+) m/z for C44H66O6Si2Na [M+Na]+ calcd 769.4296, found 769.4307.
(1R,2R,3S)-1-{(2S,5S)-5-[(E)-4-((4R,5R)-2,2-Dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-but-3-enyl]-tetrahydro-furan-2-yl}-2-(4-methoxy-benzyloxy)-3-methyl-hex-5-yn-1-ol (66)
The protiodesilylation of 65 was accomplished using a procedure analogous to that outlined for the conversion of 24 to 25: [3+2] adduct 65 (1.26 g, 1.69 mmol), DMF (1.7 mL), TBAF•3H2O (4.66 g, 14.8 mmol, added portionwise) were used. The crude product was purified by flash column chromatography to afford 66 (0.68 g, 81%) as a colorless oil: [α]25D =-4.1° (c 0.09, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.27 (d, J = 8.4 Hz, 2H), 6.87 (d, J = 8.4 Hz, 2H), 5.73-5.83 (m, 2H), 5.42 (bdd, J = 6.0, 15.2 Hz, 1H), 5.33 (d, J = 16.8 Hz, 1H), 5.22 (d, J = 10.4 Hz, 1H), 4.60 (app q, J = 10.8 Hz, 2H), 4.01-4.07 (m, 2H), 3.90 (bdt, J = 4.8, 5.2 Hz, 1H), 3.84 (bt, J = 6.8 Hz, 1H), 3.79 (s, 3H), 3.46-3.57 (m, 2H), 2.52-2.57 (m, 1H), 2.30 (ddq, J = 2.8, 7.2, 16.8 Hz, 2H), 2.08-2.20 (m, 3H), 1.99 (t, J = 2.8 Hz, 1H), 1.88-1.96 (m, 1H), 1.64-1.86 (m, 3H), 1.46-1.62 (m, 2H), 1.43 (s, 3H), 1.42 (s, 3H), 1.06 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 159.2, 136.0, 134.3, 130.6, 129.5, 126.0, 118.5, 113.8, 108.8, 83.2, 82.2, 82.2, 81.0, 80.2, 79.3, 73.7, 73.7, 69.5, 55.2, 35.1, 35.0, 31.1, 29.1, 27.7, 27.1, 27.0, 22.8, 15.0; IR (neat) 3523, 2985, 1613, 1515, 1248, 1053 cm−1; HRMS (ES+) m/z for C30H42O6Na [M+Na]+ calcd 521.2879, found 521.2879.
(1S,2R,3S)-1-{(2S,5S)-5-[(E)-4-((4R,5R)-2,2-Dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-but-3-enyl]-tetrahydro-furan-2-yl}-3-methyl-1-triethylsilanyloxy-hex-5-yn-2-ol (67)
Protection of 66 as the triethylsilyl ether was accomplished using a procedure analogous to that outlined for the conversion of 24 to 25: alcohol 66 (0.40 g, 0.80 mmol), triethylamine (0.22 mL, 1.6 mmol), dichloromethane (3 mL) and triethylsilyl trifluoromethanesulfonate (0.22 mL, 0.96 mmol) were used.
Deprotection of the p-methoxybenzyl ether from the above intermediate was accomplished using a procedure analogous to that outlined for the conversion of 24 to 25: dichloromethane (10 mL), pH 7 buffer (1 mL) and DDQ (0.37 g, 1.6 mmol) were used. Purification of the crude product by flash column chromatography afforded 67 (0.34 g, 85%) as a colorless oil: [α]25D = +18° (c 0.18, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.74-5.85 (m, 2H), 5.44 (bddd, J = 1.2, 6.0, 15.6 Hz, 1H), 5.34 (d, J = 17.6 Hz, 1H), 5.24 (d, J = 10.4 Hz, 1H), 4.02-4.10 (m, 2H), 3.81-3.88 (m, 1H), 3.74-3.81 (m, 1H), 3.67 (dd, J = 2.0, 6.8 Hz, 1H), 3.42 (ddd, J = 2.0, 7.2, 9.2 Hz, 1H), 2.46 (d, J = 8.8 Hz, 1H), 2.07-2.32 (m, 4H), 1.96 (t, J = 2.4 Hz, 1H), 1.76-1.98 (m, 3H), 1.50-1.71 (m, 3H), 1.42-1.50 (m, 1H), 1.44 (s, 3H), 1.44 (s, 3H), 1.08 (d, J = 6.8 Hz, 3H), 0.96 (t, J = 7.6 Hz, 9H), 0.59-0.75 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 135.9, 134.3, 125.9, 118.3, 108.7, 82.2, 82.2, 82.1, 81.1, 78.6, 75.1, 73.3, 69.8, 35.5, 35.1, 30.8, 29.1, 27.2, 27.0, 26.9, 22.7, 15.0, 6.9, 5.3; IR (neat) 3535, 3311, 2955, 1239, 1056, 741 cm−1; HRMS (ES+) m/z for C28H48O5SiNa [M+Na]+ calcd 515.3169, found 515.3160.
2-epi-19-epi-Amphidinolide E (4) series
Tricarbonyl[(E)-(2R,3S)-2-Methyl-hexa-3,5-dienoic acid (1R,2S)-1-((R)-{(2S,5S)-5-[(E)-4-((4R,5R)-2,2-dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-but-3-enyl]-tetrahydro-furan-2-yl}-triethylsilanyloxy-methyl)-2-methyl-pent-4-ynyl ester]iron (70)
The esterification of alcohol 67 was accomplished using a procedure analogous to that outlined for 39: alcohol 67 (0.33 g, 0.67 mmol), acid 10 (0.22 g, 0.80 mmol), triethylamine (0.22 mL, 1.6 mmol), DMAP (0.082 g, 0.67 mmol), THF (1.4 mL) and 2,4,6-trichlorobenzoyl chloride (125 μL, 0.80 mmol) were used. Purification of the crude product by flash column chromatography afforded 70 (0.418 g, 85%) as a colorless oil: [α]25D = −2.6° (c 0.54, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.75-5.83 (m, 2H), 5.41-5.47 (m, 1H), 5.35-5.40 (m, 1H), 5.34 (app d, J = 23.6 Hz, 1H), 5.25-5.30 (m, 1H), 5.23 (app d, J = 12.0 Hz, 1H), 4.89 (dd, J = 2.4, 7.6 Hz, 1H), 4.03-4.08 (m, 2H), 3.70-3.79 (m, 1H), 3.71 (dd, J = 3.2, 7.2 Hz, 1H), 3.59-3.65 (m, 1H), 2.05-2.37 (m, 6H), 1.98 (t, J = 2.0 Hz, 1H), 1.85-1.95 (m, 1H), 1.75-1.85 (m, 2H), 1.60-1.70 (m, 1H), 1.50-1.60 (m, 2H), 1.40-1.50 (m, 1H), 1.44 (s, 3H), 1.44 (s, 3H), 1.33 (d, J = 7.2 Hz, 3H), 1.00 (d, J = 6.4 Hz, 3H), 0.93-0.99 (m, 1H), 0.96 (t, J = 8.0 Hz, 9H), 0.59-0.73 (m, 6H), 0.35 (bdd, J = 2.0, 8.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 211.0, 173.6, 136.1, 134.3, 125.9, 118.4, 108.8, 87.1, 82.2, 82.2, 81.8, 80.4, 78.6, 77.2, 76.9, 74.9, 70.1, 64.0, 44.6, 40.4, 35.1, 33.0, 30.9, 29.2, 27.5, 27.0, 27.0, 22.7, 19.3, 15.6, 7.0, 5.3; IR (neat) 3312, 2877, 2050, 1982, 1732, 1380, 1239 cm−1; HRMS (ES+) m/z for C38H56FeO9SiNa [M+Na]+ calcd 763.2941, found 763.2958.
(4E,11E,13E)-(1S,6R,10R,15S,18R,19R,20S)-18-((S)-3-Iodo-1-methyl-but-3-enyl)-8,8,15-trimethyl-19-triethylsilanyloxy-7,9,17,23-tetraoxa-tricyclo[18.2.1.06,10]tricosa-4,11,13-trien-16-one (71)
The oxidative decomplexation of 70 was accomplished using a procedure analogous to that outlined for the conversion of 39 to 40: ester 70 (173 mg, 0.234 mmol), acetone (3 mL) and cerium ammonium nitrate (CAN) (0.28 g, 0.51 mmol) were used. The crude product was purified by flash column chromatography to afford the polyene product (135 mg, 96%) as a colorless oil: [α]25D = +16° (c 0.26, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.29 (dt, J = 10.4, 16.8, 1H), 6.14 (dd, J = 10.4, 15.2 Hz, 1H), 5.71-5.84 (m, 3H), 5.39-5.47 (m, 1H), 5.32 (d, J = 17.6 Hz, 1H), 5.22 (d, J = 11.6 Hz, 1H), 5.16 (d, J = 16.4, 1H), 5.05 (d, J = 10.0 Hz, 1H), 4.88 (dd, J = 2.8, 7.6 Hz, 1H), 4.01-4.07 (m, 2H), 3.61-3.77 (m, 3H), 3.20 (quint., J = 7.2 Hz, 1H), 2.26-2.37 (m, 1H), 2.04-2.26 (m, 4H), 1.96 (t, J = 2.0 Hz, 1H), 1.75-1.93 (m, 2H), 1.49-1.70 (m, 3H), 1.40-1.46 (m, 1H), 1.43 (s, 3H), 1.42 (s, 3H), 1.29 (d, J = 7.2 Hz, 3H), 0.96 (d, J = 5.6 Hz, 3H), 0.95 (t, J = 8.0 Hz, 9H), 0.56-0.72 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 173.7, 136.4, 136.2, 134.3, 132.6, 132.2, 125.9, 118.5, 117.0, 108.8, 82.2, 82.2, 82.0, 80.3, 78.6, 76.8, 74.9, 70.0, 43.1, 35.1, 33.1, 30.9, 29.2, 27.5, 27.0, 27.0, 22.7, 17.1, 15.4, 7.0, 5.3; IR (neat) 3311, 2954, 2877, 1733, 1456, 1240, 1171 cm−1; HRMS (ES+) m/z for C35H56O6SiNa [M+Na]+ calcd 623.3744, found 623.3751.
