

Abstract
Yellow 9-methyldipyrrinones can be converted readily and in high yields to symmetric linear tetrapyrroles, blue biliverdinoids, which are cleaved in half, smoothly at room temperature to afford yellow 9-H dipyrrinones, and 9-CHO dipyrrinones as their violet to orange colored adducts with the carbon acid used for the scission: thiobarbituric acid (TBA), N,N′-diethylthiobarbituric acid, barbituric acid, N,N′-dimethylbarbituric acid and Meldrum’s acid. The adducts, usually only of passing interest, are formally Knövenagel condensation products of a 9-CHO dipyrrinone with TBA and other carbon acids of this work, and a reverse Knövenagel reaction of such adducts leads to 9-CHO dipyrrinones. Under a set of improved reaction conditions the sequence thus efficiently converts 9-CH3 dipyrrinones to 9-H and 9-CHO dipyrrinones.
Keywords: Pyrroles, Biliverdinoids, Retro-Knövenagel reaction, Carbon Acids
1. Introduction
Dipyrrinones1 are the chromophores of bilirubin (Scheme 1), the yellow-orange pigment of mammalian bile and of jaundice, and they also constitute the two halves of biliverdin (Scheme 1), the blue-green biological precursor of bilirubin and the pigment of non-mammalian bile.2 In both bilirubin and biliverdin, the dipyrrinone units are connected by a single carbon, C(10). Although these pigments are not biosynthesized in nature by conjoining two dipyrrinones, bilirubin and biliverdin analogs have been prepared synthetically by coupling two 9-H dipyrrinones with formaldehyde or its equivalent, or by coupling a 9-formyldipyrrinone with a 9-H dipyrrinone ? or even by oxidative coupling of 9-methyldipyrrinones.1 The 9-H dipyrrinone precursors, as well as 9-formyldipyrrinones have been prepared by synthesis, typically from monopyrroles. 9-Methyldipyrrinones are likewise synthesized from monopyrroles, but direct conversion of these synthetically more accessible pigments to synthetically useful 9-H dipyrrinones has not been achieved.
Scheme 1.
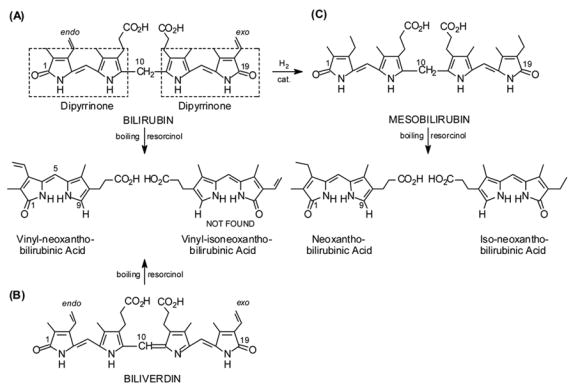
Linear representations of (A) bilirubin, (B) biliverdin and (C) mesobilirubin and their 9-H dipyrrinone products isolated from molten resorcinol.
In the mid-1920s Hans Fischer renewed his investigations of the constitutional structure of bilirubin and learned subsequently that bilirubins and biliverdins are cleaved to 9-H dipyrrinones in boiling resorcinol. Under brief reaction, bilirubin, its dimethyl ester and biliverdin dimethyl ester afforded only low yields of vinyl-neoxanthobilirubinic acid (Scheme 1) or its methyl ester ? all with an endo vinyl group.3 The “other half” of the tetrapyrrole, with the exo-vinyl group, was not recovered ? an observation that led Fischer to first assume a symmetrically-substituted linear tetrapyrrole structure for bilirubin (which he subsequently disproved). In contrast, mesobilirubin cleaves to a good yield of a mixture of neo- and iso-neo-xanthobilirubinic acids (Scheme 1) that proved difficult to separate at the time.
Some 50 years later, Manitto and Monti4 demonstrated a novel, less vigorous and high yield fragmentation of biliverdin and its symmetric analog, biliverdin-XIIIα dimethyl ester, a biliverdin analog with two endo-vinyl groups, to afford 9-H dipyrrinones from reaction with 1.5 equivalents of thiobarbituric acid (TBA) (Scheme 2) in methyl acetate at room temperature. The green-blue color of the verdin changed gradually over 6 h to purple; and poorly-soluble, magenta-colored TBA adducts of dipyrrinones were precipitated from chloroform-hexane in ~80% yield. The pale yellow filtrates yielded 9-H dipyrrinones. The reaction has several advantages, the most significant are its simplicity and high yields and the fact that the vinyl groups remain intact. Although unprecipitated TBA adducts render chromatographic separation of the 9-H dipyrrinones difficult, this cleavage reaction of biliverdin is probably the most convenient alternative to preparing not readily accessible 9-H dipyrrinones possessing vinyl groups, and it offers a convenient way to make other 9-H dipyrrinones,5,6,7 given the verdin.
Scheme 2.
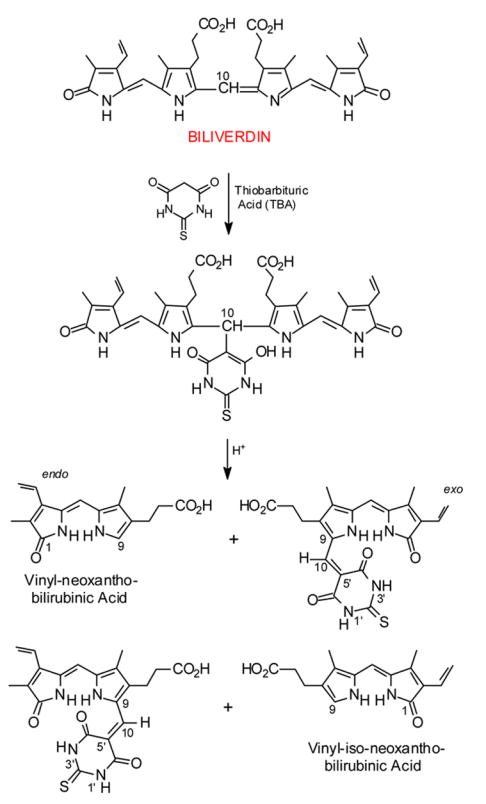
Cleavage of biliverdin into its component 9-H dipyrrinones and the complementary 9-CHO dipyrrinone thiobarbituric acid adducts (ref. 4).
When a symmetrical verdin such as biliverdin-XIIIα dimethyl ester is treated with TBA, only one 9-H dipyrrinone product is possible: vinyl-neoxanthobilirubinic acid methyl ester. In this case, as with biliverdin, one half of the verdin is “lost” to the TBA adduct, which might be viewed formally as the Knövenagel condensation product between a 9-CHO dipyrrinone and TBA. There is no evidence that a reverse Knövenagel reaction (see overview in section 2.5.) has been carried out with the adducts of Scheme 2; yet, one might imagine the adducts to be a useful source of 9-CHO dipyrrinones. In this case the TBA cleavage reaction would ultimately yield (theoretically) one equivalent of 9-H dipyrrinone and one of 9-CHO dipyrrinone, which is structurally the reverse of the verdin-forming acid-catalyzed condensation of 9-H and 9-CHO dipyrrinones. It also suggests a way for converting 9-CH3 dipyrrinones to 9-H because 9-CH3 dipyrrinones are converted smoothly and in good yields to (typically symmetrically-substituted) biliverdins.
We have reinvestigated the Manitto-Monti reaction with an eye toward (1) improving reaction conditions and product separation, (2) investigating whether the cleavage might also occur from the action of carbon acids other than TBA, and (3) converting the TBA adducts to 9-formyldipyrrinones. The verdins used in the current work are symmetric: for simplicity, etiobiliverdin-IVγ;8 and, for improved verdin and product solubility, an analog of mesobiliverdin-XIIIα with hexanoic acids replacing propionic (Fig. 1).8b The C-H acids used include TBA, its N,N′-diethyl derivative, barbituric acid (BA) and its N,N′-dimethyl derivative, and Meldrum’s acid, which exhibit acidity varying between pKa 3.75–4.83.
Figure 1.
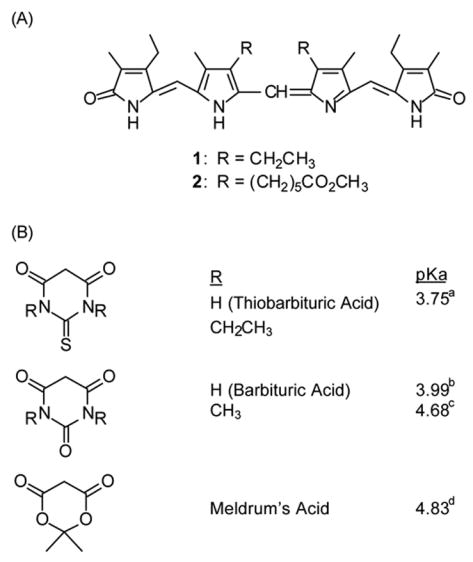
(A) The symmetric verdins and (B) the C-H acids of this work. The C-H acid pKa values may be found in the following: a ref. 10a, b ref. 10b, c ref. 10c, d ref. 10d.
2. Results and Discussion
2.1. Synthesis: 9-CH3 to 9-H Dipyrrinones
Etiobilioverdin-IVγ8 (1) and mesobiliverdin-XIIIα 8,12-bis-hexanoic acid dimethyl ester (2)8b were chosen as standard biliverdinoid reaction substrates that are readily synthesized by p-chloranil promoted self-coupling of easily available 9-CH3 dipyrrinones, kryptopyrromethenone8c,9 and xanthobilirubinic-8-hexanoic acid methyl ester,8b respectively (Scheme 3). Verdin 1 has an all-alkyl pyrrole β-periphery and possesses sufficient solubility in common organic solvents for cleavage reactions in a homogeneous phase. As described originally by Manitto and Monti,4 reaction of biliverdins with TBA suggested very low concentrations in methyl acetate (~0.6 mM), and long reaction times. In 2001, Sawamoto and Inomata6 noted greater reactant solubility and significant reaction rate acceleration by replacing methyl acetate with methanol which prompted us to use methanol solvent for reactions of verdin 1, examined in detail with TBA and with other C-H acids as projected in Scheme 3.
Scheme 3.
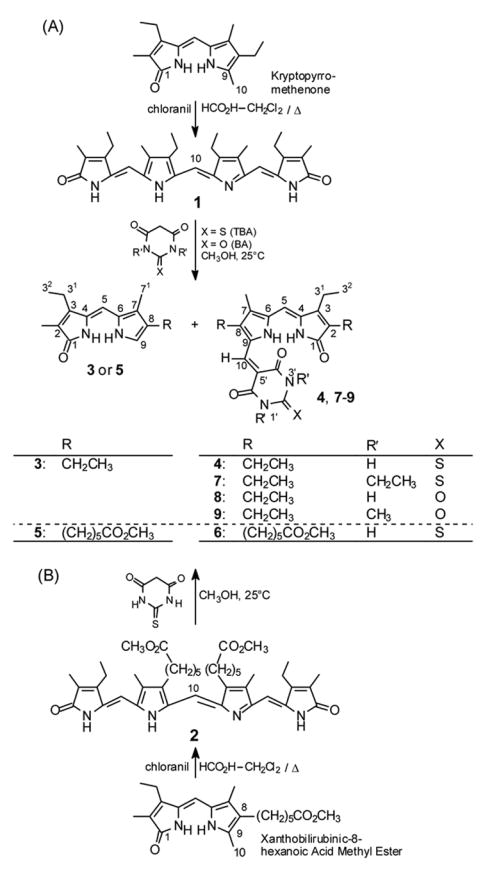
(A) Conversion of kryptopyrromethenone to etiobiliverdin-IVγ (1) and 1 to 9-H dipyrrinone 3 and adducts 4 and 7–9. (B) Conversion of xanthobilirubinic-8-hexanoic acid methyl ester to 9-H dipyrrinone 5 and TBA adduct 6.
