

Abstract
First and second generation total syntheses of mycolactones A and B are reported. The first generation total synthesis unambiguously confirmed our earlier assignment of the relative and absolute stereochemistry of mycolactones A and B. Knowledge of the chemical properties of the mycolactones accumulated through the first generation total synthesis allowed us to implement several major improvements to the original synthesis, including: (1) optimizing the choice of protecting groups, (2) eliminating the unnecessary adjustment of protecting groups, and (3) improving the overall stereoselectivity and synthetic efficiency. The second generation total synthesis consists of 21 longest linear steps, with 8.8% overall yield.
Keywords: mycolactones, total synthesis, Buruli ulcer
1. Introduction
Buruli ulcer is a severe necrotizing skin disease caused by Mycobacterium ulcerans.1 Among the diseases caused by mycobacteria, Buruli ulcer occurs with less frequency than tuberculosis and leprosy. However, the occurrence of the disease is increasing and spreading in tropical countries. Indeed, it is noted that the incidence of Buruli ulcer may exceed that of leprosy and tuberculosis in highly affected areas. Infection with Mycobacterium ulcerans, probably carried by aquatic insects,2 results in progressive necrotic lesions that, if untreated, can extend to 15% of a patient’s skin surface. Unfortunately, surgical intervention is currently the only realistic therapy for Buruli ulcer.
Most pathogenic bacteria produce toxins that play an important role(s) in disease. However, there has been no evidence thus far to suggest toxin production by Mycobacterium tuberculosis and Mycobacterium leprae, the two most-recognized pathogenic members of the genus Mycobacterium. Interestingly, the possible presence of a toxin in Mycobacterium ulcerans was hypothesized for a number of years prior to the isolation and characterization of two polyketide-derived macrolides from this bacteria by Small and co-workers in 1999.3 These macrolides were designated mycolactones A and B, and it was demonstrated that intradermal inoculation of the purified mycolactones into guinea pigs produced a lesion similar to that of Buruli ulcer in humans.
The gross structure of mycolactones A and B was elucidated by Small and co-workers via spectroscopic methods, including extensive 2D NMR experiments, to be a 12-membered macrolide bearing a highly unsaturated side-chain.4 Through the combined use of an NMR database and the preparation of model compounds, we studied and established the relative and absolute configuration of the mycolactone core,5 and then applied the newly developed universal NMR database concept in chiral solvents,6 to establish the complete structure of the mycolactones.7 Finally, the assigned structure was confirmed by total synthesis.8 Through these efforts, mycolactones A and B are now characterized as having the stereochemistry shown below, and as an approximately 3:2 mixture of Z-Δ4′,5′- and E-Δ4′,5′-geometric isomers of the unsaturated side-chain (Figure 1).
Figure 1.

Structure of mycolactones A (1) and B (2).
Mycolactones A and B constitute the major metabolites produced by West African strains of Mycobacterium ulcerans. However, several mycolactone congeners, including mycolactone C,9 mycolactone D,10 and C2’-methyl mycolactones A and B,11 have recently been isolated from clinical isolates of Mycobacterium ulcerans from Africa, Malaysia, Asia, Australia, and Mexico. In this connection, it is worthwhile noting that clinical isolates from Asia, Mexico, and Australia are less virulent than clinical isolates from Africa. In addition, mycolactone congeners have also been isolated from the frog pathogen Mycobacterium marinum12 and the fish pathogen Mycobacterium liflandii,13 respectively.
The mycolactones have attracted considerable attention from the synthetic community not only for their highly potent biological activity, but also for being the first examples of polyketide macrolides to be isolated from a human pathogen.14-18 In this paper, we report first and second generation total syntheses of mycolactones A and B.
2. Results and Discussion
First Generation Total Synthesis19
We envisioned that an obvious synthetic route to the mycolactones would proceed through esterification of the C5 hydroxyl group present in the mycolactone core with the pentaenoic acid present in the mycolactones. To demonstrate the feasibility of this approach, we decided to utilize the mycolactone core triol 3, previously synthesized for the purposes of stereochemical assignment.5 Selective protection of the C17/C19-1,3-diol of 3 was smoothly accomplished by treatment with d imethoxycyclopentane and p-TsOH, to furnish the suitably protected core 4 in good yield (Scheme 1).
Scheme 1.

Reagents: (a) 1,1-dimethoxycyclopentane, p-TsOH, benzene, 80%.
We then focused on the synthesis of a suitably protected pentaenoic acid. Considering the anticipated instability of the pentaenoate system, we chose to protect the side-chain alcohols as t-butyldimethyls ilyl (TBS) ethers. Gurjar and Cherian reported a synthesis of ethyl ester 7 of the pentaene fatty acid via Horner-Wadsworth-Emmons olefination at C8′-C9′ (Scheme 2).14 Despite the differences in the stereochemistry of triol and the protecting groups, this synthetic route appeared well suited to our needs, and we chose to adopt this route for the synthesis of tris-TBS pentaenoate 18 (see Scheme 8 for the structure 18).
Scheme 2.

Scheme 8.

Reagents: (a) 1. MeOCH2COCl, i-Pr2NEt, DMAP, CH2Cl2, 91%; 2. CH2Cl2-H2O-TFA (16:4:1), 92%; 3. TBSOTf, 2,6-lutidine, CH2Cl2, quant.; 4. K2CO3, MeOH 0 °C, 84%; (b) 1. 19, Cl3C6H2COCl, i-Pr2NEt, DMAP, benzene, 90%; 2. TBAF, THF, 80%.
Our first task was the synthesis of the tris-TBS aldehyde 12. In conjunction with the stereochemical assignment of the mycolactone unsaturated side-chain, we previously prepared all four diastereomers of 12 from D-glyceraldehyde acetonide in an optically active form.7 Worth noting is that the availability of all four diastereomers was critical for the unambiguous determination of the side-chain stereochemistry. However, once the stereochemistry was established, we could focus on the synthesis of the desired tris-TBS aldehyde 12 specifically. Although the synthetic route from D-glyceraldehyde acetonide served well for the stereochemical study, we wished to have a more efficient synthesis. For this reason, we studied an alternative synthetic route (Scheme 3). Thus, the known aldehyde 820 was subjected to Horner-Wadsworth-Emmons olefination, then catalytic asymmetric dihydroxylation with AD-mix-α,21 resulting in a 3.8:1 mixture of diastereomers,22 with the expected and desired diol 9 as the major product. The diol was protected as its bis-TBS ether, and the resulting compound was then subjected to a sequence of reduction, oxidation, and Wittig olefination, to give the corresponding α,β-unsaturated ester 10. Reduction of 10, followed by chromatographic separation of the diastereomers and then oxidation, gave aldehyde 12.
Scheme 3.

Reagents: (a) 1. NaH, (EtO)2P(O)CH2CO2Et, benzene, 64%; 2. AD-mix-α, MeSO2NH2, t-BuOH-H2O (1:1), 40h, 0 °C, 70%, d.r. = 3.8:1; (b) 1. TBSOTf, 2,6-lutidine, CH2Cl2, 0 °C, 99%; 2. DIBAL, CH2Cl2, 89%; 3. SO3•py, i-Pr2NEt, CH2Cl2-DMSO (3:2); 4. Ph3P=C(Me)CO2Et, toluene, 110 °C, 83% (2 steps). (c) DIBAL, CH2Cl2, -78 °C, followed by chromatographic separation of the diastereomers, 57%. (d) SO3•py, i-Pr2NEt, CH2Cl2-DMSO (3:2), quant.
Next, phosphonate (2′E,4′E,6′E)-17 was synthesized employing a minor modification of the procedure reported by Gurjar and Cherian.14 The allylic alcohol 14 was prepared in 4 steps from allyl alcohol in 25% overall yield (Scheme 4). Oxidation, followed by Wittig olefination, gave diene ester 15. Reduction, oxidation, and Wittig olefination provided triene ester 16. Finally, a three step sequence of deprotection, bromination, and phosphonate formation furnished (2′E,4′E,6′E)-17.
Scheme 4.

Reagents: (a) 1. TBSCl, imidazole, DMF; 2. O3, CH2Cl2, -78 °C, then Ph3P; 3. Ph3P=C(Me)CO2Et, CH2Cl2; 4. DIBAL, CH2Cl2, -78 °C, 25% (four steps); (b) 1. SO3•py, i-Pr2NEt, CH2Cl2-DMSO (3:2); 2. Ph3P=C(Me)CO2Et, benzene, 90 °C, 80% (2 steps); (c) 1. DIBAL, CH2Cl2, -78 °C; 2. SO3•py, i-Pr2NEt, CH2Cl2-DMSO (3:2); 3. Ph3P=CHCO2Me, benzene, 90 °C, 89% (3 steps) (d) 1. TBAF, THF, 87%; 2. PBr3, Et2O, 77%; 3. (EtO)3P, 90 °C, 96%.
To gain insight into the chemical behavior of the anion generated from phosphonate 17, we first studied its protonation (Scheme 5). On quenching with 2,4,6-trimethylphenol, the anion generated from (2′E,4′E,6′E)-17 with LDA at -78 °C yielded a 55:25:14:6 mixture23 of the geometric isomers of 17. An NOE experiment conducted on this mixture, yielding the results shown for the structures in Scheme 5. Based on this NOE experiment, the geometric isomers obtained from the protonation were established as (2′E,4′E,6′E)-17 (55%), (2′E,4′E,6′Z)-17 (25%), (2′E,4′Z,6′E)-17 (14%), and (2′E,4′Z,6′Z)-17 (6%). This experiment suggested that the stereochemical integrity of (2′E,4′E,6′E)-17 would likely be lost during the subsequent Horner-Wadsworth-Emmons olefination.
Scheme 5.

Reagents: (a) LDA, THF, -78 °C, then 2,4,6-trimethyl-phenol. An arrow indicates a detected, or not detected, NOE.
In the event, aldehyde 12 smoothly reacted with the anion generated from (2′E,4′E,6′E)-17, to furnish the expected product as a 73:17:7:3 mixture (Scheme 6).23 Once again, based on NOE experiments, the stereochemistry was assigned as (2′E,4′E,6′E,8′E,10′E)-18 (73%), (2′E,4′Z,6′E,8′E,10′E)-18 (17%), (2′E,4′E,6′Z,8′E,10′E)-18 (7%), and (2′E,4′Z,6′Z,8′E,10′E)-18 (3%).
Scheme 6.