The ring closing metathesis of the polyene intermediate was accomplished by using a procedure analogous to that outlined for the conversion of 39 of 40: polyene from the preceding step (40 mg, 0.066 mmol), dichloromethane (66 mL) and Grubbs’ first generation catalyst (11 mg, 0.013 mmol) were used. The crude product was purified by flash column chromatography to afford the macrocycle product (22 mg, 58%) as a colorless oil. In addition, an inseparable mixture of products thought to arrive by enyne metathesis (3.8 mg, 10%) was also isolated. Spectroscopic data for macrocycle product: [α]25D = −48° (c 0.10, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.15-6.28 (m, 2H), 5.94 (dd, J = 4.8, 14.4 Hz, 1H), 5.72 (ddd, J = 3.6, 9.6, 15.2 Hz, 1H), 5.57 (dd, J = 8.8, 14.4 Hz, 1H), 5.34 (dd, J = 8.4, 15.2 Hz, 1H), 4.74 (d, J = 8.4 Hz, 1H), 3.99-4.08 (m, 2H), 3.72 (d, J = 8.4 Hz, 1H), 3.29-3.38 (m, 1H), 3.18-3.30 (m, 2H), 2.18-2.36 (m, 4H), 1.80-2.20 (m, 2H), 1.98 (bs, 1H), 1.64-1.74 (m, 1H), 1.40-1.57 (m, 2H), 1.43 (s, 6H), 1.31 (d, J = 6.8 Hz, 3H), 1.15-1.32 (m, 2H), 1.00 (d, J = 6.4 Hz, 3H), 0.95 (t, J = 8.0 Hz, 9H), 0.53-0.70 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 172.4, 138.1, 136.0, 134.6, 128.8, 127.4, 125.9, 109.0, 83.1, 82.2, 81.5, 79.8, 77.8, 77.4, 77.2, 75.0, 70.2, 43.3, 32.2, 32.1, 29.6, 28.5, 27.2, 27.1, 22.1, 15.8, 12.0, 7.1, 5.6; IR (neat) 3309, 2934, 2874, 1727, 1458, 1378, 1238, 1052 cm−1; HRMS (ES+) m/z for C33H52O6SiNa [M+Na]+ calcd 595.3431, found 595.3439.
The stannylalumination-protonolysis of the alkyne from the preceding step was accomplished using a procedure analogous to that outlined for the conversion of 40 to 41: macrocycle alkyne (51 mg, 0.089 mmol), THF (1.5 mL), Bu3Sn-AlEt2 (1.3 mL of the 0.42M solution, 0.53 mmol) and CuCN (2 mg, 0.024 mmol) were used. Purification of the crude product by flash column chromatography afforded the vinylstannane product (40 mg, 52%) as a colorless oil: [α]25D = −138° (c 0.05, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.15-6.29 (m, 2H), 5.95 (dd, J = 4.8, 14.8 Hz, 1H), 5.67-5.78 (m, 2H), 5.57 (dd, J = 8.4, 14.0 Hz, 1H), 5.35 (dd, J = 8.0, 15.2 Hz, 1H), 5.20 (app t, 3JSn-H = 34.0 Hz, 1H), 4.58 (d, J = 6.4 Hz, 1H), 4.04 (app quint., J = 9.6 Hz, 2H), 3.68 (d, J = 8.4 Hz, 1H), 3.20-3.38 (m, 3H), 2.48 (bd, J = 18.0 Hz, 1H), 2.26-2.35 (m, 1H), 2.09-2.19 (m, 1H), 1.84-2.06 (m, 3H), 1.64-1.74 (m, 1H), 1.41-1.57 (m, 8H), 1.44 (s, 6H), 1.24-1.36 (m, 11H), 0.96 (t, J = 8.0 Hz, 9H), 0.85-0.92 (m, 15H), 0.82 (d, J = 6.8 Hz, 3H), 0.58-0.68 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 172.5, 152.9, 138.2, 136.0, 134.7, 128.8, 127.3, 126.7, 125.9, 109.0, 83.1, 82.2, 80.1, 78.6, 76.0, 44.2, 43.5, 33.2, 32.3, 29.7, 29.1, 28.5, 27.4, 27.2, 27.1, 15.5, 13.7, 12.0, 10.7, 10.5, 9.5, 7.1, 5.7; IR (neat) 2930, 1727, 1239, 1052 cm−1; HRMS (ES+) m/z for C45H80O6SiSnNa [M+Na]+ calcd 887.4644, found 887.4681.
Iododestannylation of the vinylstannane intermediate was accomplished using a procedure analogous to that outlined for the conversion of 40 to 41: vinylstannane from above (11 mg, 0.013 mmol), dichloromethane (1 mL) and NIS (3.2 mg, 0.014 mmol) were used. The crude product was purified by flash column chromatography to afford 71 (9 mg, 98%) as a colorless oil: [α]25D = −46° (c 0.07, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.15-6.29 (m, 2H), 6.09 (s, 1H), 5.89-5.98 (m, 1H), 5.76 (s, 1H), 5.68-5.76 (m, 1H), 5.58 (dd, J = 8.8, 12.4 Hz, 1H), 5.34 (dd, J = 8.0, 14.8 Hz, 1H), 4.59 (d, J = 7.2 Hz, 1H), 3.98-4.90 (m, 2H), 3.66 (d, J = 8.4 Hz, 1H), 3.20-3.37 (m, 3H), 2.61 (bd, J = 13.6 Hz, 1H), 2.26-2.38 (m, 2H), 2.08-2.17 (m, 1H), 1.84-2.02 (m, 2H), 1.62-1.74 (m, 1H), 1.40-1.54 (m, 1H), 1.43 (s, 6H), 1.34 (d, J = 6.8 Hz, 3H), 1.12-1.30 (m, 1H), 0.97 (t, J = 8.0 Hz, 9H), 0.84 (d, J = 6.8 Hz, 3H), 0.61-0.69 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 172.5, 138.2, 136.0, 134.3, 128.9, 127.5, 127.4, 126.0, 110.6, 109.0, 83.1, 82.2, 79.9, 77.6, 77.4, 75.9, 48.5, 43.4, 33.3, 32.2, 29.7, 29.6, 28.5, 27.2, 27.1, 14.7, 12.1, 7.2, 5.6; IR (neat) 2934, 1726, 1238, 1052 cm−1; HRMS (ES+) m/z for C33H53IO6SiNa [M+Na]+ calcd 723.2554, found 723.2567.
2-epi-19-epi-amphidinolide E (4)
Deprotection of 71 was accomplished using a procedure analogous to that outlined for the conversion of 41 to 2: vinyl iodide 71 (26 mg, 0.037 mmol) and AcOH, THF and water (4/1/1 ratio) (1 mL) were used. Analysis of the crude product by 1H NMR indicated a 2:1 mixture of the desired C(18) and undesired C(17) lactones.