Scheme 4.
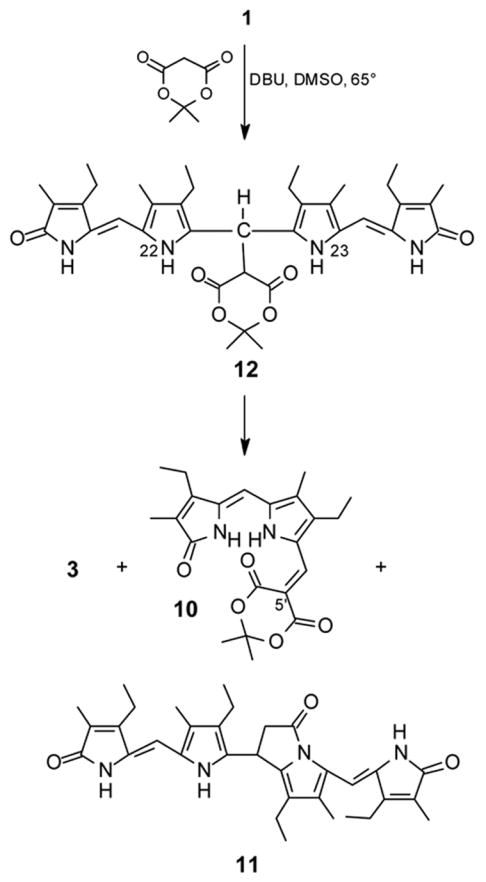
Reaction of etiobiliverdin-IVγ (1) with Meldrum’s acid (Fig. 1) to give 9-H dipyrrinone 3 (Scheme 3), adduct 10 (16% yield) and tetrapyrrole 11, with the last presumnably arising from an initially-formed tetrapyrrole adduct (12). A higher yield of 10 (46%) was obtained from refluxing methanol solvent.
The reaction mechanism postulated by Manitto and Monti (Scheme 2) involves nucleophilic addition of the TBA anion at C(10) of the verdin followed by protonation of the erstwhile verdin isopyrrole nitrogen to give a C(10) TBA substituted bilirubin intermediate that undergoes acid-catalyzed tetrapyrrole scission, formally a retro-Michael reaction. Since we did not expect complications from any second bimolecular process that might interfere with the spontaneous scission step, the reactant concentrations were increased seven-fold (to 240 mL mmol−1). Care was exercized to obtain homogeneous solutions of both the verdin and the C-H acid (2.4 equivalents) which required brief warming in some cases, then the solutions were mixed at 25°C. Optimization of the reaction time was easily achieved by following the disappearance of intense blue color of 1 or 2 by TLC. Thus, the reaction time did not exceed 2 h, but slightly longer times did not decrease the product yields. (The products 3 and 4 are stable in EtOAc for 20 h, and all reactions in CH3OH were worked up after = 2 h when TLC showed verdin disappearance.) Separation of the magenta-colored TBA adduct 4 (in 87% purified yield) from 9-H dipyrrinone analog (3) of kryptopyrromethenone (in 76% purified yield) was achieved by radial chromatography, but only on a small scale (~0.1 mmol) due to the rather low solubility of 4 in nonpolar solvents. Adduct 4 formed saturated solution in chloroform of only ~0.7 mg mL−1, and its low solubility rendered difficult its complete characterization in CDCl3 using 2D NMR techniques. The literature4–7 suggested that manipulations involving precipitation were better for working up the reaction of 1 with TBA rather than chromatography of the total crude mixture if pure 4 is not needed. This afforded a 79% yield of 3.
To overcome, at least partially, the insolubility issues encountered with 4 and to secure reliable NMR data of a TBA adduct in CDCl 3, we cleaved 2, a dimethyl bis-hexanoate analog8b of mesobiliverdin-XIIIα (Fig. 1). The reaction proceeded very well and, after chromatographic separation, TBA adduct 6 was isolated in 95% yield (recrystallization from ethyl acetate-hexane) and the 9-H dipyrrinone 5 in 76% yield (lowered by losses occurring in the last recrystallization step from CH3OH). Adduct 6, also magenta-colored, in fact exhibited enhanced solubility in CDCl3 although longer NMR acquisitions were performed at an elevated temperature in order to prevent sample crystallization.
The available literature5–7 on the Manitto-Monti4 verdin cleavage reaction focuses only on TBA as the reagent. To explore the generality of the verdin cleavage we first examined the effect of N-substitution on TBA by using 1,3-diethyl-2-thiobarbituric acid (1,3-diethyl-2-thioxo-(1H,3H,5H)pyrimidine-4,6-dione) and found that it cleaved verdin 1 to 3 and [delete +] 7, quantitatively, with adduct 7 being isolated in 92% yield (Scheme 3). A practical advantage of using the dialkylated TBA was noticed immediately: it is much more soluble than TBA in CH3OH and reacts in a somewhat shorter reaction time. Consequently, the concentrations of the reactants could be increased for convenient, large-scale preparations of dipyrrinones. The work-up and product separation by radial chromatography are also easier than in the reaction of 1 with TBA. Radial chromatographic separation of neokryptopyrromethenone 3 from the magenta-colored adduct 7 is rather easier than the separation of 3 from 4. The polarity order on silica gel is reversed relative to that observed for 3 and 4, i.e., 7 is less polar than 3 giving cleaner concentrated fractions from the chromatographic separation. Final recrystallization of these fractions yielded 92% of 7. From several experiments using diethyl TBA, a pure sample of 3 (82%) was also isolated. From the accelerated scission of 1 using diethyl TBA, one may conclude that the acidic NH protons on TBA are not involved in any crucial way in acid-base or other proton transfer reactions necessary for a successful reaction.
This investigation then led to questions as to whether a related carbon acid, barbituric acid (BA), and its N,N′-dimethyl derivative might replace TBA as potential cleavage reagents. Verdin cleavage reactions using these two carbon acids were examined in parallel. Both C-H acids reacted rapidly with etiobiliverdin-IVγ (1) under standard conditions: homogeneous solution in methanol (240 mL mmol−1) at 25°C for 2.5 hours (Scheme 3). A red-colored product (8) precipitated in 94% of the theoretical yield in >95% purity, and the filtrate afforded (after an undemanding chromatography and recrystallization) 83% yield of neokryptopyrromethenone 3. Adduct 8 is insoluble in CDCl3; therefore, its NMR spectra were acquired in only (CD3)2SO.
Reaction of 1 with dimethyl BA (Scheme 3) was also successful, but here no products precipitated, and the total crude mixture, after work-up, was separated by radial chromatography on silica gel. In contrast to diethyl TBA adduct 7, dimethyl BA adduct 9 is more polar on silica gel than neokryptopyrromethenone 3. After recrystallization, pure 9 was obtained in 88% yield as a purple solid.
Encouraged by the easy separation of 8 from 3, and predicting a further advantage in separability based on the very low solubility of BA adduct 8 in the methanol reaction medium that would facilitate the isolation of various 9-H dipyrrinones, we reacted BA with an unsymmetrical verdin, the dimethyl ester of biliverdin-IXα (Scheme 2),2 derived from natural bilirubin. The reaction proceeded rapidly, and 80–85% of the expected mixture of adducts (X=O) precipitated directly from the methanol solvent, thus indicating their increased solubility vs adduct 8, probably due to the presence of the propionic ester group. The filtrate contained a 50:50 mixture of the methyl esters of vinyl-neoxanthobilirubinic acid and vinyl-isoneoxanthobilirubinic acid (Scheme 2). These 9-H dipyrrinones are easier to obtain by this process than by the lengthy synthetic method and may be separated chromatographically.
In order to assess further the generality of the verdin scission reaction using related carbon acids, and taking note of the similar pKas of TBA (pKa 3.75)10a and BA (pKa 3.99),10b and the higher value (pKa 4.68)10c of N,N′-dimethyl BA (the pKa value of N,N′-diethyl TBA was not available), we learned that all seem to cleave the verdins of this study under the same reaction conditions: CH3OH, 25°C, 2 h. Meldrum’s acid, which has an even slightly higher pKa (4.83),10d was also found to react with verdins. However, uncatalyzed spontaneous scission of the verdin (1) did not occur in methanol at 25°C, even after 4 days. The literature indicates that Meldrum’s acid, surprisingly, does not enolize, which we link to its failure to induce spontaneous verdin scission. In order to promote reaction, triethylamine was added to remove the acidic proton at C(5′) of Meldrum’s acid, and after 3 days at room temperature, verdin 1 was converted to give a complex mixture, from which an orange, non-polar and easily crystallizable compound was isolated in 6% yield whose 1H- and 13C-NMR spectra corresponded to the expected conjugate 10 (Scheme 4).
Because dimethyl sulfoxide solvent was found to have a greater accelerating effect than methanol on the reaction rate of 1 with diethyl TBA, 1 was reacted with Meldrum’s acid in DMSO and in the presence of DBU at 65°C (reflux temperature of methanol). An orange-magenta colored pigment (characterized as 10) was isolated in 16% yield from this reaction. However, most of the verdin was consumed by conversion to a very polar yellow product, which was purified by chromatography. It is insoluble in CHCl3, sparingly soluble in CH3OH, and soluble in (CH3)2SO, and its NMR spectra in (CD3)2SO are consistent with structure 11 (Scheme 4). Transformation of 1 into 11 is feasible from an initially-formed tetrapyrrole adduct (12) that is deprotonated at N(22) or N(23), with the resulting anion attacking one of the Meldrum’s acid carbonyls. Base-catalyzed release of CO2 and (CH3)2CO from the Meldrum’s acid moiety would lead then to 11. The best results on a preparative scale for cleaving verdin 1: Meldrum’s acid (3 eq.) plus DBU (3 eq.) in refluxing methanol, gave a 46% yield of 10.
2.2. Constitutional Structures from NMR
Dipyrrinones are generally well-known in the chemical literature.1 Neokryptopyrromethenone 3, however, has been described only once ? in 1932 by H. Fischer,11 and of course no modern spectroscopic methods could have been applied. Its structure can be deduced by the method of preparation, according to the Manitto-Monti cleavage4 of the known verdin and a melting point that corresponds to that reported by Fischer.11 It was reconfirmed by its 13C NMR spectrum (Table 1), which correlates well with other known 9-H dipyrrinones, such as the tetramethyl and tetraethyl analogs.12 (The 13C (and 1H) NMR assignments of 3 were firmly established by a combination of data from 2D gHMBC and 1H{1H} NOE experiments in CDCl3.) Although the hexanoic ester analog (5) of neoxanthobilirubinic acid (Scheme 1) was unknown, its structure, too, was deduced from the known structure of the verdin precursor and the mechanism of the cleavage reaction, and was confirmed by 13C NMR.
Table 1.
1H and 13C NMR chemical shiftsa and assignments of 9-H dipyrrinones 3 and 5.
