

Abstract
The heterotrimeric G protein α-subunit Gsα is ubiquitously expressed and mediates receptor-stimulated intracellular cAMP generation. Its gene Gnas is a complex imprinted gene which uses alternative promoters and first exons to generate other gene products, including the Gsα isoform XLαs and the chromogranin-like protein NESP55, which are specifically expressed from the paternal and maternal alleles, respectively. Gsα itself is imprinted in a tissue-specific manner, being biallelically expressed in most tissues but paternally silenced in a few tissues. Gene targeting of specific Gnas transcripts demonstrates that heterozygous mutation of Gsα on the maternal (but not the paternal) allele leads to early lethality, perinatal subcutaneous edema, severe obesity, and multihormone resistance, while the paternal mutation leads to only mild obesity and insulin resistance. These parent-of-origin differences are the consequence of tissue-specific Gsα imprinting. XLαs deficiency leads to a perinatal suckling defect and a lean phenotype with increased insulin sensitivity. The opposite metabolic effects of Gsα and XLαs deficiency are associated with decreased and increased sympathetic nervous system activity, respectively. NESP55 deficiency has no metabolic consequences. Other gene targeting experiments have shown Gnas to have two independent imprinting domains controlled by two different imprinting control regions. Tissue-specific Gsα knockout models have identified important roles for Gsα signaling pathways in skeletal development, renal function, and glucose and lipid metabolism. Our present knowledge gleaned from various Gnas gene targeting models are discussed in relation to the pathogenesis of human disorders with mutation or abnormal imprinting of the human ortholog GNAS.
Keywords: G protein, Genomic Imprinting, Signal Transduction, cAMP, Obesity, Diabetes, Hormone
1. Introduction
Heterotrimeric G proteins are a group of membrane-associated complexes that are primarily involved in signaling at the plasma membrane and other intracellular sites. Each is composed of an α-subunit, which binds guanine nucleotide and mediates signals from seven transmembrane receptors to downstream effectors, as well as a βγ dimer. α, β, and γ subunits are the products of separate genes, and G proteins are generally defined by their specific α-subunit. The Gsα-subunit Gsα is a ubiquitously expressed G protein which primarily couples cell surface receptors to the stimulation of adenylyl cyclase and generation of cAMP.
Heterozygous inactivating Gsα mutations lead to Albright hereditary osteodystrophy (AHO), which is characterized by short stature, subcutaneous ossifications, brachydactyly (shortening and widening of long bones in the hands and feet), and neurobehavioral abnormalities (Spiegel & Weinstein, 2001; Spiegel & Weinstein, 2004; Weinstein, 2004). Patients who inherit AHO from their mother also develop obesity and resistance to several hormones (parathyroid hormone [PTH], thyroid stimulating hormone [TSH], gonadotropins, growth hormone) which signal through Gsα in their target tissues, a condition known as pseudohypoparathyroidism type 1a (PHP1a) (Long et al., 2006; Weinstein, 2001), while patients who inherit the same mutations on the paternal allele develop only the AHO phenotype, a condition also known as pseudopseudohypoparathyroidism (PPHP). These parent-of-origin differences are due to the fact that Gsα expression from the paternal allele is suppressed in various tissues due to genomic imprinting (Germain-Lee et al., 2002; Hayward et al., 2001; Liu et al., 2003; Mantovani et al., 2002). (Fig. 2) A small number of patients develop extensive subcutaneous ossifications which invade the deeper soft tissues (progressive osseous heteroplasia, POH) (Eddy et al., 2000; Kaplan & Shore, 2000; Shore et al., 2002; Yeh et al., 2000). A more isolated form of PTH resistance, known as pseudohypoparathyroidism type 1b (PHP1b), results from a gene imprinting defect leading tissue-specific Gsα deficiency (Bastepe et al., 2003; Liu et al., 2000a; Liu et al., 2005b; Weinstein, 2004). Somatic Gsα mutations that result in constitutive signaling may lead to endocrine tumors, fibrous dysplasia of bone, and other features of the McCune-Albright syndrome (Levine, 1999; Weinstein, 2002, 2004).
Fig. 2.
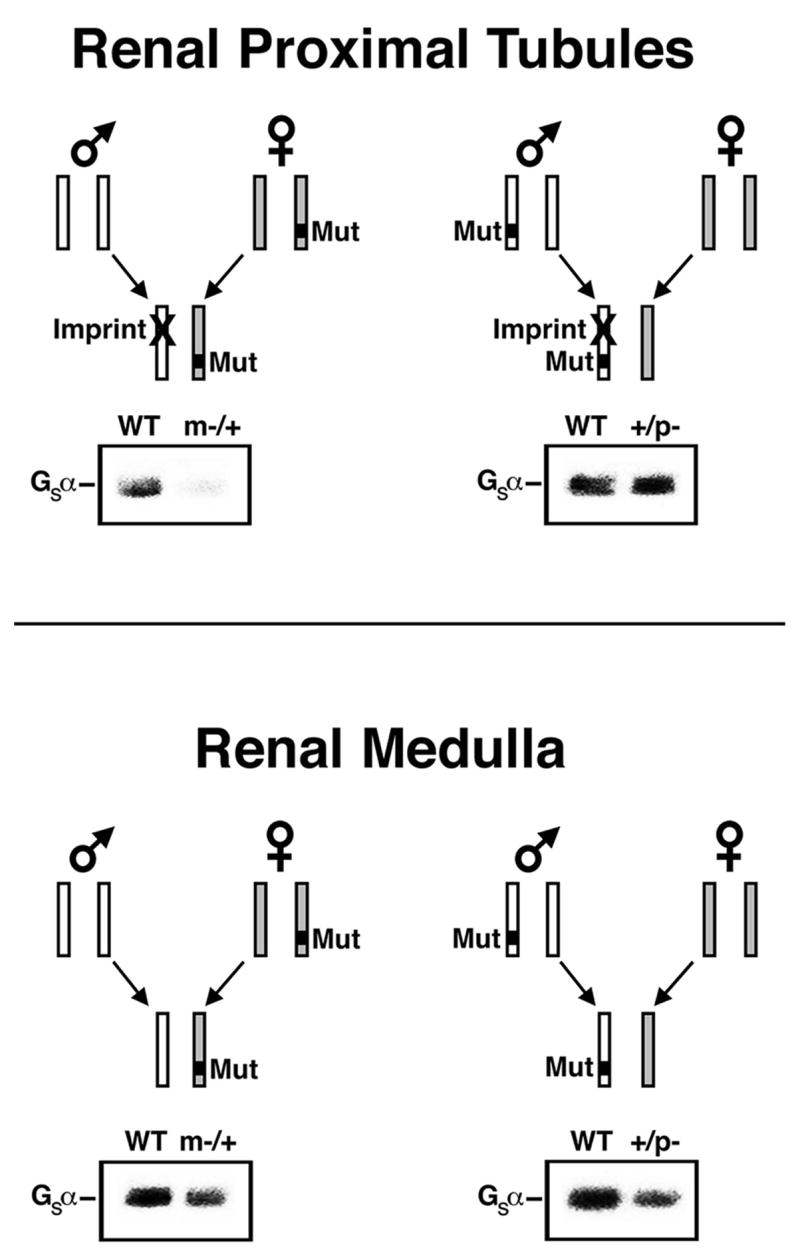
Tissue-specific Gsα imprinting and the effects of heterozygous inactivating Gsα mutations. In renal proximal tubules (upper panels) Gsα is paternally imprinted (silenced, denoted with X). Mutation (Mut) on the active maternal allele (left panel) leads to Gsα deficiency and PTH resistance while mutation on the silenced paternal allele (right panel) has little effect on Gsα expression or PTH sensitivity. This is shown in immunoblots of renal cortical membranes isolated from E2m−/+ (m−/+) and E2+/p− (+/p−) mice, respectively, and their wild type (WT) littermates (S. Yu et al., 1998). Studies in mice and humans shows Gsα to also be imprinted in thyroid, ovaries, pituitary somatotrophs, and neonatal BAT. In most other tissues (lower panels) Gsα is not imprinted and therefore heterozygous mutation leads to ~50% loss of Gsα expression (haploinsufficiency) whether or not the mutation is inherited maternally or paternally. This is demonstrated in immunoblots of renal inner medulla membranes from the same mice. Adapted from Weinstein et al., 2004.
Studies over the last decade have shown that the Gsα gene (GNAS in human, Gnas in mouse) is a complex imprinted gene that encodes multiple gene products through the use of alternative promoters and first exons (Weinstein et al., 2001). Gnas is unique in that is the only example in which oppositely imprinted gene products (some expressed from the maternal allele, others from the paternal allele) are expressed from the same transcriptional unit. This complexity has made it difficult to determine which gene products are critical for normal physiology and human disease, and how the various regulatory and promoter regions interact to maintain their normal epigenetic states. Various mouse models developed over the past decade in which regulatory or coding regions of Gnas were disrupted by gene targeting have been very important in understanding how the gene is regulated and the role of its gene products in normal physiology and disease. This review will summarize what has been learned from gene targeting studies of Gnas.
2. General organization of the Gnas gene
The mouse and human Gsα genes (Gnas and GNAS, respectively) are located within syntenic regions (distal chromosome 2 in mouse, 20q13.2–13.3 in humans) and have similar overall organization (Gejman et al., 1991; Levine et al., 1991; Peters et al., 1994; Rao et al., 1991). Both orthologs are also undergo genomic imprinting and have similar overall imprinting patterns. Genomic imprinting is an epigenetic phenomenon which results in partial or complete suppression of gene expression from one parental allele (Reik & Walter, 2001). The primary imprint ‘marks’ (generally assumed to be allele-specific differences in DNA methylation or histone modification) which distinguishes the two parental alleles are erased in primordial germ cells, reestablished in either male or female gametes, and maintained in somatic tissues throughout development. DNA methylation is the most well established imprint mark, as all imprinted genes have one or more regions in which the maternal and paternal allele are differentially methylated. In many cases DNA methylation is present on the silenced allele, especially when it is the promoter region which is silenced. However, DNA methylation may be present on the active allele when the mechanism of gene silencing is through a more indirect mechanism.
GNAS/Gnas generates multiple gene products through the use of four alternative promoters and first exons that splice onto a common exon (exon 2, Fig. 1). The most downstream alternative exon is Gsα exon 1, which generates transcripts encoding Gsα (Kozasa et al., 1988). The Gsα coding region spans 13 exons (12 exons in mice) and generates two long and two short forms of Gsα protein by alternative splicing of exon 3 (Bray et al., 1986; Kozasa et al., 1988). Use of an alternative terminal exon (N1) located between Gsα exons 3 and 4 generates a neural-specific splice variant of unknown function (Crawford et al., 1993). Typical of most ubiquitously expressed genes, the Gsα promoter resides within a CpG island (GC-rich region with a high frequency of CpG dinucleotides) which is not methylated (Hayward et al., 1998a; Kozasa et al., 1988; Liu et al., 2000b; Peters et al., 1999). Despite the lack of promoter methylation, Gsα is imprinted in a tissue-specific manner in both mice and humans, being expressed primarily from the maternal allele in several tissues, including pituitary, thyroid, renal proximal tubules, and gonads, but biallelically expressed in most tissues (Campbell et al., 1994; Davies & Hughes, 1993; Germain-Lee et al., 2002; Hayward et al., 2001; Liu et al., 2003; Mantovani et al., 2002; Weinstein et al., 2001; S. Yu et al., 1998).
Fig 1.
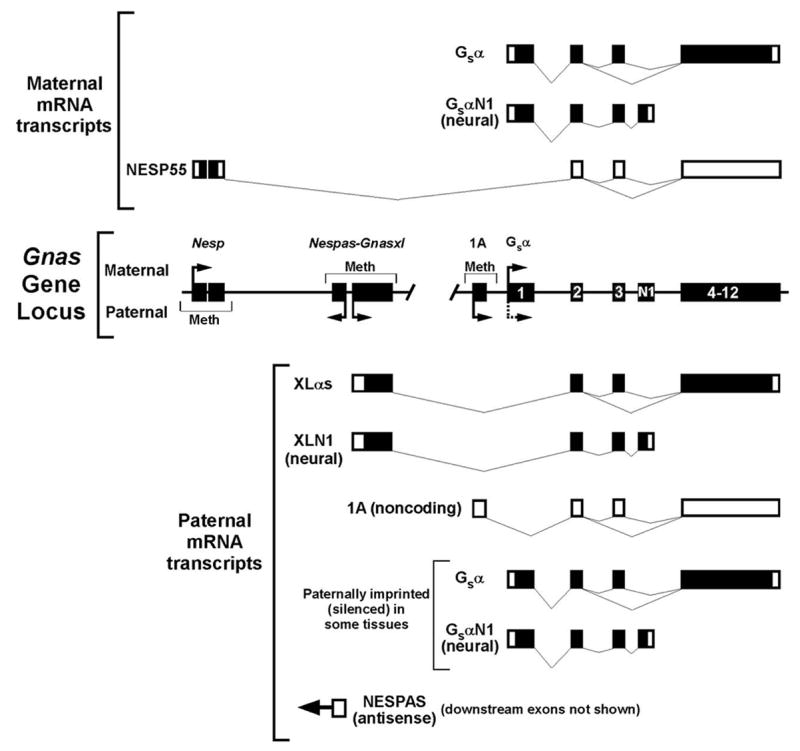
The Gnas locus and its gene products. The organization of the Gnas gene locus is shown with exons denoted as black rectangles and promoter regions Nesp, Nespas, Gnasxl, 1A, and Gsα indicated above. Gsα exons 1, 2, 3, 4–12 (as a single box) and alternative terminal exon N1 are labeled. Only Nespas exon 1 is shown. Methylated regions (Meth), transcriptionally active promoters (horizontal arrows), and resulting mRNA transcripts from the maternal and paternal alleles are shown above and below the gene locus, respectively. Coding regions within transcripts are indicated in black. The stippled horizontal arrow at the paternal Gsα promoter indicates that the promoter is silenced on the paternal allele in a tissue-specific manner.
The most upstream alternative promoter (~45 kB upstream of Gsα exon 1 and referred to as Nesp in the mouse; Fig. 1) generates transcripts that encode the neuroendocrine-specific protein of 55 kDa (NESP55) (Hayward et al., 1998b; Kelsey et al., 1999; Peters et al., 1999). NESP55 is a chromogranin-like protein which is totally unrelated to Gsα. The entire NESP55 coding sequence is within its specific upstream exon and Gsα exons 2–13 are within the 3′ untranslated region of NESP55-specific transcripts (Hayward et al., 1998b; Ischia et al., 1997). NESP55 is only transcribed from the maternal allele and its promoter region is methylated only on the paternal allele (Hayward et al., 1998b; T. Li et al., 2000; Peters et al., 1999). Imprinting of NESP55 in mice is not established until after implantation, suggesting that this region is not a primary imprinting control region for the GNAS/Gnas locus (Liu et al., 2000b).
A third alternative promoter (~30 kb upstream of Gsα exon 1 and referred to as Gnasxl in the mouse, Fig. 1) generates transcripts encoding the Gsα isoform XLαs (Hayward et al., 1998a; Kehlenbach et al., 1994; Kelsey et al., 1999; Peters et al., 1999). XLαs has a long amino-terminal extension encoded by its specific first exon (422, 367, and 551 amino acids in mice, rats, and humans, respectively) while the remainder of the protein is identical to Gsα, being encoded by Gsα exons 2–13. High levels of a neural-specific truncated form of XLαs presumed to be nonfunctional (XLN1) are generated by alternative splicing of XLαs to the alternative terminal exon N1 (Pasolli et al., 2000). The XLαs-specific first exon also has a second open reading frame for the unrelated protein Alex, which has been reported to directly interact with the XLαs protein (Freson et al., 2003; Klemke et al., 2001). Gnasxl is oppositely imprinted to Nesp, being methylated on the maternal allele and transcriptionally active only on the paternal allele (Hayward et al., 1998a; T. Li et al., 2000; Peters et al., 1999).
Within the same differentially methylated region and just upstream of the XLαs promoter is a promoter for paternally expressed antisense transcripts that traverse the NESP55 exon from the opposite direction (Nespas) (Hayward & Bonthron, 2000; T. Li et al., 2000; Williamson et al., 2002; Wroe et al., 2000). Like Gnasxl, Nespas is methylated on the maternal allele. Within the Nespas-Gnasxl promoter region is a primary DNA methylation imprint mark which is established during oogenesis and maintained throughout development (Coombes et al., 2003). Although evidence described below suggests that Nespas is the imprint control center for Nesp, Nespas and Nesp transcripts do not always colocalize during development (Ball et al., 2001).