Stille coupling of the crude mixure of lactones from above was accomplished using a procedure analogous to that outlined for the conversion of 41 to 2: CuCl (18 mg, 0.18 mmol), THF (0.5 mL), vinylstannane 6 (61 mg, 0.17 mmol) and Pd(PPh3)4 (8 mg, 0.007 mmol) were used. The crude product was purified by flash column chromatography using 10% methanol/chloroform to afford material that was still contaminated with an organotin impurity. The stannane impurity was removed by HPLC purification with 100% ethyl acetate eluent on a normal phase, Varian Dynamax Microsorb 60-8 Si, 250 × 21.4 mm column. The retention time for 2-epi-19-epi-amphidinolide E was 7.6 min. The flow rate was 18 mL/min. 2-epi-19-epi-mphidinolide E was detected using UV absorption (λ = 254 nm and 280 nm) and RI detection. Using the above conditions 6 mg (32% from 71) of pure 2-epi-19-epi-amphidinolide E was isolated: [α]25D = −51° (c 0.10, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.15-6.25 (m, 2H), 6.05 (d, J = 16.0 Hz, 1H), 5.94-6.01 (m, 1H), 5.85 (dt, J = 7.2, 15.6 Hz, 1H), 5.55-5.70 (m, 2H), 5.30 (dd, J = 7.6, 15.2 Hz, 1H), 4.98 (s, 1H), 4.88 (s, 1H), 4.74 (s, 1H), 4.70 (s, 1H), 4.69 (d, J = 10.4 Hz, 1H), 3.90-4.01 (m, 2H), 3.71-3.77 (m, 1H), 3.46-3.54 (m, 1H), 3.38-3.46 (m, 1H), 3.29-3.38 (m, 1H), 2.78 (bd, J = 6.4 Hz, 2H), 2.60 (dd, J = 3.6, 12.8 Hz, 1H), 2.22-2.41 (m, 5H), 1.73-1.96 (m, 4H), 1.72 (s, 3H), 1.36-1.70 (m, 4H), 1.34 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 172.7, 144.8, 144.0, 134.8, 134.2, 134.1, 133.1, 131.2, 129.7, 129.6, 128.4, 116.2, 110.9, 79.9, 78.2, 77.3, 76.5, 73.3, 43.2, 41.5, 36.1, 32.8, 32.3, 30.2, 29.1, 27.4, 22.7, 15.7, 13.4; IR (neat) 3401, 2931, 1724, 1238, 1047, 991 cm−1; HRMS (ES+) m/z for C30H44O6Na [M+Na]+ calcd 523.3036, found 523.3030.
Amphidinolide E (1) series
tert-Butyl-dimethyl-((E)-(R)-2-methyl-hexa-3,5-dienyloxy)-silane (75)
To a −78 °C solution of 4549 (4.19 g, 20.7 mmol) and γ-(TMS)-allyltributylstannane (11.7 g, 29.0 mmol) in CH2Cl2 (21 mL) was added BF3·OEt2 (2.89 mL, 22.8 mmol). The reaction was stirred for 30 min, then quenched at −78 °C with sat. aq. NaHCO3 (60 mL) and extracted with CH2Cl2 (40 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. The residual was dissolved in THF (20 mL) and treated with KOt-Bu (2.0 g, 18 mmol). The reaction was stirred for 3 h at room temperature and then poured into a separatory funnel containing Et2O (100 mL) and sat. aq. NaHCO3 (200 mL). The aqueous layer was extracted with Et2O (50 mL × 3). The organic phase was washed with brine, dried over anhydrous MgSO4, filtered and concentrated. Purification of the crude product by flash column chromatography afforded 75 (2.75 g, 59%) as a colorless oil: [α]25D = +11° (c 4.8, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.31 (dt, J = 10.4, 16.8 Hz, 1H), 6.08 (dd, J = 10.4, 15.2 Hz, 1H), 5.64 (dd, J = 7.2, 15.2 Hz, 1H), 5.11 (d, J = 17.2 Hz, 1H), 4.98 (d, J = 10.0 Hz, 1H), 3.51 (dd, J = 6.0, 9.6 Hz, 1H), 3.41 (dd, J = 6.8, 9.6 Hz, 1H), 2.37 (sept., J = 6.8 Hz, 1H), 1.01 (d, J = 6.8 Hz, 3H), 0.89 (s, 9H), 0.04 (s, 6H); 13C NMR (100MHz, CDCl3) δ3 137.6, 137.4, 130.5, 115.2, 67.9, 39.3, 25.9, 18.3, 16.4, −5.3, −5.4; IR (neat) 2957, 1799, 1472, 1256, 1115, 1088, 837 cm−1; HRMS (EI+) m/z for C9H17OSi [M–C4H9]+ calcd 169.1049, found 169.1044.
Tricarbonyl[(E)-(2S,3R)-2-Methyl-hexa-3,5-dienoic acid (1R,2R)-1-((R)-{(2S,5S)-5-[(E)-4-((4R,5R)-2,2-dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-but-3-enyl]-tetrahydro-furan-2-yl}-triethylsilanyloxy-methyl)-2-methyl-pent-4-ynyl ester]iron (76)
The esterification of alcohol 25 to acid 9 was accomplished using a procedure analogous to that outlined for 39: alcohol 25 (120 mg, 0.244 mmol), acid 9 (105 mg, 0.390 mmol), triethylamine (0.12 mL, 0.854 mmol), DMAP (30 mg, 0.244 mmol), THF (0.5 mL) and 2,4,6-trichlorobenzoyl chloride (61 μL, 0.390 mmol) were used. Purification of the crude product by flash column chromatography afforded 76 (179 mg, 99%) as a colorless oil: [α]25D = +13° (c 0.18, CHCl3); 1H NMR (400MHz, CDCl3) δ 5.74-5.81 (m, 2H), 5.43 (app dd, J = 4.8, 8.4 Hz, 2H), 5.33 (d, J = 16.8 Hz, 1H), 5.21-5.26 (m, 1H), 5.22 (d, J = 11.6 Hz, 1H), 4.69 (dd, J = 1.2, 9.2 Hz, 1H), 4.04 (app q, J = 6.8 Hz, 2H), 3.72 (quint., J = 5.6, 1H), 3.67 (dd, J = 1.6, 7.6 Hz, 1H), 3.60 (app q, J = 8.0 Hz, 1H), 1.99-2.32 (m, 7H), 1.94 (t, J = 2.8 Hz, 1H), 1.73-1.88 (m, 3H), 1.50-1.66 (m, 2H), 1.38-1.48 (m, 1H), 1.44 (s, 3H), 1.43 (s, 3H), 1.31 (d, J = 6.8 Hz, 3H), 1.08 (d, J = 6.8 Hz, 3H), 0.98 (t, J = 8.0 Hz, 9H), 0.97 (app d, J = 8.0 Hz, 1H), 0.59-0.76 (m, 6H), 0.32 (bdd, J = 1.6, 9.2 Hz, 1H); 13C NMR (100MHz, CDCl3) δ 211.2, 173.7, 136.1, 134.3, 126.0, 118.5, 108.9, 87.5, 82.2, 82.2, 82.2, 80.7, 78.5, 77.4, 77.2, 75.4, 69.7, 63.9, 44.5, 40.4, 35.2, 33.3, 30.9, 29.2, 27.8, 27.1, 26.9, 22.3, 19.2, 16.1, 7.1, 5.4; IR (neat) 3310, 2049, 1978, 1732, 1238, 1170 cm−1; HRMS (ES+) m/z for C38H56FeO9SiNa [M+Na]+ calcd 763.2941, found 763.2944.
(E)-(R)-2-Methyl-hexa-3,5-dienoic acid (1R,2R)-1-((R)-{(2S,5S)-5-[(E)-4-((4R,5R)-2,2-dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-but-3-enyl]-tetrahydro-furan-2-yl}-triethylsilanyloxy-methyl)-2-methyl-pent-4-ynyl ester (72)
The oxidative decomplexation of 76 was accomplished using a procedure analogous to that outlined for the conversion of 39 to 40: 76 (45 mg, 0.061 mmol), acetone (1 mL) and cerium ammonium nitrate (CAN) (67 mg, 0.122 mmol) were used. The crude product was purified by flash column chromatography to afford 72 (35 mg, 95%) as a colorless oil: [α]25D = −1.0° (c 0.10, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.29 (dt, J = 10.0, 16.8, 1H), 6.15 (dd, J = 10.4, 15.2 Hz, 1H), 5.72-5.82 (m, 3H), 5.42 (bdd, J = 1.6, 6.0, 15.6 Hz, 1H), 5.32 (d, J = 16.4 Hz, 1H), 5.22 (dd, J = 1.2, 10.4 Hz, 1H), 5.17 (d, J = 17.6, 1H), 5.06 (d, J = 10.0 Hz, 1H), 4.70 (dd, J = 2.0, 8.8 Hz, 1H), 4.04 (app q, J = 7.2 Hz, 2H), 3.71 (quint., J = 5.6 Hz, 1H), 3.67 (dd, J = 2.0, 7.2 Hz, 1H), 3.59 (q, J = 6.4 Hz, 1H), 3.21 (quint., J = 7.2 Hz, 1H), 1.98-2.36 (m, 5H), 1.94 (t, J = 2.4 Hz, 1H), 1.74-1.88 (m, 2H), 1.57-1.66 (m, 1H), 1.46-1.56 (m, 1H), 1.43 (s, 3H), 1.42 (s, 3H), 1.36-1.45 (m, 2H), 1.29 (d, J = 6.8 Hz, 3H), 1.08 (d, J = 6.4 Hz, 3H), 0.95 (t, J = 6.8 Hz, 9H), 0.57-0.72 (m, 6H); 13C NMR (100MHz, CDCl3) δ 173.7, 136.3, 136.2, 134.3, 132.5, 132.3, 125.8, 118.5, 117.0, 108.8, 82.4, 82.2, 82.2, 80.4, 78.4, 77.4, 75.3, 69.6, 42.8, 35.2, 33.3, 30.9, 29.2, 27.0, 26.9, 22.2, 17.0, 16.1, 7.0, 5.3; IR (neat) 3310, 2953, 2876, 1733, 1378, 1239 cm−1; HRMS (ES+) m/z for C35H56O6SiNa [M+Na]+ calcd 623.3744, found 623.3748.