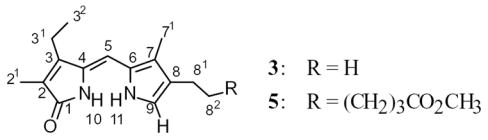
| 13C NMR | 1H NMR | ||||
|---|---|---|---|---|---|
| Carbon | 3 | 5b | 3 | 5c | |
| 1 | C = O | 174.3 | 174.3 | ? | ? |
| 2 | =C- | 123.1 | 123.2 | ? | ? |
| 21 | CH 3 | 8.0 | 8.1 | 1.96 (s) | 1.96 (s) |
| 3 | =C- | 148.4 | 148.4 | ? | ? |
| 31 | CH 2 | 18.0 | 18.0 | 2.56 (q, J=7.6) | 2.55 (t, J=7.6) |
| 32 | CH 3 | 14.9 | 14.9 | 1.19 (t, J=7.6) | 1.18 (t, J=7.6) |
| 4 | =C- | 128.3 | 128.3 | ? | ? |
| 5 | =CH- | 101.3 | 101.3 | 6.17 (s) | 6.16 (s) |
| 6 | =C- | 124.4 | 124.3 | ? | ? |
| 7 | =C- | 123.4 | 123.6 | ? | ? |
| 71 | CH 3 | 9.4 | 9.5 | 2.17 (s) | 2.13 (s) |
| 8 | =C- | 126.4 | 124.5 | ? | ? |
| 81 | CH 2 | 18.5 | 25.1 | 2.46 (t, J=7.6) | 2.43 (t, J=7.6) |
| 82 | CH 2/CH 3 | 14.6 | 30.0 | 1.21 (t, J=7.6) | 1.58 (m) |
| 9 | =C- | 120.2 | 120.8 | 6.84 (d, J=2.8) | 6.82 (d, J=2.8) |
| 10 | NH | ? | ? | 11.06 (br. s) | 11.04 (br. s) |
| 11 | NH | ? | ? | 10.46 (br. s) | 10.47 (br. s) |
Chemical shifts (δ, ppm) downfield from (CH3)4Si at 25°C in 3 × 10−3 M for 1H NMR and 2 × 10−2 M for 13C NMR solutions in CDCl3. J-values are in Hz;
24.9 ppm (84-CH2), 29.0 ppm (83-CH2), 34.1 ppm (85-CH2), 51.4 ppm (OCH3), 174.3 ppm (8 6-CO);
1.40 ppm (m, 83-CH2), 1.68 ppm (m, 84-CH2), 2.33 (t, J=7.5, 85-CH2), 3.67 ppm (OCH3);
In contrast to 3, the magenta-colored TBA adduct 4 has very low solubility in CDCl3 but it is sufficiently soluble in (CD3)2SO for NMR studies. As for 3, the 13C (and 1H) NMR assigments were firmly established in 4 by a combination of 2D gHMBC and 1H{1H} NOE experiments. Like those of 3 and 5, the 13C NMR spectra of 4 and 6 in (CD3)2SO correlate well (Table 2) with their structures assigned on the basis of the verdin cleavage mechanism of Manitto and Monti.4 One can readily detect the characteristic dipyrrinone carbon resonances, that of the C(10) methine (originally C(10) in the verdin), and the carbons of the TBA. The dipyrrinone resonances are shifted, relative to 9-H dipyrrinones, especially those of the pyrrole ring and C(4), due to field effects and conjugation across the C-10 methine between the TBA and the dipyrrinone. Remarkably, excellent correlation with only small differences in chemical shifts is found across a wide range of adducts produced in this work, from N,N′-diethyl TBA, BA, N,N′-dimethyl BA and Meldrum’s acid (7–10, respectively), whose formation is discussed below.
Table 2.
13C-NMR chemical shifts (δ)a and assignments of adducts 4 and 6–10 in (CD3)2SO.
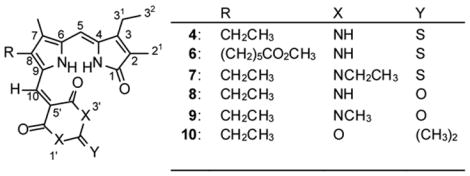
| Carbon | 4 | 6b | 7c | 8 | 9d | 10e | |
|---|---|---|---|---|---|---|---|
| 1 | C = O | 173.2 | 173.2 | 173.2 | 173.2 | 173.2 | 173.3 |
| 2 | =C- | 129.1 | 129.1 | 129.4 | 128.1 | 128.1 | 128.9 |
| 21 | CH 3 | 8.3 | 8.3 | 8.3 | 8.3 | 8.2 | 8.2 |
| 3 | =C- | 147.5 | 147.5 | 147.6 | 147.5 | 147.5 | 147.6 |
| 31 | CH 2 | 17.0 | 17.0 | 17.1 | 17.0 | 17.0 | 17.0 |
| 32 | CH 3 | 14.4 | 14.4 | 14.4 | 14.4 | 14.4 | 14.3 |
| 4 | =C- | 141.8 | 141.7 | 142.5 | 140.7 | 141.0 | 141.2 |
| 5 | =CH- | 94.3 | 94.3 | 94.1 | 94.6 | 94.5 | 94.2 |
| 6 | =C- | 140.4 | 140.3 | 141.4 | 138.4 | 138.8 | 139.5 |
| 7 | =C- | 125.6 | 125.9 | 125.9 | 124.6 | 124.7 | 124.6 |
| 77 | CH 3 | 8.9 | 9.0 | 8.9 | 8.8 | 8.8 | 8.7 |
| 8 | =C- | 144.2 | 142.6 | 145.1 | 143.1 | 143.6 | 144.2 |
| 81 | CH 2 | 17.4 | 24.1 | 17.5 | 17.4 | 17.4 | 17.3 |
| 82 | CH 3 | 16.1 | ? | 16.1 | 16.1 | 16.1 | 16.0 |
| 9 | =C- | 129.2 | 129.7 | 129.7 | 128.6 | 128.8 | 127.3 |
| 10 | =CH- | 132.7 | 132.8 | 133.8 | 133.2 | 133.8 | 134.3 |
| 2′ | C=S(O) | 176.8 | 176.8 | 177.4 | 150.1 | 151.0 | ? |
| 4′ | C=O | 162.7 | 162.7 | 160.9 | 164.5 | 162.9 | 163.9 |
| 5′ | =C- | 103.6 | 103.6 | 103.3 | 103.3 | 103.0 | 96.9 |
| 6′ | C=O | 162.9 | 162.9 | 161.1 | 165.3 | 163.3 | 164.0 |
In ppm downfield from (CH3)4Si for 2 ×10−2 M solutions in (CD3)2SO at 25°C.
30.7 ppm (δ-CH2), 28.1 ppm (γ-CH2), 23.8 ppm (β-CH2), 33.1 ppm (α-CH2), 173.2 ppm (α CO2CH3), 51.1 ppm (α-CO2CH3).
12.1 ppm (1′-CH2CH3), 43.1 ppm (1′-CH2CH3), 12.2 ppm (3′-CH2CH3), 43.2 ppm (3′-CH2CH3).
28.1 ppm (1′-CH3), 28.4 ppm (3′-CH3).
26.6 ppm (2′-CH3), 103.5 ppm (2′-C).
In the 1H NMR spectra, of particular relevance to the current study is firm assignment of the most deshielded proton NMR signals of 3 and 5 which can later be correlated with those of adducts 4, 6–9. The NMR chemical shift assignments of 3 (Table 1) are in agreement with previous extensive studies on dipyrrinones by NMR.1a,7,13,14 In CDCl 3 solvent, which promotes hydrogen bonding, the most downfield 1H NMR signal of 3 at 11.06 ppm is attributed to the lactam NH and the more shielded pyrrole NH resonates at 10.46 ppm ? both values are in the usual reported range for intermolecularly hydrogen-bonded dimers.7 The observed doublet at 6.84 ppm is assigned to C(9)-H proton, spin-coupled (3J=2.8 Hz) to the pyrrole NH whose three-bond proximity is confirmed by gHMBC. The singlet at 6.17 ppm belongs to C(5)-H methine hydrogen (for the numbering scheme, see Table 1), which is correlated by NOEs to the C(3)-ethyl and C(7)-methyl and thus supports a syn-(Z)-configuration of the exocyclic C(4)-C(5) double bond of 3, as confirmed by X-ray crystallography of similar dipyrrinones.15
It is now well-known that DMSO-d6 solvent disrupts the hydrogen-bonding network that stabilizes dimeric structures of dipyrrinones, which may be recognized by NH signals that undergo a cross-over resulting in more deshielded pyrrole NH from hydrogen bonding to solvent.1,14 Accordingly, in DMSO-d6 3 showed a pyrrole NH signal at 10.46 ppm, lactam NH at 9.69 ppm, C(9)-H at 6.71 ppm, and C(5)-CH at 5.95 ppm. The almost equal rate of deuterium exchange of the lactam and pyrrole NHs when D2O was added to a DMSO-d6 solution of 3 is much faster than in CDCl3, in which the pyrrole NH exchanges much more slowly than the lactam.16
In the 1H NMR spectra of adducts 4 and 6, the NH dipyrrinone chemical shifts are especially interesting (Table 3). In CDCl3 a very strongly deshielded nucleus exhibits a signal near 14 ppm that we assign to the pyrrole NH, and a more upfield signal near 8 ppm that we assign to the lactam NH. As will be explained later in this work, the assignments of these signals were confirmed as belonging to the dipyrrinone unit by 1H{1H} NOE measurements and their rates of N-H to N-D exchange with D2O.16 (The various assignments of the 1H NMR signals of the adducts contributed to the assignments of their 13C NMR chemical shifts by a combination of 1D and 2D experiments.) Adduct 4 is not very soluble in CDCl3; adduct 6, prepared by reaction of TBA with verdin 2, is significantly more soluble. Their 1H NMR spectra in CDCl3 were obtained with difficulty from non-homogeneous solutions, but with somewhat improved solubility in CD2Cl2 (and (CD3)2SO), one finds the 1H{1H} NOE correlations shown in Fig. 2. These data show that the dipyrrinone component of 4 (as well as 6) adopts a syn-Z conformation found in typical dipyrrinones; viz. NOE correlations show that the lactam and pyrrole NHs lie close to one another and that the C(5)-H lies proximal to the C(3)-ethyl and C(7)-methyl. The conformation about the C(9)-C(10) bond connecting the dipyrrinone and TBA is one where the C(10)-H lies close to the dipyrrinone C(8)-ethyl in 4. The conformation defined by the NOEs is thus one with a close proximity of a TBA C=O to the pyrrole NH, and with the expectation of a strong hydrogen bond between them, one can expect an unusually strong deshielding of the pyrrole NH.
Table 3.
Comparison of 1H-NMR chemical shiftsa for adducts 4, 6–10 and 14, 15.
| Compound | Pyrrole NH | Lactam NH | 10-H | TBA NH |
|---|---|---|---|---|
| 4 | 14.08 | 8.37 | 8.00 | 9.60, 9.65 |
| 13.45 | 9.93 | 7.92 | 12.23, 12.36 | |
| 6 | 13.89 | 8.95 | 7.90 | 10.24, 10.59 |
| 13.46 | 9.93 | 7.91 | 12.23, 12.36 | |
| 7 | 14.35 | 8.15 | 8.24 | ? |
| 13.36 | 9.92 | 8.02 | ? | |
| 8 | 13.45 | 9.81 | 7.96 | 11.09, 11.25 |
| 9 | 14.13 | 8.06 | 8.24 | ? |
| 13.30 | 9.84 | 8.04 | ? | |
| 10 | 13.33 | 7.56 | 8.15 | ? |
| 12.57 | 9.93 | 7.94 | ? | |
| 14a | 13.45 | ? | 8.06 | 8.89, 9.22 |
| 13.43 | ? | 7.88 | 12.16, 12.20 | |
| 14b | 13.54 | ? | 8.20 | ? |
| 13.22 | ? | 8.03 | ? | |
| 15a | 8.53 | ? | 8.44 | 8.77, 8.83 |
| 11.94 | ? | 8.19 | 11.87, 11.99 | |
| 15b | 8.61 | ? | 8.50 | ? |
| 12.06 | ? | 8.32 | ? |
δ, ppm downfield from (CH3)4Si for 3 × 10−3 M solutions in CDCl3 and (CD3)2SO at 25°C. The data from (CD3)2SO are shown in italics.