Reagents: (a) LDA, 12, THF, -78 °C to rt, 94%; (b) LiOH, THF-MeOH-H2O (4:1:1), quant.
Worth noting, in reference to the 1H NMR characteristics established for the four geometric isomers of 18, we examined the 1H NMR data reported for natural mycolactones A and B, thereby revealing that the sample of natural mycolactones contains a small amount (<5%) of a third geometric isomer whose spectroscopic characteristics match well those of the (2′E,4′Z,6′Z,8′E,10′E)-isomer of 18.
The pentaenoate system present in the product 18 was expected to be prone to cis/trans-isomerization. Indeed, on photolysis (acetone-d6, Tungsten lamp), the 73:17:7:3 mixture yielded a new mixture consisting of (2′E,4′E,6′E,8′E,10′E)-18 (36%), (2′E,4′Z,6′E,8′E,10′E)-18 (52%), (2′E,4′E,6′Z,8′E,10′E)-18 (4%), (2′E,4′Z,6′Z,8′E,10′E)-18 (5%), and two minor isomers (3% combined).23 The two minor isomers appeared to be (2′E,4′E,6′E,8′E,10′Z)-18 and (2′E,4′Z,6′E,8′E,10′Z)-18. The two major isomers were found to be readily interconvertible, and the 3:2 ratio appeared to represent the steady-state ratio for this system.4,14
The product methyl pentaenoates were found to chromatographically inseparable. However, upon hydrolysis to the corresponding acids, (2′E,4′E,6′E,8′E,10′E)-19 could be separated from (2′E,4′Z,6′E,8′E,10′E)-19 by silica gel column chromatography. Thus, it was possible to obtain the mycolactones A and B enriched with the C4’ Z geometrical isomer (vide infra).
To complete the total synthesis, 4 was coupled with 19 under Yamaguchi esterification conditions,24 to furnish the protected mycolactones 20 in excellent yield (Scheme 7). Interestingly, attempted esterification under the conditions of EDCI/DMAP or BOP/DMAP did not give the desired product 20.
Scheme 7.
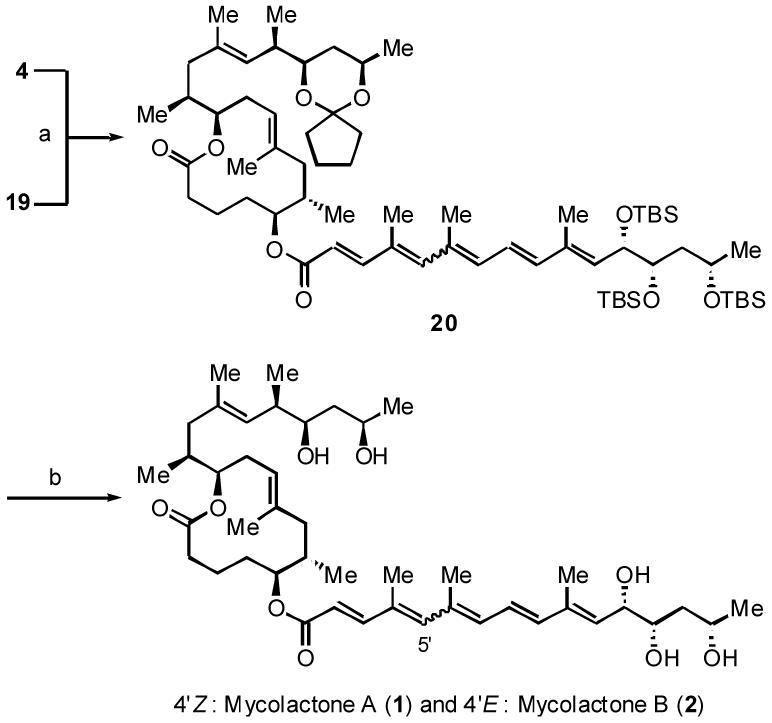
Reagents: (a) Cl3C6H2COCl, i-Pr2NEt, DMAP, benzene, 90%; (b) 1. TBAF, THF, 81%; 2. AcOH-H2O-THF (2:1:2), 67% with one recycle.
Attempted global-deprotection of 20 with HF•py in MeCN did give synthetic mycolactones A and B in 5-10% yield, but the product was accompanied by a complex mixture of side-products. 1H NMR analysis suggested that these by-products might be formed via sequential oxy-Michael additions of the alcohol moieties to the pentaenoate system. To suppress these putative side-reaction(s), stepwise deprotection was then tested. In the event, treatment with TBAF removed the three TBS groups, to furnish the corresponding triol in 81% yield. The cyclopentylidene ketal was then hydrolyzed with aqueous acetic acid in THF. Through extensive studies, the optimal ratio of AcOH:H2O:THF was found to be 2:1:2. However, even under the optimized conditions, by-product formation became significant when the reaction was allowed to go to completion. For this reason, the deprotection was quenched at approximately 60% completion, and the recovered triol was recycled. After one recycle, synthetic mycolactones A and B were isolated in 67% yield as an approximately 3:2 mixture of 4′-Z (mycolactone A) and 4′-On comparison of 1H NMR (Figure 2), 13C NMR (Table 1), and TLC [silica gel, CHCl3-MeOH-H2O (90:10:1)], the synthetic mycolactones A and B were found to be superimposable on the natural mycolactones A and B, respectively. Rigorously speaking, however, these comparisons could not eliminate the possibility that the synthetic mycolactones A and B might be the remote diastereomers25 of the natural products. In order to exclude this possibility, the 1H NMR characteristics in a chiral NMR solvent were studied. As shown in Graph 1, both the synthetic and natural mycolactones A and B exhibited the identical Δδ profile in (R)- and (S)-N,α-dimethylbenzylamines (DMBAs).6 Through these comparisons, we concluded that the synthetic mycolactones A and B are indeed identical to the natural mycolactones A and B. In addition, the synthetic mycolactones A and B exhibited biological properties identical to those of the natural products.26
Figure 2.
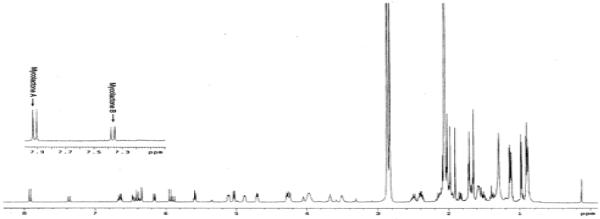
1H NMR spectrum (600 MHz, acetone-d6) of synthetic mycolactones A and B.
Table 1.
13C NMR chemical shifts observed for natural4 and synthetic mycolactones A and B (125 MHz, acetone-d6).
| Mycolactone A | Mycolactone B | |||
|---|---|---|---|---|
| Carbon | Natural | Synthetic | Natural | Synthetic |
| 1 | 173.3 | 173.3 | 173.3 | 173.3 |
| 2 | 35.9 | 35.9 | 35.9 | 35.9 |
| 3 | 20.8 | 20.7 | 20.8 | 20.7 |
| 4 | 31.4 | 31.5 | 31.4 | 31.5 |
| 5 | 79.3 | 79.3 | 79.3 | 79.3 |
| 6 | 32.8 | 32.7 | 32.8 | 32.7 |
| 7 | 46.4 | 46.3 | 46.4 | 46.3 |
| 8 | 137.2 | 137.3 | 137.2 | 137.3 |
| 9 | 123.8 | 123.8 | 123.8 | 123.8 |
| 10 | 29.3 | a | 29.3 | a |
| 11 | 76.3 | 76.2 | 76.3 | 76.2 |
| 12 | 35.4 | 35.3 | 35.4 | 35.3 |
| 13 | 44.3 | 44.3 | 44.3 | 44.3 |
| 14 | 133.9 | 133.4 | 133.9 | 133.4 |
| 15 | 131.2 | 131.2 | 131.2 | 131.2 |
| 16 | 40.5 | 40.4 | 40.5 | 40.4 |
| 17 | 76.9 | 76.9 | 76.9 | 76.9 |
| 18 | 43.8 | 43.8 | 43.8 | 43.8 |
| 19 | 68.9 | 68.9 | 68.9 | 68.9 |
| 20 | 24.6 | 24.6 | 24.6 | 24.6 |
| 21 | 20.5 | 20.5 | 20.5 | 20.5 |
| 22 | 15.9 | 15.9 | 15.9 | 15.9 |
| 23 | 15.0 | 14.9 | 15.0 | 14.9 |
| 24 | 16.2 | 16.2 | 16.2 | 16.2 |
| 25 | 17.1 | 17.1 | 17.1 | 17.1 |
| 1′ | 166.9 | 166.9 | 166.9 | 166.9 |
| 2′ | 119.6 | 119.6 | 117.4 | 117.4 |
| 3′ | 143.1 | 143.1 | 151.1 | 151.1 |
| 4′ | 132.1 | 132.1 | 133.2 | 133.2 |
| 5′ | 141.8 | 141.8 | 144.3 | 144.3 |
| 6′ | 134.7 | 134.8 | 135.3 | 135.3 |
| 7′ | 134.8 | 134.9 | 136.1 | 136.1 |
| 8′ | 125.1 | 125.1 | 125.1 | 125.1 |
| 9′ | 139.9 | 139.9 | 139.9 | 140.3 |
| 10′ | 137.2 | 137.2 | 137.2 | 137.2 |
| 11′ | 134.6 | 134.6 | 134.6 | 134.6 |
| 12′ | 72.4 | 72.3 | 72.4 | 72.3 |
| 13′ | 75.7 | 75.7 | 75.7 | 75.7 |
| 14′ | 41.9 | 41.8 | 41.9 | 41.8 |
| 15′ | 67.7 | 67.6 | 67.7 | 67.6 |
| 16′ | 24.2 | 24.2 | 24.2 | 24.2 |
| 17′ | 21.0 | 21.0 | 14.3 | 14.3 |
| 18′ | 17.6 | 17.6 | 17.1 | 17.1 |
| 19′ | 13.3 | 13.4 | 13.3 | 13.3 |
overlapped with the solvent signals.
Graph 1.

Δδ-Values of 1H NMR chemical shifts in (R)- and (S)-DMBAs-d13. Each graph shows the Δδ in chemical shift for natural and synthetic mycolactones A and B, with Δδ being [δ(R-DMBA) - Δ(S-DMBA)].
Second Generation Total Synthesis
Based on the knowledge accumulated through our first generation total synthesis of the mycolactones, we recognized that improvements could be made in two major areas. First, the acid-promoted deprotection of the cyclopentylidene ketal proved to be more problematic than originally anticipated. In contrast, mycolactones A and B appeared to be stable to TBAF-promoted TBS-deprotection conditions. These observations immediately suggested that the cyclopentylidene protecting group present in the C17/C19-diol 20 should be replaced by TBS ethers. Thus, TBAF-promoted desilylation should allow us to accomplish a more efficient global deprotection.
We were interested in demonstrating the effectiveness of this approach experimentally. For this purpose, we utilized the cyclopentylidene protected core 4 employed in the first synthesis. Thus, the protecting groups of 4 were adjusted in 4 steps to obtain the requisite TBS-protected core 21 (Scheme 8). Yamaguchi esterification of 21 with 19 proceeded to pentakis-TBS protected mycolactone. As anticipated, global deprotection conditions (TBAF, THF, rt) furnished mycolactones A and B in excellent yield.
As was previously mentioned, we decided to utilize the core triol 4 for our first generation total synthesis of the mycolactones, since 4 was already in hand from our previous work on the stereochemistry assignment. However, having identified suitable protecting groups for the alcohols at C17 and C19, we were in a position to implement specific improvements for the synthesis of the mycolactone core; in particular, we were anxious to develop a shorter synthetic route with a greater degree of selectivity and efficiency.
Having demonstrated the feasibility of global TBAF-promoted TBS-deprotection, we wished to implement the TBS-protecting groups from the very beginning of synthesis, thereby improving the overall efficiency of the synthesis (Scheme 9). Thus, olefin 23 was prepared from the known aldehyde 2220 via Brown crotylboration27 and subsequent protection of the alcohol as the TBS-silylether. Olefin 23 was then oxidatively cleaved, followed by treatment with dimethyl (diazomethyl)phosphonate (DAMP) and t-BuOK,28 to provide the terminal alkyne 24 in good yield. The alkyne was then methylated using n-BuLi and MeI. Hydrozirconation,29 followed by iodine quench, furnished the bis-TBS protected vinyl iodide 25.
Scheme 9.

Reagents: (a) 1. Z-2-butene, t-BuOK, n-BuLi, (-)-Ipc2BOMe, BF3•OEt2, THF, -78 °C, then H2O2, NaOH, 78%; 2. TBSCl, imidazole, DMF, quant.; (b) 1. O3, CH2Cl2, -78 °C, then Ph3P, 94%: 2. t-BuOK, DAMP, THF, 84%; (c) 1. n-BuLi, MeI, THF, -78 °C→rt, 93%; 2. Cp2ZrHCl, THF, 50 °C, then I2, THF, 65%.
In the interest of the overall efficiency of the synthesis, we sought to protect the alcohol at C5 with a protecting group orthogonal to the TBS-group, and therefore employed a p-methoxybenzyl (PMB) group. Thus, the C1-C7 alkyliodide with the carboxylate group at C1 was synthesized from aldehyde 26 using Brown crotylboration27,30 to install the C5 and C6 stereocenters (Scheme 10).
Scheme 10.

Reagents: (a) Z-2-butene, t-BuOK, n-BuLi, (+)-Ipc2BOMe, BF3•OEt2, THF, -78 °C, then H2O2, NaOH, 80%; (b) 1. NaH, PMBBr, DMF, 89%; 2. TBAF, THF, 96%; 3. SO3•py, i-Pr2NEt, CH2Cl2-DMSO (3:2), 94%; 4. NaClO2, NaH2PO4, 2-methyl-2-butene, t-BuOH/H2O, 5. MeI, DBU, CH3CN, 84% for 2 steps; (c) 1. OsO4, NMO, acetone-H2O (1:1), 80%; 2. Pb(OAc)4, benzene; 3. NaBH4, MeOH, 0 °C, 83% (2 steps); (d) Ph3P, imidazole, I2, CH2Cl2, 92%.
The synthesis of the C8-C13 building block is outlined in Scheme 11. Using the procedure reported by Seebach, (S)-diethyl malate (31) was alkylated with MeI and LDA in THF, to give alcohol 32, along with its syn diastereomer (stereoselectivity=8:1).31 Reduction of 32 using LiAlH4 provided the known triol 3332 in excellent yield. The trio l 33 was selectively protected to give TBS ether 34 via a dibutyltin ketal intermediate.33 The resulting diol 34 was first converted to epoxide 35 and then coupled with propyne under Yamaguchi conditions.34 Cleavage of the TBS ether and subsequent protection of the resulting 1,3-diol gave cyclopentylidene ketal 36. The alkyne moiety of 36 was then converted to vinyliodide 37 by hydrozirconation, followed by iodine quench. The vinyl iodide thus obtained was found to be identical to a sample synthesized by the previous route.5
Scheme 11.