(4E,11E,13E)-(1S,6R,10R,15R,18R,19R,20S)-18-((R)-3-Iodo-1-methyl-but-3-enyl)-8,8,15-trimethyl-19-triethylsilanyloxy-7,9,17,23-tetraoxa-tricyclo[18.2.1.06,10]tricosa-4,11,13-trien-16-one (77)
The ring closing metathesis of 72 was accomplished using a procedure analogous to that outlined for the conversion of 39 to 40: polyene 72 (63 mg, 0.105 mmol), dichloromethane (105 mL) and Grubbs’ first generation catalyst (17 mg, 0.021 mmol) were used. The crude product was purified by flash column chromatography to afford the macrocycle product (44 mg, 73%) as a colorless oil. In addition, an inseparable mixture of products thought to arrive by enyne metathesis (4 mg, 10%) was also isolated. Spectroscopic data for the macrocycle product: [α]25D = −34° (c 0.21, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.15-6.26 (m, 2H), 5.72 (ddd, J = 3.6, 9.6, 15.2 Hz, 1H), 5.49-5.57 (m, 2H), 5.33 (app dd, J = 8.4, 15.6 Hz, 1H), 4.55 (app dd, J = 1.6, 9.6 Hz, 1H), 4.02 (app dt, J = 8.4, 26.0 Hz, 2H), 3.71 (app dd, J = 1.6, 8.4 Hz, 1H), 3.20-3.35 (m, 3H), 2.19-2.36 (m, 3H), 1.82-2.03 (m, 3H), 1.95 (t, J = 2.8 Hz, 1H), 1.62-1.70 (m, 1H), 1.50-1.59 (m, 1H), 1.38-1.49 (m, 1H), 1.43 (s, 3H), 1.42 (s, 3H), 1.23 (d, J = 6.8 Hz, 3H), 1.15-1.28 (m, 2H), 1.06 (d, J = 6.4 Hz, 3H), 0.96 (t, J = 7.6 Hz, 9H), 0.57-0.73 (m, 6H); 13C NMR (100MHz, CDCl3) δ 174.2, 138.4, 135.5, 131.3, 127.6, 125.7, 109.0, 83.0, 82.9, 82.3, 79.9, 78.5, 77.2, 75.1, 69.3, 44.2, 33.2, 32.0, 29.6, 28.6, 27.2, 27.1, 27.0, 22.5, 16.9, 15.6, 7.1, 5.6; IR (neat) 3310, 2950, 2874, 1732, 1378, 1237, 1170, 1053 cm−1; HRMS (ES+) m/z for C33H52O6SiNa [M+Na]+ calcd 595.3431, found 595.3442.
Stannylalumination-protonolysis of the macrocycle alkyne was accomplished using a procedure analogous to that outlined for the conversion of 40 to 41: macrocycle alkyne from the preceding step 17 (44 mg, 0.077 mmol), THF (1 mL), Bu3Sn-AlEt2 (1.1 mL of the 0.41M solution, 0.45 mmol) and CuCN (2 mg, 0.022 mmol) were used. Purification of the crude product by flash column chromatography afforded the vinylstannane (38 mg, 58%) as a colorless oil: [α]25D = −34.5° (c 0.11, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.15-6.26 (m, 2H), 5.72 (app ddd, J = 3.6, 9.6, 15.2 Hz, 1H), 5.63 (app t, 3JSn-H = 70 Hz, 1H), 5.50-5.90 (m, 2H), 5.34 (dd, J = 8.8,15.2 Hz, 1H), 5.14 (dt, J = 2.4 Hz, 3JSn-H = 31.6 Hz, 1H), 4.50 (d, J = 10.0 Hz, 1H), 4.02 (app dt, J = 8.8, 24.0 Hz, 2H), 3.75 (d, J = 8.8 Hz, 1H), 3.15-3.35 (m, 3H), 2.33 (d, J = 13.2 Hz, 2H), 1.82-2.06 (m, 4H), 1.60-1.70 (m, 1H), 1.40-1.57 (m, 8H), 1.44 (s, 3H), 1.43 (s, 3H), 1.25-1.36 (m, 7H), 1.22 (d, J = 6.8 Hz, 3H), 1.18-1.24 (m, 2H), 0.96 (t, J = 8.0 Hz, 9H), 0.93-0.99 (m, 1H), 0.85-0.93 (m, 14H), 0.81 (d, J = 6.4 Hz, 3H), 0.55-0.72 (m, 6H); 13C NMR (100MHz, CDCl3) δ 174.2, 154.1, 138.5, 136.0, 135.8, 131.1, 127.5, 126.7, 125.7, 109.0, 83.1, 82.4, 80.1, 79.6, 77.2, 75.0, 45.4, 44.4, 33.0, 32.1, 29.7, 29.2, 29.1, 28.5, 27.4, 27.2, 27.1, 16.9, 14.8, 13.7, 9.6, 7.2, 5.7; IR (neat) 2954, 2931, 2873, 1732, 1237, 1170, 1053 cm−1; HRMS (ES+) m/z for C45H80O6SiSnNa [M+Na]+ calcd 887.4644, found 887.4655.
Iododestannylation of the vinylstannane intermediate was accomplished using a procedure analogous to that outlined for the conversion of 40 to 41: vinylstannane from the preceding step (121 mg, 0.140 mmol), dichloromethane (2 mL) and NIS (38 mg, 0.17 mmol) were used. The crude product was purified by flash column chromatography to afford 77 (94 mg, 96%) as a colorless oil: [α]25D = −63° (c 0.13, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.15-6.28 (m, 2H), 6.04 (s, 1H), 5.73 (s, 1H), 5.72 (ddd, J = 4.0, 10.4, 14.8 Hz, 1H), 5.53 (app dt, J = 9.2, 14.4 Hz, 2H), 5.33 (dd, J = 8.8, 15.2 Hz, 1H), 4.56 (d, J = 10.0 Hz, 1H), 4.02 (app dt, J = 8.8, 28.4 Hz, 2H), 3.73 (d, J = 8.8 Hz, 1H), 3.20-3.37 (m, 3H), 2.28-2.48 (m, 3H), 2.00 (dd, J = 10.0, 13.6 Hz, 3H), 1.82-1.95 (m, 2H), 1.62-1.70 (m, 1H), 1.49-1.58 (m, 1H), 1.43 (s, 3H), 1.42 (s, 3H), 1.31-1.46 (m, 2H), 1.22 (d, J = 6.8 Hz, 3H), 1.16-1.20 (m, 1H), 0.98 (t, J = 8.0 Hz, 9H), 0.85 (d, J = 6.4 Hz, 3H), 0.56-0.75 (m, 6H); 13C NMR (100MHz, CDCl3) δ 174.4, 138.3, 135.9, 135.5, 131.3, 127.7, 127.1, 125.7, 111.4, 109.0, 83.1, 82.3, 80.0, 78.6, 77.2, 75.0, 48.0, 44.1, 32.7, 32.0, 29.5, 28.5, 27.2, 27.0, 16.9, 14.5, 7.2, 5.7; IR (neat) 2950, 2874, 1731, 1378, 1237, 1170, 1053 cm−1; HRMS (ES+) m/z for C33H53IO6SiNa [M+Na]+ calcd 723.2554, found 723.2559.
Amphidinolide E (1)
Deprotection of 77 was accomplished using a procedure analogous to that outlined for the conversion of 41 to 2: vinyl iodide 77 (15 mg, 0.021 mmol) and AcOH, THF and water (4/1/1) (0.5 mL) were used. The crude product was purified by flash column chromatography to afford only the C18 lactone (9 mg, 77%) as a colorless oil: [α]25D = −14.2° (c 0.12, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.10-6.28 (m, 2H), 6.05 (s, 1H), 5.74 (s, 1H), 5.50-5.69 (m, 3H), 5.26 (dd, J = 8.0, 15.2 Hz, 1H), 4.68 (d, J = 9.2 Hz, 1H), 3.91 (app dt, J = 8.8, 28.8 Hz, 2H), 3.68-3.74 (m, 1H), 3.55 (app q, J = 7.6 Hz, 1H), 3.36-3.44 (m, 1H), 3.22-3.31 (m, 1H), 3.35-2.57 (m, 4H), 2.23-2.31 (m, 1H), 2.05 (dd, J = 10.0, 14.0 Hz, 1H), 1.72-1.93 (m, 3H), 1.29-1.62 (m, 5H), 1.25 (d, J = 6.8 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H); 13C NMR (100MHz, CDCl3) δ 174.4, 135.0, 134.9, 134.1, 131.5, 129.5, 127.3, 110.7, 79.8, 78.1, 77.6, 77.2, 77.1, 76.6, 73.3, 48.4, 44.0, 33.2, 32.5, 29.9, 29.0, 27.1, 17.5, 14.7; IR (neat) 3428, 2932, 2873, 1729, 1167, 990 cm−1; HRMS (ES+) m/z for C24H35IO6Na [M+Na]+ calcd 569.1376, found 569.1369.