Figure 2.
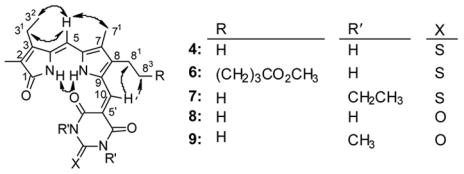
Nuclear Overhauser Effect (NOE) enhancements observed for adducts 4 and 6–9 and represented by curved arrows. A dashed arrow indicates a weak NOE.
A further examination of the distinction between the lactam and pyrrole NHs, and their assignments, was accomplished with 4 in CDCl3 by deuterium exchange with added D2O. A fast exchange was clearly indicated for the more shielded NH (lactam) vs. the more deshielded NH (pyrrole), with extremely slow exchange attributed to intramolecular hydrogen bonding (Fig. 3). Quantitatively similar results were found with 6 in CD2Cl2 and CDCl3. Thus, the most deshielded signal in CD2Cl2 did not exchange considerably with deuterium, confirming not only that the signal belongs to pyrrole NH but also the persistence of intramolecular hydrogen bonding in CD2Cl2. Even in (CD3)2SO solvent, exchange of the more deshielded NH was very slow; whereas, that of the more shielded NH was fast ? a behavior without parallel in simple dipyrrinones like 3.14
Figure 3.
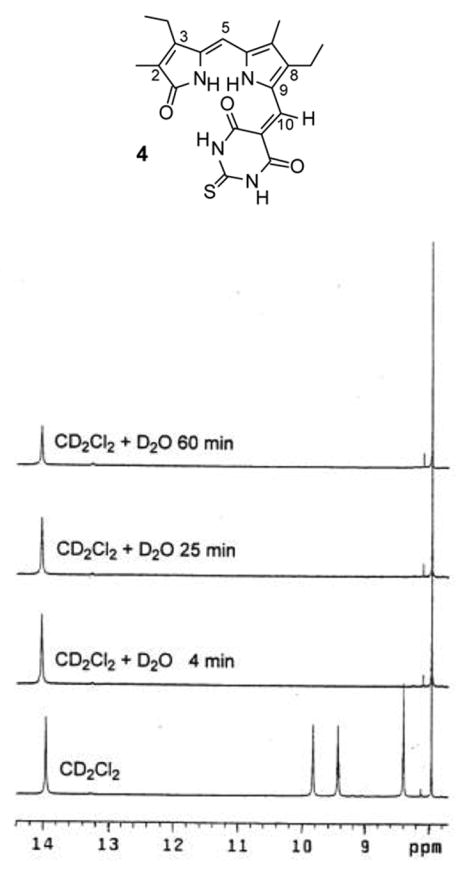
Timewise progression of deuterium exchange of the NHs of 3.5 mM 4 in CD2Cl2 solution at 25°C.
The D2O exchange experiments showed, however, a subtle difference between 4 and 7 in (CD3)2SO only. Upon adding D2O to a solution of 7 in CDCl3, the lactam NH signal at 8.15 ppm decreased in intensity to ~10% of its initial value; whereas, the C(10)-H (as a reference point at 8.24 ppm) and the pyrrole NH signal at 14.35 ppm remained unperturbed. In contrast, in (CD3)2SO solvent both NHs (pyrrole at 13.36 ppm and lactam at 9.92 ppm) of 7 were replaced by deuterium within 4 min. This rapid exchange of the pyrrole NH of 7 contrasts with the much slower rate of exchange found in 4 where the pyrrole NH is not as mobile, even in (CD3)2SO.
Diethyl TBA adduct 7 was characterized by the usual NMR experiments in CDCl3 and (CD3)2SO solvents. The carbon signal assignments (Table 2) followed those made earlier by 2D NMR spectroscopy on 4 and 6. In both solvents, 7 showed identical NOE enhancements (Fig. 2), indicating a conformational preference similar to that in 4 (and 6).
In general agreement with these findings, BA adduct 8 exhibited a resonance at the lower field (13.45 ppm) that did not exchange with D2O, but the other three NHs were completely exchanged with deuterium within 4 min in (CD3)2SO solvent. Nuclear Overhauser effect experiments on 8 confirmed the syn-Z configuration at C(4)=C(5) double bond of the dipyrrinone portion and a conformation around C(9)-C(10) single bond rendering the C(10)-methine hydrogen proximal to C(8)-CH2CH3 (as shown in Fig. 2). Taken collectively, the NMR properties of BA adduct 8 are very similar (except solubility in CHCl3) to the TBA adducts, and clearly the pigment exists in a conformation supporting strong intramolecular hydrogen bonding involving the pyrrole NH.
The NMR characteristics of 1,3-dimethyl BA analog 9 are identical to those of 8, with additional data being available in CDCl3 solvent. The pyrrole NH of 9 is even more deshielded in CDCl3, to 14.13 ppm vs 13.30 pm in (CD3)2SO; whereas, the lactam NH of 9 is more shielded in CDCl3 to 8.06 ppm vs 9.84 ppm in (CD3)2SO. The NOE correlations of 9 in CDCl3 are identical to those found in (CD3)2SO for 8 (Fig. 2). The rate of deuterium exchange in 9 is in line with those found in TBA derivatives like 4, 6 and 7, thus reaffirming that the pyrrole NH is the most deshielded proton, which becomes strongly electron-deficient from participation in strong intramolecular hydrogen bonding.
The strongly deshielded NH signal at 13.33 ppm of 10 in CDCl3 remained intact following addition of D2O (as it did in (CD3)2SO) ; whereas, the signal at 7.56 ppm was diminished significantly within 4 min, thereby suggesting that the latter is the lactam NH and the former is the pyrrole NH. Consequently, the conformation in the Meldrum’s acid conjugate (10) corresponds to that found in 4 and 8 in both polar and nonpolar solvents.
2.3. Reaction Mechanism
The mechanism and kinetics of the cleavage reaction were studied by 1H NMR. On the basis of its favorable solubiltiy, 1,3-diethyl TBA was judged to be an excellent candidate for examining the cleavage reaction kinetics. Perdeuterated methanol solvent was used in the initial study. The reaction concentration for NMR experiments was kept rather low and equal to the concentration used for preparative syntheses (~4.2 μmol.mL−1), and diethyl TBA was decreased from 2.5 eq. to 1.2 eq. in order to slow the transformation. Separate solutions of diethyl TBA and verdin 1 were prepared, and their 1H-NMR spectra obtained (Fig. 4). Verdin 1, as expected, did not exhibit any NH signals because the protons were exchanged with deuterium from CD3OD solvent. The 1H NMR chemical shifts followed during the experiment were from protons at C(5) and C(15) at 6.09 ppm and from the C(10)-H at 6.86 ppm, which was remarkably quite broad in CD3OD. (An explanation for such broadening might invoke spin-spin coupling to ND deuterium with a small 3JH-D.) The 1H NMR of diethyl TBA in CD3OD was even more interesting because the TBA methylene C(5′) protons did not appear at all, and two sets of N-ethyl groups were found in a 60:40 ratio. Such data can be explained by enolization that leads to rapid and total deuterium exchange at C(5′) and the significant presence of the enol form in CD3OD. In contrast, diethyl TBA in CD2Cl2 shows only one species in solution, with the C(5′)-CH2 at 3.70 ppm and a correct integral for two protons, and with one set of ethyl group signals. In (CD3)2CO solvent, diethyl TBA also exhibited the presence of two species (ratio 80:20) and a minor broad feature at 3.85 ppm. These observations suggest different amounts of the enol form of diethyl TBA are present in different solvents and a possible link to the faster reaction of diethyl TBA observed in more polar solvents, presumably via the enol. The verdin scission rate is even faster in (CD3)2SO than in CD3OD, and much slower in ethyl acetate.
Figure 4.
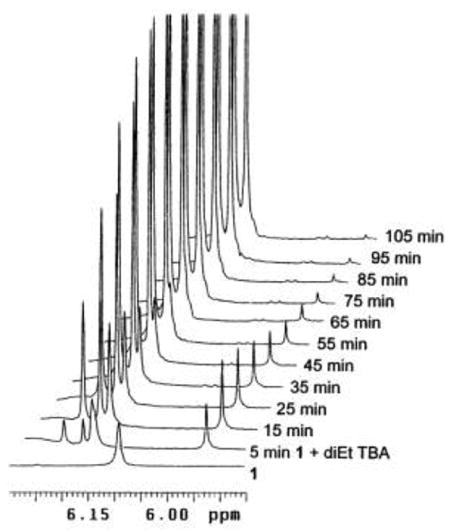
Timewise 1H NMR scans in the expanded region of the C(5)-H methine hydrogens of 3 (6.18 ppm), 7 (6.21 ppm) and C(10)-CH (5.96 ppm) of transient 13 during reaction between etiobiliverdin-IVγ (1) and 1,3-diethyl TBA in CD3OD at 20°C (Scheme 5). N,N′-Diethyl-TBA does not exhibit NMR signals in the region shown.
The two separate solutions of 1 and diethyl TBA in CD3OD were mixed in an NMR tube, and the 1H NMR spectra (Fig. 4) of the resulting mixture were acquired in 5 min intervals at 20°C. Of course, each resulting spectrum is an average for the acquisition time of 5 min. After only 25–30 spectra, the reaction was complete. In the final spectrum, obtained after 125 min, the appearance of the expected 1:1 mixture of 3 and 7 (Scheme 3) is rather clean, and all signals can be accounted for, including some from unreacted diethyl TBA and those from wet (HDO)CD3OD. Conspicuously missing, however, is the C(9)-H signal of the neokryptopyrromethenone (3) product at 6.75 ppm whose integral is only 9% of each integral for C(10)-H at 8.18 ppm and C(5)-H at 6.21 ppm for adduct 7 or at 6.18 ppm for 3. This unexpected result clearly shows that a deuteration step occurs during the verdin fragmentation. In controlled experiments a purified sample of 3 in contact with CD3OD and sonicated for one hour exhibited the correct integral for exactly one proton at C(9). Similarly, exchange at C(9) by deuterium in 3 did not happen when CD2Cl2 solvent was used for measuring the kinetics of the scission of verdin 1. These data indicate that the 9-H or 9-D of 3 is incorporated at a final step in the mechanism.