Reagents: (a) LDA, MeI, THF, -78 °C, 80%, d.r. = 8:1; (b) LiAlH4, THF, reflux; (c) 1. n-Bu2SnO, MeOH, reflux; 2. TBSCl, CHCl3, 70% for 3 steps; (d) 1-(p-toluenesulfonyl)imidazole, NaH, THF, 88%; e) 1. n-BuLi, propyne, BF3•OEt2, THF, -78 °C, 96%; 2. TBAF, THF; 3. cyclopentanone, p-TsOH, benzene, 76%; (f) Cp2ZrHCl, THF, 50 °C, then I2, 62%.
With the three building blocks 25, 30, and 37 in hand, we next focused on the coupling reactions. Negishi coupling35 was used to form the C7-C8 bond, i.e., 30 + 37 → 38. Considering the presence of an electrophilic ester group in 30, we chose to prepare the alkylzinc iodide species via zinc insertion by an active Zn-Cu couple,36 instead of via transmetallation from Li to Zn. Additionally, Pd(PPh3)4 (10 mol%) was found the most effective catalyst for this case. In the event, the coupling of 30 (1.4 equiv.) and 37 proceeded smoothly in the presence of 6-8 equivalents of LiCl in N-methylpyrrolidinone (NMP),37 to furnish 38 in 83% yield (Scheme 12).
Scheme 12.

Reagents: (a) Zn, Cu(OAc)2, Pd(PPh3)4, LiCl, NMP, 60 °C 83%; (b) 1. CH2Cl2-H2O-TFA (16:4:1), 90%; 2. TIPSCl, imidazole, DMF, quant.; 3. LiOH, 4:1:1 THF/MeOH/H2O, 81%; (c) Cl3C6H2COCl, i-Pr2NEt, benzene, then DMAP, benzene 96%; (d) 1. HF•py-py-CH3CN, 90% 2. Ph3P, imidazole, I2, CH2Cl2, 98%; (e) Zn, Cu(OAc)2, 25, Pd(PPh3)4, LiCl, NMP, 60 °C 80%; (f) DDQ, CH2Cl2/H2O, 91%.
The coupled product 38 was transformed to the seco-acid 39 in better than 70% overall yield for 3 steps: (1) acid-promoted deprotection of the cyclopentylidene group, (2) selective protection of the resultant primary alcohol as the triisopropylsilyl (TIPS) ether, and (3) base-promoted hydrolysis of the methyl ester. Yamaguchi macrolactonization of 39 then furnished the desired macrolactone 40 in 96% yield. It is worthwhile noting that the lactone was designed to serve as a protecting group for the C11 alcohol during the following steps.
Deprotection of the silyl ether in 40, followed by iodination, gave alkyl iodide 41. Under the Negishi coupling conditions optimized for the case of the coupling of 30 with 37, alkyl iodide 41 was coupled with 25 (1.5 equiv.), to give 42 in 80% yield. The PMB group was then cleaved with DDQ, to furnish the bis-TBS protected core 21.
As demonstrated in Scheme 8, 21 was coupled with 19 and then subjected to the tetra-n-butylammonium fluoride (TBAF) promoted global deprotection, to furnish a 3:2 mixture of the mycolactones A and B in 72% overall yield. Upon comparison of spectroscopic data and TLC, the mycolactones A and B thus synthesized were found to be identical to the synthetic mycolactones A and B obtained via the first route as well as to the authentic natural products.
As was previously discussed, the pentaenoate system present in the mycolactones is readily prone to cis/trans-isomerization. This isomerization appeared to be facile under the acidic conditions used for deprotection of the cyclopentylidene ketal in the first generation total synthesis (Scheme 7). Interestingly, under the conditions of TBAF-promoted global deprotection used in the second generation total synthesis (Scheme 12), the cis/trans-isomerization seemed to be controllably slow in the dark, thereby suggesting the possibility that either mycolactone A free of mycolactone B, or mycolactone B free of mycolactone A, could be obtained, if pure Z-Δ4’,5’ or E-Δ4’,5’ pentaenoic acid was used. In order to test this possibility, a 10:1 mixture of Z-Δ4’,5’ and E-Δ4’,5’ pentaenoic acids, obtained by silica gel column chromatography (vide ante), was coupled with 21 and then subjected to TBAF-deprotection in the dark, to furnish a 6:1 mixture of mycolactone A and mycolactone B (Figure 3). Apparently, during this synthetic operation, the stereochemical integrity of the pentaenoic acid was compromised to some extent. We would attribute, at least partly, the observed cis/trans-isomerization to the exposure of these substrates to light. If this is the case, this experiment indicates the possibility of obtaining pure mycolactone A and/or B through this route. In this connection, we specifically reference the recent work by Negishi and co-workers, disclosing a stereoselective synthesis of both protected (2′E,4′E,6′E,8′E,10′E)- and (2′E,4′Z,6′E,8′E,10′E)-pentaenoic acids.17,38
Figure 3.