Stille coupling of the C18 lactone from above was accomplished using a procedure analogous to that outlined for the conversion of 41 to 2: vinyl iodide lactone from the preceding step (20 mg, 0.037 mmol), CuCl (20 mg, 0.201 mmol), THF (0.5 mL), vinylstannane 6 (68 mg, 0.183 mmol) and Pd(PPh3)4 (8.5 mg, 0.00732 mmol) were used. The crude product was purified by flash column chromatography using 10% methanol/chloroform to afford material that was still contaminated with an organotin impurity. The stannane impurity was removed by HPLC purification with 100% ethyl acetate eluent on a normal phase, Varian Dynamax Microsorb 60-8 Si, 250 × 21.4 mm column. The retention time for amphidinolide E was 9.5 min. The flow rate was 18 mL/min. Amphidinolide E was detected using UV absorption (λ = 254 nm and 280 nm) and RI detection. Using the above conditions 10.6 mg (59%) of pure synthetic amphidinolide E was isolated: [α]25D = −86° (c 0.08, CHCl3); 1H NMR (400MHz, CDCl3) δ 6.10-6.28 (m, 2H) (H4 and H5), 6.05 (d, J = 15.2 Hz, 1H) (H22), 5.58-5.75 (m, 3H) (H3, H10, H23), 5.53 (dd, J = 8.8, 14.4, 1H) (H6), 5.27 (dd, J = 7.6, 14.4 Hz, 1H) (H9), 4.98 (s, 1H) (H29), 4.87 (s, 1H) (H29), 4.75 (s, 1H) (H26), 4.71 (s, 1H) (H26), 4.66 (d, J = 9.2 Hz, 1H) (H18), 3.95 (t, J = 8.4 Hz, 1H) (H8), 3.89 (t, J = 8.8 Hz, 1H) (H7), 3.68-3.74 (m, 1H) (H17), 3.52-3.60 (m, 1H) (H16), 3.36-3.45 (m, 1H) (H13), 3.21-3.30 (m, 1H) (H2), 2.71-2.84 (m, 2H) (H24), 2.20-2.45 (m, 6H) (H20a, H19, H11a and –OH x 3), 1.75-1.94 (m, 3H) (H11b, H12a, H20b), 1.72 (s, 3H) (H27), 1.51-1.68 (m, 1H) (H15a, overlapping w/water), 1.21-1.51 (m, 4H) (H12b, H14a, H14b, H15b), 1.25 (d, J = 6.8 Hz, 3H) (H30), 0.92 (d, J = 6.8 Hz, 3H) (H29); 13C NMR (100MHz, CDCl3) δ 174.4, 144.4, 144.0, 135.1, 135.0, 134.1, 133.3, 131.4, 131.4, 129.4, 127.9, 115.7, 110.7, 79.9, 78.3, 78.0, 77.6, 76.7 (overlapping w/chloroform), 73.2, 44.1, 41.2, 36.0, 32.6, 32.3, 29.9, 28.9, 27.1, 22.5, 17.5, 15.3; IR (neat) 3439, 2929, 1731, 1454, 1168, 990 cm−1; HRMS (ES+) m/z for C30H44O6Na [M+Na]+ calcd 523.3036, found 523.3038.
19-epi-Amphidinolide E (3) series
Tricarbonyl[(E)-(2S,3R)-2-Methyl-hexa-3,5-dienoic acid (1R,2S)-1-((R)-{(2S,5S)-5-[(E)-4-((4R,5R)-2,2-dimethyl-5-vinyl-[1,3]dioxolan-4-yl)-but-3-enyl]-tetrahydro-furan-2-yl}-triethylsilanyloxy-methyl)-2-methyl-pent-4-ynyl ester]iron (78)
Esterification of alcohol 67 with acid 9 was accomplished using a procedure analogous to that outlined for 39: alcohol 67 (0.34 g, 0.68 mmol), acid 9 (0.29 g, 1.09 mmol), triethylamine (0.35 mL, 2.4 mmol), DMAP (83 mg, 0.68 mmol), THF (1.5 mL) and 2,4,6-trichlorobenzoyl chloride (0.17 mL, 1.09 mmol) were used. Purification of the crude product by flash column chromatography afforded 78 (0.501 g, 99%) as a colorless oil: [α]25D = +13° (c 0.25, CHCl3); 1H NMR (400 MHz, CDCl3) δ 5.74-5.84 (m, 2H), 5.39-5.49 (m, 2H), 5.34 (d, J = 17.6 Hz, 1H), 5.20-5.28 (m, 2H), 4.86 (d, J = 8.4 Hz, 1H), 4.02-4.09 (m, 2H), 3.68-3.78 (m, 2H), 3.61-3.68 (m, 1H), 2.06-2.40 (m, 6H), 1.99 (t, J = 2.4 Hz, 1H), 1.72-1.90 (m, 3H), 1.46-1.70 (m, 3H), 1.45 (s, 3H), 1.44 (s, 3H), 1.32 (d, J = 6.8 Hz, 3H), 0.89-1.02 (m, 13H), 0.60-0.77 (m, 6H), 0.33 (bdd, J = 1.6, 8.4 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 211.3, 173.8, 136.2, 134.4, 126.1, 118.6, 108.9, 87.5, 82.4, 82.3, 81.7, 80.7, 78.7, 77.1, 75.3, 70.4, 64.1, 44.6, 40.5, 35.3, 32.9, 31.0, 29.3, 27.8, 27.2, 27.1, 22.6, 19.4, 15.9, 7.2, 5.5; IR (neat) 3311, 2936, 2049, 1979, 1731 cm−1; HRMS (ESI-TOF+) m/z for C38H56FeO9SiH [M+H]+ calcd 741.3116, found 741.3101.
(4E,11E,13E)-(1S,6R,10R,15R,18R,19R,20S)-18-((S)-3-Iodo-1-methyl-but-3-enyl)-8,8,15-trimethyl-19-triethylsilanyloxy-7,9,17,23-tetraoxa-tricyclo[18.2.1.06,10]tricosa-4,11,13-trien-16-one (79)
The oxidative decomplexation of 78 was accomplished using a procedure analogous to that outlined for the conversion of 39 to 40: ester 78 (0.45 g, 0.60 mmol), acetone (6 mL) and cerium ammonium nitrate (CAN) (0.66 g, 1.2 mmol) were used. The crude product was purified by flash column chromatography to afford the polyene product (0.30 g, 82%) as a colorless oil: [α]25D = −7.3° (c 1.6, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.31 (dt, J = 10.4, 16.8, 1H), 6.15 (dd, J = 10.8, 15.6 Hz, 1H), 5.74-5.84 (m, 3H), 5.43 (bdd, J = 5.6, 14.4 Hz, 1H), 5.33 (d, J = 17.2 Hz, 1H), 5.23 (d, J = 10.4 Hz, 1H), 5.17 (d, J = 16.4, 1H), 5.06 (d, J = 10.0 Hz, 1H), 4.86 (dd, J = 2.4, 8.0 Hz, 1H), 4.02-4.09 (m, 2H), 3.60-3.77 (m, 1H), 3.22 (quint., J = 7.2 Hz, 1H), 2.31-2.41 (m, 1H), 2.04-2.26 (m, 4H), 1.98 (t, J = 2.8 Hz, 1H), 1.72-1.91 (m, 2H), 1.58-1.69 (m, 1H), 1.39-1.57 (m, 3H), 1.44 (s, 3H), 1.44 (s, 3H), 1.30 (d, J = 6.8 Hz, 3H), 0.99 (d, J = 6.8 Hz, 3H), 0.96 (t, J = 8.0 Hz, 9H), 0.57-0.74 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 173.8, 136.5, 136.2, 134.4, 132.7, 132.3, 126.0, 118.5, 117.1, 108.9, 82.3, 82.3, 81.9, 80.5, 78.6, 77.0, 75.2, 70.2, 43.0, 35.3, 33.0, 31.0, 29.3, 27.6, 27.1, 27.0, 22.7, 17.2, 15.7, 7.1, 5.4; IR (neat) 3309, 2953, 1983, 1734, 1239, 1169, 1054 cm−1; HRMS (ESI-TOF+) m/z for C35H56O6SiNa [M+Na]+ calcd 623.3744, found 623.3728.
Ring closing metathesis of the polyene was accomplished using a procedure analogous to that outlined for the conversion of 39 to 40: polyene from the preceding step (124 mg, 0.206 mmol), dichloromethane (206 mL) and Grubbs’ first generation catalyst (17 mg, 0.021 mmol) were used. The crude product was purified by flash column chromatography to afford the macrocycle product (67 mg, 57%) as a colorless oil. In addition, an inseparable mixture of products thought to arrive by enyne metathesis (6 mg, 5%) was also isolated. Spectroscopic data for the macrocycle product: [α]25D = −44° (c 0.25, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.14-6.26 (m, 2H), 5.71 (ddd, J = 3.2, 9.6, 14.4 Hz, 1H), 5.48-5.58 (m, 2H), 5.32 (dd, J = 8.8, 15.2 Hz, 1H), 4.76 (d, J = 8.0 Hz, 1H), 4.01 (app dt, J = 8.4, 25.2 Hz, 2H), 3.73 (d, J = 8.4 Hz, 1H), 3.18-3.34 (m, 3H), 2.15-2.36 (m, 4H), 1.98 (t, J = 2.0 Hz, 1H), 1.82-1.96 (m, 2H), 1.61-1.70 (m, 1H), 1.48-1.57 (m, 1H), 1.38-1.48 (m, 1H), 1.42 (s, 3H), 1.42 (s, 3H), 1.10-1.28 (m, 2H), 1.22 (d, J = 6.8 Hz, 3H), 0.98 (d, J = 6.4 Hz, 3H), 0.95 (t, J = 8.0 Hz, 9H), 0.54-0.72 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 174.2, 138.5, 136.1, 135.8, 131.3, 127.7, 125.8, 109.1, 83.2, 82.5, 81.4, 80.1, 77.9, 75.1, 70.4, 44.4, 32.1, 29.6, 28.7, 27.3, 27.2, 27.1, 22.2, 17.0, 16.0, 7.3, 5.8; IR (neat) 3310, 2935, 1732, 1237, 1170 cm−1; HRMS (ESI-TOF+) m/z for C33H52O6SiH [M+H]+ calcd 573.3606, found 573.3596.