Certain signals grew fast over time: 8.18 ppm, 6.21 ppm and 6.18 ppm belonging to the C(10)-H, C(5)-H of adduct 7, and to the C(5)-H of dipyrrinone 3, respectively. The rapidly increasing, weak signal at 6.75 ppm is that of C(9)-H from 3. The data show that the broadened C(10)-H signal of 1 moves quickly from 6.86 ppm to about 6.4 ppm, then slowly disappears. The initially sharp C(5,15)-H singlet of 1 found at ~6.2 ppm after mixing with diethyl TBA also slowly decreases in intensity at the base of emerging peaks at 6.18 and 6.21 ppm. Of particular interest is the range of chemical shifts which is shown expanded in Fig. 4. A new signal appears at 5.96 ppm, immediately after mixing the component solutions, grows in intensity, reaches maximum at about 15 min and then its intensity slowly decreases. This signal is consistent with accumulation of an intermediate of rubin-type 13 (Scheme 5) according to the previously proposed mechanism.4 Presumably, in addition to the C(10)-H there should have been another emergent signal for the C(5′)-H of diethyl TBA, which is absent because it had been exchanged with deuterium (from CD3OD solvent) even before the reaction started, as pointed out above. Moreover, if a C(5′)-H were present instead of D in 13, then the C(10)-H would be spin-spin coupled (3J) to the erstwhile C(5′)-H, which is not observed.
Scheme 5.
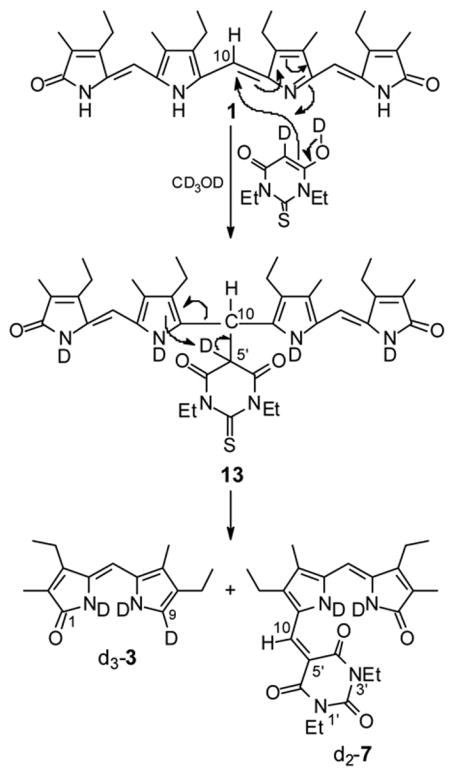
Mechanism of verdin cleavage by diethyl TBA in CD3OD.
After these results had been interpreted, it seemed worthwhile examining the reaction in several different solvents of varying polarity (and thus different enol content in the diethyl TBA) where the reaction might be slower and accumulation of a rubin type intermediate akin to 13 even more detectable. Reactions in non-deuterated solvents on the microscale were run with similar stoichiometry and concentrations as in CD3OD. Strikingly, the reaction between 1 and diethyl TBA in (CH3)2SO was complete in less than 15 min. In CH2Cl2, the reaction was sluggish but was complete after 2.5 h at 45°C (at reflux). In an NMR experiment conducted at 20 °C in CD2Cl2, the appearance of the final spectrum after 3 h was very clean, and all signals were easily assigned. However, the initial spectra are rather complex, suggesting multiple species/equilibria. Although no signals for intermediate 13 could be firmly assigned in CD2Cl2, the experiment allowed one to conclude that deuterium is not incorporated at C(9) in 3 (the signal at 6.83 ppm is as intense as those at 5.98 ppm and 6.16 ppm, but is with lower height due to coupling to the pyrrole NH).
Acetone-d6 was also used as a solvent in the cleavage reaction, and in this solvent diethyl TBA alone gave an indication for two species in equilibrium. Etiobiliverdin 1 did not show the lactam NH, lost presumably by exchange with HDO present in the solvent. Similarly, in the final spectrum, the acidic “protons” of products 3 and 7 are barely visible. Although the reaction in acetone seemed slower than in methanol or DMSO on microscale, it was complete after 3 h at 20°C. Again, the C(9)-H signal of 3 at 6.78 ppm showed a decreased intensity due to D exchange. Immediately after mixing, two new intense peaks appeared at 6.0–6.1 ppm, then their intensity gradually decreased. They might be attributed to the C(10)-H as in 13, and if correct, the intermediate is apparently formed very quickly (there is no intensity increase similar to that found in CD3OD), followed by a slower fragmentation step.
2.4. Synthesis and Characterization of Model Monopyrrole Conjugates of Thiobarbituric Acid
The strongly deshielded pyrrole NH observed in the 1H NMR of dipyrrinone-TBA conjugates such as 4 and 6 (Table 3), and the failure of their pyrrole NH chemical shift crossing that of lactam NH in (CD3)2SO (the pyrrole NH of 4 appears in CDCl3 at δ = 14.08 and moves to 13.45 ppm in (CD3)2SO) suggested building simpler model compounds with but one pyrrole ring. In one model compound, the conjugate is at the α-position of the pyrrole in order to mimic the arrangement in 4 and be able to participate in intramolecular hydrogen bonding. In a second, it would be attached at the pyrrole β-position, thereby excluding such hydrogen bonding. The syntheses (Scheme 6) of two such model compounds (14 and 15) relied on a Knövenagel condensation between TBA or diethyl TBA with 3,4-diethyl-5-methylpyrrole-2-carbaldehyde,17 and with 2,5-dimethylpyrrole-3-carbaldehyde,18 where the latter was synthesized by a Vilsmeier reaction of 2,5-dimethylpyrrole. Both aldehydes reacted smoothly with TBA (1.1 eq.) in ethanol at 55°C during 3 h without the need of base catalysis.19 The precipitated individual products were separated by filtration and, rather unexpectedly, both TBA derivatives 14a and 15a were found to exhibit extremely low solubility in CHCl3 and CH3OH, which excluded chromatographic purification. Therefore, the syntheses were repeated with diethyl TBA to yield adducts (14b and 15b) with more favorable solubility.
Scheme 6.
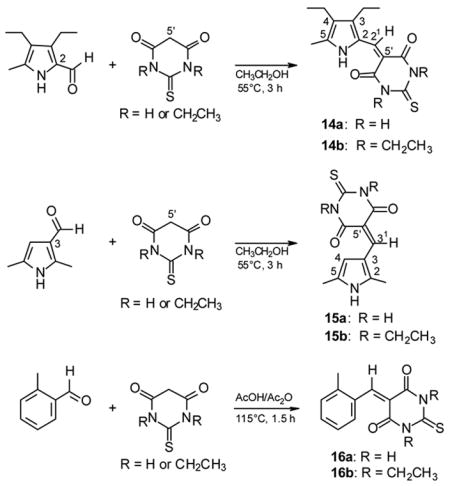
Knövenagel condensation of monopyrrole aldehydes and o-tolualdehyde with TBA and diethyl TBA.
These conjugates were characterized by NMR in (CD3)2SO. It is noteworthy that the carbon chemical shifts of the TBA carbonyls at C(4′) and C(6′) are very close (162.90 ppm and 162.94 ppm) in 14a but are rather different in 15a (160.42 ppm and 163.29 ppm). The 1H NMR chemical shifts of interest confirmed all expectations. The β-derivative (15a) which is incapable of intramolecular hydrogen bonding exhibited all NH signals of almost identical chemical shifts in (CD3)2SO: pyrrole NH at 11.94 ppm and TBA NHs at 11.87 ppm and 11.99 ppm (differentiated by larger broadening of the pyrrole NH). In (CD3)2SO, however, the α-derivative (14a), showed a pyrrole NH at 13.43 ppm, deshielded by 1.2 ppm relative to the TBA NHs at 12.16 ppm and 12.20 ppm. This difference is even more pronounced in CDCl3 solvent, for non-homogeneous solutions. The β-derivative 15a showed a pyrrole NH signal at 8.53 ppm and TBA NHs at 8.77 ppm and 8.83 ppm; whereas, the hydrogen bonded pyrrole NH of α-derivative 14a is at 13.45 ppm (very similar to that in 4 and 6) and the TBA NHs are at 8.89 ppm and 9.22 ppm. The pyrrole NH signal of 14a in both (CD3)2SO and CDCl3 is unexchangeable with deuterium (D2O) over 15–20 min but the TBA protons are completely exchanged within 5 min. In contrast, all of the NHs of 15a exchanged rapidly with deuterium in (CD3)2SO. The significantly more soluble diethyl TBA derivatives 14b and 15b exhibited similar trends.
Taken collectively, the 1H NMR chemical shifts and exchange rates indicate that the pyrrole NH of 14 is engaged in strong intramolecular hydrogen bonding to TBA carbonyl oxygen, in agreement with the observations on dipyrrinone adducts. However, an alternative rationalization for the strong deshielding of the pyrrole NH might come simply from lying in an anisotropic deshielding region of a TBA C=O. In order to disprove this possibility, the Knövenagel products (16) of o-tolualdehyde and TBA or diethyl TBA were prepared (Scheme 6), and their 1H NMR spectra were measured. In 16 the benzene o-H, which lies in a C=O deshielding cone, was observed to be a maximum of only 0.4–0.5 ppm more deshielded (16b in CDCl3) than the m or p-Hs.
Nuclear Overhauser effect spectra of 15 in (CD3)2SO indicated that the C(3′)-H bridging methine hydrogen is oriented preferentially toward pyrrole C(2)-CH3 methyl group (as drawn in Scheme 6) and not toward pyrrole C(4) position, in which conformation the C(2)-CH3 would buttress a carbonyl. In accord with the conclusion above, the NOE in 14 indicated a spatial proximity between the C(21)-H and the C(3)-CH2 (Scheme 6).
2.5. Converting Adducts to 9-CHO Dipyrrinones
Most of the chemistry of TBA derivatives is concerned with their rich functionality that is often used for further building of more complex heterocyclic systems.20 Very limited information is scattered about on the possibility of breaking the 5′-exocyclic double bond, e.g., in Scheme 7. Addition of nucleophiles, such as carbanions derived from malononitrile, ethyl methyl ketone, cyclopentanone and camphor, to this type of double bond, has been described,21 as well as addition of secondary amines,21b and even addition of water in the special case of an electron-withdrawing p-nitrophenyl substituent on the double bond (the condensation product of TBA with p-nitrobenzaldehyde)21c ? in all cases without further cleavage or degradation. TBA adducts such as 4, 6–9 and 17 (and their analogs of Scheme 2) could be viewed as Knövenagel condensation products19,22 between an aromatic aldehyde and C-H acidic β-dicarbonyl component. Surprisingly, the term “retro-Knövenagel reaction”23 has not been often used in the literature, although a mild hydrolytic cleavage of barbituric acid derivatives (retro-Knövenagel) has been demonstrated in a monolayer with 2,4,6-triaminopyrimidine inserted from the aqueous phase.23a Alkaline solvolysis of arylidene barbituric acids has been reported and their kinetics examined spectrophotometrically, without isolation of the resulting benzaldehydes.24 Inspired by the removal of the malononitrile protecting group25 from 2-formyl and 3-formyl pyrroles with strong aqueous alkali, formally a retro-Knövenagel reaction, we anticipated that high temperature and concentrated aqueous hydroxide would be a prerequisite for such energy unfavorable process.
Scheme 7.
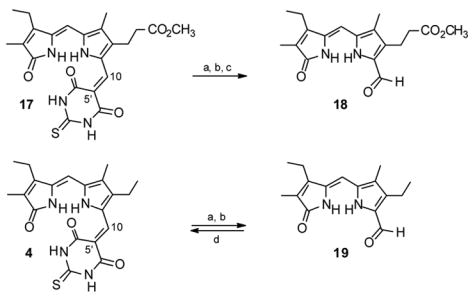
Retro-Knövenagel reactions leading to aldehydes 18 and 19. Reagents and conditions: (a) aq. NaOH/Δ, (b) HCl, (c), CH2N2, (d) TBA, piperidine.