1H NMR spectrum (500 MHz, acetone-d6) of the synthetic mycolactones enriched with mycolactone A.
3. Conclusion
Mycolactones A and B have been synthesized through two different routes. Our first generation total synthesis unambiguously confirmed the relative and absolute stereochemistry predicted via an NMR database approach. Knowledge regarding the chemical properties of the mycolactones accumulated through the first generation total synthesis allowed us to implement several improvements to the original synthesis, including: (1) optimizing the choice of protecting groups, (2) eliminating the unnecessary adjustment of protecting groups, and (3) improving the overall stereoselectivity and synthetic efficiency. The second generation of total synthesis consists of 21 steps in the longest linear sequence, with 8.8% overall yield.
As was previously noted, several mycolactone congeners have been isolated from various clinical isolates of Mycobacterium ulcerans and also from the frog pathogen Mycobacterium marinum and the fish pathogen Mycobacterium liflandii. Structurally, all of these congeners appear to contain the same core structure of mycolactones A and B, but different unsaturated side-chains. However, given the fact that these congeners were obtained in very minute amounts, an unambiguous structural determination is challenging. In this connection, we should specifically emphasize that our second generation total synthesis offers an appealing opportunity to study their structure as well as their biological profile, particularly because of its overall efficiency and flexibility. Indeed, the effectiveness of this approach has recently been demonstrated in the case of mycolactone C.39
4. Experimental Section
General Procedures and Methods
NMR spectra were recorded on Varian Inova spectrometers (400, 500, and 600 MHz). Chemical shifts are reported in parts per million (ppm) and coupling constants in Hz. For 1H and 13C spectra, the central residual solvent peak (methanol, acetone, benzene) was used as the internal reference (3.30, 2.05, 7.15 ppm, and 49.0, 29.8, 128.0 ppm respectively). Analytical thin layer chromatography (TLC) was performed with E. Merck pre-coated TLC plates, silica gel 60F-254, layer thickness 0.25 mm. Flash chromatography separations were performed on E. Merck kieselgel 60 (230-400 mesh) silica gel. Reagents and solvents are commercial grade and were used as supplied, with the following exceptions: Benzene, ether, and THF were distilled from sodium benzophenone ketyl. Dichloromethane was distilled from calcium hydride, and toluene was distilled from sodium. All reactions were conducted under an argon atmosphere unless otherwise noted. Reaction vessels and apparatus were flame-dried or oven-dried and allowed to cool under an inert atmosphere.
First Generation of Total Synthesis
Experimental details outlined in Schemes 1-7 are given in the Supporting Information of references 5, 7, and 8.
Second Generation of Total Synthesis
Synthesis outlined in Scheme 8
To a stirred solution of alcohol 4 (23.2 mg, 0.047 mmol) in CH2Cl2 (3.0 mL) at 0 °C was added i-Pr2NEt (25 μL, 0.142 mmol), DMAP (11.5 mg, 0.095 mmol), and methoxyacetyl chloride (13 μL, 0.142 mmol). The resulting solution was stirred at 0 °C for 1 h, then diluted with CH2Cl2 (10 mL) and water (10 mL). The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo. Flash chromatography (4:1 hexanes/EtOAc) provided the methoxyacetate (24.2 mg, 91%).
Dichloromethane saturated with aqueous TFA was prepared by shaking CH2Cl2, TFA (8 mL), and H2O (2 mL) in a separatory funnel. The layers were allowed to separate for 30 seconds, and the organic layer was used. The organic layer (5 mL) was added to the above ester (57.2 mg, 0.102 mmo l), and the resulting solution stirred for 3.5 h. CH2Cl2 (10 mL) and saturated aqueous sodium bicarbonate (15 mL) were then added. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were washed with water (30 mL) and brine (30 mL), dried over Na2SO4, and concentrated in vacuo. Flash chromatography (4:1 hexanes/EtOAc, then 2:1 EtOAc/hexanes) provided the corresponding diol (46.0 mg, 92%).
To a stirred solution of the above diol (46.0 mg, 0.093 mmol) in CH2Cl2 (6.0 mL) at 0 °C were added 2,6-lutidine (44 μL, 0.372 mmol) and TBSOTf (87 μL, 0.372 mmol). The resulting solution was stirred for 90 min, then saturated aqueous ammonium chloride (10 mL) was added. The layers were separated, and the aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with water and brine, dried over Na2SO4, and concentrated in vacuo. Flash chromatography (4:1 hexanes/EtOAc) afforded the methoxyacetate (67.1 mg, quant.).
To a stirred solution of the methoxyacetate (70.0 mg, 0.096 mmol) in MeOH (7.0 mL) at 0 °C was added K2CO3 (35.0 mg, 0.25 mmol). The resulting solution was stirred at 0 °C for 2h, then at room temperature for 5h. The reaction mixture was the diluted with CH2Cl2 (15 mL) and saturated aqueous ammonium chloride (15 mL). The layers were separated, and the aqueous layer extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with brine (2 × 30 mL), dried over Na2SO4, and concentrated in vacuo. Flash chromatography (8:1 hexanes/EtOAc) provided 21 (52.9 mg, 84%). 1H NMR (CD3COCD3, 500 MHz,) δ 5.23 (d, J = 9.0, 1H), 5.04 (d, J = 11.5, 1H), 4.91 (ddd, J = 12.5, 5.5, 2.5, 1H), 3.98 (q, J = 6.5, 1H), 3.74 (td J = 5.5, 4.5, 1H), 3.36 (m, 1H), 2.59 (m, 1H), 2.49 (dt, J = 14.0, 11.5, 1H), 2.31 (ddd, J = 13.5, 8.0, 3.0, 1H), 2.08-2.18 (m, 3H), 1.91 (m, 3H), 1.76 (dd, J = 13.0, 10.0, 1H), 1.65 (s, 6H), 1.42-1.62 (m, 7H), 1.16 (d, J = 6.0, 3H), 0.96 (d, J = 6.5, 3H), 0.92 (s, 9H), 0.91 (m, 3H), 0.90 (s, 9H), 0.87 (d, J = 6.5, 3H), 0.091 (s, 3H), 0.086 (s, 3H), 0.08 (s, 6H).
Mycolactones A and B via penta-TBS protection (1 and 2)
To a stirred solution of pentaene acid 19 (89.6 mg, 0.132 mmol) in benzene (1 mL) were added i-Pr2NEt (92 μL, 0.53 mmol), Cl3C6H2COCl (42.6 μL, 0.26 mmol), and DMAP (80.8 mg, 0.66 mmol). Alcohol 21 (43.1 mg, 0.066 mmol) was then added in benzene (1.5 mL, 0.5 mL wash). The resulting solution was stirred for 20h. Benzene (5 mL) and saturated aqueous sodium bicarbonate (5 mL) were then added. The layers were separated, and the aqueous layer was extracted with benzene (3 × 5 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, and concentrated in vacuo. Flash chromatography (20:1 hexanes/EtOAc, followed by 4:1 hexanes/EtOAc) gave the protected mycolactones (78.0 mg, 90%). This protected mycolactones exist as a 2:3 mixture of Z:E isomers at Δ4′,5′ (mycolactone numbering). 1H NMR (CD3COCD3, 600 MHz, Z-Δ4′,5′ isomer) δ 7.93 (d, J = 15.6, 1H), 6.66 (dd, J = 15.0, 11.4, 1H), 6.44 (d, J = 15.0, 1H), 6.34 (s, 1H), 6.19 (d, J = 11.4, 1H), 5.94 (d, J = 15.6, 1H), 5.66 (d, J = 9.0, 1H), 5.23 (d, J = 9.0, 1H), 5.15 (br d, J = 10.2, 1H), 4.88 (m, 1H), 4.71 (sextet, J = 4.2, 1H), 4.58 (dd, J = 9.0, 3.6, 1H), 3.96-4.06 (m, 2H), 3.76-3.80 (m, 1H), 3.72-3.76 (m, 1H), 2.55-2.62 (m, 1H), 2.47-2.55 (m, 1H), 2.37-2.45 (m, 1H), 2.14-2.19 (m, 1H), 2.08-2.13 (m, 1H), 2.09 (s, 3H), 1.92-2.05 (m, 5H), 1.98 (s, 3H), 1.95 (s, 3H), 1.84-1.92 (m, 2H), 1.76-1.82 (m, 1H), 1.71 (s, 3H), 1.56-1.72 (m, 6H), 1.66 (s, 3H), 1.17 (d, J = 6.0 Hz, 3H), 1.16 (d, J = 6.6 Hz, 3H), 0.92 (s, 9H), 0.91 (s, 9H), 0.86-0.91 (overlapped 3 singlets and 3 doublets, 36H), 0.08-0.10 (overlapped 8 singlets, 24H), 0.04 (s, 3H), 0.03 (s, 3H). 1H NMR (CD3COCD3, 600 MHz, E-Δ4′,5′ isomer) δ 7.37 (d, J = 15.0 Hz, 1H), 6.68 (dd, J = 15.0, 11.4 Hz, 1H), 6.49 (d, J = 15.0, 1H), 6.48 (s, 1H), 6.40 (d, J = 11.4 Hz, 1H), 5.89 (d, J = 15.0 Hz, 1H), 5.67 (d, J = 9.0 Hz, 1H), 5.23 (d, J = 9.0, 1H), 5.15 (br d, J = 10.2, 1H), 4.88 (m, 1H), 4.71 (sextet, J = 4.2, 1H), 4.58 (dd, J = 9.0, 3.6, 1H), 3.96-4.06 (m, 2H), 3.76-3.80 (m, 1H), 3.72-3.76 (m, 1H), 2.55-2.62 (m, 1H), 2.47-2.55 (m, 1H), 2.37-2.45 (m, 1H), 2.14-2.19 (m, 1H), 2.08-2.13 (m, 1H), 2.09 (s, 3H), 1.92-2.05 (m, 5H), 1.98 (s, 3H), 1.94 (s, 3H), 1.84-1.92 (m, 2H), 1.76-1.82 (m, 1H), 1.72 (s, 3H), 1.56-1.72 (m, 6H), 1.66 (s, 3H), 1.17 (d, J = 6.0 Hz, 3H), 1.16 (d, J = 6.6 Hz, 3H), 0.92 (s, 9H), 0.91 (s, 9H), 0.86-0.91 (overlapped 3 singlets and 3 doublets, 36H), 0.08-0.10 (overlapped 8 singlets, 24H), 0.04 (s, 3H), 0.03 (s, 3H). MS (ES) m/z 1332 (M + NH4+).
To a stirred solution of the pentakis-TBS protected mycolactone precursor (6.6 mg, 5.03 μmol) in THF (1 mL) was added TBAF (1 M solution in THF, 75 μL, 75 μmol). The solution was stirred at room temperature for 8 h. The reaction mixture was concentrated. Flash chromatography (100:1 EtOAc/MeOH, followed by 90:10:1 CHCl3/MeOH/H2O) gave mycolactones (3.0 mg, 80%).
Synthesis outlined in Scheme 9
Preparation of vinyl iodide 25
To a solution of t-BuOK (6.56 g, 58.5 mmol) in THF (400 mL) at -78 °C was added Z-2-butene (8.2 g, 146 mmol) followed by n-BuLi in hexanes (2.16 M, 27.1 mL, 58.5 mmol). The bright yellow suspension was stirred at -78 °C for 5 min, -45 °C for 10 min, and then -78 °C for 15 min. A solution of (-)-Ipc2BOMe (21.58 g, 68.2 mmol) in Et2O (50 mL) was then added via cannula. The colorless solution was allowed to stir at -78 °C for 30 min, and then BF3•Et2O (9.5 mL, 75 mmol) was added followed immediately by a solution of the known aldehyde 2220 (9.86 g, 48.7 mmol) in THF (50 mL, 10 mL wash). The solution was stirred at -78 °C for 3 h and then quenched by addition of 3 N NaOH (130 mL). H2O2 (30%, 65 mL) was then added carefully with stirring, and the resulting mixture was stirred vigorously at room temperature for 12 h. The mixture was then diluted with EtOAc and washed with H2O, and brine. The organic phase was then dried over anhydrous MgSO4 and concentrated in vacuo. Flash chromatography (39:5 hexanes 1/EtOAc) gave alcohol (9.87 g, 78% yield). H NMR (CDCl3, 400 MHz) δ 5.78 (ddd, J = 17.6, 10.4, 7.6, 1H), 5.02 (br d, J = 17.6, 1H), 5.01 (br d, J = 10.4, 1H), 4.03 (ddq, J = 9.6, 4.0, 5.6, 1H), 3.60 (dddd, J = 9.6, 5.6, 2.0, 1.2, 1H), 3.41 (d, J = 1.2, OH), 2.21 (apparent sextet, J = 7.0, 1H), 1.59 (ddd, J = 14.4, 4.0, 2.0, 1H), 1.43 (dd, J = 14.4, 9.6, 1H), 1.16 (d, J = 6.0, 1H), 1.01 (d, J = 6.8, 1H), 0.88 (s, 9H), 0.10 (s, 3H), 0.08 (s, 3H); 13C NMR (CDCl3, 400 MHz) d 141.0, 114.6, 74.6, 70.3, 43.7, 42.6, 25.8 (3C), 24.5, 17.8, 14.9, -4.0, -4.8.