A fifth alternative promoter and first exon (exon 1A, also referred to as exon A/B in humans) located ~2.5 kb upstream of Gsα exon 1 (Fig. 1) is methylated on the maternal allele and generates paternal-specific transcripts that are presumably untranslated (Ishikawa et al., 1990; Liu et al., 2000a; Liu et al., 2000b). The exon 1A region is also a primary imprinting control center as its maternal-specific methylation is established in oocytes and maintained throughout development in mice (Liu et al., 2000b). In PHP1b patients who have renal PTH resistance presumably due to Gsα deficiency in renal proximal tubules, maternal-specific exon 1A methylation is absent (Bastepe et al., 2001; Jan de Beur et al., 2003; Liu et al., 2000a; Liu et al., 2005b). This suggests that exon 1A controls tissue-specific Gsα imprinting via the presence of one or more negatively regulatory cis-acting elements that are both tissue-specific and methylation-sensitive (therefore only leading to suppression of Gsα expression on the unmethylated paternal allele) (Fig. 3). Promoter competition between the exon 1A and Gsα exon 1 promoters is unlikely to be the mechanism for tissue-specific Gsα imprinting as exon 1A-specific transcripts are expressed in virtually all tissues with a similar tissue distribution pattern as Gsα (Liu et al., 2000b). Deletions identified in autosomal dominant forms of PHP1b suggest that cis-acting elements within the upstream NESP55 imprinted region and the nearby STX16 gene may be critical for the establishment and/or maintenance of exon 1A maternal-specific methylation (Bastepe et al., 2003; Bastepe et al., 2005; Linglart et al., 2005; Liu et al., 2005b).
Fig. 3.

Role of the 1A differentially methylated region in tissue-specific Gsα imprinting and the mechanism of abnormal Gsα expression in PHP1b patients and mice with 1A deletion. The model predicts that the 1A region has a cis-acting silencer element which binds a tissue-specific repressor (R). In renal proximal tubules (upper panels) the repressor is present and binds to the unmethylated silencer on the paternal allele, leading to silencing of the paternal Gsα promoter. The repressor is unable to bind to the maternal allele due to methylation of the silencer, allowing the maternal Gsα promoter to remain active. In most other tissues (lower panels) the repressor is not expressed and therefore the paternal Gsα promoter is unaffected and Gsα is biallelically expressed. Loss of maternal 1A methylation in PHP1b allows the repressor to bind to both alleles, leading to biallelic Gsα silencing, Gsα deficiency, and PTH resistance in proximal tubules (down arrow). In most other tissues the repressor is not expressed and therefore Gsα expression is unaffected (N). Maternal 1A deletion in mice (1Am−/+) has no effect on Gsα expression. In contrast, paternal 1A deletion (1A+/p−) results in loss of Gsα imprinting in proximal tubules due to loss of the silencer, leading to Gsα overexpression (up arrow) and increased PTH sensitivity, but has no effect on Gsα expression in other tissues. Although shown here as a silencer, the negative cis-acting element involved in tissue-specific Gsα imprinting may be an insulator. Loss of tissue-specific Gsα imprinting explains why paternal 1A deletion rescues the phenotype produced from disruption of the maternal Gsα allele. Reproduced from Liu et al., 2005a.
3. Protein products encoded by Gnas
3.1. Gsα
Gsα couples seven transmembrane receptors for a wide variety of extracellular factors, including glycoprotein and peptide hormones, catecholamines and other biogenic amines, and neurotransmitters, to adenylyl cyclase and is required for receptor-stimulated intracellular cAMP generation (Weinstein et al., 2004; Weinstein et al., 2001). Gsα is ubiquitously expressed and in almost all tissues is the G protein mediating receptor-stimulated cAMP responses. The related G protein Golfα, which is encoded by an independent gene, also stimulates adenylyl cyclase but has a much more restricted tissue distribution (Jones & Reed, 1989).
Gsα is targeted to the inner leaflet of both plasma and intracellular membranes by palmitoylation at its amino terminus (Degtyarev et al., 1993; Linder et al., 1993; Mumby et al., 1994). It has been suggested that in addition to its role in signal transduction, it may also play a role in membrane trafficking (Bomsel & Mostov, 1992; Zheng et al., 2001). In the basal state Gsα has GDP bound to its guanine nucleotide binding pocket and combines with βγ dimers to form a heterotrimer. Activated receptors promote GDP release, allowing ambient GTP to bind to Gsα. Upon GTP binding Gsα switches to an active conformation, leading to depalmitoylation and release from βγ and the plasma membrane and direct activation of adenylyl cyclase and other effectors (Allen et al., 2005; Huang et al., 1999; Wedegaertner et al., 1996; J. Z. Yu & Rasenick, 2002). Gsα is deactivated by an intrinsic GTPase activity that hydrolyzes bound GTP to GDP. Evidence suggests that the GTPase cycle of Gsα may be influenced by an RGS protein (Zheng et al., 2001) and by adenylyl cyclase itself (Scholich et al., 1999).
Like all Gα subunits, Gsα has two structural domains, a Ras-like GTPase domain, which includes the sites for guanine nucleotide binding and effector interaction, and a helical domain which forms a cleft in which guanine nucleotides bind (Sunahara et al., 1997; Tesmer et al., 1997). Interactions between the two domains may be important to prevent constitutive guanine nucleotide release (Mixon et al., 1995; D. R. Warner et al., 1998). Alternative splicing of exon 3 leads to two long and two short forms of Gsα with helical domains of slightly differing length (Bray et al., 1986; Kozasa et al., 1988) which are all biologically active with only subtle biochemical differences between them (Jones et al., 1990; Seifert et al., 1998).
GTP binding alters the conformation of three regions (switch 1, 2 and 3 regions) within the GTPase domain (Lambright et al., 1994; Noel et al., 1993). Interactions between the switch 2 and 3 regions, as well as between switch 3 region and the helical domain, are important to maintain the active conformation (Grishina & Berlot, 1998; Iiri et al., 1997; Q. B. Li & Cerione, 1997; Marsh et al., 1998; D. R. Warner et al., 1999; D.R. Warner & Weinstein, 1999; D. R. Warner et al., 1998) Two highly conserved residues (Arg201 and Gln227 in the long form of Gsα) are critical for catalyzing the GTPase reaction (Coleman et al., 1994; Graziano & Gilman, 1989; Landis et al., 1989; Sondek et al., 1994). Post-translational modification (ADP-ribosylation) of Arg201 by cholera toxin or missense mutations encoding substitutions of Arg201 or Gln227 lead to constitutive activation by disrupting the GTPase ‘turn-off’ mechanism. Such somatic mutations have been identified in sporadic endocrine tumors (pituitary, thyroid, adrenal), fibrous dysplasia of bone, and the McCune-Albright syndrome (Lyons et al., 1990; Weinstein, 2002, 2004). The amino terminus and switch 2 regions interact directly with β-subunit to form the heterotrimer (Lambright et al., 1996; Wall et al., 1995), while the carboxyl terminus is important for receptor interactions (Conklin et al., 1996; Grishina & Berlot, 2000; Mazzoni et al., 2000; Schwindinger et al., 1994; Simonds et al., 1989; Sullivan et al., 1987).
cAMP, the major second messenger resulting from Gsα signaling, mediates its effects through direct binding and stimulation of several molecules, including protein kinase A (PKA), cAMP-regulated guanine nucleotide exchange factors (cAMP-GEFs) (de Rooij et al., 1998; Kawasaki et al., 1998), and ion channels (Sudlow et al., 1993; Wainger et al., 2001). PKA is a serine/threonine kinase which activates physiological changes and gene expression through direct phosphorylation of enzymes involved in intermediary metabolism and transcription factors such as the cAMP response element binding protein (CREB) (Montminy, 1997). cAMP-GEFs are expressed at high levels in neuroendocrine cells and may lead to mitogenesis through activation of the ras family member Rap1 (Cass & Meinkoth, 1998; Mei et al., 2002; Miller et al., 1997; Vossler et al., 1997; Wang et al., 2006). There is evidence that Gsα may also directly activate effectors other than adenylyl cyclase, including cardiac Ca2+ channels (Mattera et al., 1989; Yatani et al., 1988) and Src tyrosine kinases (Ma et al., 2000) and may be activated by various tyrosine kinase receptors, including the epidermal growth factor (EGF) (Nair et al., 1990; Nair & Patel, 1993; Poppleton et al., 1996; H. Sun et al., 1997) and basic fibroblast growth factor receptors (Krieger-Brauer et al., 2000)
3.2. XLαs
XLαs is a Gsα isoform only produced from the Gnas paternal allele which differs from Gsα in that the first 47 amino acids of Gsα are replaced by a large amino terminal domain with multiple repeat regions (the XL domain) that is encoded by the XLαs-specific first exon (Abramowitz et al., 2004; Kehlenbach et al., 1994; Pasolli et al., 2000). The XL domain targets XLαs to the plasma membrane (Pasolli et al., 2000). Some, but not all, evidence suggests XLαs may also be localized to the Golgi (Pasolli et al., 2000; Ugur & Jones, 2000). XLαs can bind to βγ and can activate adenylyl cyclase (Klemke et al., 2000). Although one study in native cells did not show evidence for receptor signaling by XLαs (Klemke et al., 2000), a subsequent study showed that XLαs transfected into Gsα-null cells mediated stimulation of adenylyl cyclase by various receptors with the same efficacy and potency as Gsα (Bastepe et al., 2002). However whether XLαs has an important physiological function in humans is unclear, as PPHP patients with paternal GNAS mutations predicted to disrupt XLαs expression do not have a specific phenotype.
XLαs has a more restricted tissue distribution than Gsα, being primarily expressed in neuroendocrine tissues (Kehlenbach et al., 1994; Pasolli & Huttner, 2001; Pasolli et al., 2000). In rats, XLαs is present in the melanotrophs within the pars intermedia of the posterior lobe, and to a lesser extent in brain, adrenal, heart, and pancreatic islets. In mouse neonates XLαs was shown to be expressed in brown and white adipose tissue (BAT, WAT), adrenal medulla, intermediate lobe of the pituitary, brain, stomach, heart, pancreas, and kidney (Plagge et al., 2004). In situ hybridization performed in these mice showed XLαs in the brain to be localized to the hypothalamus, locus coeruleus, as well as the laterodorsal tegmental, hypoglossal, trigeminal, ambiguous, medullary reticular, raphe obscurus and facial nuclei. XLαs may play a specific role during early development, as XLαs expression in adipose tissue was shown to be lost during the first two weeks of mouse postnatal development (Xie et al., 2006). Low levels of XLαs expression have also been reported in endochondral growth plates of developing bone (Bastepe et al., 2004).
The XLαs-specific first exon also generates small amounts of an unrelated basic protein named Alex by translation from an alternative reading frame, and this protein may bind to XLαs (Abramowitz et al., 2004; Freson et al., 2003; Klemke et al., 2001). One study suggested that polymorphisms within the XLαs-specific exon may generate alternative forms of Alex and XLαs which fail to interact (Freson et al., 2003). In the central nervous system the XLαs promoter also produces large amounts of a truncated form of XLαs (XLN1) by splicing to the alternative terminal exon N1 (Pasolli et al., 2000). Although XLN1 is missing most of the elements that are critical for signaling, it may potentially act as a dominant negative regulator of Gsα signaling (Plagge et al., 2004; Xie et al., 2006)
3.3. NESP55
Rather than being a Gα subunit, NESP55 is a chromogranin-like protein located within large dense core granules of secretory cells and is presumed to be a cosecretagogue (Ischia et al., 1997). In rodents NESP55 transcripts begin to be expressed in somites and vasculature during midgestation (Ball et al., 2001), and in adults is primarily expressed in neuroendocrine tissues, including the adrenal medulla, pituitary, hypothalamus, and other midbrain and brainstem regions (Bauer et al., 1999a; Bauer et al., 1999b; Ischia et al., 1997; Lovisetti-Scamiform et al., 1999). It has been suggested that a proteolytic cleavage product of NESP55 may be an endogenous antagonist of serotonin 1b receptors (Ischia et al., 1997). However loss of NESP55 expression in humans is not associated with an obvious phenotype (Kim et al., 2000; Liu et al., 2000a), so the normal role for NESP55, if any, is presently unknown.
4. Overview of Gnas knockout mouse models
4.1. Mice with disruption of Gnas exon 2 (E2−)
The initial Gnas knockout model was generated by insertion of a neomycin-resistance cassette into Gnas exon 2 (S. Yu et al., 1998) (germline Gnas knockout models are summarized in Table 1). Similar to all other subsequent germline Gsα knockout lines (Cattanach et al., 2000; M. Chen et al., 2005a; Germain-Lee et al., 2005; Skinner et al., 2002), homozygous Gsα mutations were embryonically lethal with failure to develop beyond gestational day 10.5, as might be expected for total deficiency of a key signaling molecule that is ubiquitously expressed. In fact, heterozygotes were also lethal in several inbred genetic backgrounds and in the CD-1 outbred strain had only 20% survival to weaning even though they were born at the expected Mendelian ratios (S. Yu et al., 1998).
Table 1.
Germline Gnas knockout and related mouse models
| Model | Gene Defect | Transcripts Disrupted | Phenotype | References |
|---|---|---|---|---|
| E2m−/+ | Gsα exon 2 insertion (maternal) |
|
|
Ecelbarger et al., 1999
S. Yu et al., 2001 S. Yu et al., 2000 S. Yu et al., 1998 |
| PatDp distal 2 | Paternal duplication/maternal deletion of distal chrom 2 |
|
|
Cattanach & Kirk, 1985
Williamson et al., 1998 |
| E1m−/+ | Gsα exon 1 deletion (maternal) |
|
|
M. Chen et al., 2005a
Germain-Lee et al., 2005 Liu et al., 2005a |
| Oed | Gsα exon 6 Val159Glu point (maternal) mutation |
|
|
Cattanach et al., 2000
Skinner et al., 2002 |
| E2+/p− | Gsα exon 2 insertion (paternal) |
|
|
M. Chen et al., 2004
S. Yu et al., 2001 S. Yu et al., 2000 S. Yu et al., 1998 |
| MatDp distal 2 | Maternal duplication/paternal deletion of distal chrom 2 |
|
|
Cattanach & Kirk, 1985
Williamson et al., 1998 |
| E1+/p− | Gsα exon 1 deletion (paternal) |
|
|
M. Chen et al., 2005a
Germain-Lee et al., 2005 |
| Sml | Gsα exon 6 Val159Glu point mutation (maternal) |
|
|
Cattanach et al., 2000
Skinner et al., 2002 |
| Gnasxl+/p− | XLαs exon 1 deletion |
|
|
Plagge et al., 2004
Xie et al., 2006 |
| NESP55 KO | Nesp deletion |
|
|
Plagge et al., 2005 |
| 1A+/p−Δ1A-DMR | Deletion of 1A region (paternal) |
|
|
Williamson et al., 2004
Liu et al., 2005a |
| ΔNAS-DMR (paternal) | Deletion within Nespas promoter |
|
|
Williamson et al., 2006 |
Most strikingly, heterozygotes in which the mutant allele was inherited from the mother (E2m−/+) had a very different phenotype than those in which the mutant allele was inherited from their father (E2+/p−) (S. Yu et al., 1998). E2m−/+ neonates had wide, square shaped bodies, increased birth weights, and marked subcutaneous edema which resolved within 1–2 days. In addition, they had increased convex skin folds in the dorsal neck due to increased interscapular brown adipose tissue (BAT) and inset ears. Most of the mice developed neurological abnormalities, including ataxia, tremor, and imbalance, associated with delayed maturation of the cerebellar cortex with persistence of superficial granular cells, which eventually led to respiratory difficulty and premature death. Other features included thymic cortical atrophy, delayed kidney maturation, and delayed growth. With respect to the thymic cortical atrophy, thymic cortical hyperplasia has been reported in McCune-Albright syndrome due to constitutively activating Gsα mutation (Danon & Crawford, 1987; Shenker et al., 1993). E2m−/+ mice that survived to adulthood had low levels of energy expenditure and developed obesity (S. Yu et al., 2000).