Preliminary experiments on the TBA adduct 17 of 9-formylneoxanthobilirubinic acid (18, Scheme 7), available from TBA cleavage of mesobiliverdin-XIIIα dimethyl ester, indicated that ethanol co-solvent was necessary for solution homogeneity; however, the lower (~70°C) reflux temperature of aqueous ethanolic sodium hydroxide did not give satisfactory results. Evaporation of the ethanol from the reaction mixture by heating led to an increased reaction temperature (to ~90°C) and to cleavage of the C(5′)-C(10) double bond in 17. The pale yellow aldehyde 18 product was isolated in 26% yield after acidification, followed by separation of the crude dark acid by filtration and esterification with diazomethane. The necessity to re-esterify the propionic acid side chain might have had a deleterious effect on the overall yield; therefore, the retro-Knövenagel reaction was also performed on adduct 4, and this cleanly afforded aldehyde 19 in 44% yield. Further improvement (to 61% yield of 19) was achieved when diethylene glycol co-solvent (8% v/v) in refluxing aqueous NaOH was used. Similarly, cleavage of the BA residue of 8 was accomplished in an initial mixture of ethanol-diethylene glycol-aq. NaOH and, after evaporation of all ethanol at reflux, the reaction provided 19 in 66% yield. The structure of known26 aldehyde 19 was confirmed by its NMR spectra and, more importantly, it was converted back to magenta-colored adduct 4 (77% yield, identical in all aspects to that isolated from verdin cleavage) by a typical Knövenagel condensation with TBA in refluxing chloroform in the presence of piperidine catalyst. Despite the lower yields for aldehydes 18 and 19, which can also be synthesized efficiently by a one-step ester deprotection-formylation procedure from the corresponding 9-carbo-tert-butoxy esters,27 their recovery from pigments 4, 8 and 17 proved the concept that both the 9-H and 9-CHO dipyrrinones of biliverdins can be recovered.
2.6. UV-Visible Spectra of Adducts
Magenta-colored TBA adducts 4 and 6 show a strong absorption (ε~60,000) near 560–550 nm, with weaker absorption (ε ~40,000) near 525 (shoulder) and 325 nm (Table 4). The influence of ethyl groups (spectra from diethyl TBA adduct 7) is only small. However, the spectra were found to be sensitive to replacing the C=S group of TBA. The BA and dimethyl BA adducts (8 and 9, respectively) showed an approximately 30 nm hypsochromic shift in the longest wavelength band and a hypochromic shift of approximately 15,000–20,000 ε units. A similar hypsochromic shift was found for the 520 nm band, but with a much smaller drop in ε. Similarly, the band near 325 nm was shifted to ~315 nm and ε dropped by ~10,000 units. The trend toward hypsochromic wavelength shifts is seen further in Meldrum’s acid derivative 10. The influence of solvent polarity and protic vs aprotic on the λmax and ε values is not large.
Table 4.
Comparison of UV-vis spectral data of adducts 4, 6–10, 14 and 15.a
| Pigment | εmax (λmax, nm)
|
||||
|---|---|---|---|---|---|
| C6H6 | CHCl3 | CH3CN | CH3OH | (CH3)2SO | |
| 4 | 62100 (564) | 67000 (563) | 55300 (552) | 58500 (552) | 53700 (558) |
| 36600 (527) | 38700 (526) | 41400 (518) sh | 42600 (518) sh | 41300 (526) sh | |
| 44000 (326) | 52900 (324) | 47900 (321) | 47000 (321) | 41000 (326) | |
| 6 | 63800 (564) | 64000 (563) | 57700 (552) | 60400 (552) | 55300 (558) |
| 36800 (527) | 39000 (525) | 42500 (519) sh | 43800 (521) sh | 42300 (526) sh | |
| 44300 (326) | 52500 (324) | 50400 (321) | 48800 (321) | 41100 (326) | |
| 7 | 62200 (567) | 68500 (566) | 60700 (557) | 62300 (555) | 58800 (564) |
| 38800 (531) sh | 40400 (530) sh | 41600 (522) sh | 45100 (522) sh | 42300 (528) sh | |
| 39400 (328) | 44000 (327) | 42100 (323) | 42600 (323) | 40200 (329) | |
| 8b | 44700 (535) | 48500 (537) | 42500 (526) | 45100 (527) | 41400 (531) |
| 34300 (503) sh | 34000 (505) | 35300 (497) sh | 36900 (498) sh | 35200 (501) | |
| 38200 (317) | 45900 (315) | 43100 (311) | 43300 (313) | 37900 (316) | |
| 9 | 48700 (539) | 52300 (539) | 46800 (528) | 48300 (528) | 43200 (534) |
| 34700 (508) sh | 35700 (505) sh | 38300 (499) sh | 39800 (500) sh | 37000 (505) sh | |
| 39900 (317) | 46500 (316) | 44500 (312) | 44300 (313) | 38600 (316) | |
| 10 | 41500 (523) | 42600 (521) | 41200 (513) | 41500 (512) | 40200 (519) |
| 32600 (494) | 33800 (490) sh | 34800 (487) sh | 36900 (486) sh | 34200 (491) sh | |
| 40600 (311) | 47600 (310) | 46800 (307) | 45700 (308) | 40300 (311) | |
| 14ab | 118700 (460) | 126300 (460) | 122300 (455) | 130200 (456) | 121800 (461) |
| 43800 (439) sh | 47500 (439) | 47500 (434) sh | 50000 (436) sh | 43400 (439) sh | |
| 6100 (324) | 5900 (312) | 4300 (312) | 5700 (316) | 7700 (306) sh | |
| 15ab | 43200 (427) | 39900 (431) | 42200 (425) | 44000 (428) | 43400 (431) |
| 11900 (357) | 11900 (360) | 12300 (353) | 12500 (360) | 11700 (360) | |
| 5900 (311) sh | 5900 (308) sh | 6500 (310) sh | 5700 (312) sh | 6400 (313) sh | |
| 14b | 125400 (464) | 130000 (464) | 126600 (459) | 136300 (459) | 124200 (464) |
| 41000 (440) sh | 45600 (442) sh | 48700 (439) sh | 50500 (439) sh | 44900 (442) sh | |
| 6100 (328) | 5700 (328) | 5600 (309) | 5000 (312) | 5600 (313) | |
| 15b | 42900 (428) | 42700 (430) | 42400 (428) | 44100 (423) | 42200 (437) |
| 10900 (357) | 11300 (358) | 11100 (357) | 11700 (363) | 11000 (364) | |
| 6600 (314) sh | 6300 (309) sh | 6700 (308) | 5900 (305) | 6400 (305) | |
Concentration range 8.1 ×10−6–3.6 ×10−5 of solutions containing 2% v/v CHCl3.
Solutions containing 2% v/v (CH3)2SO.
The data may be compared with those from adducts 14 and 15 which possess only one pyrrole ring. Solutions of both 14 and 15 in (CH3)2SO are yellow (somewhat deeper yellow than that found in a typical dipyrrinone), but 15 is a dark, red-orange solid and 14 is a yellow solid.
Anticipating an absorption band near 450 nm, we found it near 460 nm for 14 and near 430 nm for 15. Strikingly, however, the ε value of this band in 14 exceeded 100,000, while that from 15 was close to 40,000 (Fig. 5). The 460 nm band of 14 is very sharp and very intense, e.g., 14a in methanol ε = 130,200 (456 nm). In contrast, 15a in methanol has ε = 44,000 (428 nm), an ε value that is still higher than from a typical dipyrrinone. The observed strong intensity of 14 is very much related to its narrow shape. In comparing the integrated intensities of 14 and 15, and also those of the dipyrrinone adduct 4 and xanthobilirubinic acid methyl ester, one finds nearly identical values, with that of 14 being only ~10% higher. Thus, the observed difference in ε values due to TBA attachment to an α- vs a β-pyrrole position in 14 vs 15 is clarified; the dipole strengths are actually quite similar.
Figure 5.

(A) UV-visible absorption spectra of dipyrrinone TBA adduct 4 (magenta), BA adduct 8 (red), Meldrum’s acid adduct 10 (blue) and monopyrrole TBA adducts 14a (green) and 15a (yellow) in CHCl3 at ~10−5 M concentrations. (B) Comparison of UV-visible absorption spectra of monopyrrole-TBA conjugates 14a (green) and 15a (yellow), dipyrrinone-TBA adduct 4 (magenta) and methyl xanthobilirubinate (X, an analog of 5 (yellow-green)) in methanol. The integrated absorption coefficients in L.mol−1 cm−2, defined by , are shown below the graph B.
In contrast to the long wavelength band in 14 or 15, which is shifted by ~100 nm from that of the dipyrrinone adducts of TBA, the shorter wavelength band lies (in 14) at nearly the same wavelength (near 325 nm) but is shrunk considerably in intensity, down to ε ~6,000. Apparently the intensity of the last is very much related to the presence of the dipyrrinone unit, as is the long wavelength band hypsochromic shift of ~100 nm.
3. Concluding Comments
The current work elaborates on the mechanism and usefulness of the smooth cleavage of biliverdins by selected carbon acids to 9-H dipyrrinones and the carbon acid (Knövenagel) adducts of 9-CHO dipyrrinones. The reaciton is improved by changes in solvent from the originally used ethyl acetate to methanol or DMSO and by choosing from among thiobarbituric acid (TBA), diethyl TBA, barbituric acid (BA), dimethyl BA and Meldrum’s acid ? depending on the requirements of product isolation. For example, it is easy to isolate the adduct in high yield (>90%) when BA in CH3OH is used to cleave the verdin, as the product precipitates almost entirely and in high purity (>95%) from the reaction mixture. From this carbon acid, the 9-H dipyrrinone may be isolated relatively easily and in high yield by chromatography of the mother liquor. The adduct, which shows tight intramolecular hydrogen bonding between the dipyrrinone pyrrole NH and a proximal C=O of the carbon acid, will undergo a retro-Knövenagel reaction, from which a 9-CHO dipyrrinone may be isolated in 26–66% yield.