To a solution of the alcohol (5.069 g, 19.65 mmol) in DMF (100 mL) were added imidazole. (3.345 g, 49.13 mmol), and TBSCl (5.924 g, 39.30 mmol). The solution was stirred for 15 h. Then water was added and the mixture was extracted with Et2O twice. The combined extracts were washed with H2O, brine, dried over anhydrous MgSO4 and concentrated in vacuo. Flash chromatography (49:1 hexanes/EtOAc) gave the silylether 23 (7.318 g, 19.67 mmol, quant.). 1H NMR (CDCl3, 500 MHz) d 5.90 (ddd, J = 17.5, 10.5, 6.5, 1H), 5.00 (br d, J = 10.5, 1H), 4.99 (br d, J = 17.5, 1H), 3.90 (apparent sextet J = 6.5, 1H), 3.68 (dddd, J = 6.5, 6.0, 4.0, 1H), 2.29-2.36 (m, 1H),1.60 (ddd, J = 13.5, 6.5, 6.5, 1H), 1.48 (ddd, J = 13.5, 6.5, 6.5, 1H), 1.13 (d, J = 6.5, 1H), 0.94 (d, J = 7.0, 1H), 0.89 (s, 9H), 0.88 (s, 9H), 0.05 (s, 3H), 0.044 (s, 3H), 0.039 (s, 3H), 0.035 (s, 3H); 13C NMR (CDCl3, 400 MHz) d 141.4, 113.8, 72.9, 76.9, 43.8, 42.2, 25.9 (6C), 23.7, 18.11, 18.08, 13.7, -4.31, -4.33, -4.4, -4.7.
A solution of 23 (7.318 g, 19.67 mmol) at -78 °C in CH2Cl2 (190 mL) was saturated with ozone until a blue color persisted. The solution was then purged with argon until the blue color dissipated, and Ph3P (5.419 g, 20.66 mmol) was added. The solution was 3 warmed to room temperature and stirred for 12 h, then concentrated in vacuo. Flash chromatography (39:1 hexanes/EtOAc) gave the aldehyde (6.910 g, 94% yield) as a colorless oil. 1H NMR (CDCl3, 500 MHz) δ 9.74 (br s, 1H), 4.37 (ddd, J = 8.4, 6.0, 3.0, 1H), 3.83 (apparent sextet, J = 6.5, 1H), 2.48 (dq, J = 3.0, 6.0, 1H), 1.59-1.71 (m, 2H), 1.16 (d, J = 6.0, 3H), 1.05 (d, J = 6.5, 3H), 0.89 (s, 9H), 0.85 (s, 9H), 0.07 (s, 3H), 0.06 (s, 3H), 0.04 (s, 3H), 0.02 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 205.0, 68.7, 65.6, 50.5, 44.2, 25.8 (3C), 25.7 (3C), 24.0, 18.0, 17.9, 6.9, -4.1, -4.2, -4.7, -4.8.
To a solution of DAMP (2.451g, 16.34 mmol) in THF (90 mL) at -78 °C was added t-BuOK (95%, 1.930 g, 16.34 mmol). To the resulting yellow solution was added a solution of the aldehyde (6.112 g, 16.34 mmol) in THF (20 mL, 5 mL wash), and the resulting solution was stirred at -78 °C for 1 h and then warmed to 0 °C for 1 h. The mixture was then diluted with saturated aqueous NaHCO3 and EtOAc. The organic phase was washed with saturated aqueous NaHCO3, dried over MgSO4 and concentrated in vacuo . Flash chromatography (40:1 hexanes/EtOAc) gave the alkyne 24 (5.083 g, 84% yield). 1H NMR (CDCl, 500 MHz) δ 3.98 (apparent sextet, J = 6.5, 1H), 3.72 (apparent q, J = 5.5, 1H), 2.56-2.64 (m, 1H), 2.20 (d, J = 2.5, 1H), 1.83 (ddd, J = 13.0, 6.5, 5.5, 1H), 1.56 (ddd, J = 13.0, 6.5, 5.5, 1H), 1.13(d, J = 5.5, 3H), 1.12 (d, J = 6.5, 3H), 0.90 (s, 9H), 0.89 (s, 9H), 0.09 (s, 3H), 0.06 (three overlapped singlets, 9H).
To a solution of the alkyne 24 (3.653 g, 9.87 mmol) in THF (100 mL) at -78 °C were added n-BuLi (2.38 M in hexanes, 4.98 mL, 11.84 mmol) and MeI (1.0 mL, 15.99 mmol). The solution was stirred and warmed to room temperature for 1 h. The solution was then diluted with saturated aqueous NaHCO3 and EtOAc. The organic phase was washed with saturated aqueous NaHCO3, brine, dried over MgSO4 and concentrated in vacuo. Flash chromatography (20:1 hexanes/EtOAc) gave the methyl alkyne (3.541 g, 93% yield). [α]D23 +10.8 (c 1.28, CHCl3). 1H NMR (CDCl3, 500 MHz) δ 3.98 (apparent sextet, J = 6.0, 1H), 3.68 (apparent q, J = 6.0, 1H), 2.46-2.54 (m, 1H), 1.82 (ddd, J = 13.5, 6.0, 6.0, 1H), 1.77 (d, J = 2.5, 3H), 1.54 (ddd, J = 13.5, 6.0, 6.0, 1H), 1.14 (d, J = 6.0, 3H), 1.07 (D, J = 6.0, 3H), 0.90 (overlapped 2 singlets, 18H), 0.08 (s, 3H), 0.07 (s, 3H), 0.061 (s, 3H), 0.057 (s, 3H). HRMS (ES) 385.2954 (M + H+), calcd. 385.2958.
A solution of the methyl alkyne (1.627 g, 4.24 mmol) in THF (10.5 mL) was added to Cp2ZrHCl (2.189 g, 8.49 mmol) via cannula. The mixture was protected from light and stirred at 50 °C for 2 h. The resulting dark red suspension was cooled to room temperature. A solution of I2 (2.150 g, 8.47 mmol) in THF (7 mL) was added via cannula. The dark brown mixture was stirred at room temperature for 30 min and then poured into a 1:1 mixture of saturated aqueous Na2S2O3 and saturated aqueous NaHCO3. The mixture was diluted with EtOAc and the organic phase was separated and washed with saturated aqueous Na2S2O3, saturated aqueous NaHCO3 and brine. The organic solution was dried over anhydrous MgSO4 and concentrated in vacuo. Flash chromatography (100:1 hexanes/EtOAc) gave the vinyl iodide 25 (1.413 g, 65% yield). 1H NMR (CDCl3, 500 MHz) δ 6.13 (br d, J = 9.5, 1H), 3.87 (apparent sextet, J = 6.0, 1H), 3.66 (ddd, J = 7.0, 5.5, 4.0, 1H), 2.52 (ddq, J = 9.5, 4.0, 6.5, 1H), 2.38 (d, J = 1.5, 3H), 1.53-1.64 (m, 2H), 1.13 (d, J = 6.0, 3H), 0.91 (d, J = 7.0, 3H), 0.89 (s, 9H), 0.88 (s, 9H), 0.056 (s, 3H), 0.054 (s, 3H), 0.04 (two overlapped singlets, 6H); 13C NMR (CDCl3, 125 MHz) δ 145.0, 93.1, 71.9, 65.8, 44.9, 40.3, 27.9, 25.90 (3C), 25.86 (3C), 24.1, 18.1 (2C), 14.1, -4.20 (2C), -4.6, -4.7.
Synthesis outlined in Scheme 10
To a solution of t-BuOK (6.56 g, 58.5 mmol) in THF (400 mL) at -78 °C was added Z-2-butene (8.2 g, 146 mmol) followed by n-BuLi in hexanes (2.16 M, 27.1 mL, 58.5 mmol). The bright yellow suspension was stirred at -78 °C for 5 min, -45 °C for 10 min, and then -78 °C for 15 min. A solution of (+)-Ipc2 BOMe (21.58 g, 68.2 mmol) in Et2O (50 mL) was then added via cannula. The colorless solution was allowed to stir at -78 °C for 30 min, and then BF3•Et2O (9.5 mL, 75 mmol) was added followed immediately by a solution of aldehyde 26 (9.86 g, 45.6 mmol) in THF (50 mL, 10 mL wash). The solution was stirred at -78 °C for 3 h and then quenched by addition of 3 N NaOH (130 mL). Then, H2O2 (30%, 65 mL) was added carefully with stirring, and the resulting mixture was stirred vigorously at room temperature for 12 h. The mixture was then diluted with EtOAc (1 L) and washed with H O (500 mL), and brine (500 mL). The organic phase was then dried over anhydrous MgSO4, concentrated in vacuo. Flash chromatography (39:1 hexanes/EtOAc) gave alcohol 27 (9.87 g, 80% yield) as colorless oil. 1H NMR (CDCl3, 400 MHz) δ 5.79 (m, 1H), 5.05-5.11 (m, 2H), 3.61 (t, J = 5.6, 2H), 3.49 (m, 1H), 2.27 (apparent sextet, J = 6.8, 1H), 1.46-1.58 (m, 4H), 1.33-1.41 (m, 2H), 1.02 (d, J = 6.8, 3H), 0.89 (s, 9H), 0.04 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 141.3, 115.4, 74.9, 63.4, 43.7, 33.9, 33.0, 26.2 (3C), 22.6, 18.6, 14.3, -5.0 (2C).
To a solution of alcohol 27 (2.985 g, 10.97 mmol) in DMF (80 mL) at 0 °C was added NaH (1.317 g, 60%, 32.92 mmo l). The mixture was stirred at 0 °C for 30 min. Then p-methoxybenzyl bromide (3.308 g, 16.46 mmol) was added and the mixture was stirred at room temperature for 14 h. The mixture was diluted with EtOAc and washed with saturated aqueous NH4Cl. The aqueous phase was extracted with EtOAc twice. The combined organic phase was washed with H2O, brine, dried over anhydrous MgSO4 and concentrated in vacuo. Flash chromatography (20:1 hexanes/EtOAc) gave the PMB ether (3.818 g, 89% yield). [α]D23 -32.9 (c 1.70, CHCl3). 1H NMR (CDCl, 400 MHz) δ 7.27 (d, J = 8.4, 2H), 6.87 (d, J = 8.4, 2H), 5.85 (ddd, J = 17.2, 10.4, 7.2, 1H), 5.04 (br d, J = 17.2, 1H), 5.01 (br d, J = 10.4, 1H), 4.49 (d, J = 10.8, 1H), 4.44 (d, J = 10.8, 1H), 3.80 (s, 3H), 3.60 (t, J = 6.4, 2H), 3.21-3.27 (m, 1H), 2.47 (apparent sextet, J = 7.2, 1H), 1.46-1.55 (m, 4H), 1.28-1.38 (m, 2H), 1.04 (d, J = 7.2, 3H), 0.90 (s, 9H), 0.05 (s, 6H); 13C NMR (CDCl3, 100 MHz) δ 159.0, 141.1, 131.1, 129.3 (2C), 114.2, 113.7 (2C), 82.6, 71.4, 63.2, 55.2, 40.8, 33.0, 30.9, 26.0 (3H), 21.9, 18.3, 15.6, -5.3 (2C). MS (ES) 394 (M + H+).
To a solution of the PMB ether (3.019 g, 7.70 mmol) in THF (70 mL) was added TBAF (1M in THF, 11.5 mL, 11.5 mmo l). The solution was stirred at room temperature for 1.5 h. The solution was concentrated in vacuo. Flash chromatography (2:1 hexanes/EtOAc) gave the alcohol (2.045 g, 96% yield). [α]D23 -43.8 (c 0.55, CHCl3). 1H NMR (CDCl3, 500 MHz) δ 7.27 (d, J = 8.5, 2H), 6.87 (d, J = 8.5, 2H), 5.85 (ddd, J = 17.5, 11.0, 8.0, 1H), 5.05 (br d, J = 17.5, 1H), 5.02 (br d, J = 11.0, 1H), 4.50 (d, J = 11.0, 1H), 4.43 (d, J = 11.0, 1H), 3.80 (s, 3H), 3.61 (t, J = 6.0, 2H), 3.22-3.27 (m, 1H), 2.49 (apparent sextet, J = 6.5, 1H), 1.44-1.58 (m, 4H), 1.32-1.42 (m, 2H), 1.04 (d, J = 6.5, 3H); 13C NMR (CDCl3, 100 MHz) δ 159.1, 140.9, 131.0, 129.4 (2C), 114.3, 113.7 (2C), 82.5, 71.5, 62.9, 55.3, 40.7, 32.8, 30.7, 21.7, 15.6. MS (ES) m/z 279 (M + H+).
To a solution of the alcohol (1.539 g, 5.53 mmol) in CH2Cl2 (78 mL) and DMSO (39 mL) were added i-PrN2Et (6.3 mL, 36.17 mmol) and sulfur trioxide pyridine complex (3.940 g, 24.75 mmol). The solution was stirred at room temperature for 1 h. The solution was diluted with Et2O and washed with water, brine, dried over MgSO4 and concentrated in vacuo. Flash chromatography (4:1 hexanes/EtOAc) gave the aldehyde (1.436 g, 94% yield). 1H NMR (CDCl3, 500 MHz) δ 9.73 (d, J = 2.0, 1H), 7.27 (d, J = 8.5, 2H), 6.87 (d, J = 8.5, 2H), 5.83 (ddd, J = 17.5, 10.5, 6.7, 1H), 5.04 (br d, J = 17.5, 1H), 5.03 (br d, J = 10.5, 1H), 4.52 (d, J = 11.0, 1H), 4.42 (d, J = 11.0, 1H), 3.80 (s, 3H), 3.25 (ddd, J = 8.7, 6.7, 4.7, 1H), 2.51 (apparent sextet, J = 6.7, 1H), 2.39(t, J = 7.5, 2H), 1.74-1.84 (m, 1H), 1.58-1.68 (m, 1H), 1.42-1.55 (m, 2H), 1.04 (d, J = 6.7, 3H); 13C NMR (CDCl3, 125 MHz) δ 202.6, 159.1, 140.5, 130.8, 129.4 (2C), 114.6, 113.7 (2C), 82.1, 71.4, 55.2, 43.9, 40.5, 30.3, 18.2, 15.7. MS (ES) m/z 277 (M + H+).
To a solution of the aldehyde (1.044 g, 3.78 mmol) in t-BuOH (80 mL) was added 2-methyl-2-butene (19 mL). Then NaClO2 (3.152 g) and NaH2PO4 (3.152 g) in H2O (32 mL) was added during 10 min. The mixture was stirred at room temperature for 30 min. The volatile component was evaporated under vacuum. The residue is diluted with H2O and extracted with EtOAc twice. The aqueous phase was acidified to pH = 3 with 1N HCl. The acidified aqueous phase was extracted with EtOAc three times. The combined organic phase was washed with water, brine, dried over MgSO4 and concentrated. The residue was used directly for the next step without further purification.