In contrast to E2m−/+ neonates, E2+/p− neonates did not have subcutaneous edema but had low birth weights, narrow bodies, arched backs, and concavity of the dorsal neck due to reduced BAT mass. Most failed to suckle, developed hypoglycemia, and died within hours after birth. In contrast to E2m−/+ mice, E2+/p− mice had markedly reduced adiposity (S. Yu et al., 2000). Both E2m−/+ and E2+/p− had slight reductions in body length.
E2− mice have been used to develop Gsα-null cells by immortalization of cells derived from 9.5 day-old E2m−/p− embryos (Bastepe et al., 2002). Transfection studies with these cells have proven useful in defining the role of Gsα in signaling and its regulation. Using these cells, XLαs was shown to have identical receptor-adenylyl cyclase signaling properties to Gsα (Bastepe et al., 2002). These cells were also used to determine the carboxy-terminal residues required for coupling to prostacyclin receptors (Zhang et al., 2006), to establish that Gsα is required for β2-adrenergic receptor stimulation of MAP kinase at low but not at high agonist concentrations (Y. Sun et al., 2007), and to define the role of Gsα in sensitization of adenylyl cyclase isoforms resulting from chronic D2 dopamine receptor stimulation (Vortherms et al., 2006). Embryonic stem cells have been generated from E2m−/p− blastocysts and used to generate E2m−/p−:wild-type chimeras to show that Gsα signaling is required for normal regulation of chondrocyte differentiation within embryonic growth plates (Bastepe et al., 2004).
4.2. PatDp and MatDp distal 2 mice
The different phenotypes observed in the maternal and paternal heterozygotes provided strong evidence for imprinting of the Gnas locus and suggested that the E2m−/+ and E2+/p− phenotypes result from loss of maternal and paternal-specific Gnas transcripts, respectively. Support from this model derives from the fact that mice with paternal duplication/maternal deletion of the distal chromosome 2 region which includes Gnas (PatDp distal 2) have features in common with E2m−/+ mice, while mice with maternal duplication/paternal deletion of the same region (MatDp distal 2) develop features in common with E2+/p− mice (Cattanach & Kirk, 1985; Peters et al., 1994; S. Yu et al., 1998). Due to the fact that PatDp and MatDp distal 2 mice have only paternally and maternally derived alleles of Gnas, respectively, they have been used to establish the imprinting of Nesp, Nespas, and Gnasxl in mice (Kelsey et al., 1999; Peters et al., 1999; Wroe et al., 2000).
Like E2m−/+ mice, PatDp distal 2 mice were reported to have subcutaneous edema at birth, with a similar body habitus and hyperactivity, but no survival beyond several days (Cattanach & Kirk, 1985). Detailed studies showed that the edema began to develop on gestational day e11.5 (Williamson et al., 1998). As this is five days before the embryonic kidneys begin to function, the edema is not likely to be the result of abnormal renal excretion but rather may be due to a defect in placental fluid transport as the placentas of PatDp distal 2 embryos had reduced wet weight. PatDp embryos also have slightly increased bone length compared to normals (Williamson et al., 1998). Conversely, MatDp distal 2 mice had similarities to E2+/p− mice, including similar body habitus, failure to suckle, hypoactivity, and death within hours after birth (Cattanach & Kirk, 1985). Neither PatDp or MatDp distal 2 mice survived beyond several days. The differences in survival between E2− and PatDp/MatDp distal 2 mice is likely due to differences in the genetic backgrounds in which the mice were studied. It is possible that effects in the latter mice are due to overexpression of Gnas products (eg. XLαs or NESP55 overexpression in PatDp or MatDp distal 2 mice, respectively, due to the presence of two paternal or maternal Gnas alleles) or to potential effects on other unknown genes within the duplicated chromosomal region which may also be imprinted.
4.3. Mice with a point mutation in Gnas exon 6 (Oed-Sml)
Another Gnas mutation, a point mutation in Gsα exon 6 leading to a single amino acid substitution (Val159 to Glu) was generated in mice by chemical mutagenesis using ethylnitrosourea (Cattanach et al., 2000; Skinner et al., 2002). Similar to E2− mice, homozygotes were embryonically lethal. This mutation has been referred to as Oed-Sml because maternal transmission of the mutation leads to subcutaneous edema while paternal transmission leads to a small phenotype. Oed mice are similar to E2m−/+ and PatDp distal 2 mice in that they have subcutaneous edema at birth, increased BAT mass, early death due to respiratory abnormalities, and initial growth retardation followed by significant weight gain. Like E2m−/+ mice, Oed mice were shorter than normal. Unlike E2m−/+ mice, Oed mice had reduced prenatal survival. Oed mice were also reported to have small hearts (microcardia) in the neonatal period which normalized by adulthood, and followup studies of PatDp distal 2 mice also showed evidence for neonatal microcardia (Cattanach et al., 2000). Oed mice did not display the hyperkinetic behavior observed in PatDp distal 2 mice. Sml mice had features similar to E2+/p− and MatDp distal 2 mice, including substantial early lethality, decreased BAT mass, hypoglycemia, growth retardation, and a lean adult phenotype (Cattanach et al., 2000; Plagge et al., 2004; Skinner et al., 2002). However, the abnormal body habitus, hypokinetic behavior and abnormal suckling observed in E2+/p− and MatDp distal 2 mice was not observed in Sml mice. Differences between Oed-Sml and the other mouse models may be due to differences in genetic background or differential effects on Gnas gene products as mentioned above.
4.4. Transcript-specific and tissue-specific Gnas knockouts
Based upon the findings in E2−, Oed-Sml, and PatDp and MatDp distal 2 mice, it can be concluded that loss of one or more maternal Gnas gene products leads to subcutaneous edema during gestation, increased BAT mass, obesity, and increased lethality while loss of paternal Gnas gene products leads to failure to suckle, perinatal lethality, growth retardation, and a lean phenotype in adulthood. However, because these mutations occur in exons that are not transcript-specific, it is difficult to assign specific phenotypic features to the loss of specific Gnas transcripts. To address this issue, subsequent knockout models which specifically disrupt the expression of Gnas products Gsα, XLαs, or NESP55 through disruption or deletion of their specific first exons have been generated, and are discussed in detail below (transcript-specific phenotypes summarized in Tables 1 and 2). Specific deletions of the regions presumed to be important for Gnas imprinting have also been generated.
Table 2.
Phenotype consequences resulting from loss of specific Gnas transcripts in mice
| Gsα (homozygous) | Embryonic lethality |
| Gsα (maternal) | Perinatal subcutaneous edema |
| Early postnatal lethality in a significant proportion | |
| Mildly reduced body length | |
| Severe obesity | |
| Severe glucose intolerance, insulin resistance | |
| Hypertriglyceridemia | |
| PTH resistance | |
| TSH resistance (mild, variable depending on genetic background | |
| Gsα (paternal) | Mildly increased adiposity, normal weight |
| Mild glucose intolerance, insulin resistance | |
| No sucking defect or perinatal lethality | |
| XLαs | Perinatal lethality associated with sucking defect and hypoglycemia |
| Severely reduced adiposity | |
| Markedly increased glucose tolerance and insulin sensitivity | |
| Increased energy expenditure and sympathetic activity levels | |
| Increased lipid metabolism in adipose tissue | |
| No increase in sensitivity of adipose tissue to sympathetic stimulation | |
| NESP55 | Increased reactivity to novel environment |
| No obvious effects on development, survival, or metabolism |
It is difficult to decipher the effects of Gsα deficiency in specific tissues using germline Gsα knockout models as homozygotes are embryonically lethal and Gsα is a ubiquitously expressed protein which is subject to imprinting. In order to examine the role of Gsα signaling pathways in specific tissues, floxed Gsα knockout mice have been generated in which loxP recombination sites have been placed on either side of Gsα exon 1 (E1fl). The presence of the loxP sites themselves in E1fl/fl mice has no effect on Gsα expression or phenotype (M. Chen et al., 2005b). Homozygous or heterozygous tissue-specific Gsα knockouts are generated by mating E1fl/fl or E1fl/+ mice to tissue-specific Cre recombinase transgenic lines in which Cre transgenes are driven by gene promoters with tissue-specific expression (tissue-specific Gsα knockout models are summarized in Table 3).
Table 3.
Tissue-specific Gsα knockout models
| Cre-Promoter | Tissue | Phenotype | References |
|---|---|---|---|
| Collagen Ia1 | Osteoblasts, osteocytes |
|
Sakamoto et al., 2005b |
| ?Extraskeletal tissues | Perinatal subcutaneous edema | ||
| Collagen 2a1 | Chondrocytes |
|
Sakamoto et al., 2005a |
| Albumin | Liver |
|
M. Chen et al., 2005b |
| Renin | Juxtaglomerular (JGA) cells |
|
L. Chen et al., 2007 |
| Renal collecting ducts |
|
5. Use of Gnas gene targeting in miceto study Gnas imprinting
Because PatDp and MatDp distal 2 mice carry only paternal and maternally derived Gnas alleles, respectively, they were used to first establish the allele-specific patterns of transcription activity and methylation of the Nesp, Gnasxl, and Nespas, promoters, which are each methylated on one allele and transcriptionally active on the opposite allele as described above (Kelsey et al., 1999; Peters et al., 1999; Wroe et al., 2000). By Southern analysis using restriction enzymes that were sensitive to DNA methylation it was established that the exon 1A region is methylated exclusively on the maternal allele (Liu et al., 2000a; Liu et al., 2000b). Examination of histone modifications in mice with paternal or maternal exon 1A deletions showed this region to be heterochromatic on the maternal allele and euchromatic on the paternal allele (Sakamoto et al., 2004). Exon 1A-specific transcripts were detected in E2m−/+, but not in E2+/p− mice, consistent with these transcripts being only transcribed from the unmethylated promoter on the paternal allele (Liu et al., 2000b), and similar paternal-specific expression of these transcripts was confirmed in humans (Liu et al., 2000a). Other studies have confirmed these imprinting patterns of the Gnas upstream promoter regions (T. Li et al., 2000).
Unlike the Nesp, Gnasxl, Nespas, and exon 1A promoters, in which one parental allele is fully silenced in all tissues due to promoter methylation, the Gsα promoter is unmethylated on both parental alleles and Gsα expression from the paternal allele is suppressed in a tissue-specific manner. Moreover, the imprinting may only be partial, with paternal-specific expression being present, but at a lower level than expression from the maternal allele. Gsα mRNA and protein expression was significantly reduced in proximal tubules of E2m−/+ while Gsα expression was minimally affected in E2+/p− mice, consistent with Gsα being expressed primarily from the maternal allele in renal proximal tubules (S. Yu et al., 1998) (Fig. 2). Consistent with this finding, E2m−/+, but not E2+/p− mice, have PTH resistance in proximal tubules (S. Yu et al., 1998). These initial findings were confirmed in a Gsα-specific knockout model in which Gsα exon 1 was deleted (Germain-Lee et al., 2005; Schwindinger et al., 1997) and PTH resistance was also observed in Oed mice (Williamson et al., 2004).
The Gsα knockout model generated by Germain-Lee and her colleagues also had Gsα imprinting in the thyroid with associated TSH resistance and elevated serum TSH levels in the maternal heterozygote (Germain-Lee et al., 2005), while TSH levels were normal in E2m−/+ mice (S. Yu et al., 2000) and another Gsα-specific knockout model (M. Chen et al., 2005a). These differences are probably due to the fact that the model with TSH resistance was in the SvEv genetic background while the latter two were in a CD-1 background, indicating strain-specific differences in thyroid-specific Gsα imprinting or other physiological variables which regulate TSH sensitivity. Gsα expression studies in E2− mice also showed evidence for Gsα imprinting within the brain (S. Yu et al., 1998). Overall these findings are consistent with what is observed in AHO patients, in whom maternal, but not paternal inheritance of Gsα mutations lead to renal PTH resistance and partial TSH resistance (Weinstein, 2004; Weinstein et al., 2001). As PTH resistance in these patients develops during the first one or two years of life and renal tubules continue to mature during this period, it is possible that Gsα imprinting is temporally regulated and is only present in mature proximal tubule cells (Weinstein, 2001).
Initial studies showing low Gsα expression in BAT and WAT of adult E2m−/+ but not E2+/p− suggested that Gsα is also imprinted in BAT and WAT (S. Yu et al., 2000; S. Yu et al., 1998). These findings have been refuted by studies in adult Gsα-specific knockout models in which Gsα levels were reduced to a similar extent in WAT from maternal and paternal heterozygotes (Germain-Lee et al., 2005) and with only a small difference in Gsα BAT expression between maternal and paternal heterozygotes (M. Chen et al., 2005a). One study also showed no evidence for Gsα imprinting in human WAT (Mantovani et al., 2004). We suspect that the differential Gsα expression observed in E2m−/+ and E2+/p− mice may result from secondary changes in Gsα expression resulting from different levels of metabolic activity in adipose tissues of these mice (S. Yu et al., 2000) rather than imprinting. Gsα expression studies in neonatal BAT of various Gnas knockout models have also suggested that Gsα is imprinted in this tissue (Plagge et al., 2004; Williamson et al., 2004; Williamson et al., 2006), but these studies must also be interpreted with caution as BAT undergoes rapid cellular changes in this period which are affected by Gnas perturbations. One study examining allele-specific expression using a polymorphism within the Gsα transcript showed strong evidence for imprinting in neonatal BAT (Williamson et al., 2006). It is likely that Gsα is imprinted in neonatal BAT, but that this imprinting is lost by the time animals reach adulthood. It is clear that temporal changes in Gnas regulation occur in adipose tissue, as XLαs is expressed in neonatal BAT and WAT but this expression is lost within 1–2 weeks (Xie et al., 2006).
Studies in E2− and Gsα-specific knockout models showed that in many other tissues (liver, heart, renal inner and outer medulla, muscle, lung) disruption of the maternal or paternal Gsα allele resulted in similar (~50%) decreases in Gsα expression levels, indicating that Gsα is not imprinted in these tissues (M. Chen et al., 2005a; Ecelbarger et al., 1999; Germain-Lee et al., 2005; S. Yu et al., 2000; S. Yu et al., 1998) (Fig. 2). Although one study using PatDp and MatDp distal 2 mice suggested that Gsα was imprinted in an opposite manner in renal glomeruli with silencing of the maternal allele (Williamson et al., 1996), there was no evidence for Gsα imprinting in E2− mice (S. Yu et al., 1998). Tissue-specific Gsα imprinting likely explains why PHP1a patients with maternal Gsα mutations develop resistance to some hormones (eg. PTH, TSH) but not to other hormones that also signal through Gsα (eg. adrenocorticotropic hormone, vasopressin). It is likely that in tissues where Gsα is not imprinted, Gsα haploinsufficiency is still capable of mediating hormone action without an obvious physiological effect (Weinstein et al., 2000). Gsα imprinting within the renal proximal tubule, but not within the more distal tubule segments likely explains why PHP1a patients show evidence for PTH resistance in the proximal tubule but maintain responsiveness to PTH in the distal nephron (Stone et al., 1993). Analysis of histone methylation in mice with paternal or maternal deletion of the Gsα promoter showed that tissue-specific differences in allele-specific expression were associated with corresponding tissue-specific differences in the extent of histone 3 lysine 4 methylation (a marker of transcriptional activity) within the paternal Gsα promoter (Sakamoto et al., 2004).
5.1. Mice with deletion of the exon 1A imprinting control region (1A−)
In mice the maternal-specific methylation of exon 1A is established during oogenesis and maintained throughout pre- and postimplantation development, features consistent with this region being an primary imprinting control center (Liu et al., 2000b). Evidence that the exon 1A region is important for Gsα imprinting comes from the observation that the maternal-specific methylation imprint in this region is lost in PHP1b patients, who have renal PTH resistance but lack the AHO phenotype and maintain normal Gsα levels in peripheral blood cells (Liu et al., 2000a).