Stannylalumination-protonolysis of the macrocycle alkyne was accomplished using a procedure analogous to that outlined for the conversion of 40 to 41: macrocycle alkyne from above (32 mg, 0.056 mmol), THF (1 mL), Bu3Sn-AlEt2 (0.80 mL of the 0.42M solution, 0.34 mmol) and CuCN (1 mg, 0.011 mmol) were used. Purification of the crude product by flash column chromatography afforded the vinylstannane (31 mg, 64%) as a colorless oil: [α]25D = −119° (c 0.10, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.15-6.28 (m, 2H), 5.67-5.76 (m, 2H), 5.53 (app dd, J = 9.6, 14.4 Hz, 2H), 5.33 (dd, J = 8.8, 15.6 Hz, 1H), 5.20 (t, 3JSn-H = 32.0 Hz, 1H), 4.57 (d, J = 7.6 Hz, 1H), 4.02 (app dt, J = 8.8, 23.2 Hz, 2H), 3.72 (app q, J = 12.4 Hz, 1H), 3.19-3.36 (m, 3H), 2.49 (bd, J = 13.6 Hz, 1H), 2.28-2.36 (m, 1H), 2.10-2.20 (m, 1H), 1.83-2.24 (m, 3H), 1.56-1.71 (m, 1H), 1.40-1.56 (m, 9H), 1.43 (s, 3H), 1.43 (s, 3H), 1.25-1.36 (m, 7H), 1.22 (d, J = 6.8 Hz, 3H), 0.96 (t, J = 7.6 Hz, 9H), 0.85-0.92 (m, 14H), 0.81 (d, J = 6.8 Hz, 3H), 0.59-0.69 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 174.3, 152.9, 138.6, 136.2, 136.0, 131.3, 127.6, 126.6, 125.8, 109.1, 83.2, 82.5, 80.3, 80.0, 77.5, 75.9, 44.4, 44.0, 33.1, 32.3, 29.7, 29.4, 29.3, 28.7, 27.5, 27.3, 27.2, 17.1, 15.7, 13.8, 9.6, 7.4, 5.8; IR (neat) 2955, 1732, 1171, 1054 cm−1; HRMS (ESI-TOF+) m/z for C45H80O6SiSnH [M+H]+ calcd 865.4819, found 865.4825.
Iododestannylation of vinylstannane was accomplished using a procedure analogous to that outlined for the conversion of 40 to 41: vinylstannane from the preceding step (54 mg, 0.063 mmol), dichloromethane (2 mL) and NIS (17 mg, 0.075 mmol) were used. The crude product was purified by flash column chromatography to afford 79 (34 mg, 77%) as a colorless oil: [α]25D = −110° (c 0.11, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.15-6.28 (m, 2H), 6.09 (s, 1H), 5.76 (s, 1H), 5.72 (ddd, J = 3.6, 9.6, 15.2 Hz, 1H), 5.48-5.58 (m, 2H), 5.33 (dd, J = 8.8, 15.2 Hz, 1H), 4.60 (d, J = 7.6 Hz, 1H), 4.02 (app dt, J = 8.4, 25.2 Hz, 2H), 3.68 (d, J = 8.8 Hz, 1H), 3.30-3.38 (m, 1H), 3.19-3.30 (m, 2H), 2.63 (dd, J = 4.4, 13.6 Hz, 1H), 2.26-2.39 (m, 2H), 2.12 (dd, J = 9.2, 13.6 Hz, 1H), 1.84-2.10 (m, 2H), 1.36-1.55 (m, 2H), 1.43 (s, 3H), 1.43 (s, 3H), 1.11-1.27 (m, 3H), 1.23 (d, J = 7.2 Hz, 3H), 0.97 (t, J = 7.6 Hz, 9H), 0.84 (d, J = 6.8 Hz, 3H), 0.60-0.71 (m, 6H); 13C NMR (100 MHz, CDCl3) δ 174.3, 138.6, 136.2, 135.7, 131.5, 127.7, 127.5, 125.8, 110.7, 109.2, 83.1, 82.5, 80.1, 77.7, 77.6, 75.9, 48.5, 44.3, 33.3, 32.2, 29.6, 28.7, 27.3, 27.2, 27.2, 17.0, 15.0, 7.4, 5.8; IR (neat) 2981, 1731, 1377, 1170, 1053 cm−1; HRMS (ESI-TOF+) m/z for C33H53IO6SiH [M+H]+ calcd 701.2729, found 701.2723.
19-epi-amphidinolide E (3)
Deprotection of 79 was accomplished using a procedure analogous to that outlined for the conversion of 41 to 2: vinyl iodide 79 (34 mg, 0.049 mmol) and AcOH, THF and water (4/1/1) (1 mL) were used. The crude product was purified by flash column chromatography to afford only the C18 lactone (24 mg, 90%) as a colorless oil: [α]25D = −90° (c 0.18, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.10-6.26 (m, 2H), 6.08 (s, 1H), 5.76 (s, 1H), 5.49-5.69 (m, 3H), 5.26 (dd, J = 8.0, 15.2 Hz, 1H), 4.75 (d, J = 8.0 Hz, 1H), 3.91 (app dt, J = 8.8, 26.0 Hz, 2H), 3.63-3.68 (m, 1H), 3.58-3.60 (m, 1H), 3.34-3.46 (m, 1H), 3.21-3.30 (m, 1H), 2.66 (dd, J = 3.6, 13.6 Hz, 1H), 2.33-2.50 (m, 4H), 2.20-2.29 (m, 1H), 2.14 (app dd, J = 9.6, 14.0 Hz, 1H), 1.70-1.93 (m, 3H), 1.54-1.64 (m, 1H), 1.32-1.49 (m, 3H), 1.26 (d, J = 6.4 Hz, 3H), 0.89 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.2, 135.0, 134.3, 131.6, 131.6, 129.6, 127.6, 110.5, 80.0, 78.3, 77.7, 76.2, 73.4, 48.3, 44.1, 33.9, 32.9, 30.1, 29.1, 27.2, 17.6, 15.0; IR (neat) 3430, 2933, 1731, 1169, 990 cm−1; HRMS (ES+) m/z for C24H35IO6Na [M+Na]+ calcd 569.1376, found 569.1367.
Stille coupling of the C18 lactone from was accomplished using a procedure analogous to that outlined for the conversion of 41 to 2: vinyl iodide from above (24 mg, 0.044 mmol), CuCl (24 mg, 0.24 mmol), THF (0.5 mL), vinylstannane 6 (82 mg, 0.22 mmol) and Pd(PPh3)4 (10 mg, 0.0087 mmol) were used. The crude product was purified by flash column chromatography using 10% methanol/chloroform to afford material that was still contaminated with an organotin impurity. The stannane impurity was removed by HPLC purification with 100% ethyl acetate eluent on a normal phase, Varian Dynamax Microsorb 60-8 Si, 250 × 21.4 mm column. The retention time for 19-epi-amphidinolide E was 7.5 min. The flow rate was 18 mL/min. 19-epi-Amphidinolide E was detected using UV absorption (λ = 254 nm and 280 nm) and RI detection. Using the above conditions 15 mg (68%) of pure 19-epi-amphidinolide E (3) was isolated: [α]25D = −7.8° (c 0.50, CHCl3); 1H NMR (400 MHz, CDCl3) δ 6.10-6.28 (m, 2H), 6.05 (d, J = 15.6 Hz, 1H), 5.87 (dt, J = 6.8, 16.0 Hz, 1H), 5.49-5.70 (m, 3H), 5.27 (dd, J = 8.0, 15.2 Hz, 1H), 4.98 (s, 1H), 4.88 (s, 1H), 4.73 (s, 1H), 4.70 (s, 1H), 4.68 (d, J = 10.4 Hz, 1H), 3.95 (t, J = 8.4 Hz, 1H), 3.88 (t, J = 8.8 Hz, 1H), 3.78 (app q, J = 8.8 Hz, 1H), 3.35-3.44 (m, 1H), 3.21-3.31 (m, 1H), 2.77 (d, J = 6.8 Hz, 2H), 2.62 (dd, J = 3.6, 13.2 Hz, 1H), 2.24-2.46 (m, 5H), 1.74-1.94 (m, 4H), 1.71 (s, 3H), 1.53-1.64 (m, 1H), 1.22-1.51 (m, 3H), 1.25 (d, J = 6.8 Hz, 3H), 0.81 (d, J = 6.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.3, 144.8, 143.9, 135.4, 135.1, 134.3, 133.1, 131.6, 129.6, 128.5, 116.3, 111.0, 80.1, 78.2, 78.0, 77.7, 76.8, 73.4, 44.2, 41.6, 36.0, 32.5, 32.2, 30.0, 29.1, 27.1, 22.7, 17.5, 15.7; IR (neat) 3400, 2931, 1731, 1169, 989 cm−1; HRMS (ESI-TOF+) m/z for C30H44O6Na [M+Na]+ calcd 523.3036, found 523.3024.