To a solution of the crude acid in CH3CN (10 mL) was added DBU (0.57 mL, 3.81 mmol) with good stirring. Then MeI (0.26 mL, 4.18 mmol) was added and the solution was stirred at room temperature for 14 h. Water was added and the mixture was extracted with Et2O. The combined extracts were washed with saturated aqueous Na2S2O3, H2O and brine. The organic phase was dried over MgSO4 and concentrated in vacuo. Flash chromatography (10:1 hexanes/EtOAc) gave the methyl ester 28 (0.967 g, 84% yield for 2 steps). [α]D23 -26.3 (c 0.40, CHCl3). 1H NMR (CDCl3, 500 MHz) δ 7.27 (d, J = 9.0, 2H), 6.87 (d, J = 9.0, 2H), 5.83 (ddd, J = 17.5, 11.0, 8.0, 1H), 5.04 (br d, J = 17.5, 1H), 5.02 (br d, J = 11.0, 1H), 4.50 (d, J = 11.5, 1H), 4.43 (d, J = 11.5, 1H), 3.80 (s, 3H), 3.66 (s, 3H), 3.24 (ddd, J = 7.2, 5.6, 4.4,1H), 2.49 (apparent sextet, J = 7.2, 1H), 2.22-2.32 (m, 2H), 1.71-1.83 (m, 1H), 1.59-1.70 (m, 1H), 1.44-1.58 (m, 2H), 1.04 (d, J = 7.2, 3H); 13C NMR (CDCl3, 125 MHz) δ 174.1, 159.1, 140.7, 130.9, 129.4 (2 C), 114.5, 113.7 (2C), 82.1, 71.4, 55.3, 51.4, 40.6, 34.1, 30.4, 21.0, 15.7. MS (ES) m/z 324 (M + NH4+).
To a solution of the methyl ester 28 in 1:1 mixture of acetone/water (24 mL) was added NMO (1.133 g, 9.67 mmo l) and OsO4 (0.3 M in toluene, 1.09 mL, 3.27 mmol). The solution was stirred at room temperature for 2 h. Saturated aqueous Na2S2O3 and EtOAc were added. The mixture stirred for 1 h. The aqueous phase was separated and extracted with EtOAc three times. The combined organic phase was washed with brine, dried over Na2SO4 and concentrated in vacuo. Flash chromatography (1:1 hexanes/EtOAc) gave the diastereomeric mixture of diol (0.881 g, 80% yield).
To a solution of the diol (0.881 g, 2.59 mmol) in benzene (16 mL) was added Pb(OAc)4 (1.814 g, 4.09 mmol). The mixture was stirred at room temperature for 3 h. Saturated aqueous NaHCO3 was added and the mixture was extracted with EtOAc three times. The combined organic phase was washed with brine, dried over MgSO4, and concentrated in vacuo. The crude aldehyde was reduced directly without further purification.
To a solution of the crude aldehyde in MeOH (15 mL) at 0 °C was added NaBH4 (0.147 g, 3.89 mmol). The solution was stirred at 0 °C for 1 h. Saturated aqueous NH4Cl was added and the mixture was extracted with EtOAc three times. The combined organic phase was washed with brine, dried over Na2SO4 and concentrated in vacuo. Flash chromatography (3:1 hexanes/EtOAc) gave the alcohol 29 (0.662 g, 83% yield for 2 steps). [α]D23 +0.8 (c 1.77, CHCl3). 1H NMR (CDCl3, 500 MHz) δ 7.26 (d, J = 9.0, 2H), 6.87 (d, J = 9.0, 2H), 4.52 (d, J = 11.0, 1H), 4.47 (d, J = 11.0, 1H), 3.79 (s, 3H), 3.66 (s, 3H), 3.63-3.67 (m, 1H), 3.57 (dd, J = 10.5, 5.0, 1H), 3.49 (ddd, J = 7.0, 5.0, 4.0, 1H), 2.24-2.37 (m, 2H), 2.04-2.12 (m, 1H), 1.71-1.80 (m, 1H), 1.57-1.67 (m, 2H), 1.47-1.55 (m, 1H), 0.87 (d, J = 7.0, 3H); 13C NMR (CDCl3, 100 MHz) δ 173.9, 159.2, 130.3, 129.5 (2C), 113.8 (2C), 81.4, 71.4, 65.8, 55.2, 51.5, 36.7, 33.9, 29.3, 21.6, 11.9. MS (ES) m/z 311 (M + H+).
To a solution of the alcohol 29 (1.999 g, 6.448 mmol) in CH2Cl2 (70 mL) were added imidazole (1.317 g, 19.35 mmo l), Ph3P (3.551 g, 13.54 mmol), and I (3.436 g, 13.54 mmo l). The solution was stirred at room temperature for 2 h. The solution was diluted with EtOAc and washed with saturated aqueous Na2S2O3, saturated aqueous NaHCO3 and brine. The organic phase was dried over MgSO4, concentrated in vacuo. Flash chromatography (9:1 hexanes/EtOAc) gave the alkyl iodide 30 (2.481 g, 92% yield). 1H NMR (CDCl3, 400 MHz) δ 7.25 (d, J = 8.8, 2H), 6.87 (d, J = 8.8, 2H), 4.49 (d, J = 11.2, 1H), 4.44 (d, J = 11.2, 1H), 3.79 (s, 3H), 3.67 (s, 3H), 3.43 (ddd, J = 6.8, 5.2, 4.0, 1H), 3.35 (dd, J = 9.6, 6.0, 1H), 3.06 (dd, J = 9.6, 7.6, 1H), 2.28-2.34 (m, 2H), 1.87-1.97 (m, 1H), 1.43-1.77 (m, 4H), 1.02 (d, J = 6.8, 3H); 13C NMR (CDCl3, 100 MHz) δ 173.8, 159.1, 130.6, 129.3 (2C), 113.8 (2C), 80.7, 71.9, 55.2, 51.5, 39.0, 33.9, 30.1, 21.0, 15.5, 12.3.
Synthesis outlined in Scheme 11
To a suspension of LiAlH4 (2.765g, 72.86 mmol) in THF (65 mL) was added slowly a solution of an 8:1 diastereomeric mixture of methylated diethyl (S)-malate 3231 (3.770 g, 18.48 mmol) in THF (35 mL). The mixture was refluxed for 2 h and was cooled to room temperature. Water (2.77 mL), 15% aqueous NaOH (2.77 mL) and water (8.31 mL) were added slowly with care sequentially. The mixture was filtered through celite and washed with Et2O (300 mL). The filtrate was concentrated in vacuo to give an 8:1 diastereomeric mixture of triols 33 (2.101 g). 1H NMR (CD3OD, 500 MHz) δ 3.61-3.69 (m, 2H), 3.50-3.55 (m, 3H), 1.74-1.82 (m, 1H), 0.95 (d, J = 6.0, 3H).
A solution of the crude triol 33 in MeOH (100 mL) was treated with n-Bu2SnO (4.358 g, 17.51 mmol). The mixture was refluxed overnight to give a clear solution. The solution was concentrated in high vacuum to give white solid. The solid in CHCl3 (100 mL) was treated with TBSCl (3.167 g, 21.01 mmol) for 20 min. Acetonitrile (100 mL) was added and the solution was concentrated to about 30 mL. Hexanes (100 mL) was added and then was extracted with CH3CN (100 mL × 3). The combined acetonitrile extracts were concentrated. Flash chromatography (2:1 hexanes/EtOAc) gave an 8:1 diastereomeric mixture of 1,2-diols 34 (3.431g, 79% yield from the diethyl ester). 1H NMR (CDCl, 500 MHz) δ 4.10 (br s, OH), 3.74 (dd, J = 10.5, 4.0, 1H), 3.68 (br dd, J = 11.0, 2.0, 1H), 3.61 (dd, J = 10.5, 9.0, 1H), 3.54-3.64 (m, 2H), 2.49 (br s, OH), 1.82-1.92 (m, 1H), 0.90 (s, 9H), 0.85 (d, J = 7.0, 3H), 0.09 (two overlapped singlets, 6H); 13C NMR (CDCl3, 100 MHz) δ 76.7, 68.1, 64.9, 36.9, 25.8 (3C), 18.1, 13.3, -5.6, -5.7. MS (ES) m/z 235 (M + H+).
To a solution of the 8:1 diastereomeric mixture of 1,2-diols 34 (8.009 g, 34.23 mmol) in THF (200 mL) at 0 °C was added NaH (60% in mineral oil, 0.904 g, 37.65 mmol). The mixture was stirred at 0 °C for 20 min. Then 1-(p-toluenesulfonyl)imidazole (8.453 g, 37.65 mmol) was added slowly with caution. The cooling bath was removed and the mixture was stirred at room temperature for 14 h. The reaction was quenched by addition of saturated aqueous NH4Cl. The mixture was extracted with Et2O three times. The combined organic extracts were washed with water, brine, dried over MgSO4 and concentrated. Flash chromatography (20:1 hexanes/EtOAc) gave a 8:1 diastereomeric mixture of epoxides 35 (6.501g, 88% yield). 1H NMR (C6D6, 500 MHz) δ 3.55 (d, J = 6.0, 2H), 2.67 (ddd, J = 7.0, 4.0, 3.0, 1H), 2.35 (dd, J = 5.5, 4.0, 1H), 2.15 (dd, J = 5.5, 3.0, 1H), 1.28-1.40 (m, 1H), 0.95 (s, 9H), 0.84 (d, J = 7.0, 3H), 0.03 (s, 3H), 0.02 (s, 3H).
To a solution of propyne (4.397 g, 109.73 mmol) in THF (290 mL) at -78 °C was added n-BuLi (2.38 M in hexanes, 39.51 mL, 94.04 mmol). The solution was stirred for 10 min. A solution of the 8:1 diastereomeric mixture of epoxides 35 (6.771 g, 31.35 mmol) in THF (20 mL, 6 mL wash) was added, followed by BF3•OEt2 (11.92 mL, 94.04 mmol). The solution was stirred at -78 ° C for 2 h. The reaction was quenched by addition of saturated NH4Cl and the mixture was extracted with EtOAc three times. The combined organic phase was washed with saturated NaHCO3, brine, dried over MgSO4 and concentrated. Flash chromatography (8:1 hexanes/EtOAc) gave an 8:1 diastereomeric mixture of homopropargylic alcohols (7.712 g, 96% yield). 1H NMR (C6D6, 500 MHz) δ 3.69 (dd, J = 10.0, 5.0, 1H), 3.62-3.66 (m, 1H), 3.51-3.57 (m, 1H), 2.90 (d, J = 4.5, OH), 2.41-2.47 (m, 1H), 2.33-2.41 (m, 1H), 1.85-1.91 (m, 1H), 1.48 (t, J = 2.5, 3H), 0.91 (s, 9H), 0.85 (d, J = 7.0, 3H), -0.00 (s, 3H), -0.01 (s, 3H).
To a solution of the 8:1 diastereomeric mixture of homopropargylic alcohols (7.712 g, 30.12 mmol) in THF (150 mL) was added TBAF (1.0 M in THF, 36.15 mL, 36.15 mmol). Brine was added and the mixture was extracted with Et2O three times. The combined organic phase was washed with brine, dried over Na2SO4 and concentrated. Flash chromatography (1:2 hexanes/EtOAc) gave a 8:1 diastereomeric mixture of diols (4.107 g, 96% yield). 1H NMR (CDCl, 500 MHz) δ 3.66-3.74 (m, 1H), 3.56-3.65 (m, 2H), 3.25 (br s, OH), 3.20 (br d, J = 4.0, OH), 2.42-2.48 (m, 1H), 2.28-2.35 (m, 1H), 1.83 (dq, J = 4.0, 6.0, 1H), 1.79 (t, J = 2.5, 3H), 0.86 (d, J = 7.0, 3H); 13C NMR (CDCl3, 125 MHz) δ 78.6, 75.3, 74.8, 67.3, 39.1, 25.9, 13.6, 3.5.
To a solution of the 8:1 diastereomeric mixture of diols (4.300 g, 30.28 mmol) in benzene (160 mL) were added cyclopentanone (40 mL, 452 mmol), TsOH (576 mg, 3.03 mmol), and MgSO4 (10 g). The mixture was stirred for 72 h, filtered and concentrated in vacuo. Flash chromatography using EtOAc/hexanes (1:9) as eluent to provide cyclopentylidene-protected diol 36 (5.150 g, 76% yield). [α]D23 -5.2 (c 1.95, CHCl3). 1H NMR (400 MHz, C6D6) δ 3.62 (dd, J = 5.1, 11.4 Hz, 1 H), 3.30 (ddd, J = 4.4, 5.9, 9.9 Hz, 1 H), 3.20 (t, J = 11.4, 1 H), 2.31-2.46 (m, 2 H), 2.05-2.16 (m, 2 H), 1.88-1.98 (m, 1 H), 1.73-1.84 (m, 2 H), 1.52-1.64 (m, 4 H), 1.54 (t, J = 2.6 Hz, 3 H), 0.45 (d, J = 6.6 Hz, 3 H); 13C NMR (100 MHz, C6D6) δ 110.4, 77.0, 75.9, 75.9, 67.2, 40.4, 33.6, 30.8, 24.7, 24.2, 22.8, 12.3, 3.4. HRMS (ES) m/z 209.1544 (M + H+), calcd. 209.1541.
A solution of alkyne 36 (588.0 mg, 2.87 mmol) in THF (7.2 mL) was added to Cp2ZrHCl (1.480 g, 5.74 mmol) via cannula. The mixture was protected from light and stirred at 50 °C for 1 h. The resulting dark red suspension was cooled to room temperature, and a solution of I2 (1.454 g, 5.73 mmol) in THF (5 mL) was added via cannula. The dark brown mixture was stirred for 30 min and quenched with 10% aq. Na2S2O3 he and saturated aqueous NaHCO3 (30 mL each). The mixture was diluted with EtOAc and separated. The organic phase was washed with 10% aqueous Na2S2O3, saturated aqueous NaHCO3 and brine. The organic phase was then dried over anhydrous MgSO4 and concentrated in vacuo. Flash chromatography (19:1 hexanes/EtOAc) gave vinyl iodide 37 (596.2 mg, 62% yield). 1H NMR (500 MHz, C6D6) δ 6.41 (ddq, J = 7.5, 7.0, 1.5, 1H), 3.56 (dd, J = 11.5, 5.0, 1H), 3.11 (apparent t, J = 11.5), 3.07 (ddd, J = 11.0, 8.0, 3.5), 2.14 (d, J = 1.5, 3H), 1.90-2.06 (m, 4H), 1.70-1.77 (m, 1H), 1.60-1.66 (m, 1H), 1.48-1.58 (m, 3H), 0.26 (d, J = 6.0, 3H).