We have proposed a model in which the exon 1A region has one or more cis-acting regulatory elements (eg. silencer elements) which suppress activity of the Gsα promoter and which are both methylation-sensitive and tissue-specific (Fig. 3) (Liu et al., 2000a; Weinstein et al., 2001). Methylation of the silencer prevents binding of a trans-acting repressor protein allowing the maternal Gsα promoter to remain active in all tissues. Suppression of the paternal Gsα allele would occur only in those tissues where the repressor is expressed (eg. renal proximal tubules). In PHPIb the maternal-specific methylation is absent, allowing the repressor to bind to both alleles leading to Gsα deficiency in renal proximal tubules and PTH resistance. In most other tissues where Gsα is normally not imprinted (eg. peripheral blood cells) Gsα expression levels will not be affected because the repressor is not present in these tissues. It is possible that the cis-element is an insulator, rather than a repressor. The imprinting mechanism is not likely to depend on the exon 1A promoter activity or its transcripts, as the tissue distribution of exon 1A expression does not correlate with the tissue distribution of Gsα imprinting (Liu et al., 2000b).
Two independent groups have generated models in which a significant proportion of the exon 1A differentially methylated region was deleted (Liu et al., 2005a; Williamson et al., 2004). In both cases imprinting of the upstream Nesp, Gnasxl, and Nespas imprinted regions was unaffected, indicating that imprinting of these regions is independent of the exon 1A imprinting control center and that the imprinting of the Gnas gene is dependent on at least two independent imprinting control centers. This is consistent with findings in PHP1b patients showing that imprinting of exon 1A may be disrupted without affecting the NESP, NESPAS, or XLαs upstream regions and that a control element within the linked STX16 gene appears to be important for imprinting of exon 1A, but not the other upstream regions (Bastepe et al., 2003; Linglart et al., 2005; Liu et al., 2000a; Liu et al., 2005b).
The Gsα imprinting model proposed above would predict that maternal deletion of exon 1A should have no effect on Gsα expression, while paternal exon 1A deletion should result in Gsα overexpression in tissues where Gsα is normally imprinted due to loss of the cis-acting regulatory elements required for silencing of the paternal Gsα promoter (Fig. 3). As predicted, Liu and colleagues showed that paternal exon 1A deletion (1A+/p−) resulted in Gsα overexpression in renal proximal tubules (where Gsα is normally imprinted) but not in liver (where Gsα is not imprinted), while maternal deletion (1Am−/+) had no effect on Gsα expression in either tissue (Liu et al., 2005a). Consistent with increased Gsα expression in renal proximal tubules, both paternal exon 1A deletion models showed evidence for increased PTH sensitivity with reduced serum PTH levels (Liu et al., 2005a; Williamson et al., 2004). Similarly, Williamson and colleagues showed that paternal, but not maternal deletion of exon 1A (Δ1A-DMR) resulted in Gsα overexpression in neonatal BAT, but had little effect on allele-specific Gsα expression in brain (Williamson et al., 2004). Moreover, paternal exon 1A deletion reversed the subcutaneous edema, preweaning lethality, and other gross phenotypic changes observed in neonatal Gsα-specific knockout and Oed mice, respectively (Liu et al., 2005a; Williamson et al., 2004). In these latter models mutation of the maternal Gsα allele leads to severe Gsα deficiency in tissues where Gsα is normally imprinted. The presence of the exon 1A deletion on the paternal allele reverses the imprinting, allowing Gsα to be expressed in these tissues and the phenotypes to be rescued.
5.2. Mice with deletion of the Nespas imprinting control region
A portion of the Nespas promoter region was identified as a likely primary imprinting control region based upon the fact that its maternal-specific methylation is established during oogenesis in mice and maintained through development (Coombes et al., 2003). To determine the role of this region in Gnas imprinting, a mouse model was generated in which 1.6 kilobases including the Nespas promoter and first exon were deleted (ΔNAS-DMR) (Williamson et al., 2006). Maternal transmission of ΔNAS-DMR produced no phenotype in offspring while paternal heterozygotes were born at the expected frequency but died by postnatal day 2.
Paternal ΔNAS-DMR deletion resulted in loss of paternal Nesp imprinting, with loss of paternal-specific methylation of the Nesp promoter and biallelic expression of Nesp transcripts, while the maternal ΔNAS-DMR deletion had no effect on Nesp imprinting. This provides strong evidence that Nesp imprinting, which is established during postimplantation development (Liu et al., 2000b) and therefore is not a primary imprinting event, is dependent on the Nespas imprinting control center. This is consistent with the fact that the imprinting status of the NESP55 and NESPAS promoters are virtually always concordant in PHP1b patients (Liu et al., 2005b). While antisense transcripts have been shown to be important for imprinting of other genes (Sleutels et al., 2002), it is unclear whether Nespas transcripts themselves are required for Nesp imprinting, as studies show that the tissue-specific expression of Nespas and Nesp do not correlate with each other (Ball et al., 2001; T. Li et al., 2000).
The ΔNAS-DMR model suggests that Gnasxl imprinting is not dependent upon Nespas, as the deletion on either allele did not affect Gnasxl methylation or allele-specific expression. This may well also be the case in humans, as the imprinting status of the NESPAS promoter and XLαs exon 1 are different in a significant number of PHP1b patients (Liu et al., 2005b). However XLαs expression was significantly reduced in mice with the paternal deletion, as the deleted region appears to have enhancer activity for the Gnasxl promoter (Williamson et al., 2006). The severe phenotype in paternal ΔNAS-DMR mice is likely to result from XLαs deficiency, as the phenotype is reminiscent of that observed in XLαs-specific knockout mice (see below) (Plagge et al., 2004).
The presence of the ΔNAS-DMR deletion on the maternal-allele had no effect on maternal-specific methylation of exon 1A nor any effect on expression of Gsα or exon 1A-specific transcripts, and therefore Nespas is not required to establish maternal-specific exon 1A methylation. Rather, deletion mutations identified in familial PHP1b patients suggest that elements required to establish or maintain this methylation may be located within the closely linked STX16 gene (Bastepe et al., 2003; Linglart et al., 2005) and in the vicinity of the NESP55 upstream region (Bastepe et al., 2005). In contrast, when the deletion was on the paternal allele it resulted in partial methylation of the exon 1A region on the paternal allele, associated with reduced paternal expression of exon 1A transcripts. This was associated with loss of Gsα imprinting, with Gsα overexpression in neonatal BAT but no effect on Gsα expression in liver. Moreover, the presence of the ΔNAS-DMR deletion on the paternal allele partially rescued the edematous phenotype seen in mice with the maternal Gsα Oed mutation due to relaxation of Gsα imprinting on the paternal allele. These observations provide further evidence for the important role of exon 1A for Gsα imprinting. These findings also suggest that the Nespas region may in some way be important to prevent the exon 1A region from being methylated, although how important this is in the normal imprinting process and the mechanisms involved need to be further studied. The effects on exon 1A and Gsα imprinting and expression were not due to reduced XLαs expression, as there were no similar changes in XLαs knockout mice (Plagge et al., 2004).
6. Transcript-specific Gnas knockout mouse models
6.1. Gsα-specific knockout models
In order to determine the effects of deleting Gsα specifically without affecting the expression of the other Gnas transcripts, two groups have generated models in which the Gsα-specific exon 1 was deleted (M. Chen et al., 2005a; Germain-Lee et al., 2005). In both models homozygotes were embryonically lethal, confirming that total Gsα deficiency is sufficient to impair gestational development.
In one model (E1−) mice were studied in the CD-1 genetic background so they could be directly compared to E2− mice (M. Chen et al., 2005a). Similar to E2m−/+ mice, E1m−/+ mice were born with subcutaneous edema, confirming that this manifestation is secondary to loss of Gsα expression from the maternal allele (see Table 2 for summary of phenotypic manifestations due to loss of specific Gnas gene products). As this manifestation is absent in E1+/p− mice, the edema seen in several models with disruption of the maternal Gsα allele presumably results from severe Gsα deficiency in one or more tissues where Gsα normally undergoes imprinting. Support from this comes from the fact that the edema phenotype is rescued by loss of paternal Gsα imprinting resulting from deletion or methylation of the exon 1A imprinting control region on the paternal allele (Liu et al., 2005a; Williamson et al., 2004; Williamson et al., 2006). Gsα imprinting in placenta may account for this phenotype, as several genes have been shown to be imprinted in placenta and abnormal placental function may lead to abnormal water balance.
However in several respects, E1m−/+ and E2m−/+ are not alike, as E1m−/+ mice lack the neurological phenotype present in E2m−/+ mice and have better survival. Moreover adult E1m−/+ mice have more severe obesity, hypertriglyceridemia, and insulin resistance (M. Chen et al., 2005a), whereas E2m−/+ mice have reduced triglyceride levels and increased insulin sensitivity (S. Yu et al., 2001; S. Yu et al., 2000). These differences may result from disruption of other maternal-specific Gnas products in E2m−/+ mice. Disruption of NESP55 is unlikely to account for these differences, as its expression is not disrupted in E2m−/+ mice and NESP55 deficiency does not appear to produce a major phenotype in mouse (Plagge et al., 2005) or humans (Kim et al., 2000; Liu et al., 2000a). Whether the differences observed between E2m−/+ and E1m−/+ mice result from disruption of other potential maternal Gnas transcripts (Holmes et al., 2003) or from genetic drift in the outbred CD-1 background over time is unknown.
Comparison of E1+/p− and E2+/p− mice shows very dramatic differences, as E1+/p− do not exhibit the suckling defect and perinatal lethality observed in E2+/p− mice (M. Chen et al., 2005a). In addition, adult E1+/p− mice have mildly increased fat mass, insulin resistance, and hypertriglyceridemia, whereas adult E2+/p− mice are very lean with increased sensitivity and hypotriglyceridemia (M. Chen et al., 2004; S. Yu et al., 2001; S. Yu et al., 2000). Studies in XLαs-specific knockout mice (Plagge et al., 2004; Xie et al., 2006) confirm that the severe neonatal and adult phenotype in E2+/p− mice results from disruption of XLαs expression from the paternal allele (see below). The more severe phenotype observed in E1m−/+ compared to E1+/p− mice (subcutaneous edema, increased lethality, more severe metabolic phenotype) presumably results from the effects of tissue-specific Gsα imprinting, as reversal of paternal Gsα imprinting has been shown to rescue some of the features of the E1m−/+ phenotype (Liu et al., 2005a; Williamson et al., 2004; Williamson et al., 2006).
Another model with deletion of Gsα exon 1 was studied in a different genetic background (SvEv or SvEv/CD-1) (Germain-Lee et al., 2005). Studies in these mice confirmed tissue-specific Gsα imprinting, and the presence of PTH and TSH resistance in the maternal heterozygote. Both maternal and paternal heterozygotes had greater preweaning lethality than their counterparts studied in the CD-1 genetic background (M. Chen et al., 2005a). Similar to E1− mice, the maternal heterozygotes weighed more than their paternal counterparts and many of the early features seen in E2m−/+ and E2+/p− mice were absent. There were other differences besides lethality between the two Gsα-specific knockout models. Germain-Lee do not report any perinatal subcutaneous edema in the maternal heterozygotes. There was also a tendency for the paternal heterozygotes (particularly the males) to weigh less than their normal littermates in the Germain-Lee study, which may be partly accounted for by reduced body length and differences in genetic background. The Germain-Lee study also provided evidence that maternal heterozygotes (both male and female) have reduced fertility, perhaps due to gonadotropin resistance as seen in PHP1a patients (Mantovani et al., 2002; Namnoum et al., 1998), and that female maternal heterozygotes show poor mothering behavior. Their study showed no obvious skeletal phenotype or changes in bone remodeling based upon histomorphometric studies. These mice were also used to show that loss of Gsα signaling leads to reduced ethanol consumption and increased sensitivity to the sedative effects of ethanol (Wand et al., 2001).
6.2. XLαs-specific knockout model
XLαs-specific knockout mice were generated in which 60 base pairs within XLαs exon 1 were deleted (Gnasxl−) (Plagge et al., 2004). This deletion had no effect on Gnas imprinting and only resulted in XLαs deficiency with an associated phenotype when present on the paternal allele as XLαs is only paternally expressed. Overall Gnasxl+/p− mice had a phenotype very similar to that of E2+/p−, MatDp distal 2, and to some extent Sml mice, with failure to suckle associated with hypoglycemia and neonatal lethality. Like the other models, these mice had markedly reduced lipid accumulation in BAT and WAT and maintained a lean phenotype through adulthood (Plagge et al., 2004; Xie et al., 2006). It was proposed that XLαs deficiency may result in reduced suckling by a direct effect on orofacial muscle activity, as XLαs is expressed in the facial, hypoglossal, and trigeminal nuclei, which provide motor innervation to the orofacial muscles (Plagge et al., 2004). It was also suggested that loss of XLαs in the locus coeruleus and the cholinergic laterodorsal tegmental nucleus may underlie the observed hypoactivity. Based upon the findings in Gnasxl+/p− mice and the lack of similar findings in E1+/p− mice, it is clear that the severe phenotype in E2+/p− and MatDp distal 2 is due to XLαs deficiency, which appears to have a dominant effect.
Studies of adult Gnasxl+/p− mice in a CD-1 genetic background (Xie et al., 2006) showed them to have a metabolic phenotype very similar to that of E2+/p− mice (M. Chen et al., 2004; S. Yu et al., 2001; S. Yu et al., 2000), including reduced adiposity, increased energy expenditure, hypolipidemia, and greater than normal glucose tolerance and insulin sensitivity. Gene expression studies in BAT, WAT, muscle, and liver suggested that the phenotype results from increased lipid oxidation in adipose tissue, particularly in BAT. Several lines of evidence showed that these changes were due to BAT overstimulation due to increased sympathetic nervous system activity, rather than cell-autonomous effects on adipocytes (Xie et al., 2006). First, similar to E2+/p− mice (S. Yu et al., 2000), Gnasxl+/p− mice had very elevated levels of urinary norepinephrine, consistent with increased sympathetic nervous system activity. Second, Gnasxl+/p− mice did not show evidence for increased responsiveness to a β3 adrenergic agonist which specifically activates adipose tissue. Finally, XLαs is not expressed in adipose tissue by two weeks of age (Xie et al., 2006), making it unlikely that disruption of XLαs expression would result in a cell-autonomous change in adipocytes.
It therefore appears most likely that XLαs is involved in regulation of sympathetic output from the central nervous system. Whether this occurs by acting at sites in the central nervous system that are distinct from Gsα or whether it or its alternative splice form XLN1 directly inhibit Gsα signaling remains to be determined. A role for XLN1 may explain why the Sml phenotype is not as severe, as the Sml exon 6 mutation alters the coding sequence of Gsα and XLαs, but not that of XLN1 (Cattanach et al., 2000; Plagge et al., 2004; Skinner et al., 2002). Although paternal deletion of 20q13 including GNAS has been associated with feeding difficulties, poor growth, and abnormal adipose tissue distribution in humans (Genevieve et al., 2005), it remains unclear if XLαs has an important role in humans as PPHP patients with paternal GNAS mutations that disrupt XLαs do not produce a similar phenotype.
6.3. NESP55-specific knockout model
Mice with specific deficiency of NESP55 were generated by deleting 26 bp at the NESP55 start codon (Plagge et al., 2005). NESP55 knockout mice were reported to have a grossly normal phenotype with no lethality, obvious changes in body habitus, or decrease in fertility. Therefore, NESP55 deficiency does not contribute to the severe phenotypes observed in E2m−/+, PatDp distal 2, and Oed mice. This is not unexpected, as the E2− and Oed mutations do not disrupt the NESP55 coding regions and NESP55 expression is unaffected in E2m−/+ mice (T. Xie, unpublished results). Moreover, loss of NESP55 expression due to biallelic methylation of the NESP55 promoter in PHP1b patients (Liu et al., 2000a) or due to deletion within the coding region (Kim et al., 2000) is not associated with an obvious phenotype. Behavioral studies in NESP55 knockout mice showed these mice to have increased reactivity to a novel environment that was not due to general changes in locomoter activity or anxiety, and was not associated with gross differences in monoamine concentrations within the pons or prefrontal cortex.