4. Experimental Section
4.1. General Procedures
Nuclear magnetic resonance (NMR) spectra were obtained on a Varian Unity Plus 500 MHz spectrometer in CDCl3 solvent (unless otherwise specified) at 25°C. Chemical shifts were reported in δ ppm referenced to the residual CHCl3 1H signal at 7.26 ppm and 13C signal at 77.0 ppm. A combination of heteronuclear multiple bond correlation (HMBC) spectra and 1H{1H} NOE data were used to assign 1H and 13C NMR spectra. UV-visible spectra were recorded on a Perkin-Elmer Lambda-12 spectrophotometer. Melting points were taken on a Mel Temp capillary apparatus and are corrected. Combustion analyses were carried out by Desert Analytics, Tucson, AZ. Analytical thin layer chromatography was carried out on J. T. Baker silica gel IB-F plates (125 μ layers). Radial chromatography was carried out on Merck silica gel PF254 with gypsum preparative layer grade, using a Chromatotron (Harrison Research, Palo Alto, CA). Spectral data were obtained in spectral grade solvents (Aldrich or Fisher). Deuterated chloroform and dimethylsulfoxide were from Cambridge Isotope Laboratories. (4Z)-3,8-Diethyl-2,7,9-trimethyl-10H-dipyrrin-1-one (kryptopyrromethenone)8c,9 and etiobiliverdin-IVγ (1)8 (4Z)-3-ethyl-8-(5-carbomethoxypentyl)-2,7,9-trimethyl-10H-dipyrrin-1-one,8b mesobiliverdin-XIIIα-8,12-dihexanoic acid dimethyl ester (2),8b 3,4-diethyl-5-methylpyrrole-2-carbaldehyde,17 and 2,5-dimethylpyrrole-3-carbaldehyde18 were prepared as described in the literature. Barbituric, 1,3-dimethylbarbituric, thiobarbituric, 1,3-diethylthiobarbituric acids were from Aldrich and used as received, Meldrum’s acid was synthesized according to a literature procedure,28 and biliverdin-IXa dimethyl ester was obtained by esterification of bilirubin followed by oxidation.2
4.2. General procedure for biliverdin cleavage
To a solution of 249 mg (0.5 mmol) of etiobiliverdin-IV? (1)8 in 60 mL of methanol (obtained within 45–60 min) was added a solution of 1.2 mmol of carbon acid (thiobarbituric acid, TBA; diethyl TBA; barbituric acid, BA; dimethyl BA) in 60 mL of methanol, and the mixture was stirred at 25°C for 1.5–2.5 h. Then the solvent was evaporated under vacuum (rotovap), and the residue was purified by radial chromatography. In reactions using TBA and diethyl TBA, radial chromatography was performed after filtration of the magenta-colored, partially insoluble adducts. The adduct from reaction of the verdin with BA was separated by filtration, and after evaporation the filtrate was chromatographed to yield 9-H dipyrrinones. Using the same stoichiometry, reaction conditions and work-up, verdin 2 and biliverdin dimethyl ester were reacted and worked up similarly.
4.2.1. 3,8-Diethyl-2,7-dimethyl-(10H)-dipyrrin-1-one (3)
Neokryptopyrromethenone (3) was isolated (eluent CH2Cl2:AcOH:CH3OH = 100:2:1 to 100:2:3) in 76% yield following cleavage of etiobiliverdin-IV? (1). It had mp 202–204°C after crystallization from CH3OH-CH2Cl2 (Lit. mp 197°C11); 1H NMR ((CD3)2SO) d: 1.08 (3H, t, J=7.6 Hz), 1.09 (3H, t, J=7.6 Hz), 1.77 (3H, s), 2.02 (3H, s), 2.33 (2H, q, J=7.6 Hz), 2.49 (2H, q, J=7.6 Hz), 5.95 (1H, s), 6.71 (1H, d, J=2.5 Hz), 9.69 (1H, s), 10.46 (1H, s) ppm; 13C NMR ((CD3)2SO) d: 8.0, 9.0, 14.6, 14.8, 17.1, 18.0, 97.7, 118.8, 121.0, 123.4, 123.8, 125.3, 128.5, 147.3, 172.0 ppm; NMR data in CDCl3 are in Table 1.
Anal. Calcd for C15H20N2O (244.3): C, 73.73; H, 8.25; N, 11.47.
Found: C, 73.45; H, 8.13; N, 11.64.
4.2.2. 3,8-Diethyl-2,7-dimethyl-9-(4′,6′-dioxo-2′-thioxo-(1′H,3′H,5′H)-pyrimidin-5′-ylidene)methyl-(10H)-dipyrrin-1-one (4)
This pigment adduct was obtained (eluent CH2Cl2:AcOH:CH3OH = 100:2:1 to 100:2:3) in 87% yield from 1. It had mp 334–336°C after crystallization from EtOAc-hexane; 1H NMR d: 1.17 (3H, t, J=7.6 Hz), 1.21 (3H, t, J=7.6 Hz), 1.98 (3H, s), 2.16 (3H, s), 2.55 (2H, q, J=7.6 Hz), 2.71 (2H, q, J=7.6 Hz), 5.94 (1H, s), 8.00 (1H, s), 8.37 (1H, br s), 9.60 (1H, br s), 9.65 (1H, br s), 14.08 (1H, br s) ppm; 1H NMR ((CD3)2SO) d: 1.11 (3H, t, J=7.6 Hz), 1.12 (3H, t, J=7.6 Hz), 1.85 (3H, s), 2.13 (3H, s), 2.57 (2H, q, J=7.6 Hz), 2.67 (2H, q, J=7.6 Hz), 6.16 (1H, s), 7.92 (1H, s), 9.93 (1H, s), 12.23 (1H, s), 12.36 (1H, s), 13.45 (1H, s) ppm; 13C NMR ((CD3)2SO) data are in Table 2.
Anal. Calcd for C20H22N4O3S (398.5): C, 60.28; H, 5.57; N, 14.06.
Found: C, 59.96; H, 5.69; N, 13.91.
4.2.3. 2,7-Dimethyl-3-ethyl-8-(5-methoxycarbonylpentyl)-(10H)-dipyrrin-1-one (5)
Neo-dipyrrinone 5 was isolated (eluent CHCl3:CH2Cl2:AcOH:CH3OH = 50:50:2:1 to 50:50:2:2.5) in 76% yield from cleavage of mesobiliverdin-XIIIα-bis-hexanoate (2). It had mp 129–130°C (from CH3OH-CH2Cl2); 1H NMR ((CD3)2SO) d: 1.08 (3H, t, J=7.6 Hz), 1.29 (2H, m), 1.46 (2H, m), 1.54 (2H, m), 1.77 (3H, s), 2.02 (3H, s), 2.28 (2H, t, J=7.5 Hz), 2.31 (2H, t, J=7.6 Hz), 2.50 (2H, q, J=7.6 Hz), 3.57 (3H, s), 5.95 (1H, s), 6.72 (1H, d, J=2.8 Hz), 9.70 (1H, s), 10.48 (1H, s) ppm; 13C NMR ((CD3)2SO) d: 8.0, 9.1, 14.8, 17.1, 24.3, 24.6, 28.3, 29.5, 33.2, 51.1, 97.7, 119.5, 121.2, 123.3, 123.4, 123.7, 128.5, 147.3, 172.0, 173.3 ppm; 1H and 13C -NMR data in CDCl3 are in Table 1.
Anal. Calcd for C20H28N2O3 (344.4): C, 69.74; H, 8.19; N, 8.13.
Found: C, 70.00; H, 8.19; N, 8.40.
4.2.4. 2,7-Dimethyl-3-ethyl-8-(5-methoxycarbonylpentyl)-9-(4′,6′-dioxo-2′-thioxo-(1 ′H,3′H,5′H)-pyrimidin-5′-ylidene)methyl-(10H)-dipyrrin-1-one (6)
Isolated (eluent CHCl3:CH2Cl2:AcOH:CH3OH = 50:50:2:1 to 50:50:2:2.5) in 95% yield from 2, the desired adduct pigment (6) had mp 223–225°C after crystallization from EtOAc-hexane; 1H NMR d 1.21 (3H, t, J=7.6 Hz), 1.36 (2H, m), 1.52 (2H, m), 1.64 (2H, m), 1.94 (3H, s), 2.15 (3H, s), 2.30 (2H, t, J=7.4 Hz), 2.54 (2H, q, J=7.6 Hz), 2.66 (2H, t, J=7.4 Hz), 3.66 (3H, s), 5.94 (1H, s), 7.90 (1H, s), 8.95 (1H, br s), 10.24 (1H, br s), 10.59 (1H, br s), 13.89 (1H, br s) ppm; 1H NMR ((CD3)2SO) d: 1.11 (3H, t, J=7.6 Hz), 1.31 (2H, m), 1.48 (2H, m), 1.55 (2H, m), 1.85 (3H, s), 2.12 (3H, s), 2.28 (2H, t, J=7.3 Hz), 2.57 (2H, q, J=7.6 Hz), 2.65 (2H, t, J=7.4 Hz), 3.56 (3H, s), 6.16 (1H, s), 7.91 (1H, s), 9.93 (1H, s), 12.23 (1H, s), 12.36 (1H, s), 13.46 (1H, s) ppm; 13C NMR d: 8.7, 9.5, 14.3, 17.9, 24.6, 24.7, 28.9, 31.2, 33.9, 51.5, 94.1, 102.6, 126.9, 130.5, 131.4, 133.6, 142.4, 142.5, 144.5, 147.3, 163.0, 163.2, 173.7, 174.0, 175.4 ppm; 13C NMR ((CD3)2SO) data are in Table 2.
Anal. Calcd for C25H30N4O5S (498.6): C, 60.22; H, 6.07; N, 11.24.
Found: C, 59.94; H, 5.83; N, 11.16.
4.2.5. 3,8-Diethyl-2,7-dimethyl-9-(1′,3′-diethyl-4′,6′-dioxo-2′-thioxo-(1′H,3′H,5′H)-pyrimidin-5′-ylidene)methyl-(10H)-dipyrrin-1-one (7)
Obtained (eluent CH2Cl2:CH3OH = 100:0.5 to 100:2) in 92% yield from 1, this adduct pigment had mp 206–207°C after crystallization from EtOAc-hexane; 1H NMR d: 1.20 (3H, t, J=7.6 Hz), 1.21 (3H, t, J=7.6 Hz), 1.33 (3H, t, J=7.0 Hz), 1.35 (3H, t, J=7.0 Hz), 1.93 (3H, s), 2.16 (3H, s), 2.55 (2H, q, J=7.6 Hz), 2.75 (2H, q, J=7.6 Hz), 4.60 (2H, q, J=7.0 Hz), 4.67 (2H, q, J=7.0 Hz), 5.95 (1H, s), 8.15 (1H, br s), 8.24 (1H, s), 14.35 (1H, br s) ppm; 1H NMR ((CD3)2SO) d: 1.12 (3H, t, J=7.6 Hz), 1.13 (3H, t, J=7.6 Hz), 1.20 (3H, t, J=7.0 Hz), 1.24 (3H, t, J=7.0 Hz), 1.85 (3H, s), 2.14 (3H, s), 2.58 (2H, q, J=7.6 Hz), 2.69 (2H, q, J=7.6 Hz), 4.45 (2H, q, J=7.0 Hz), 4.50 (2H, q, J=7.0 Hz), 6.21 (1H, s), 8.02 (1H, s), 9.92 (1H, s), 13.36 (1H, s) ppm; 13C NMR d: 8.4, 9.2, 12.3, 12.4, 14.4, 16.1, 18.0, 18.1, 43.8, 44.0, 93.8, 104.5, 126.0, 129.3, 130.9, 135.7, 140.3, 141.0, 145.6, 147.4, 161.9, 162.1, 172.6, 177.9 ppm; 13C NMR ((CD3)2SO) data are in Table 2.
Anal. Calcd for C24H30N4O3S (454.6): C, 63.41; H, 6.65; N, 12.33.
Found: C, 63.39; H, 6.83; N, 12.33.
4.2.6. 3,8-Diethyl-2,7-dimethyl-9-(2′,4′,6′-trioxo-(1′H,3′H,5′H)-pyrimidin-5′-lidene)methyl- (10H)-dipyrrin-1-one (8)
Obtained in 94% yield from 1 and barbituric acid, this pigment adduct had mp 347–349°C (dec) after crystallization from (CH3)2SO-CH3OH + CHCl3; 1H NMR ((CD3)2SO) d: 1.109 (3H, t, J=7.6 Hz), 1.114 (3H, t, J=7.6 Hz), 1.84 (3H, s), 2.12 (3H, s), 2.56 (2H, q, J=7.6 Hz), 2.66 (2H, q, J=7.6 Hz), 6.13 (1H, s), 7.96 (1H, s), 9.81 (1H, s), 11.09 (1H, s), 11.25 (1H, s), 13.45 (1H, s) ppm; 13C NMR ((CD3)2SO) data are in Table 2.