Synthesis outlined in Scheme 12
Active Zn-Cu couple was prepared from Zn (1.396 g, 21.34 mmol) and Cu(OAc)2•H2O (85.2 mg, 0.43 mmol) following literature procedure36 and was dried under vacuum for 30 min. Alkyl iodide 30 (1.794 g, 4.27 mmol) in a 15:1 mixture of benzene/DMF (15.0 mL) was added to the Zn-Cu couple. The mixture was heated in a 55 °C oil bath for 1 h with stirring to give the alkylzinc iodide. Anhydrous LiCl (1.09 mg, 25.61 mmo l) (dried with flame) and Pd(PPh3)4 (352.3 mg, 0.30 mmol) in a 50 mL flask was degassed for four times. NMP (12.0 mL) was added, followed by addition of the vinyl iodide 37 (1.025 g, 3.05 mmo l) in NMP (4.0 mL). Then the colorless alkylzinc iodide solution (the excess Zn was removed as much as possible) was added via cannula. The reaction mixture was degassed once and was stirred at room temperature for 15 min and then at 55 °C overnight. The cooled reaction mixture was poured into a mixture of saturated aqueous NaHCO3 and EtOAc. The mixture was extracted with EtOAc four times. The combined extracts were washed with H2O, brine, dried over Na2SO4 and concentrated. Flash chromatography (10:1 hexanes/EtOAc) afforded the coupled product 38 (1.265 g, 83% yield). [α] 23D +4.9 (c 0.73, CHCl3). 1H NMR (CDCl3, 500 MHz) δ 7.27 (d, J = 9.0, 2H), 6.87 (d, J = 9.0, 2H), 5.26 (dd, J = 7.0, 6.0, 1H), 4.47 (d, J = 11.5, 1H), 4.42 (d, J = 11.5, 1H), 3.79 (s, 3H), 3.71 (dd, J = 11.0, 4.5, 1H), 3.66 (s, 3H), 3.39 (t, J = 11.0, 1H), 3.32-3.40 (m, 1H), 3.25 (ddd, J = 7.0, 3.5, 3.5, 1H), 2.28-2.36 (m, 3H), 2.20-2.25 (m, 1H), 2.08-2.16 (m, 1H), 1.86-1.94 (m, 2H), 1.70-1.85 (m, 6H), 1.57-1.68 (m, 5H), 1.57 (s, 3H), 1.46-1.54 (m, 2H), 0.82 (d, J = 6.8, 3H), 0.74 (d, J = 6.8, 3H); 13C NMR (CDCl3, 100 MHz) δ 174.0, 159.0, 135.0, 131.2, 129.2 (2C), 122.2, 113.7 (2C), 110.1, 82.0, 77.2, 71.4, 67.5, 55.2, 51.4, 42.7, 40.2, 34.1, 33.9, 33.3, 31.6, 30.6, 30.1, 24.3, 22.5, 21.5, 16.1, 14.5, 12.7. HRMS (ES) m/z 503.3361 (M + H+), calcd. 503.3372.
A mixture of CH2Cl2 (200 mL), H2O (50 mL) and TFA (12.5 mL) was shaken vigorously. The CH2Cl2 layer (150 mL) was used to dissolve 38 (1.437 g, 2.86 mmol), which was stirred for 5.5 h at room temperature. The solution was poured into saturated aqueous NaHCO3 and diluted with EtOAc. The mixture was extracted with EtOAc four times. The combined extracts were washed with brine, dried over Na2SO4, and concentrated. Flash chromatography (1:1 hexanes/EtOAc) gave the diol (1.119g, 90% yield). [α] 23D +2.6 (c 1.08, CHCl3). 1H NMR (CDCl3, 600 MHz) δ 7.25 (d, J = 9.0, 2H), 6.87 (d, J = 9.0, 2H), 5.13 (apparent t, J = 7.2, 1H), 4.46 (d, J = 11.4, 1H), 4.40 (d, J = 11.4, 1H), 3.80 (s, 3H), 3.70 (dd, J = 11.4, 3.6, 1H), 3.67 (s, 3H), 3.63 (dd, J = 11.4, 7.2, 1H), 3.50 (apparent dt, J = 3.0, 8.4, 1H), 3.23 (ddd, J = 7.4, 3.7, 3.7, 1H), 2.18-2.32 (m, 5H), 1.97-2.03 (m, 1H), 1.84 (dd, J = 13.2, 9.0, 1H), 1.69-1.77 (m, 1H), 1.63 (s, 3H), 1.43-1.62 (m, 4H), 0.89 (d, J = 6.6, 3H), 0.82 (d, J = 6.6, 3H); 13C NMR (CDCl3, 100 MHz) δ 174.0, 159.0, 137.9, 130.8, 129.3 (2C), 121.4, 113.6 (2C), 82.2, 77.0, 71.4, 67.6, 55.1, 51.4, 42.9, 39.6, 34.2, 33.9, 32.8, 29.4, 21.5, 16.0, 15.3, 13.8. HRMS (ES) m/z 437.2879 (M + H+), calcd. 437.2903.
To a solution of the diol (1.119 g, 2.56 mmol) in DMF (25 mL) were added imidazole (0.384 g, 5.64 mmol) and TIPSCl (0.70 mL, 3.27 mmol). The solution was stirred at room temperature for 5 h and then at 0 °C for 26 h. The mixture was diluted with water and was extracted with Et2O three times. The combined extracts were washed with brine, dried over Na2SO4 and concentrated. Flash chromatography (3:1 hexanes/EtOAc) gave the silyl ether quantitatively. [α] 23D +5.5 (c 1.09, CHCl3). 1H NMR (CDCl3, 500 MHz) δ 7.27 (d, J = 8.5, 2H), 6.87 (d, J = 8.5, 2H), 5.27 (apparent t, J = 7.0, 1H), 4.47 (d, J = 11.0, 1H), 4.43 (d, J = 11.0, 1H), 3.88 (dd, J =9.5, 4.0, 1H), 3.80 (s, 3H), 3.69 (dd, J = 9.5, 7.5, 1H), 3.67 (s, 3H), 3.60 (apparent dt, J = 4.5, 7.0, 1H), 3.24 (apparent dt, J = 7.0, 3.5, 1H), 2.19-2.33 (m, 5H), 1.88-1.96 (m, 1H), 1.80 (dd, J = 12.5, 9.5, 1H), 1.70-1.82 (m, 1H), 1.57-1.67 (m, 1H), 1.60 (s, 3H), 1.44-1.57 (m, 3H), 1.06-1.16 (m, 3H), 1.05-1.09 (m, 18H), 0.90 (d, J = 6.5, 3H), 0.82 (d, J = 6.5, 3H); 13C NMR (CDCl3, 100 MHz) δ 174.0, 159.0, 135.6, 131.2, 129.2 (2C), 122.3, 113.7 (2C), 82.1, 76.3, 71.4, 68.4, 55.2, 51.4, 42.8, 39.3, 34.1, 33.8, 33.3, 30.0, 21.6, 17.9 (6C), 16.1, 14.7, 13.7, 11.7 (3C). HRMS (ES) m/z 593.4230 (M + H+), calcd. 593.4237.
To a solution of the silyl ether (1.6297 g, 2.75 mmol) in a 4:1:1 mixture of THF/MeOH/H O (144 mL) was added aqueous solution of LiOH (1.0 M, 24 mL). The solution was stirred at room temperature for 5 h. Saturated aqueous NH4Cl was added and the mixture was extracted with EtOAc three times. The combined extracts were washed with brine, dried over Na2SO4 and concentrated. Flash chromatography (1:1 hexanes/EtOAc) gave the seco-acid 39 (1.205 g, 81% yield). 1H NMR (CDCl, 500 MHz) δ 7.27 (d, J = 8.5, 2H), 6.87 (d, J = 8.5, 2H), 5.25 (apparent t, J = 7.0, 1H), 4.48 (d, J = 11.5, 1H), 4.43 (d, J = 11.5, 1H), 3.89 (dd, J =10.0, 4.0, 1H), 3.79 (s, 3H), 3.69 (dd, J = 10.0, 7.0, 1H), 3.62 (apparent dt, J = 4.5, 7.0, 1H), 3.26 (apparent dt, J = 6.5, 4.5, 1H), 2.30-2.37 (m, 2H), 2.21-2.30 (m, 3H), 1.88-1.96 (m, 1H), 1.80 (dd, J = 12.5, 9.5, 1H), 1.71-1.82 (m, 1H), 1.47-1.68 (m, 4H), 1.60 (s, 3H), 1.06-1.16 (m, 3H), 1.05-1.09 (m, 18H), 0.90 (d, J = 7.5, 3H), 0.82 (d, J = 7.0, 3H); 13C NMR (CDCl3, 100 MHz) δ 178.7, 159.0, 135.7, 131.1, 129.3 (2C), 122.3, 113.7 (2C), 81.9, 76.5, 71.4, 68.4, 55.2, 42.9, 39.2, 34.0, 33.7, 33.3, 30.0, 21.3, 17.9 (6C), 16.1, 14.8, 13.7, 11.7 (3C).
To a solution of the seco-acid 39 (1.078 g, 1.86 mmol) in benzene (19 mL) were added i-Pr2NEt (1.95 mL, 11.18 mmo l) and Cl3C6H2COCl (0.90 mL, 5.59 mmo l). The solution was stirred for 50 min at room temperature and then was added to a solution of DMAP (0.683 g, 5.59 mmo l) in benzene (660 mL) via syringe pump over 10 h. The resulting solution was stirred at room temperature for 8 h. The solution was washed with saturated aqueous NaHCO3, brine, dried over Na2S2O4 and concentrated. Flash chromatography (10:1 hexanes/EtOAc) gave the macrolactone 40 (0.999 g, 96% yield). [α]D23 -19.8 (c 1.06, CHCl3). 1H NMR (CDCl3, 600 MHz) δ 7.27 (d, J = 9.0, 2H), 6.87 (d, J = 9.0, 2H), 5.07 (ddd, J = 12.0, 6.0, 3.0, 1H), 5.00 (br d, J = 10.2), 4.50 (d, J = 11.4, 1H), 4.31 (d, J = 11.4, 1H), 3.80 (s, 3H), 3.66 (dd, J = 9.9, 5.1, 1H), 3.54 (dd, J = 9.9, 6.3, 1H), 3.12 (ddd, J = 8.4, 4.2, 4.2, 1H), 2.43-2.51 (m, 2H), 2.03-2.11 (m, 2H), 1.90-1.95 (m, 1H), 1.85-1.91 (m, 3H), 1.67-1.81 (m, 2H), 1.65 (s, 3H), 1.58-1.64 (m, 1H), 1.40-1.48 (m, 1H), 1.03-1.13 (m, 21H), 1.03(d. J = 7.2, 3H), 0.98(d. J = 7.2, 3H); 13C NMR (CDCl3, 100 MHz) δ 173.4, 159.0, 137.1, 131.1, 129.4 (2C), 121.9, 113.6 (2C), 83.1, 73.6, 70.8, 65.2, 55.2, 45.7, 40.4, 35.9, 32.6, 30.6, 29.0, 20.5, 19.3, 18.0 (6C), 15.7, 12.8, 11.9 (3C). HRMS (ES) m/z 561.3985 (M + H+), calcd. 561.3975.
To a solution of 40 (0.460 g, 0.82 mmol) in CH3CN (48 mL) was added a mixture of CH3CN (48 mL), HF•pyridine (2.3 mL) and pyridine (2.3 mL). The solution was stirred at 0 °C for 120 h. The solution was poured into saturated aqueous NaHCO3 and the mixture was extracted with EtOAc three times. The combined extracts were washed with brine, dried over Na2SO4 and concentrated. Flash chromatography (10:1 hexanes/EtOAc followed by 1:1 hexanes/EtOAc) gave the alcohol (0.300 g, 90% yield). [α]D23 -32.9 (c 1.48, CHCl3). 1H NMR (CDCl, 500 MHz) δ 7.27 (d, J = 9.0, 2H), 6.88 (d, J = 9.0, 2H), 4.99 (br d, J = 10.0, 1H), 4.86 (ddd, J = 12.0, 9.5, 3.0, 1H), 4.50 (d, J = 11.0, 1H), 4.30 (d, J = 11.0, 1H), 3.80 (s, 3H), 3.56 (dd, J = 12.0, 4.0, 1H), 3.46 (br dd, J = 12.0, 2.8, 1H), 2.99-3.14 (m, 1H), 2.54 (ddd, J = 13.0, 5.0, 3.5, 1H), 2.42 (ddd, J = 14.0, 11.5, 11.5, 1H), 2.19 (br d, J = 11.5, 1H), 2.11 (ddd, J= 13.0, 13.0, 3.5, 1H), 1.68-1.96 (m, 6H), 1.67 (s, 3H), 1.56-1.65 (m, 1H), 1.44-1.53 (m, 1H), 1.06 (d. J = 7.3, 3H), 1.04 (d. J = 7.3, 3H); 13C NMR (CDCl3, 100 MHz) δ 175.0, 159.0, 137.4, 130.9, 129.3 (2C), 121.7, 113.6 (2C), 83.1, 74.3, 70.7, 63.9, 55.2, 45.6, 40.2, 35.9, 32.4, 31.5, 28.8, 20.6, 19.0, 15.7, 13.6. HRMS (ES) m/z 405.2640 (M + H+), calcd. 405.2641.
To a solution of the alcohol (0.605 g, 1.50 mmol) in CH2Cl2 (30 mL) were added imidazole (0.305 g, 4.49 mmo l), Ph3P (0.824 g, 3.14 mmol) and I2 (0.797 g, 3.14 mmo l). The solution was stirred at room temperature for 30 min and diluted with saturated aqueous Na2S2O3 solution. The mixture was extracted with EtOAc three times. The combined extracts were washed with brine, dried over Na2SO4 and concentrated. Flash chromatography (10:1 hexanes/EtOAc) gave the alkyl iodide 41 (0.753 g, 98% yield). 1H NMR (CDCl3, 500 MHz) δ 7.26 (d, J = 8.5, 2H), 6.87 (d, J = 8.5, 2H), 4.97 (br d, J = 10.5, 1H), 4.93 (ddd, J = 12.0, 7.0, 3.0, 1H), 4.50 (d, J = 11.0, 1H), 4.30 (d, J = 11.0, 1H), 3.80 (s, 3H), 3.29 (dd, J = 10.0, 4.0, 1H), 3.11 (ddd, J = 8.0, 4.0, 4.0, 1H), 3.00 (dd, J = 10.0, 8.5, 1H), 2.48 (ddd, J = 12.0, 5.0, 3.5, 1H), 2.41 (ddd, J = 14.0, 11.5, 11.5, 1H), 2.12 (br d, J = 11.5, 1H), 2.08 (ddd, J= 12.0, 12.0, 3.5, 1H), 1.84-1.93 (m, 3H), 1.68-1.81 (m, 3H), 1.63 (s, 3H), 1.56-1.63 (m, 1H), 1.37-1.46 (m, 1H), 1.09 (d. J = 7.0, 3H), 1.02 (d. J = 7.0, 3H).