7. Role of Gnas in development and physiology based upon mouse knockout models
7.1. Glucose and energy metabolism
Studies in E2− mice showed them to have opposite changes in energy metabolism depending on parental inheritance (S. Yu et al., 2000). E2m−/+ mice developed obesity with increased lipid accumulation in BAT and WAT, reduced energy expenditure at ambient temperature and activity levels. These changes are most likely due to reduced sympathetic activity, as these mice had normal energy expenditure at the thermoneutral temperature 30° when sympathetic activity is minimized, normal metabolic and biochemical responsiveness to a β3-adrenergic agonist, and reduced urine excretion of norepinephrine and its metabolites (S. Yu et al., 2000). In contrast, E2+/p− mice had a severely lean phenotype with increased levels of activity and energy expenditure, presumably due to increased sympathetic activity as they had increased urine excretion of norepinephrine (S. Yu et al., 2000). Leptin levels varied in proportion to adiposity in both groups of mice. Despite the opposite effects on energy metabolism, both groups of mice had hypolipidemia and improved glucose tolerance and insulin sensitivity, although the effects were greater in E2+/p− mice (M. Chen et al., 2004; S. Yu et al., 2001; S. Yu et al., 2000). Isolated skeletal muscles from both E2m−/+ and E2+/p− mice showed similar increases in insulin-stimulated glucose uptake without any changes in the expression of the glucose transporter GLUT4 (S. Yu et al., 2001). E2+/p− have increased insulin sensitivity in liver, muscle, and adipose tissue (M. Chen et al., 2004; S. Yu et al., 2001).
Studies in transcript-specific Gnas knockouts have begun to decipher the role of individual gene products in metabolic regulation. E1− (Gsα-specific knockout) mice develop increased fat mass, glucose intolerance, insulin resistance, and hypertriglyceridemia, but the effects are much greater in E1m−/+ than in E1+/p− mice (M. Chen et al., 2005a). Another Gsα-specific knockout model also showed greater obesity in mice with maternal inheritance of the mutation (Germain-Lee et al., 2005). These findings mimic the pattern of inheritance in humans, as obesity is a more prominent feature of PHP1a (who have maternal Gsα mutations) as compared to PPHP patients (who have paternal Gsα mutations) (Long et al., 2006). Therefore the obesity phenotype resulting from heterozygous Gsα mutation appears to be an imprinted phenotype, similar to multihormone resistance. The central nervous system appears to be a good candidate site for this effect, as E1m−/+ mice were shown to have altered balance between food intake and energy expenditure rates, which are both regulated in the central nervous system, and Gsα is known to mediate pathways involved in energy balance (eg. melanocortin signaling through MC4 receptors).
E1+/p− mice had mild obesity and insulin resistance, in contrast to the lean phenotype with increased insulin sensitivity observed in E2+/p− mice, thus implicating that the E2+/p− metabolic phenotype was due to loss of an alternative paternal-specific Gnas transcript. As discussed above, Gnasxl+/p− mice had a metabolic phenotype that was very similar to that of E2+/p− mice (Plagge et al., 2004; Xie et al., 2006), confirming that XLαs deficiency is the underlying cause of the E2+/p− phenotype. Studies in E2+/p− and Gnasxl+/p− outlined above implicate XLαs (or XLN1) as a negative regulator of sympathetic nervous system activity in the central nervous system, either by directly counteracting Gsα function or by acting in brain regions distinct from those of Gsα (Xie et al., 2006). Studies in NESP55 knockout mice suggests that NESP55 does not play an important role in metabolic regulation (Plagge et al., 2005).
Tissue-specific knockouts are also useful to examine the role of Gsα signaling in specific tissues on metabolic regulation. Mice with liver-specific Gsα deficiency due to homozygous Gsα deletion (LGsKO) were generated by mating Efl/fl mice with albumin-promoter-Cre transgenic mice (M. Chen et al., 2005b). Consistent with Gsα mediating the effects of glucagon, which counterregulates insulin in the liver, these mice had increased hepatic glycogen synthesis and had reduced expression of hepatic genes involved in gluconeogenesis, such as phosphoenolpyruvate carboxykinase and glucose-6-phosphatase. Although LGsKO mice were hypoglycemic and hypoinsulinemic at baseline, they maintained normal glucose and insulin levels after prolonged fasting, probably due to prolonged breakdown of increased hepatic glycogen stores and increased extrahepatic gluconeogenesis. LGsKO mice did not lower their triglyceride levels upon fasting due to inappropriate persistence of lipogenesis. Hepatic glucagon resistance and/or hypoglycemia led to markedly elevated circulating glucagon and glucagon-like peptide 1 (GLP-1) levels and pancreatic α-cell hyperplasia. LGsKO mice also had increased levels of pancreatic insulin secretion, possibly due to increased GLP-1 levels, and increased insulin sensitivity in skeletal muscle and WAT. These mice also had reduced adiposity, although the mechanism for this is unclear.
7.2. Skeletal development
Heterozygous Gsα mutations in AHO patients are associated with several skeletal abnormalities, including short stature, ectopic ossifications, and brachydactyly, in which specific long bones in the hands and feet are shortened due to premature chondrocyte differentiation and fusion of the growth plates, consistent with a defect in endochondral ossification. Similar skeletal abnormalities have not been reported in the analogous genetic mouse models (E1− or E2−) (M. Chen et al., 2005a; Germain-Lee et al., 2005; S. Yu et al., 1998).
Endochondral ossification is a process of ordered chondrocyte differentiation and osseous replacement at growth plates leading to longitudinal bone growth (Kronenberg, 2003). The epiphyseal growth plate consists of three zones (from the end toward the metaphysis): the periarticular zone, the proliferative zone, and the hypertrophic zone. The periarticular zone consists of immature chondrocytes which proliferate and supply cells which form ordered columns of proliferating chondrocytes in the proliferative zone. At the other end of the columns cells undergo hypertrophic differentiation and these cells form the scaffold for ossification which occurs at the end of the growth plate towards the diaphysis (center) of the bone. Periarticular chondrocytes secrete PTH-related peptide (PTHrP) which works in a paracrine manner to inhibit chondrocyte differentiation and expression of Indian hedgehog (Ihh). Once cells reach a certain distance from the periarticular zone where PTHrP levels are below a critical level, cells begin to differentiate (prehypertrophic chondrocytes) and secrete Ihh. Ihh positively feeds back on the periarticular zone to stimulate PTHrP secretion and has independent effects on chondrocyte differentiation.
PTHrP binds to the PTH/PTHrP receptor (PPR) (also referred to as PTH1R), which stimulates both Gs-cAMP and Gq/11-phospholipase C pathways (Bringhurst et al., 1993; Schwindinger et al., 1998). Evidence suggests that these two signaling pathways may have opposite effects on cellular proliferation and differentiation (Gs-cAMP stimulating proliferation and inhibiting differentiation, Gq/11 having opposite effects) (Guo et al., 2002; Guo et al., 2001). The Gs-cAMP action appears to be dominant as deficiency of either PTHrP or PPR results in severe chondrodysplasia due to premature hypertrophic differentiation of growth plate chondrocytes in both mice and humans (Jobert et al., 1998; Karaplis et al., 1994; Lanske et al., 1996; Vortkamp et al., 1996).
Two related models using targeted disruption of Gnas have provided direct evidence that Gsα plays an important role in regulating chondrocyte differentiation. In one model Gsα-null embryonic stem cells were generated from E2m−/p− blastocysts and injected into normal blastocysts to generate E2m−/p−:wild-type chimeras (Bastepe et al., 2004). Examination of growth plates showed mutant chondrocytes to undergo premature hypertrophic differentiation, which can be rescued by transfection of a Gsα-expressing construct. Similar studies using E2m−/+ or E2+/p− cells showed only a partial effect, which was somewhat greater using E2+/p− cells. Expression studies showed no differences in Gsα expression between E2m−/+ and E2+/p− chondrocytes, and it was suggested that E2+/p− mice may show a greater effect due to disruption of XLαs, which is expressed at low levels in chondrocytes. While the columns of proliferative chondrocytes were elongated in PPR−/−:wild-type chimeras (Chung et al., 1998), this was not observed in E2m−/p−:wild-type chimeras. One potential explanation for this difference that was proposed is that PTHrP stimulation of Gq-phospholipase C is only preserved in the latter model.
In the second model, mice with chondrocyte-specific Gsα deficiency were generated by mating E1fl:fl mice with transgenic mice in which the Cre recombinase gene was driven by the collagen 2a1 gene promoter (Sakamoto et al., 2005a). Similar to PTHrP and PPR knockout mouse models (Karaplis et al., 1994; Kobayashi et al., 2002; Lanske et al., 1996), these mice died soon after birth with a severe skeletal abnormality including shortened limbs and domed skull and had severe shortening of the growth plated due to premature chondrocyte differentiation. While PPR and Ihh expression in prehypertrophic chondrocytes were unaffected, PTHrP expression in periarticular chondrocytes was increased presumably because Ihh-secreting prehypertrophic chondrocytes were in closer proximity to these cells. These findings confirm the critical role for Gsα in mediating the effects of PTHrP on chondrocyte differentiation in the growth plate. These mice also developed ectopic chondrocyte differentiation under the perichondrium in the anterior surface of proximal metaphysis of the tibia near the end of gestation. This effect is unlikely to be due to disruption of PTHrP signaling, as PTHrP is not expressed within this area and this phenotype was not observed in mice with chondrocyte-specific PPR deficiency (Kobayashi et al., 2002). Consistent with the lack of a skeletal phenotype in E1− or E2− heterozygotes, mice with heterozygous deletion of Gsα exon 1 in chondrocytes did not demonstrate an obvious phenotype.
The role of Gsα signaling in osteoblasts and osteocytes was examined by mating E1fl:fl mice with mice with a collagen Ia1-Cre recombinase transgene (BGsKO) (Sakamoto et al., 2005b). All BGsKO mice died soon after birth and had subcutaneous edema similar to other maternal Gsα knockout models. It is presumed that this effect was due to extraskeletal Cre expression leading to Gsα deficiency in other tissues. BGsKO had severe hypoplasia of the anterior craniofacial bones, often associated with cleft palate, suggesting an important role for Gsα in craniofacial skeletal development, along with premature endochondral ossification of Meckel’s cartilage, a feature also observed in PTHrP knockout mice (Ishii-Suzuki et al., 1999).
BGsKO mice showed no effects on early skeletal development, including formation of the initial cartilaginous template or bone collar, nor were there effects on the growth plates. However, mice had shortening of long bones associated with reduced formation of primary spongiosa, which is the precursor for trabecular bone. In contrast, the cortical bone was thickened due to reduced osteoclast resorption at the endosteal surface resulting from reduced expression of the osteoclast-stimulating factor receptor activator of nuclear factor-κB (RANKL). These differential effects on cortical and trabecular bone with low bone remodeling are similar to those observed in PTH and PPR knockout mice (Lanske et al., 1999; Miao et al., 2002) and opposite to those seen in mice with osteoblast-specific expression of a constitutively activated PPR (Calvi et al., 2001), indicating that Gsα mediates the differential effects of PTH on cortical and trabecular bone. Osteoblasts in BGsKO mice had normal levels of alkaline phosphate, an early marker of osteoblast differentiation, but reduced levels of later differentiation markers, suggesting that Gsα pathways are important for the later stages of osteoblast differentiation in vivo.
7.3. Renal function
Gsα-cAMP pathways mediate different receptor signaling pathways and physiological responses along the nephron, and the effects of heterozygous Gsα deficiency in mice is further complicated by the fact that Gsα imprinting is variable in different segments of the nephron (Ecelbarger et al., 1999; Weinstein et al., 2000; S. Yu et al., 1998). In the proximal tubule Gsα mediates the action of PTH to reduce phosphate reabsorption, leading to increased renal phosphate wasting, and to increase conversion of 25 hydroxyvitamin D to 1, 25 dihydroxyvitamin D, the active form which stimulates calcium absorption from the gut and calcium release from bone. PHP1a and PHP1b patients develop hypocalcemia, hyperphosphatemia, and low circulating 1, 25 dihydroxyvitamin D levels due to Gsα deficiency and loss of PTH signaling in renal proximal tubules. Studies in E2− mice showed Gsα to be imprinted in renal proximal tubules and E2m−/+, but not E2+/p− mice, develop renal PTH resistance due to loss of the active maternal Gsα allele (S. Yu et al., 1998). This pattern was mimicked in Gsα-specific knockout mice (Germain-Lee et al., 2005). These results explain why PHP1a patients with maternal Gsα mutations develop PTH resistance while PPHP patients with paternal Gsα mutations do not. Moreover, paternal deletion of 1A in 1A+/p− and paternal Δ1A-DMR mice leads to loss of Gsα paternal imprinting, Gsα overexpression in renal proximal tubules, and increased PTH sensitivity (Liu et al., 2005a; Williamson et al., 2004). Overall these findings show that Gsα is a rate-limiting signaling component, consistent with results observed in the kidney cell line HEK293 when Gsα levels were altered (Yang et al., 1997).
In contrast to what is observed in proximal tubules, there is no evidence for Gsα imprinting in more distal portions of the nephron, including the loop of Henle, distal convoluted tubules, and collecting ducts (Ecelbarger et al., 1999; S. Yu et al., 1998). Expression of the cAMP-responsive Na-K-2Cl cotransporter gene in the outer medulla (primarily thick ascending limb) was similarly reduced by ~50% in both E2m−/+ and E2+/p− mice, consistent with reduced levels of hormone-stimulated cAMP generation in this renal segment (Ecelbarger et al., 1999). This was accompanied by low levels of adenylyl cyclase type VI, suggesting a potential feed-forward mechanism for regulation of this enzyme. Reduced expression of the cotransporter would be predicted to lead to reduced countercurrent multiplication by the loop of Henle and reduced medullary solute concentrations. As predicted, E2m−/+ mice had reduced urine concentration after a single bolus of vasopressin (Ecelbarger et al., 1999). More chronic vasopressin stimulation resulting from 48 hours of water deprivation resulted in correction of the urinary concentrating defect and normalization of Na-K-2Cl cotransporter gene expression (Ecelbarger et al., 1999; S. Yu et al., 1998), indicating that chronic hormone stimulation can raise cAMP levels sufficiently to elicit a maximal physiological response. In the inner medulla (primarily collecting ducts) hormone-stimulated cAMP, adenylyl cyclase type VI, and aquaporin 2 levels were all unaffected in E2m−/+ and E2+/p− mice, suggesting that Gsα is not a rate-limiting signaling component in this segment of the nephron (Ecelbarger et al., 1999).
Mice in which Gsα was deleted in juxtaglomerular cells were generated by mating E1fl/fl mice with mice in which Cre recombinase was under the control of the endogenous renin promoter (L. Chen et al., 2007). These mice had markedly reduced basal and stimulated plasma renin levels, along with reduced blood pressure, aldosterone levels, and glomerular filtration rate. Studies in juxtaglomerular cells confirmed that renin production was markedly reduced, confirming that renin production is dependent on one or more receptor pathways which are mediated by Gsα. Unexpectedly, these mice also had Cre expression and Gsα deficiency in the renal medulla, which led to vasopressin resistance and a urinary concentrating defect.
8. Conclusion
Gnas is a unique gene in that it has multiple promoters that are oppositely imprinted, resulting in maternally, paternally, and biallelically expressed gene products. This provides an explanation for the unique observation that disruption of the maternal or paternal alleles of Gnas leads to distinct, and in many respects opposite, phenotypes. One prominent theory for why genomic imprinting occurs is the parent-offspring conflict theory, which hypothesizes that for multiparous species the paternal genome promotes increased allocation of resources to the offspring in order to confer survival advantage, while the maternal genome promotes the opposite effect to conserve maternal resources for future litters (Haig, 2004; Moore & Haig, 1991). This theory predicts that disruption of paternally expressed imprinted genes inhibits fetal and early postnatal growth, while disruption of maternally expressed imprinted genes promotes growth. Studies in Gnas knockout mouse models fits these predictions, as E2m−/+ and PatDp distal 2 mice have increased body mass, while E2+/p− and MatDp distal 2 mice have reduced body mass.
Studies in Gnas knockout mouse models have confirmed the tissue-specific nature of Gsα imprinting, which provides a molecular mechanism for the different parent-of-origin effects in PHP1a and PPHP, respectively, and have confirmed the important role of the 1A region in tissue-specific Gsα imprinting, providing a molecular model for the pathogenesis of PHP1b. Future tissue-specific Gsα knockout models will define the tissue where disruption of the maternal Gsα allele leads to obesity in PHP1a patients and mice with maternal Gsα mutations in the germline.