Acknowledgments
This work is supported by a grant from the National Institutes of Health (GM38436). Preliminary studies at the University of Michigan were supported by GM38907. We thank Dr. Troy Ryba for assistance with the HPLC purification of amphidinolide E, and Prof. Jun-ichi Kobayashi for providing comparative spectroscopic data for the natural product.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For reviews on the amphidinolides: Kobayashi J, Ishibashi M. Chem Rev. 1993;93:1753.Chakraborty TK, Das S. Curr Med Chem: Anti-Cancer Agents. 2001;1:131. doi: 10.2174/1568011013354660.Kobayashi J, Shimbo K, Kubota T, Tsuda M. Pure Appl Chem. 2003;75:337.Kobayashi J, Tsuda M. Nat Prod Rep. 2004;21:77. doi: 10.1039/b310427n.
- 2.(a) Lam HW, Pattenden G. Angew Chem, Int Ed Engl. 2002;41:508. doi: 10.1002/1521-3773(20020201)41:3<508::aid-anie508>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]; (b) Maleczka RE, Jr, Terrell LR, Geng F, Ward JS., III Org Lett. 2002;4:2841. doi: 10.1021/ol0262284. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Chisholm JD, Wrobleski SJ, Jung M. J Am Chem Soc. 2002;124:12420. doi: 10.1021/ja027883+. [DOI] [PubMed] [Google Scholar]; (d) Trost BM, Harrington PE. J Am Chem Soc. 2004;126:5028. doi: 10.1021/ja049292k. [DOI] [PubMed] [Google Scholar]; (e) Trost BM, Wrobleski ST, Chisholm JD, Harrington PE, Jung M. J Am Chem Soc. 2005;127:13589. doi: 10.1021/ja0533646. [DOI] [PubMed] [Google Scholar]; (f) Trost BM, Harrington PE, Chisholm JD, Wrobleski ST. J Am Chem Soc. 2005;127:13598. doi: 10.1021/ja053365y. [DOI] [PubMed] [Google Scholar]
- 3.Williams DR, Kissel WS. J Am Chem Soc. 1998;120:11198. [Google Scholar]
- 4.Williams DR, Meyer KG. J Am Chem Soc. 2001;123:765. doi: 10.1021/ja005644l. [DOI] [PubMed] [Google Scholar]
- 5.(a) Williams DR, Myers BJ, Mi L. Org Lett. 2000;2:945. doi: 10.1021/ol0000197. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Papillon JPN. J Am Chem Soc. 2004;126:13618. doi: 10.1021/ja045449x. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Papillon JPN, Nussbaumer T. J Am Chem Soc. 2005;127:17921. doi: 10.1021/ja055967n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Fürstner A, Aissa C, Riveiros R, Ragot J. Angew Chem, Int Ed Engl. 2002;41:4763. doi: 10.1002/anie.200290042. [DOI] [PubMed] [Google Scholar]; (b) Aissa C, Riveiros R, Ragot J, Fürstner A. J Am Chem Soc. 2003;125:15512. doi: 10.1021/ja038216z. [DOI] [PubMed] [Google Scholar]; (c) Ghosh AK, Liu C. J Am Chem Soc. 2003;125:2374. doi: 10.1021/ja021385j. [DOI] [PubMed] [Google Scholar]; (d) Ghosh AK, Liu C. Strategies Tactics Org Synth. 2004;5:255. [Google Scholar]; (e) Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2004;126:998. doi: 10.1021/ja039716v. [DOI] [PubMed] [Google Scholar]; (f) Colby EA, O’Brien KC, Jamison TF. J Am Chem Soc. 2005;127:4297. doi: 10.1021/ja042733f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) O’Brien KC, Colby EA, Jamison TF. Tetrahedron. 2005;61:6243. [Google Scholar]; (h) Deng LS, Huang XP, Zhao G. J Org Chem. 2006;71:4625. doi: 10.1021/jo0605086. [DOI] [PubMed] [Google Scholar]
- 7.(a) Ghosh AK, Gong G. J Am Chem Soc. 2004;126:3704. doi: 10.1021/ja049754u. [DOI] [PubMed] [Google Scholar]; (b) Ghosh AK, Gong G. J Org Chem. 2006;71:1085. doi: 10.1021/jo052181z. [DOI] [PubMed] [Google Scholar]
- 8.Lepage O, Kattnig E, Fürstner A. J Am Chem Soc. 2004;126:15970. doi: 10.1021/ja044130+. [DOI] [PubMed] [Google Scholar]
- 9.Fürstner A, Kattnig E, Lepage O. J Am Chem Soc. 2006;128:9194. doi: 10.1021/ja061918e. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kobayashi J, Ishibashi M, Murayama T, Takamatsu M, Iwamura M, Ohizumi Y, Sasaki T. J Org Chem. 1990;55:3421. [Google Scholar]; (b) Kubota T, Tsuda M, Kobayashi J. J Org Chem. 2002;67:1651. doi: 10.1021/jo016326n. [DOI] [PubMed] [Google Scholar]
- 11.Kim CH, An HJ, Shin WK, Yu W, Woo SK, Jung SK, Lee E. Angew Chem Int Ed. 2006;45:8019. doi: 10.1002/anie.200603363. [DOI] [PubMed] [Google Scholar]
- 12.Gurjar MK, Mohapatra S, Phalgune UD, Puranik VG, Mohapatra DK. Tetrahedron Lett. 2004;45:7899. [Google Scholar]
- 13.Marshall JA, Schaaf G, Nolting A. Org Lett. 2005;7:5331. doi: 10.1021/ol0523493. [DOI] [PubMed] [Google Scholar]
- 14.(a) Micalizio GC, Roush WR. Org Lett. 2000;2:461. doi: 10.1021/ol9913082. [DOI] [PubMed] [Google Scholar]; (b) Micalizio GC, Roush WR. Org Lett. 2001;3:1949. doi: 10.1021/ol0160250. [DOI] [PubMed] [Google Scholar]; (c) Shotwell JB, Roush WR. Org Lett. 2004;6:3865. doi: 10.1021/ol048381z. [DOI] [PubMed] [Google Scholar]; (d) Tinsley JM, Roush WR. J Am Chem Soc. 2005;127:10818. doi: 10.1021/ja051986l. [DOI] [PubMed] [Google Scholar]; (e) Mertz E, Tinsley JM, Roush WR. J Org Chem. 2005;70:8035. doi: 10.1021/jo0511290. [DOI] [PubMed] [Google Scholar]; (f) Tinsley JM, Mertz E, Chong PY, Rarig RAF, Roush WR. Org Lett. 2005;7:4245. doi: 10.1021/ol051719k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Lambert WT, Roush WR. Org Lett. 2005;7:5501. doi: 10.1021/ol052321r. [DOI] [PubMed] [Google Scholar]
- 15.For reviews of reactions of allylsilanes: Masse CE, Panek JS. Chem Rev. 1995;95:1293.Chabaud L, James P, Landais Y. Eur J Org Chem. 2004:3173.