Active Zn-Cu couple was prepared from Zn (0.462 g, 7.07 mmol) and Cu(OAc)2•H2O (0.028 g, 0.14 mmol) following literature procedure36 and was dried under vacuum for 30 min. The alkyl iodide 41 (0.727 g, 1.41 mmo l) in a 15:1 mixture of benzene/DMF (5.0 mL) was added to the Zn-Cu couple. The mixture was heated in a 55 °C oil bath for 1 h with stirring to give the alkylzinc iodide. Anhydrous LiCl (0.360 g, 8.48 mmol) (dried with flame) and Pd(PPh3)4 (0.245 g, 0.21 mmol) in a 25 mL flask was degassed for four times. NMP (8.0 mL) was added, followed by addition of the vinyl iodide 25 (1.085 g, 2.12 mmo l). The colorless alkylzinc iodide solution (the excess Zn was removed as much as possible) was added via cannula. The reaction mixture was degassed once and was stirred at room temperature for 1 h 20 min and then at 50 °C for 15 h. The cooled reaction mixture was poured into a mixture of saturated aqueous NaHCO3 and EtOAc. The mixture was extracted with EtOAc three times. The combined extracts were washed with H2O, brine, dried over Na2SO4 and concentrated. Medium-pressure chromatography (50:1 hexanes/EtOAc, Biotage) gave the protected core structure 42 (0.876 g, 80% yield). [α]D23 -9.2 (c 1.08, CHCl3). 1H NMR (CD3COCD3, 600 MHz) δ 7.28 (d, J = 9.0, 2H), 6.89 (d, J = 9.0, 2H), 5.23 (br d, J = 9.0, 1H), 5.01 (br d, J = 8.4, 1H), 4.87 (ddd, J = 11.4, 5.4, 3.0, 1H), 4.52 (d, J = 10.8, 1H), 4.30 (d, J = 10.8, 1H), 3.98 (apparent sextet, J = 6.0, 1H), 3.78 (s, 3H), 3.74 (ddd, J = 6.0, 6.0, 4.2, 1H), 3.15 (ddd, J = 7.8, 4.2, 3.0, 1H), 2.55-2.62 (m, 1H), 2.44-2.50 (m, 2H), 2.17 (dd, J = 13.8, 5.4, 1H), 1.99-2.10 (m, 2H), 1.92-1.98 (m, 1H), 1.87-1.92 (m, 2H), 1.60-1.80 (m, 7H), 1.67 (s, 3H), 1.66 (s, 3H), 1.41-1.49 (m, 1H), 1.16 (d, J = 6.0, 3H), 1.00 (d, J = 6.6, 3H), 0.92 (s, 9H), 0.92 (d, J = 6.0, 3H), 0.90 (s, 9H), 0.88 (d. J = 6.6, 3H), 0.091 (s, 3H), 0.086 (s, 3H), 0.08 (s, 6H); 13C NMR (CD3COCD3, 100 MHz) δ 173.1, 159.7, 137.2, 132.3, 132.2, 131.0, 129.7, 122.9, 114.0, 82.9, 75.9, 73.5, 70.9, 66.4, 55.2, 46.1, 45.7, 43.7, 37.6, 36.1, 35.6, 33.2, 30.1, 29.8, 26.1(3C), 26.0 (3C), 24.3, 20.2, 19.7, 18.4, 18.3, 16.0, 15.6, 15.4, 14.7, -4.0, -4.3, -4.4, -4.7. HRMS (ES) m/z 773.5577 (M + H+), calcd. 773.5572.
To a solution of the protected core structure 42 (60.1 mg, 0.078 mmo l) in a 18:1 mixture of CH2Cl2/H2O (19 mL) at 0 °C was added DDQ (26.5 mg, 0.117 mmol). The solution was stirred at 0 °C for 2 h. Saturated aqueous NaHCO3 was added and the mixture was extracted with CH2Cl2 three times. The combined extracts were washed with saturated aqueous NaHCO3, H2O, brine and dried over Na2SO4. The solution was concentrated. Flash chromatography (15:1 hexanes/EtOAc) gave the core structure alcohol 21 (46.1 mg, 91% yield). [α]D23 -29.2 (c 0.92, CHCl3). 1H NMR (CD3COCD3, 600 MHz) δ 5.23 (br d, J = 9.0, 1H), 5.04 (br d, J = 10.8, 1H), 4.90 (ddd, J = 11.4, 3.6, 2.4, 1H), 3.98 (apparent sext, J = 6.0, 1H), 3.74 (ddd, J = 6.0, 6.0, 4.2, 1H), 3.34-3.38 (m, 1H) 3.15 (d, J = 5.4, OH), 2.55-2.61 (m, 1H), 2.49 (ddd, J = 14.4, 11.7, 11.7, 1H), 2.31 (ddd, J = 13.2, 6.8, 3.0, 1H), 2.16 (dd, J = 13.2, 4.8, 1H), 2.06-2.14 (m, 2H), 1.88-1.97 (m, 3H), 1.76-1.82 (m, 2H), 1.65 (s, 6H), 1.51-1.66 (m, 5H), 1.42-1.48 (m, 1H), 1.16 (d, J = 6.0, 3H), 0.96 (d, J = 7.2, 3H), 0.92 (s, 9H), 0.90 (d, J = 6.0, 3H), 0.896 (s, 9H), 0.87 (d. J = 6.6, 3H), 0.089 (s, 3H), 0.083 (s, 3H), 0.08 (s, 6H); 13C NMR (CD3COCD3, 100 MHz) δ 173.9, 137.3, 132.3, 131.0, 123.1, 76.4, 73.5, 73.4, 66.4, 46.4, 45.7, 43.6, 37.6, 35.7, 35.6, 35.0, 34.9, 30.2, 26.1 (3C), 26.0 (3C), 24.3, 20.4, 18.7, 18.4, 18.2, 16.0, 15.7, 15.3, 14.8, -4.0, -4.3, -4.5, -4.7. MS (ES) m/z 671 (M + NH4+).
Acknowledgements
We thank Professor P. L. C. Small at the University of Tennessee for a generous gift of natural mycolactones and also for performing biological tests. We are grateful to the National Institutes of Health (CA 22215) and to the Eisai Research Institute for generous financial support. A.B.B. gratefully acknowledge supports in the form of an American Cancer Society postdoctoral fellowship (PF-99-117-01-CDD, partially funded by the National Fisheries Institute).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.For a general review on Buruli ulcer, see:; (a) Asiedu K, Scherpbier R, Ravinglione M. Buruli ulcer: Mycobacterium ulcerans infection. World Health Organization; Geneva, Switzerland: 2000. [Google Scholar]; (b) Rohr J. Angew. Chem. Int. Ed. 2000;39:2847. doi: 10.1002/1521-3773(20000818)39:16<2847::aid-anie2847>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 2.Marsollier L, Robert R, Aubry J, Saint Andre J-P, Kouakou H, Legras P, Manceau A-L, Mahaza C, Carbonnelle B. Appl. Environ. Microbiol. 2002;68:4623. doi: 10.1128/AEM.68.9.4623-4628.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.George KM, Chatterjee D, Gunawardana G, Welty D, Hayman J, Lee R, Small PLC. Science. 1999;283:854. doi: 10.1126/science.283.5403.854. [DOI] [PubMed] [Google Scholar]
- 4.Gunawardana G, Chatterjee D, George KM, Brennan P, Whittern D, Small PLC. J. Am. Chem. Soc. 1999;121:6092. [Google Scholar]
- 5.Benowitz AB, Fidanze S, Small PLC, Kishi Y. J. Am. Chem. Soc. 2001;123:5128. doi: 10.1021/ja0105414. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kobayashi Y, Hayashi N, Tan C-H, Kishi Y. Org. Lett. 2001;3:2245. doi: 10.1021/ol010108z. [DOI] [PubMed] [Google Scholar]; (b) Hayashi N, Kobayashi Y, Kishi Y. Org. Lett. 2001;3:2249. doi: 10.1021/ol010109r. [DOI] [PubMed] [Google Scholar]; (c) Kobayashi Y, Hayashi N, Kishi Y. Org. Lett. 2001;3:2253. doi: 10.1021/ol010110q. [DOI] [PubMed] [Google Scholar]
- 7.Fidanze S, Song F, Szlosek-Pinaud M, Small PLC, Kishi Y. J. Am. Chem. Soc. 2001;123:10117. doi: 10.1021/ja011824z. [DOI] [PubMed] [Google Scholar]
- 8.Song F, Fidanze S, Benowitz AB, Kishi Y. Org. Lett. 2002;4:647. doi: 10.1021/ol0172828. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hong H, Gates PJ, Staunton J, Stinear T, Cole ST, Leadlay PF, Spencer JB. Chem. Commun. 2003:2822. doi: 10.1039/b308163j. [DOI] [PubMed] [Google Scholar]; (b) Mve-Obiang A, Lee RE, Portaels F, Small PLC. Infect. Immun. 2003;71:774. doi: 10.1128/IAI.71.2.774-783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.See ref. 9b.
- 11.Hong H, Spencer JB, Porter JL, Leadlay PF, Stinear T. Chem Bio Chem. 2005;6:643. doi: 10.1002/cbic.200400339. [DOI] [PubMed] [Google Scholar]
- 12.(a) Hong H, Stinear T, Skelton P, Spencer JB, Leadlay PF. Chem. Commun. 2005:4306. doi: 10.1039/b506835e. [DOI] [PubMed] [Google Scholar]; (b) Mve-Obiang A, Lee RE, Umstot ES, Trott KA, Grammer TC, Parker JM, Ranger BS, Grainger R, Mahrous EA, Small PLC. Infect. Immun. 2005;73:3307. doi: 10.1128/IAI.73.6.3307-3312.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ranger BS, Mahrous EA, Mosi L, Adusumilli S, Lee RE, Colorni A, Phodes M, Small PLC. Infect. Immun. 2006;74:6037. doi: 10.1128/IAI.00970-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gurjar MK, Cherian J. Heterocycles. 2001;55:1095. [Google Scholar]
- 15.Tong Z, Ma S, Fuchs PL. J. Sulfur Chem. 2004;25:1. [Google Scholar]
- 16.van Summeren RP, Feringa BL, Minnaard AJ. Org. Biomol. Chem. 2005;3:2524. doi: 10.1039/b505980a. [DOI] [PubMed] [Google Scholar]
- 17.Yin N, Wang G, Qian M, Negishi E. Angew. Chem. Int. Ed. 2006;45:2916. doi: 10.1002/anie.200600012. [DOI] [PubMed] [Google Scholar]
- 18.Alexander MD, Fontaine SD, La Clair JJ, DiPasquale AG, Rheingold AL, Burkart MD. Chem. Commun. 2006:4602. doi: 10.1039/b609408b. [DOI] [PubMed] [Google Scholar]
- 19.A preliminary report of this work has been reported in Ref. 8.
- 20.Paterson I, Craw PA. Tetrahedron Lett. 1989;30:5799. [Google Scholar]
- 21.(a) Jacobsen EN, Markó I, Mungall WS, Schröder G, Sharpless KB. J. Am. Chem. Soc. 1988;110:1968. [Google Scholar]; (b) Sharpless KB, Amberg W, Bennani YL, Crispino GA, Hartung J, Jeong K-S, Kwong H-L, Morikawa K, Wang Z-M, Xu D, Zhang X-L. J. Org. Chem. 1992;57:2768. [Google Scholar]
- 22.Corey EJ, Jardine PD, Virgil S, Yuen P-W, Connel RD. J. Am. Chem. Soc. 1989;111:9243. [Google Scholar]
- 23.The ratio was estimated from the peak intensity in the 1H NMR spectrum of crude product
- 24.(a) Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989. [Google Scholar]; (b) Hikota M, Sakurai Y, Horita K, Yonemitsu O. Tetrahedron Lett. 1990;31:6367. [Google Scholar]
- 25.Remote diastereomers are referred to as the diastereomers due to the stereocenter(s) present outside a self-contained box(es); see:Kobayashi K, Tan C-H, Kishi Y. Helv. Chim. Acta. 2000;83:2562.Boyle CD, Harmange J-C, Kishi Y. J. Am. Chem. Soc. 1994;116:4995.
- 26.Mycolactones are known to exhibit intriguing biological properties; see:George KM, Pascopella L, Welty DM, Small PLC. Infect. Immun. 2000;68:877. doi: 10.1128/iai.68.2.877-883.2000.Dobos KM, Small PL, Deslauriers M, Quinn FD, King CH. Infect. Immun. 2001;69:7182. doi: 10.1128/IAI.69.11.7182-7186.2001.
- 27.(a) Brown HC, Bhat KS. J. Am. Chem. Soc. 1986;108:5919. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]; (b) Brown HC, Bhat KS, Randad RS. J. Org. Chem. 1989;54:1570. [Google Scholar]
- 28.(a) Seyferth D, Marmor RS, Hilbert P. J. Org. Chem. 1971;36:1379. [Google Scholar]; (b) Gilbert JC, Weerasooriya U. J. Org. Chem. 1982;47:1837. [Google Scholar]; (c) Brown DG, Velthuisen EJ, Commerford JR, Brisbois RG, Hoye TR. J. Org. Chem. 1996;61:2540. [Google Scholar]
- 29.(a) Hart DW, Schwartz J. J. Am. Chem. Soc. 1974;96:8115. [Google Scholar]; (b) Schwartz J, Labinger JA. Angew. Chem. Int. Ed. 1976;15:333. [Google Scholar]
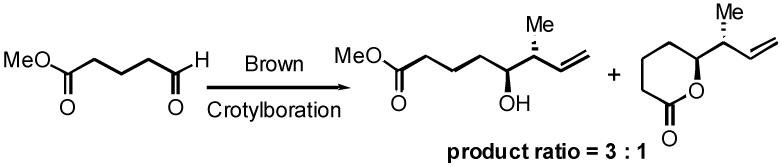
-
30.With use of the C1-carboxylate aldehyde (shown below) for Brown crotylboration, the two steps required for the oxidation of the primary alcohol to the acid could be eliminated. However, the crotylboration gave a 3:1 mixture of the desired product and the δ-lactone.
- 31.Seebach D, Wasmuth D. Helv. Chim. Acta. 1980;63:197. [Google Scholar]
- 32.Guindon Y, Yoakim C, Gorys V, Ogilvie WW, Delorme D, Renaud J, Robinson G, Lavallée J-F, Slassi A, Jung G, Rancourt J, Durkin K, Liotta D. J. Org. Chem. 1994;59:1166. [Google Scholar]
- 33.Leigh DA, Martin RP, Smart JP, Truscello AM. J. Chem. Soc., Chem. Commun. 1994:1373. [Google Scholar]
- 34.Yamaguchi M, Hirao I. Tetrahedron Lett. 1983;24:391. [Google Scholar]
- 35.Negishi E, Valente LF, Kobayashi M. J. Am. Chem. Soc. 1980;102:3298. [Google Scholar]
- 36.Legoff E. J. Org. Chem. 1964;29:2048. [Google Scholar]
- 37.Han X, Stoltz BM, Corey EJ. J. Am. Chem. Soc. 1999;121:7600. [Google Scholar]
- 38.To adopt this synthesis for our purpose, it would be necessary to adjust the C12’-OH protecting group, i.e., from the MOM ether to the TBS ether
- 39.Judd TC, Bischoff A, Kishi Y, Adusumilli S, Small PLC. Org. Lett. 2004;6:4901. doi: 10.1021/ol0479996. [DOI] [PubMed] [Google Scholar]