Abbreviations
- Gsα
stimulatory G protein α-subunit
- AHO
Albright hereditary osteodystrophy
- XLαs
extra-large isoform of Gsα
- NESP55
neuroendocrine-specific protein of 55 kDa
- E2−
mice with disruption of Gnas exon 2
- PatDp and MatDp distal 2
paternal or maternal duplication of distal chromosome 2, respectively
- Oed-Sml
maternal or paternal Gnas exon 6 mutation, respectively
- E1−
mice with deletion of Gsα exon 1
- 1A−
Δ1A-DMR, mouse models with deletion of exon 1A differentially methylated region
- PTH
parathyroid hormone
- TSH
thyroid stimulating hormone
- cAMP
cyclic adenosine monophosphate
- PKA
cAMP-dependent protein kinase
- PHP1a
PHP1b, pseudohypoparathyroidism types 1a and 1b
- PPHP
pseudopseudohypoparathyroidism
- POH
progressive osseous heteroplasia
- XLN1
neural-specific truncated form of XLαs
- Alex
alternative protein transcribed from XLαs exon 1
- E1fl/fl
mice with loxP recombination sites surrounding Gsα exon 1
- BAT
WAT, brown and white adipose tissue
- ΔNAS-DMR
mice with deletion of Nespas promoter region
- PTHrP
PTH-related peptide
- Ihh
Indian hedgehog
- PPR
PTH/PTHrP receptor, BGsKO, osteoblast/osteocyte-specific Gsα knockout
- LGsKO
liver-specific Gsα knockout
- GLP-1
glucagon-like peptide 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abramowitz J, Grenet D, Birnbaumer M, Torres HN, Birnbaumer L. XLαs, the extra-long form of the α-subunit of the Gs G protein, is significantly longer than suspected, and so is its companion Alex. Proc Natl Acad Sci USA. 2004;101:8366–371. doi: 10.1073/pnas.0308758101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JA, Yu JZ, Donati RJ, Rasenick MM. β-adrenergic receptor stimulation promotes Gαs internalization through lipid rafts: a study in living cells. Mol Pharmacol. 2005;67:1493–1504. doi: 10.1124/mol.104.008342. [DOI] [PubMed] [Google Scholar]
- Ball ST, Williamson CM, Hayes C, Hacker T, Peters J. The spatial and temporal expression pattern of Nesp and its antisense Nespas, in mid-gestation mouse embryos. Mech Develop. 2001;100:79–81. doi: 10.1016/s0925-4773(00)00490-1. [DOI] [PubMed] [Google Scholar]
- Bastepe M, Frohlich LF, Hendy GN, Indridason OS, Josse RG, Koshiyama H, et al. Autosomal dominant pseudohypoparathyroidism type Ib is associated with a heterozygous microdeletion that likely disrupts a putative imprinting control element of GNAS. J Clin Invest. 2003;112:1255–1263. doi: 10.1172/JCI19159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastepe M, Frohlich LF, Linglart A, Abu-Zahra HS, Tojo K, Ward LM, et al. Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypoparathyroidism type Ib. Nat Genet. 2005;37:25–27. doi: 10.1038/ng1487. [DOI] [PubMed] [Google Scholar]
- Bastepe M, Gunes Y, Perez-Villamil B, Hunzelman J, Weinstein LS, Jüppner H. Receptor-mediated adenylyl cyclase activation through XLαs, the extra-large variant of the stimulatory G protein alpha subunit. Mol Endocrinol. 2002;16:1912–1919. doi: 10.1210/me.2002-0054. [DOI] [PubMed] [Google Scholar]
- Bastepe M, Pincus JE, Sugimoto T, Tojo K, Kanatani M, Azuma Y, et al. Positional dissociation between the genetic mutation responsible for pseudohypoparathyroidism type Ib and the associated methylation defect at exon A/B: evidence for a long-range regulatory element within the imprinted GNAS1 locus. Hum Mol Genet. 2001;10:1231–1241. doi: 10.1093/hmg/10.12.1231. [DOI] [PubMed] [Google Scholar]
- Bastepe M, Weinstein LS, Ogata N, Kawaguchi H, Juppner H, Kronenberg HM, et al. Stimulatory G protein directly regulates hypertrophic differentiation of growth plate cartilage in vivo. Proc Natl Acad Sci USA. 2004;101:14794–14799. doi: 10.1073/pnas.0405091101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer R, Ischia R, Marksteiner J, Kapeller I, Fischer-Colbrie R. Localization of neuroendocrine secretory protein 55 messenger RNA in the rat brain. Neuroscience. 1999a;91:685–694. doi: 10.1016/s0306-4522(98)00668-x. [DOI] [PubMed] [Google Scholar]
- Bauer R, Weiss C, Marksteiner J, Doblinger A, Fischer-Colbrie R, Laslop A. The new chromogranin-like protein NESP55 is preferentially localized in adrenaline-synthesizing cells of the bovine and rat adrenal medulla. Neurosci Lett. 1999b;263:13–16. doi: 10.1016/s0304-3940(99)00091-9. [DOI] [PubMed] [Google Scholar]
- Bomsel M, Mostov K. Role of heterotrimeric G proteins in membrane traffic. Mol Biol Cell. 1992;3:1317–1328. doi: 10.1091/mbc.3.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray P, Carter A, Simons C, Guo V, Puckett C, Kamholz J, et al. Human cDNA clones for four species of Gαs signal transduction protein. Proc Natl Acad Sci USA. 1986;83:8893–8897. doi: 10.1073/pnas.83.23.8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringhurst FR, Juppner H, Guo J, Urena P, Potts JT, Jr, Kronenberg HM, et al. Cloned, stably expressed parathyroid hormone (PTH)/PTH-related peptide receptors activate multiple messenger signals and biological responses in LLC-PK1 kidney cells. Endocrinology. 1993;132:2090–2098. doi: 10.1210/endo.132.5.8386606. [DOI] [PubMed] [Google Scholar]
- Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, et al. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107:277–286. doi: 10.1172/JCI11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell R, Gosden CM, Bonthron DT. Parental origin of transcription from the human GNAS1 gene. J Med Genet. 1994;31:607–614. doi: 10.1136/jmg.31.8.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass LA, Meinkoth JL. Differential effects of cyclic adenosine 3′,5′-monophosphate on p70 ribosomal S6 kinase. Endocrinology. 1998;139:1991–1998. doi: 10.1210/endo.139.4.5880. [DOI] [PubMed] [Google Scholar]
- Cattanach BM, Kirk M. Differential activity of maternally and paternally derived chromosome regions in mice. Nature. 1985;315:496–498. doi: 10.1038/315496a0. [DOI] [PubMed] [Google Scholar]
- Cattanach BM, Peters J, Ball S, Rasberry C. Two imprinted gene mutations: three phenotypes. Hum Mol Genet. 2000;9:2263–2273. doi: 10.1093/oxfordjournals.hmg.a018917. [DOI] [PubMed] [Google Scholar]
- Chen L, Kim SM, Oppermann M, Faulhaber-Walter R, Huang Y, Mizel D, et al. Regulation of renin in mice with Cre recombinase-mediated deletion of G protein Gsα in juxtaglomerular cells. Am J Physiol. 2007;292:F27–F37. doi: 10.1152/ajprenal.00193.2006. [DOI] [PubMed] [Google Scholar]
- Chen M, Gavrilova O, Liu J, Xie T, Deng C, Nguyen AT, et al. Alternative Gnas gene products have opposite effects on glucose and lipid metabolism. Proc Natl Acad Sci USA. 2005a;102:7386–7391. doi: 10.1073/pnas.0408268102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Gavrilova O, Zhao WQ, Nguyen A, Lorenzo J, Shen L, et al. Increased glucose tolerance and reduced adiposity in the absence of fasting hypoglycemia in mice with liver-specific Gsα deficiency. J Clin Invest. 2005b;115:3217–3227. doi: 10.1172/JCI24196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Haluzik M, Wolf NJ, Lorenzo J, Dietz KR, Reitman ML, et al. Increased insulin sensitivity in paternal Gnas knockout mice Is associated with increased lipid clearance. Endocrinology. 2004;145:4094–4102. doi: 10.1210/en.2004-0038. [DOI] [PubMed] [Google Scholar]
- Chung UI, Lanske B, Lee K, Li E, Kronenberg H. The parathyroid hormone/parathyroid hormone-related peptide receptor coordinates endochondral bone development by directly controlling chondrocyte differentiation. Proc Natl Acad Sci USA. 1998;95:13030–13035. doi: 10.1073/pnas.95.22.13030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman DE, Berghuis AM, Lee E, Linder ME, Gilman AG, Sprang SR. Structures of active conformations of Giα1 and the mechanism of GTP hydrolysis. Science. 1994;265:1405–1412. doi: 10.1126/science.8073283. [DOI] [PubMed] [Google Scholar]
- Conklin BR, Herzmark P, Ishida S, Voyno-Yasenetskaya TA, Sun Y, Farfel Z, et al. Carboxyl-terminal mutations of Gqα and Gsα that alter the fidelity of receptor activation. Mol Pharmacol. 1996;50:885–890. [PubMed] [Google Scholar]
- Coombes C, Arnaud P, Gordon E, Dean W, Coar EA, Williamson CM, et al. Epigenetic properties and identification of an imprint mark in the Nesp-Gnasxl domain of the mouse Gnas imprinted locus. Mol Cell Biol. 2003;23:5475–5488. doi: 10.1128/MCB.23.16.5475-5488.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JA, Mutchler KJ, Sullivan BE, Lanigan TM, Clark MS, Russo AF. Neural expression of a novel alternatively spliced and polyadenylated Gsα transcript. J Biol Chem. 1993;268:9879–9885. [PubMed] [Google Scholar]
- Danon M, Crawford JD. The McCune-Albright syndrome. Engeb Inn Med Kinderheilkd. 1987;55:81–115. doi: 10.1007/978-3-642-71052-0_3. [DOI] [PubMed] [Google Scholar]
- Davies SJ, Hughes HE. Imprinting in Albright’s hereditary osteodystrophy. J Med Genet. 1993;30:101–103. doi: 10.1136/jmg.30.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Degtyarev MY, Spiegel AM, Jones TLZ. The G protein αs subunit incorporates [3H]palmitic acid and mutation of cysteine-3 prevents this modification. Biochemistry. 1993;32:8057–8061. doi: 10.1021/bi00083a001. [DOI] [PubMed] [Google Scholar]
- Ecelbarger CA, Yu S, Lee AJ, Weinstein LS, Knepper MA. Decreased Na-K-2Cl cotransporter expression in mice with heterozygous disruption of the gene for heterotrimeric G-protein Gsα. Am J Physiol. 1999;277:F235–44. doi: 10.1152/ajprenal.1999.277.2.F235. [DOI] [PubMed] [Google Scholar]
- Eddy MC, Jan de Beur SM, Yandow SM, McAlister WH, Shore EI, Kaplan FS, et al. Deficiency of the α-subunit of the stimulatory G protein and severe extraskeletal ossification. J Bone Miner Res. 2000;15:2074–2083. doi: 10.1359/jbmr.2000.15.11.2074. [DOI] [PubMed] [Google Scholar]
- Freson K, Jaeken J, Van Helvoirt M, de Zegher F, Wittevrongel C, Thys C, et al. Functional polymorphisms in the paternally expressed XLαs and its cofactor ALEX decrease their mutual interaction and enhance receptor-mediated cAMP formation. Hum Mol Genet. 2003;12:1121–1130. doi: 10.1093/hmg/ddg130. [DOI] [PubMed] [Google Scholar]
- Gejman PV, Weinstein LS, Martinez M, Spiegel AM, Cao Q, Hsieh WT, et al. Genetic mapping of the Gs-α subunit gene (GNAS1) to the distal long arm of chromosome 20 using a polymorphism detected by denaturing gradient gel electrophoresis. Genomics. 1991;9:782–783. doi: 10.1016/0888-7543(91)90377-q. [DOI] [PubMed] [Google Scholar]
- Genevieve D, Sanlaville D, Faivre L, Kottler ML, Jambou M, Gosset P, et al. Paternal deletion of the GNAS imprinted locus (including Gnasxl) in two girls presenting with severe pre- and post-natal growth retardation and intractable feeding difficulties. Eur J Hum Genet. 2005;13:1033–1039. doi: 10.1038/sj.ejhg.5201448. [DOI] [PubMed] [Google Scholar]
- Germain-Lee EL, Ding C, Deng Z, Crane JL, Saji M, Ringel MD, et al. Paternal imprinting of Gαs in the human thyroid as the basis of TSH resistance in pseudohypoparathyroidism type 1a. Biochem Biophys Res Commun. 2002;296:67–72. doi: 10.1016/s0006-291x(02)00833-1. [DOI] [PubMed] [Google Scholar]
- Germain-Lee EL, Schwindinger W, Crane JL, Zewdu R, Zweifel LS, Wand G, et al. A mouse model of Albright hereditary osteodystrophy generated by targeted disruption of exon 1 of the Gnas gene. Endocrinology. 2005;146:4697–4709. doi: 10.1210/en.2005-0681. [DOI] [PubMed] [Google Scholar]
- Graziano MP, Gilman AG. Synthesis in Escherichia coli of GTPase-deficient mutants of Gsα. J Biol Chem. 1989;264:15475–15482. [PubMed] [Google Scholar]
- Grishina G, Berlot CH. Mutations at the interface of Gsα impair receptor-mediated activation by altering receptor and guanine nucleotide binding. J Biol Chem. 1998;273:15053–15060. doi: 10.1074/jbc.273.24.15053. [DOI] [PubMed] [Google Scholar]
- Grishina G, Berlot CH. A surface-exposed region of Gsα in which substitutions decrease receptor-mediated activation and increase receptor affinity. Mol Pharmacol. 2000;57:1081–1092. [PubMed] [Google Scholar]
- Guo J, Chung UI, Kondo H, Bringhurst FR, Kronenberg HM. The PTH/PTHrP receptor can delay chondrocyte hypertrophy in vivo without activating phospholipase C. Dev Cell. 2002;3:183–194. doi: 10.1016/s1534-5807(02)00218-6. [DOI] [PubMed] [Google Scholar]
- Guo J, Lanske B, Liu BY, Divieti P, Kronenberg HM, Bringhurst FR. Signal-selectivity of parathyroid hormone (PTH)/PTH-related peptide receptor-mediated regulation of differentiation in conditionally immortalized growth-plate chondrocytes. Endocrinology. 2001;142:1260–1268. doi: 10.1210/endo.142.3.8001. [DOI] [PubMed] [Google Scholar]
- Haig D. Genomic imprinting and kinship: how good is the evidence? Annu Rev Genet. 2004;38:553–585. doi: 10.1146/annurev.genet.37.110801.142741. [DOI] [PubMed] [Google Scholar]
- Hayward BE, Barlier A, Korbonits M, Grossman AB, Jacquet P, Enjalbert A, et al. Imprinting of the Gsα gene GNAS1 in the pathogenesis of acromegaly. J Clin Invest. 2001;107:R31–R36. doi: 10.1172/JCI11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward BE, Bonthron DT. An imprinted antisense transcript at the human GNAS1 locus. Hum Mol Genet. 2000;9:835–841. doi: 10.1093/hmg/9.5.835. [DOI] [PubMed] [Google Scholar]
- Hayward BE, Kamiya M, Strain L, Moran V, Campbell R, Hayashizaki Y, et al. The human GNAS1 gene is imprinted and encodes distinct paternally and biallelically expressed G proteins. Proc Natl Acad Sci USA. 1998a;95:10038–10043. doi: 10.1073/pnas.95.17.10038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward BE, Moran V, Strain L, Bonthron DT. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc Natl Acad Sci USA. 1998b;95:15475–15480. doi: 10.1073/pnas.95.26.15475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes R, Williamson C, Peters J, Denny P, Wells C. A comprehensive transcript map of the mouse Gnas imprinted complex. Genome Res. 2003;13:1410–1415. doi: 10.1101/gr.955503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Duncan JA, Gilman AG, Mumby SM. Persistent membrane association of activated and depalmitoylated G protein α subunits. Proc Natl Acad Sci USA. 1999;96:412–417. doi: 10.1073/pnas.96.2.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iiri T, Farfel Z, Bourne HR. Conditional activation defect of a human Gsα mutant. Proc Natl Acad Sci USA. 1997;94:5656–5661. doi: 10.1073/pnas.94.11.5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ischia R, Lovisetti-Scamihorn P, Hogue-Angeletti R, Wolkersdorfer M, Winkler H, Fischer-Colbrie R. Molecular cloning and characterization of NESP55, a novel chromogranin-like precursor of a peptide with 5-HT1B receptor antagonist activity. J Biol Chem. 1997;272:11657–11662. doi: 10.1074/jbc.272.17.11657. [DOI] [PubMed] [Google Scholar]