- 16.(a) Panek JS, Yang M. J Am Chem Soc. 1991;113:9868. [Google Scholar]; Panek JS, Beresis R. J Org Chem. 1993;58:809. [Google Scholar]; (c) Beresis R, Panek JS. Bioorg Med Chem Lett. 1993;3:1609. [Google Scholar]; (d) Peng ZH, Woerpel KA. Organic Letters. 2002;4:2945. doi: 10.1021/ol026343e. [DOI] [PubMed] [Google Scholar]; (e) Peng ZH, Woerpel KA. Org Lett. 2001;3:675. doi: 10.1021/ol0069960. [DOI] [PubMed] [Google Scholar]; (f) Roberson CW, Woerpel KA. J Am Chem Soc. 2002;124:11342. doi: 10.1021/ja012152f. [DOI] [PubMed] [Google Scholar]; (g) Peng ZH, Woerpel KA. J Am Chem Soc. 2003;125:6018. doi: 10.1021/ja034865z. [DOI] [PubMed] [Google Scholar]
- 17.Va P, Roush WR. J Am Chem Soc. 2006;128:15960. doi: 10.1021/ja066663j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Va P, Roush WR. Org Lett. 2007;9:307. doi: 10.1021/ol0628098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Stille JK, Groh BL. J Am Chem Soc. 1987;109:813. [Google Scholar]; (b) Han X, Stoltz BM, Corey EJ. J Am Chem Soc. 1999;121:7600. [Google Scholar]
- 20.Sarabia F, Sanchez-Ruiz A. J Org Chem. 2005;70:9514. doi: 10.1021/jo0516032. [DOI] [PubMed] [Google Scholar]
- 21.Johnson WS, Werthemann L, Bartlett WR, Brocksom TJ, Li TT, Faulkner DJ, Petersen MR. J Am Chem Soc. 1970;92:741. [Google Scholar]
- 22.Roush WR, Koyama K, Curtin ML, Moriarty KJ. J Am Chem Soc. 1996;118:7502. [Google Scholar]
- 23.(a) Roush WR, Halterman RL. J Am Chem Soc. 1986;108:294. [Google Scholar]; (b) Roush WR, Hoong LK, Palmer MAJ, Park JC. J Org Chem. 1990;55:4109. [Google Scholar]; (c) Roush WR, Hoong LK, Palmer MAJ, Straub JA, Palkowitz AD. J Org Chem. 1990;55:4117. [Google Scholar]; (d) Roush WR, Palkowitz AD, Ando K. J Am Chem Soc. 1990;112:6348. [Google Scholar]
- 24.Schmid CR, Bradley DA. Synthesis. 1992:587. [Google Scholar]
- 25.Parikh JR, Doering WvE. J Am Chem Soc. 1967;89:5505. [Google Scholar]
- 26.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769. [Google Scholar]
- 27.(a) Roush WR, Grover PT. Tetrahedron Lett. 1990;31:7567. [Google Scholar]; (b) Roush WR, Grover PT. Tetrahedron. 1992;48:1981. [Google Scholar]
- 28.Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512. [Google Scholar]
- 29.Hudrlik PF, Peterson D. J Am Chem Soc. 1975;97:1464. [Google Scholar]
- 30.[3+2] annulations involving aldehydes with α-chelating groups promoted by SnCl4 typically afford 2,5-trans-THF products.14a Since, aldehyde 5 has no α-chelating group, we anticipated 2,5-cis product 24 to predominate.
- 31.Heitzman CL, Lambert WT, Mertz E, Shotwell JB, Tinsley JM, Va P, Roush WR. Org Lett. 2005;7:2405. doi: 10.1021/ol0506821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horita K, Yoshioka T, Tanaka T, Oikawa Y, Yonemitsu O. Tetrahedron. 1986;42:3021. [Google Scholar]
- 33.Acid 13 was synthesized by hydrolysis of the chiral auxiliary in oxazolidinone 36 (64% yield).
- 34.Hikota M, Sakurai Y, Horita K, Yonemitsu O. Tetrahedron Lett. 1990;31:6367. [Google Scholar]
- 35.For use of EDCI·MeI: Mergott DJ, Frank SA, Roush WR. Proc Natl Acad Sci U S A. 2004;101:11955. doi: 10.1073/pnas.0401247101.For use of PyBroP: Coste J, Frerot E, Jouin P. J Org Chem. 1994;59:2437.For use of DCC: Xia Z, Smith CD. J Org Chem. 2001;66:3459. doi: 10.1021/jo005783l.
- 36.(a) Otera J, Danoh N, Nozaki H. J Org Chem. 1991;56:5307. [Google Scholar]; (b) Orita A, Sakamoto K, Hamada Y, Mitsutome A, Otera J. Tetrahedron. 1999;55:2899. [Google Scholar]; (c) Orita A, Mitsutome A, Otera J. J Org Chem. 1998;63:2420. doi: 10.1021/jo9800412. [DOI] [PubMed] [Google Scholar]
- 37.(a) Kita Y, Maeda H, Omori K, Okuno T, Tamura Y. Synlett. 1993:273. [Google Scholar]; (b) Kita Y, Maeda H, Omori K, Okuno T, Tamura Y. J Chem Soc Perkin Trans. 1993;1:2999. [Google Scholar]; (c) Trost BM, Chisholm JD. Org Lett. 2002;4:3743. doi: 10.1021/ol026726c. [DOI] [PubMed] [Google Scholar]
- 38.For reviews of enolate alkylations: Evans DA. In: Asymmetric Synthesis. Morrison JD, editor. Vol. 3. Academic Press; New York: 1984. p. 1.Caine D. In: Comprehensive Organic Synthesis. Pattenden G, editor. Vol. 3. Pergamon Press; Oxford: 1991. p. 1.
- 39.Takacs JM, Jaber MR, Swanson BJ, Mehrman SJ. Tetrahedron: Asymmetry. 1998;9:4313. [Google Scholar]
- 40.Acid 9 has been previously synthesized in racemic form: Donaldson WA, Craig R, Spanton S. Tetrahedron Letters. 1992;33:3967.Wasicak JT, Craig RA, Henry R, Dasgupta B, Li H, Donaldson WA. Tetrahedron. 1997;53:4185.
- 41.For reviews on olefin metathesis: Gradillas A, Perez-Castells J. Angew Chem Int Ed. 2006;45:6086. doi: 10.1002/anie.200600641.Nicolaou KC, Bulger PG, Sarlah D. Angew Chem, Int Ed. 2005;44:4490. doi: 10.1002/anie.200500369.Fürstner A. Angew Chem, Int Ed. 2000;39:3012.
- 42.For recent examples of diene or triene forming ring closing metatheses: Biswas K, Lin H, Njardarson JT, Chappell MD, Chou TC, Guan Y, Tong WP, He L, Horwitz SB, Danishefsky SJ. J Am Chem Soc. 2002;124:9825. doi: 10.1021/ja0262333.Evano G, Schaus JV, Panek JS. Org Lett. 2004;6:525. doi: 10.1021/ol036284k.Fürstner A, Nevado C, Tremblay M, Chevrier C, Teply F, Aissa C, Waser M. Angew Chem, Int Ed. 2006;45:5837. doi: 10.1002/anie.200601860.
- 43.(a) Fürstner A, Dierkes T. Org Lett. 2000;2:2463. doi: 10.1021/ol006122d. [DOI] [PubMed] [Google Scholar]; (b) Evans PA, Cui J, Gharpure SJ, Polosukhin A, Zhang H-R. J Am Chem Soc. 2003;125:14702. doi: 10.1021/ja0384734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang YG, Kobayashi Y. Org Lett. 2002;4:4615. doi: 10.1021/ol020193q. [DOI] [PubMed] [Google Scholar]
- 45.(a) Crimmins MT, She J. J Am Chem Soc. 2004;126:12790. doi: 10.1021/ja0455852. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Frederiksen MU, Papillon JPN, Harrington PE, Shin S, Shireman BT. J Am Chem Soc. 2005;127:3666. doi: 10.1021/ja042435i. [DOI] [PubMed] [Google Scholar]
- 46.Ono K, Nagata T, Nishida A. Synlett. 2003:1207. [Google Scholar]
- 47.Sharma S, Oehlschlager AC. J Org Chem. 1989;54:5064. [Google Scholar]
- 48.(a) De Riccardis F, Minale L, Riccio R, Giovannitti B, Iorizzi M, Debitus C. Gazz Chim Ital. 1993;123:79. [Google Scholar]; (b) Finamore E, Minale L, Riccio R, Rinaldo G, Zollo F. J Org Chem. 1991;56:1146. [Google Scholar]
- 49.(a) Roush WR, Palkowitz AD, Ando K. J Am Chem Soc. 1990;112:6348. [Google Scholar]; (b) Kalesse M, Chary KP, Quitschalle M, Burzlaff A, Kasper C, Scheper T. Chem--Eur J. 2003;9:1129. doi: 10.1002/chem.200390130. [DOI] [PubMed] [Google Scholar]
- 50.Fuwa H, Okamura Y, Natsugari H. Tetrahedron. 2004;60:5341. [Google Scholar]
- 51.Harada N, Nakanishi K. Circular Dichroic Spectrometry: Exciton Coupling in Organic Stereochemistry. University Science Books; Mill Valley, CA: 1983. [Google Scholar]
- 52.Lo LC, Berova N, Nakanishi K, Schlingmann G, Carter GT, Borders DB. J Am Chem Soc. 1992;114:7371. [Google Scholar]
- 53.Keck GE, Romer DR. J Org Chem. 1993;58:6083. [Google Scholar]
- 54.Fürstner has postulated the formation of a ketene intermediate during an attempted Yamaguchi macrocyclization. This postulated ketene intermediate ultimately led to the formation of an undesired substituted phenol: Fürstner A, Aissa C, Chevrier C, Teply F, Nevado C, Tremblay M. Angew Chem, Int Ed. 2006;45:5832. doi: 10.1002/anie.200601859.. Although it is possible that acid 13 (Table 2) could have suffered from a similar side-reaction pathway, the expect side product(s) (ortho-methylphenol or acylated ortho-methylphenol) was not observed
- 55.(a) Hodous BL, Ruble JC, Fu GC. J Am Chem Soc. 1999;121:2637. [Google Scholar]; (b) Wiskur SL, Fu GC. J Am Chem Soc. 2005;127:6176. doi: 10.1021/ja0506152. [DOI] [PubMed] [Google Scholar]
- 56.Curran DP, Zhang Q, Lu H, Gudipati V. J Am Chem Soc. 2006;128:9943. doi: 10.1021/ja062469l. [DOI] [PubMed] [Google Scholar]