- Ishii-Suzuki M, Suda N, Yamazaki K, Kuroda T, Senior PV, Beck F, et al. Differential responses to parathyroid hormone-related protein (PTHrP) deficiency in the various craniofacial cartilages. Anat Rec. 1999;255:452–457. doi: 10.1002/(SICI)1097-0185(19990801)255:4<452::AID-AR10>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Bianchi C, Nadal-Ginard B, Homcy CJ. Alternative promoter and 5′ exon generate a novel Gsα mRNA. J Biol Chem. 1990;265:8458–8462. [PubMed] [Google Scholar]
- Jan de Beur S, Ding C, Germain-Lee EL, Cho J, Maret A, Levine MA. Discordance between genetic and epigenetic defects in pseudohypoparathyroidism type 1b revealed by inconsistent loss of maternal imprinting at GNAS1. Am J Hum Genet. 2003;73:314–322. doi: 10.1086/377136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobert AS, Zhang P, Couvineau A, Bonaventure J, Roume J, LeMerrer M, et al. Absence of functional receptors for parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J Clin Invest. 1998;102:34–40. doi: 10.1172/JCI2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Masters SB, Bourne HR, Reed RR. Biochemical characterization of three stimulatory GTP-binding proteins. The large and small forms of Gs and the olfactory-specific G-protein, Golf. J Biol Chem. 1990;265:2671–2676. [PubMed] [Google Scholar]
- Jones DT, Reed RR. Golf: an olfactory neuron specific-G protein involved in odorant signal transduction. Science. 1989;244:790–795. doi: 10.1126/science.2499043. [DOI] [PubMed] [Google Scholar]
- Kaplan FS, Shore EI. Progressive osseous heteroplasia. J Bone Miner Res. 2000;15:2084–2094. doi: 10.1359/jbmr.2000.15.11.2084. [DOI] [PubMed] [Google Scholar]
- Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VLJ, Kronenberg HM, et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, et al. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- Kehlenbach RH, Matthey J, Huttner WB. XLαs is a new type of G protein. Nature. 1994;372:804–809. doi: 10.1038/372804a0. [DOI] [PubMed] [Google Scholar]
- Kelsey G, Bodle D, Miller HJ, Beechey CV, Coombes C, Peters J, et al. Identification of imprinted loci by methylation-sensitive representational difference analysis: application to mouse distal chromosome 2. Genomics. 1999;62:129–138. doi: 10.1006/geno.1999.6022. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Gonen D, Hanna GL, Leventhal BL, Cook EH., Jr Deletion polymorphism in the coding region of the human NESP55 alternative transcript of GNAS1. Mol Cell Probes. 2000;14:191–194. doi: 10.1006/mcpr.2000.0300. [DOI] [PubMed] [Google Scholar]
- Klemke M, Kehlenbach RH, Huttner WB. Two overlapping reading frames in a single exon encode interacting proteins-a novel way of gene usage. EMBO J. 2001;20:3849–3860. doi: 10.1093/emboj/20.14.3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemke M, Pasolli HA, Kehlenbach RH, Offermanns S, Schultz G, Huttner WB. Characterization of the extra-large G protein α-subunit XLαs. II. Signal transduction properties. J Biol Chem. 2000;275:33633–33640. doi: 10.1074/jbc.M006594200. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Chung U-i, Schipani E, Starbuck M, Karsenty G, Katagiri T, et al. PTHrP and Indian hedgehog control differentiation of growth plate chondrocytes at multiple steps. Development. 2002;129:2977–2986. doi: 10.1242/dev.129.12.2977. [DOI] [PubMed] [Google Scholar]
- Kozasa T, Itoh H, Tsukamoto T, Kaziro Y. Isolation and characterization of the human Gs alpha gene. Proc Natl Acad Sci USA. 1988;85:2081–2085. doi: 10.1073/pnas.85.7.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieger-Brauer HI, Medda P, Kather H. Basic fibroblast growth factor utilized both types of component subunits of Gs for dual signaling in human adipocytes. Stimulation of adenylyl cyclase vis Gαs and inhibition of NADPH oxidase by Gβγs. J Biol Chem. 2000;275:35920–35925. doi: 10.1074/jbc.M002490200. [DOI] [PubMed] [Google Scholar]
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- Lambright DG, Noel JP, Hamm HE, Sigler PB. Structural determinants for activation of the α-subunit of a heterotrimeric G protein. Nature. 1994;369:621–628. doi: 10.1038/369621a0. [DOI] [PubMed] [Google Scholar]
- Lambright DG, Sondek J, Bohm A, Skiba NP, Hamm HE, Sigler PB. The 2.0A crystal structure of a heterotrimeric G protein. Nature. 1996;379:297–299. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340:692–696. doi: 10.1038/340692a0. [DOI] [PubMed] [Google Scholar]
- Lanske B, Amling M, Neff L, Guiducci J, Baron R, Kronenberg HM. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. J Clin Invest. 1999;104:399–407. doi: 10.1172/JCI6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanske B, Karaplis AC, Lee K, Luz A, Vortkamp A, Pirro A, et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth [see comments] Science. 1996;273:663–666. doi: 10.1126/science.273.5275.663. [DOI] [PubMed] [Google Scholar]
- Levine MA. Clinical implications of genetic defects in G proteins: oncogenic mutations in Gαs as the molecular basis for the McCune-Albright syndrome. Arch Med Res. 1999;30:522–531. doi: 10.1016/s0188-4409(99)00075-2. [DOI] [PubMed] [Google Scholar]
- Levine MA, Modi WS, O’Brien SJ. Mapping of the gene encoding the α subunit of the stimulatory G protein of adenylyl cyclase (GNAS1) to 20q13.2-q13.3 in human by in situ hybridization. Genomics. 1991;11:478–479. doi: 10.1016/0888-7543(91)90164-a. [DOI] [PubMed] [Google Scholar]
- Li QB, Cerione RA. Communication between switch II and switch III of the transducin α subunit is essential for target activation. J Biol Chem. 1997;272:21673–21676. doi: 10.1074/jbc.272.35.21673. [DOI] [PubMed] [Google Scholar]
- Li T, Vu TH, Zeng ZL, Nguyen BT, Hayward BE, Bonthron DT, et al. Tissue-specific expression of antisense and sense transcripts at the imprinted Gnas locus. Genomics. 2000;69:295–304. doi: 10.1006/geno.2000.6337. [DOI] [PubMed] [Google Scholar]
- Linder ME, Middleton P, Hepler JR, Taussig R, Gilman AG, Mumby SM. Lipid modifications of G proteins: α subunits are palmitoylated. Proc Natl Acad Sci USA. 1993;90:3675–3679. doi: 10.1073/pnas.90.8.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linglart A, Gensure RC, Olney RC, Juppner H, Bastepe M. A novel STX16 deletion in autosomal dominant pseudohypoparathyroidism type Ib redefines the boundaries of a cis-acting imprinting control element of GNAS. Am J Hum Genet. 2005;76:804–814. doi: 10.1086/429932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Chen M, Deng C, Bourc’his D, Nealon JG, Erlichman B, et al. Identification of the control region for tissue-specific imprinting of the stimulatory G protein α-subunit. Proc Natl Acad Sci USA. 2005a;102:5513–5518. doi: 10.1073/pnas.0408262102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Erlichman B, Weinstein LS. The stimulatory G protein α-subunit Gsα is imprinted in human thyroid glands: implications for thyroid function in pseudohypoparathyroidism types 1A and 1B. J Clin Endocrinol Metab. 2003;88:4336–4341. doi: 10.1210/jc.2003-030393. [DOI] [PubMed] [Google Scholar]
- Liu J, Litman D, Rosenberg MJ, Yu S, Biesecker LG, Weinstein LS. A GNAS1 imprinting defect in pseudohypoparathyroidism type IB. J Clin Invest. 2000a;106:1167–1174. doi: 10.1172/JCI10431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nealon JG, Weinstein LS. Distinct patterns of abnormal GNAS imprinting in familial and sporadic pseudohypoparathyroidism type IB. Hum Mol Genet. 2005b;14:95–102. doi: 10.1093/hmg/ddi009. [DOI] [PubMed] [Google Scholar]
- Liu J, Yu S, Litman D, Chen W, Weinstein LS. Identification of a methylation imprint mark within the mouse Gnas locus. Mol Cell Biol. 2000b;20:5808–5817. doi: 10.1128/mcb.20.16.5808-5817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long DN, McGuire S, Levine MA, Weinstein LS, Germain-Lee EL. Body mass index differences in pseudohypoparathyroidism type 1a versus pseudopseudohypoparathyroidism may implicate paternal imprinting of Gαs in the development of human obesity. J Clin Endocrinol Metab. 2006 doi: 10.1210/jc.2006-1497. published online. [DOI] [PubMed] [Google Scholar]
- Lovisetti-Scamiform P, Fischer-Colbrie R, Leitner B, Scherzer G, Winkler H. Relative amounts and molecular forms of NESP55 in various bovine tissues. Brain Res. 1999;829:99–106. doi: 10.1016/s0006-8993(99)01345-1. [DOI] [PubMed] [Google Scholar]
- Lyons J, Landis CA, Harsh G, Vallar L, Grünewald K, Feichtinger H, et al. Two G protein oncogenes in human endocrine tumors. Science. 1990;249:655–659. doi: 10.1126/science.2116665. [DOI] [PubMed] [Google Scholar]
- Ma Y, Huang J, Ali S, Lowry W, Huang X. Src tyrosine kinase is a novel direct effector of G proteins. Cell. 2000;102:635–646. doi: 10.1016/s0092-8674(00)00086-6. [DOI] [PubMed] [Google Scholar]
- Mantovani G, Ballare E, Giammona E, Beck-Peccoz P, Spada A. The Gsα gene: predominant maternal origin of transcription in human thyroid gland and gonads. J Clin Endocrinol Metab. 2002;87:4736–4740. doi: 10.1210/jc.2002-020183. [DOI] [PubMed] [Google Scholar]
- Mantovani G, Bondioni S, Locatelli M, Pedroni C, Lania AG, Ferrante E, et al. Biallelic expression of the Gsα gene in human bone and adipose tissue. J Clin Endocrinol Metab. 2004;89:6316–6319. doi: 10.1210/jc.2004-0558. [DOI] [PubMed] [Google Scholar]
- Marsh SR, Grishina G, Wilson PT, Berlot CH. Receptor-mediated activation of Gsα: evidence for intramolecular signal transduction. Mol Pharmacol. 1998;53:981–990. [PubMed] [Google Scholar]
- Mattera R, Graziano MP, Yatani A, Zhou Z, Graf R, Codina J, et al. Splice variants of the α-subunit of the G protein Gs activate both adenylyl cyclase and calcium channels. Science. 1989;243:804–807. doi: 10.1126/science.2536957. [DOI] [PubMed] [Google Scholar]
- Mazzoni MR, Taddei S, Giusti L, Rovero P, Galoppini C, D’Ursi A, et al. A Gαs carboxyl-terminal peptide prevents Gs activation by the A2A adenosine receptor. Mol Pharmacol. 2000;58:226–236. doi: 10.1124/mol.58.1.226. [DOI] [PubMed] [Google Scholar]
- Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X. Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem. 2002;277:11497–11504. doi: 10.1074/jbc.M110856200. [DOI] [PubMed] [Google Scholar]
- Miao D, He B, Karaplis AC, Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. J Clin Invest. 2002;109:1173–1182. doi: 10.1172/JCI14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Prigent S, Kupperman E, Rioux L, Park SH, Feramisco JR, et al. RalGDS functions in Ras- and cAMP-mediated growth stimulation. J Biol Chem. 1997;272:5600–5605. doi: 10.1074/jbc.272.9.5600. [DOI] [PubMed] [Google Scholar]
- Mixon MB, Lee E, Coleman DE, Berghuis AM, Gilman AG, Sprang SR. Tertiary and quaternary structural changes in Giα1. Science. 1995;270:954–960. doi: 10.1126/science.270.5238.954. [DOI] [PubMed] [Google Scholar]
- Montminy M. Transcriptional regulation by cyclic AMP. Annu Rev Biochem. 1997;66:807–822. doi: 10.1146/annurev.biochem.66.1.807. [DOI] [PubMed] [Google Scholar]
- Moore T, Haig D. Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet. 1991;7:45–49. doi: 10.1016/0168-9525(91)90230-N. [DOI] [PubMed] [Google Scholar]
- Mumby SM, Kleuss C, Gilman AG. Receptor regulation of G-protein palmitoylation. Proc Natl Acad Sci USA. 1994;91:2800–2804. doi: 10.1073/pnas.91.7.2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair BG, Parikh B, Milligan G, Patel TB. Gsα mediates epidermal growth factor-elicited stimulation of rat cardiac adenylate cyclase. J Biol Chem. 1990;265:21317–21322. [PubMed] [Google Scholar]
- Nair BG, Patel TB. Regulation of cardiac adenylyl cyclase by epidermal growth factor (EGF). Role of EGF receptor protein tyrosine kinase activity. Biochem Pharmacol. 1993;46:1239–1245. doi: 10.1016/0006-2952(93)90473-a. [DOI] [PubMed] [Google Scholar]
- Namnoum AB, Merriam GR, Moses AM, Levine MA. Reproductive dysfunction in women with Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab. 1998;83:824–829. doi: 10.1210/jcem.83.3.4652. [DOI] [PubMed] [Google Scholar]
- Noel JP, Hamm HE, Sigler PB. The 2.2 Å crystal structure of transducin-α complexed with GTPgS. Nature. 1993;366:654–663. doi: 10.1038/366654a0. [DOI] [PubMed] [Google Scholar]
- Pasolli HA, Huttner WB. Expression of the extra-large G protein α-subunit XLαs in neuroepithelial cells and young neurons during development of the rat nervous system. Neurosci Lett. 2001;301:119–122. doi: 10.1016/s0304-3940(01)01620-2. [DOI] [PubMed] [Google Scholar]
- Pasolli HA, Klemke M, Kehlenbach RH, Wang Y, Huttner WB. Characterization of the extra-large G protein α-subunit XLαs. I. Tissue distribution and subcellular localization. J Biol Chem. 2000;275:33622–33632. doi: 10.1074/jbc.M001335200. [DOI] [PubMed] [Google Scholar]
- Peters J, Beechey CV, Ball ST, Evans EP. Mapping studies of the distal imprinting region of mouse chromosome 2. Genet Res. 1994;63:169–174. doi: 10.1017/s0016672300032316. [DOI] [PubMed] [Google Scholar]
- Peters J, Wroe SF, Wells CA, Miller HJ, Bodle D, Beechey CV, et al. A cluster of oppositely imprinted transcripts at the Gnas locus in the distal imprinting region of mouse chromosome 2. Proc Natl Acad Sci USA. 1999;96:3830–3835. doi: 10.1073/pnas.96.7.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagge A, Gordon E, Dean W, Boiani R, Cinti S, Peters J, et al. The imprinted signaling protein XLαs is required for postnatal adaptation to feeding. Nat Genet. 2004;36:818–826. doi: 10.1038/ng1397. [DOI] [PubMed] [Google Scholar]
- Plagge A, Isles AR, Gordon E, Humby T, Dean W, Gritsch S, et al. Imprinted Nesp55 influences behavioral reactivity to novel environments. Mol Cell Biol. 2005;25:3019–3026. doi: 10.1128/MCB.25.8.3019-3026.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppleton H, Sun H, Fulgham D, Bertics P, Patel TB. Activation of Gsα by the epidermal growth factor receptor involves phosphorylation. J Biol Chem. 1996;271:6947–6951. doi: 10.1074/jbc.271.12.6947. [DOI] [PubMed] [Google Scholar]
- Rao VV, Schnittger S, Hansmann I. G protein Gsα (GNAS1), the probable candidate gene for Albright hereditary osteodystrophy, is assigned to human chromosome 20q12–q13.2. Genomics. 1991;10:257–261. doi: 10.1016/0888-7543(91)90508-c. [DOI] [PubMed] [Google Scholar]
- Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. 2001;2:21–32. doi: 10.1038/35047554. [DOI] [PubMed] [Google Scholar]
- Sakamoto A, Chen M, Kobayashi T, Kronenberg HM, Weinstein LS. Chondrocyte-specific knockout of the G protein Gsα leads to epiphyseal and growth plate abnormalities and ectopic chondrocyte formation. J Bone Miner Res. 2005a;20:663–671. doi: 10.1359/JBMR.041210. [DOI] [PubMed] [Google Scholar]
- Sakamoto A, Chen M, Nakamura T, Xie T, Karsenty G, Weinstein LS. Deficiency of the G-protein α-subunit Gsα in osteoblasts leads to differential effects on trabecular and cortical bone. J Biol Chem. 2005b;280:21369–21375. doi: 10.1074/jbc.M500346200. [DOI] [PubMed] [Google Scholar]
- Sakamoto A, Liu J, Greene A, Chen M, Weinstein LS. Tissue-specific imprinting of the G protein α-subunit Gsα is associated with tissue-specific differences in histone methylation. Hum Mol Genet. 2004;15:819–828. doi: 10.1093/hmg/ddh098. [DOI] [PubMed] [Google Scholar]
- Scholich K, Mullenix JB, Wittpoth C, Poppleton HM, Pierre SC, Lindorfer MA, et al. Facilitation of signal onset and termination by adenylyl cyclase. Science. 1999;283:1328–1331. doi: 10.1126/science.283.5406.1328. [DOI] [PubMed] [Google Scholar]
- Schwindinger WF, Fredericks J, Watkins L, Robinson H, Bathon JM, Pines M, et al. Coupling of the PTH/PTHrP receptor to multiple G-proteins: direct demonstration of receptor activation of Gs, Gq/11, and Gi1 by [α-32P]GTP-γ-azidoanilide photoaffinity labeling. Endocrine. 1998;8:201–209. doi: 10.1385/ENDO:8:2:201. [DOI] [PubMed] [Google Scholar]
- Schwindinger WF, Miric A, Zimmerman D, Levine MA. A novel Gsα mutant in a patient with Albright hereditary osteodystrophy uncouples cell surface receptors from adenylyl cyclase. J Biol Chem. 1994;269:25387–25391. [PubMed] [Google Scholar]
- Schwindinger WF, Reese KJ, Lawler AM, Gearhart JD, Levine MA. Targeted disruption of Gnas in embryonic stem cells. Endocrinology. 1997;138:4058–4063. doi: 10.1210/endo.138.10.5439. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K, Lee TW, Gether U, Sanders-Bush E, Kobilka BK. Different effects of Gsα splice variants on β2-adrenoreceptor-mediated signaling. The β2-adrenoreceptor coupled to the long splice variant of Gsα has properties of a constitutively active receptor. J Biol Chem. 1998;273:5109–5116. [PubMed] [Google Scholar]
- Shenker A, Weinstein LS, Moran A, Pescovitz OH, Charest NJ, Boney CM, et al. Severe endocrine and nonendocrine manifestations of the McCune-Albright syndrome associated with activating mutations of stimulatory G protein Gs. J Pediatr. 1993;123:509–518. doi: 10.1016/s0022-3476(05)80943-6. [DOI] [PubMed] [Google Scholar]
- Shore EM, Ahn J, Jan de Beur S, Li M, Xu M, Gardner RJM, et al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346:99–106. doi: 10.1056/NEJMoa011262. [DOI] [PubMed] [Google Scholar]
- Simonds WF, Goldsmith PK, Woodard CJ, Unson CG, Spiegel AM. Receptor and effector interactions of Gs. Functional studies with antibodies to the αs carboxyl-terminal decapeptide. FEBS Lett. 1989;249:189–194. doi: 10.1016/0014-5793(89)80622-2. [DOI] [PubMed] [Google Scholar]
- Skinner JA, Cattanach BM, Peters J. The imprinted oedematous-small mutation on mouse chromosome 2 identifies new roles for Gnas and Gnasxl in development. Genomics. 2002;80:373–5. doi: 10.1006/geno.2002.6842. [DOI] [PubMed] [Google Scholar]
- Sleutels F, Zwart R, Barlow DP. The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature. 2002;415:810–813. doi: 10.1038/415810a. [DOI] [PubMed] [Google Scholar]
- Sondek J, Lambright DG, Noel JP, Hamm HE, Sigler PB. GTPase mechanism of G proteins from the 1.7-Å crystal structure of transducin α-GDP-AlF4−. Nature. 1994;372:276–279. doi: 10.1038/372276a0. [DOI] [PubMed] [Google Scholar]
- Spiegel AM, Weinstein LS. Pseudohypoparathyroidism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8. New York: McGraw-Hill; 2001. pp. 4205–4221. [Google Scholar]
- Spiegel AM, Weinstein LS. Inherited diseases involving G proteins and G protein-coupled receptors. Annu Rev Med. 2004;55:27–39. doi: 10.1146/annurev.med.55.091902.103843. [DOI] [PubMed] [Google Scholar]
- Stone MD, Hosking DJ, Garcia-Himmelstine C, White DA, Rosenblum D, Worth HG. The renal response to exogenous parathyroid hormone in treated pseudohypoparathyroidism. Bone. 1993;14:727–735. doi: 10.1016/8756-3282(93)90204-n. [DOI] [PubMed] [Google Scholar]
- Sudlow LC, Huang RC, Green DJ, Gillette R. cAMP-activated Na+ cuurent of molluscum neurons is resistant to kinase inhibitors and is gated by cAMP in the isolated patch. J Neurosci. 1993;13:5188–5193. doi: 10.1523/JNEUROSCI.13-12-05188.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KA, Miller RT, Masters SB, Beiderman B, Heideman W, Bourne HR. Identification of receptor contact site involved in receptor-G protein coupling. Nature. 1987;330:758–760. doi: 10.1038/330758a0. [DOI] [PubMed] [Google Scholar]
- Sun H, Chen Z, Poppleton H, Scholich K, Mullenix J, Weipz GJ, et al. The juxtamembrane, cytosolic region of the epidermal growth factor receptor is involved in association with α-subunit of Gs. J Biol Chem. 1997;272:5413–5420. [PubMed] [Google Scholar]
- Sun Y, Huang J, Xiang Y, Bastepe M, Juppner H, Kobilka BK, et al. Dosage-dependent switch from G protein-coupled to G protein-independent signaling by a GPCR. EMBO J. 2007;26:53–64. doi: 10.1038/sj.emboj.7601502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunahara RK, Tesmer JJG, Gilman AG, Sprang SR. Crystal structure of the adenylyl cyclase activator Gsα. Science. 1997;278:1943–1947. doi: 10.1126/science.278.5345.1943. [DOI] [PubMed] [Google Scholar]
- Tesmer JJ, Sunahara RK, Gilman AG, Sprang SR. Crystal structure of the catalytic domains of adenylyl cyclase in a complex with Gsα. GTPγS. Science. 1997;278:1907–1916. doi: 10.1126/science.278.5345.1907. [DOI] [PubMed] [Google Scholar]
- Ugur O, Jones TLZ. A proline-rich region and nearby cysteine residues target XLαs to the Golgi complex region. Mol Biol Cell. 2000;11:1421–1432. doi: 10.1091/mbc.11.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vortherms TA, Nguyen CH, Bastepe M, Juppner H, Watts VJ. D2 dopamine receptor-induced sensitization of adenylyl cyclase type 1 is Gαs independent. Neuropharmacology. 2006;50:576–584. doi: 10.1016/j.neuropharm.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, DeGennaro M, Santoro B, Siegelbaum SA, Tibbs GR. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411:805–810. doi: 10.1038/35081088. [DOI] [PubMed] [Google Scholar]
- Wall MA, Coleman DE, Lee E, Iñiguez-Lluhi JA, Posner BA, Gilman AG, et al. The structure of the G protein heterotrimer Giα1β1γ2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- Wand G, Levine MA, Zweifel L, Schwindinger W, Abel T. The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J Neurosci. 2001;27:5297–5303. doi: 10.1523/JNEUROSCI.21-14-05297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Dillon TJ, Pokala V, Mishra S, Labudda K, Hunter B, et al. Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol Cell Biol. 2006;26:2130–2145. doi: 10.1128/MCB.26.6.2130-2145.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner DR, Romanowski R, Yu S, Weinstein LS. Mutagenesis of the conserved residue Glu259 of Gsα demonstrates the importance of interactions between switches 2 and 3 for activation. J Biol Chem. 1999;274:4977–4984. doi: 10.1074/jbc.274.8.4977. [DOI] [PubMed] [Google Scholar]
- Warner DR, Weinstein LS. A mutation in the heterotrimeric stimulatory guanine nucleotide binding protein α-subunit with impaired receptor-mediated activation because of elevated GTPase activity. Proc Natl Acad Sci USA. 1999;96:4268–4272. doi: 10.1073/pnas.96.8.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner DR, Weng G, Yu S, Matalon R, Weinstein LS. A novel mutation in the switch 3 region of Gsα in a patient with Albright hereditary osteodystrophy impairs GDP binding and receptor activation. J Biol Chem. 1998;273:23976–23983. doi: 10.1074/jbc.273.37.23976. [DOI] [PubMed] [Google Scholar]
- Wedegaertner PB, Bourne HR, Von Zastrow M. Activation-induced subcellular redistribution of Gsα. Mol Biol Cell. 1996;7:1225–1233. doi: 10.1091/mbc.7.8.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein LS. Editorial: The stimulatory G protein α-subunit gene: mutations and imprinting lead to complex phenotypes. J Clin Endocrinol Metab. 2001;86:4622–4626. doi: 10.1210/jcem.86.10.8007. [DOI] [PubMed] [Google Scholar]
- Weinstein LS. Other skeletal diseases resulting from G protein defects- fibrous dysplasia and McCune-Albright syndrome. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of Bone Biology. Vol. 2. San Diego: Academic Press; 2002. pp. 1165–1176. [Google Scholar]
- Weinstein LS. GNAS and McCune-Albright syndrome/fibrous dysplasia, Albright hereditary osteodystrophy/pseudohypoparathyroidism type 1A, progressive osseous heteroplasia, and pseudohypoparathyroidism type 1B. In: Epstein CJ, Erickson RP, Wynshaw-Boris A, editors. Molecular Basis of Inborn Errors of Development. 1. San Francisco: Oxford University Press; 2004. pp. 849–866. [Google Scholar]
- Weinstein LS, Liu J, Sakamoto A, Xie T, Chen M. Minireview: GNAS: Normal and Abnormal Functions. Endocrinology. 2004;145:5459–5464. doi: 10.1210/en.2004-0865. [DOI] [PubMed] [Google Scholar]
- Weinstein LS, Yu S, Ecelbarger CA. Variable imprinting of the heterotrimeric G protein Gs α-subunit within different segments of the nephron. Am J Physiol. 2000;278:F507–F514. doi: 10.1152/ajprenal.2000.278.4.F507. [DOI] [PubMed] [Google Scholar]
- Weinstein LS, Yu S, Warner DR, Liu J. Endocrine manifestations of stimulatory G protein α-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001;22:675–705. doi: 10.1210/edrv.22.5.0439. [DOI] [PubMed] [Google Scholar]
- Williamson CM, Ball ST, Nottingham WT, Skinner JA, Plagge A, Turner MD, et al. A cis-acting control region is required exclusively for the tissue-specific imprinting of Gnas. Nat Genet. 2004;36:894–899. doi: 10.1038/ng1398. [DOI] [PubMed] [Google Scholar]
- Williamson CM, Beechey CV, Papworth D, Wroe SF, Wells CA, Cobb L, et al. Imprinting of distal mouse chromosome 2 is associated with phenotypic anomalies in utero. Genet Res. 1998;72:255–265. doi: 10.1017/s0016672398003528. [DOI] [PubMed] [Google Scholar]
- Williamson CM, Schofield J, Dutton ER, Seymour A, Beechey CV, Edwards YH, et al. Glomerular-specific imprinting of the mouse Gsα gene: How does this relate to hormone resistance in Albright hereditary osteodystophy? Genomics. 1996;36:280–287. doi: 10.1006/geno.1996.0463. [DOI] [PubMed] [Google Scholar]
- Williamson CM, Skinner JA, Kelsey G, Peters J. Alternative non-coding splice variants of Nespas, an imprinted gene antisense to Nesp in the Gnas imprinting cluster. Mamm Genome. 2002;13:74–79. doi: 10.1007/s00335-001-2102-2. [DOI] [PubMed] [Google Scholar]
- Williamson CM, Turner MD, Ball ST, Nottingham WT, Glenister P, Fray M, et al. Identification of an imprinting control region affecting the expression of all transcripts in the Gnas cluster. Nat Genet. 2006;38:350–355. doi: 10.1038/ng1731. [DOI] [PubMed] [Google Scholar]
- Wroe SF, Kelsey G, Skinner JA, Bodle D, Ball ST, Beechey CV, et al. An imprinted transcript, antisense to Nesp, adds complexity to the cluster of imprinted genes at the mouse Gnas locus. Proc Natl Acad Sci USA. 2000;97:3342–3346. doi: 10.1073/pnas.050015397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Plagge A, Gavrilova O, Pack S, Jou W, Lai EW, et al. The alternative stimulatory G protein α-subunit XLαs is a critical regulator of energy and glucose metabolism and sympathetic nerve activity in adult mice. J Biol Chem. 2006;281:18989–18999. doi: 10.1074/jbc.M511752200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Lee FY, Sr, Wand GS. Increased expression of Gsα enhances activation of the adenylyl cyclase signal transduction cascade. Mol Endocrinol. 1997;11:1053–1061. doi: 10.1210/mend.11.8.9957. [DOI] [PubMed] [Google Scholar]
- Yatani A, Imoto Y, Codina J, Hamilton SL, Brown AM, Birnbaumer L. The stimulatory G protein of adenylyl cyclase, Gs, also stimulates dihydropyridine-sensitive Ca2+ channels. Evidence for direct regulation independent of phosphorylation by cAMP-dependent protein kinase or stimulation by a dihydropyridine agonist. J Biol Chem. 1988;263:9887–9895. [PubMed] [Google Scholar]
- Yeh GL, Mathur S, Wivel A, Li M, Gannon FH, Ulied A, et al. GNAS1 mutation and Cbfa1 misexpression in a child with severe congenital platelike osteoma cutis. J Bone Miner Res. 2000;15:2063–2073. doi: 10.1359/jbmr.2000.15.11.2063. [DOI] [PubMed] [Google Scholar]
- Yu JZ, Rasenick MM. Real-time visualization of a fluorescent Gαs: dissociation of the activated G protein from plasma membrane. Mol Pharmacol. 2002;61:352–359. doi: 10.1124/mol.61.2.352. [DOI] [PubMed] [Google Scholar]
- Yu S, Castle A, Chen M, Lee R, Takeda K, Weinstein LS. Increased insulin sensitivity in Gsα knockout mice. J Biol Chem. 2001;276:19994–19998. doi: 10.1074/jbc.M010313200. [DOI] [PubMed] [Google Scholar]
- Yu S, Gavrilova O, Chen H, Lee R, Liu J, Pacak K, et al. Paternal versus maternal transmission of a stimulatory G protein α subunit knockout produces opposite effects on energy metabolism. J Clin Invest. 2000;105:615–623. doi: 10.1172/JCI8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Yu D, Lee E, Eckhaus ME, Lee R, Corria Z, et al. Variable and tissue-specific hormone resistance in heterotrimeric Gs protein α-subunit (Gsα) knockout mice is due to tissue-specific imprinting of the Gsα gene. Proc Natl Acad Sci USA. 1998;95:8715–8720. doi: 10.1073/pnas.95.15.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Bastepe M, Juppner H, Ruan KH. Characterization of the molecular mechanisms of the coupling between intracellular loops of prostacyclin receptor with the C-terminal domain of the Gαs protein in human coronary artery smooth muscle cells. Arch Biochem Biophys. 2006;454:80–88. doi: 10.1016/j.abb.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Zheng B, Ma Y, Ostrom RS, Lavoie C, Gill GN, Insel PA, et al. RGS-PX1, a GAP for Gαs and sorting nexin in vesicular trafficking. Science. 2001;294:1939–1942. doi: 10.1126/science.1064757. [DOI] [PubMed] [Google Scholar]