Anal. Calcd for C20H22N4O4 (382.4): C, 62.81; H, 5.80; N, 14.65.
Calcd for C20H22N4O4 · ¼ H2O (386.9): C, 62.08; H, 5.86; N, 14.48.
Found: C, 62.34; H, 6.12; N, 14.62.
4.2.7. 3,8-Diethyl-2,7-dimethyl-9-(1′,3′-dimethyl-2′,4′,6′-trioxo-(1′H,3′H,5′H)-pyrimidin-5′-ylidene)methyl-(10H)-dipyrrin-1-one (9)
Isolated (eluent CH2Cl2:CH3OH = 100:0.5 to 100:1.5) in 88% yield from 1, this pigment adduct had mp 233–234°C after crystallization from EtOAc-hexane; 1H NMR d: 1.18 (3H, t, J=7.6 Hz), 1.20 (3H, t, J=7.6 Hz), 1.95 (3H, s), 2.15 (3H, s), 2.55 (2H, q, J=7.6 Hz), 2.73 (2H, q, J=7.6 Hz), 3.39 (3H, s), 3.43 (3H, s), 5.94 (1H, s), 8.06 (1H, br s), 8.24 (1H, s), 14.13 (1H, br s) ppm; 13C NMR d: 8.5, 9.2, 14.4, 16.2, 17.9, 18.0, 28.7, 28.8, 93.9, 103.2, 125.35, 129.1, 129.7, 135.6, 138.9, 140.1, 145.0, 147.4, 151.5, 163.7, 164.4, 172.5 ppm; 1H NMR ((CD3)2SO) d: 1.12 (6H, t, J=7.6 Hz), 1.85 (3H, s), 2.12 (3H, s), 2.57 (2H, q, J=7.6 Hz), 2.67 (2H, q, J=7.6 Hz), 3.22 (3H, s), 3.26 (3H, s), 6.16 (1H, s), 8.04 (1H, s), 9.84 (1H, s), 13.30 (1H, s) ppm; 13C NMR ((CD3)2SO) data are in Table 2.
Anal. Calcd for C22H26N4O4 (410.5): C, 64.37; H, 6.39; N, 13.65.
Found: C, 64.37; H, 6.20; N, 13.57.
4.3. 3,8-Diethyl-2,7-dimethyl-9-(2′,2′-dimethyl-4′,6′-dioxo-1′,3′-dioxan-5′-ylidene)methyl-(10H)-dipyrrin-1-one (10)
To a solution of 249 mg (0.5 mmol) etiobiliverdin-IV? (1) in 55 mL of methanol was added a solution of 432 mg (1.5 mmol) 2,2-dimethyl-1,3-dioxane-4,6-dione (Meldrum’s acid28) in 20 mL of methanol and 0.23 mL (1.5 mmol) of DBU, and the mixture was heated at reflux for 5 h. After cooling, the mixture was diluted with 200 mL of CH2Cl2 and washed with 0.5 % aq. HCl (70 mL) and water (3 × 100 mL). After drying (Na2SO4), filtration and evaporation of the solvent, the residue was purified by radial chromatography on silica gel using as gradient CH2Cl2:CHCl3:CH3OH = 80:20:0.5 to 80:20:3. Crystallization from ethyl acetate-hexane gave 91 mg (46 %) of 10 as a purple solid. It had mp 228–229°C (dec); 1H NMR d: 1.18 (3H, t, J=7.6 Hz), 1.20 (3H, t, J=7.6 Hz), 1.77 (6H, s), 1.96 (3H, s), 2.14 (3H, s), 2.54 (2H, q, J=7.6 Hz), 2.72 (2H, q, J=7.6 Hz), 5.91 (1H, s), 7.56 (1H, br s), 8.15 (1H, s), 13.33 (1H, br s) ppm; 13C NMR δ: 8.6, 9.2, 14.4, 16.0, 17.90, 17.92, 27.1, 93.5, 97.5, 104.1, 125.0, 128.7, 129.3, 136.0, 139.2, 140.1, 145.3, 147.3, 164.9, 165.6, 172.1 ppm. 13C NMR data from (CD3)2SO are in Table 2.
Anal. Calcd for C22H26N2O5: C, 66.31; H, 6.58; N, 7.03
Found: C, 65.79; H, 6.69; N, 6.93.
4.4. General Procedure for Condensation of Pyrrolecarbaldehydes with Thiobarbituric Acid
To a solution of 2.0 mmol of pyrrolecarbaldehyde in 7 mL of ethanol was added a solution of 2.1 mmol of TBA or diethyl TBA in 23 mL of ethanol, and the mixture was stirred at 55°C for 3 h. About 10–13 mL of the solvent was removed during the last one hour of the reaction by blowing a slow stream of nitrogen close to the surface. The mixture was cooled; then 3 mL of water was added slowly, and the mixture was chilled at −15°C for 1 h. The precipitated product was collected by filtration and purified by two recrystallizations from methanol (with dichloromethane co-solvent) to obtain a clear solution, which was partially evaporated by boiling to remove CH2Cl2.
4.4.1. 3,4-Diethyl-5-methyl-(4′,6′-dioxo-2′-thioxo-(1′H,3′H,5′H)-pyrimidin-5′-ylidene)methyl-(1H)-pyrrole (14a)
This product was obtained from 3,4-diethyl-5-methylpyrrole-2-carbaldehyde and TBA in 91% yield as an orange solid; mp 323–325°C (dec) (from CH3OH-CH2Cl2); 1H NMR d: 1.12 (3H, t, J=7.6 Hz), 1.21 (3H, t, J=7.6 Hz), 2.45 (3H, s), 2.48 (2H, q, J=7.6 Hz), 2.73 (2H, q, J=7.6 Hz), 8.06 (1H, s), 8.89 (1H, br s), 9.22 (1H, br s), 13.45 (1H, br s) ppm; 1H NMR ((CD3)2SO) d: 1.05 (3H, t, J=7.6 Hz), 1.12 (3H, t, J=7.6 Hz), 2.41 (3H, s), 2.44 (2H, q, J=7.6 Hz), 2.64 (2H, q, J=7.6 Hz), 7.88 (1H, s), 12.16 (1H, s), 12.20 (1H, s), 13.43 (1H, s) ppm; 13C NMR ((CD3)2SO) d: 12.6, 15.1, 16.8, 17.3, 17.4, 101.3, 126.4, 129.7, 133.3, 145.2, 146.9, 162.90, 162.94, 176.9 ppm.
Anal. Calcd for C14H17N3O2S (291.4): C, 57.71; H, 5.88; N, 14.42.
Found: C, 57.51; H, 5.89; N, 14.04.
4.4.2. 3,4-Diethyl-5-methyl-(1′,3′-diethyl-4′,6′-dioxo-2′-thioxo-(1′H,3′H,5′H)-pyrimidin-5′-ylidene)methyl-(1H)-pyrrole (14b)
Obtained from 3,4-diethyl-5-methylpyrrole-2-carbaldehyde and diethyl TBA (eluent CH2Cl2: CH3OH = 100:0.25) in 91% yield, this yellow pigment had mp 163–164.5°C after crystallization from EtOAc-hexane; 1H NMR d: 1.11 (3H, t, J=7.6 Hz), 1.20 (3H, t, J=7.6 Hz), 1.31 (3H, t, J=7.0 Hz), 1.32 (3H, t, J=7.0 Hz), 2.43 (3H,s), 2.47 (2H, q, J=7.6 Hz), 2.72 (2H, q, J=7.6 Hz), 4.58 (2H, q, J=7.0 Hz), 4.62 (2H, q, J=7.0 Hz), 8.20 (1H, s), 13.54 (1H, br s) ppm; 1H NMR ((CD3)2SO) d: 1.08 (3H, t, J=7.6 Hz), 1.16 (3H, t, J=7.6 Hz), 1.22 (6H, br), 2.45 (3H, s), 2.47 (2H, q, J=7.6 Hz), 2.68 (2H, q, J= 7.6 Hz), 4.45 (2H, br), 4.51 (2H, br), 8.03 (1H, s), 13.22 (1H, br s) ppm; 13C NMR d: 12.3, 12.5, 13.1, 15.1, 17.3, 17.4, 18.0, 43.4, 43.9, 101.9, 127.8, 130.0, 136.1, 146.2, 146.6, 161.8, 162.3, 178.2 ppm; 13C NMR ((CD3)2SO) d: 11.8, 11.9, 12.4, 14.4, 16.4, 16.6, 17.1, 42.4, 42.7, 100.8, 126.7, 129.6, 134.4, 145.7, 147.4, 160.8, 160.9, 177.4 ppm.
Anal. Calcd for C18H25N3O2S (347.5): C, 62.22; H, 7.25; N, 12.09.
Found: C, 62.42 ; H, 7.22; N, 12.19.
4.4.3. 2,5-Dimethyl-3-(4′,6′-dioxo-2′-thioxo-(1′H,3′H,5′H)-pyrimidin-5′-ylidene)methyl-(1 H)-pyrrole (15a)
Isolated from reaction of 2,5-dimethylpyrrole-3-carbaldehyde with TBA in 92% yield, the yellow pigment had mp 286–288°C (dec.) after crystallization from CH3OH; 1H NMR d: 2.27 (3H, d, 4J=0.9 Hz), 2.57 (3H, s), 7.40 (1H, br s), 8.44 (1H, s), 8.53 (1H, v br s), 8.77 (1H, br s), 8.83 (1H, br s) ppm; 1H NMR ((CD3)2SO) d: 2.16 (3H, d, 4J=0.9 Hz), 2.43 (3H, s), 7.25 (1H, br q), 8.19 (1H, s), 11.87 (1H, s), 11.94 (1H, br s), 11.99 (1H, s) ppm; 13C NMR ((CD3)2SO) d: 11.4, 12.4, 105.8, 110.7, 119.3, 129.9, 147.1, 147.9, 160.4, 163.3, 177.6 ppm.
Anal. Calcd for C11H11N3O2S (249.3): C, 53.00; H, 4.45; N, 16.86.
Calcd for C11H11N3O2S · ¼ H2O (253.8): C, 52.06; H, 4.57; N, 16.56.
Found: C, 52.19; H, 4.74; N, 16.46.
4.4.4. 2,5-Dimethyl-3-(1′,3′-diethyl-4′,6′-dioxo-2′-thioxo-(1′H,3′H,5′H)-pyrimidin-5′-ylidene)-methyl-(1H)-pyrrole (15b)
The desired compound was isolated (eluent CH2Cl2:CH3OH = 100:0.3 to 100:0.5) following reaction of 2,5-dimethylpyrrole-3-carbaldehyde with diethyl TBA in 83% yield. It had mp 198–200°C (dec.) after crystallization from EtOAc-hexane; 1H NMR (CDCl3) d: 1.31 (3H, t, J= 7.0 Hz), 1.32 (3H, t, J=7.0 Hz), 2.26 (3H, d, 4J=1.2 Hz), 2.53 (3H, s), 4.57 (2H, q, J=7.0 Hz), 4.59 (2H, q, J=7.0 Hz), 7.40 (1H, br q), 8.50 (1H, s), 8.61 (1H, br s) ppm; 1H NMR ((CD3)2SO) d: 1.18 (3H, t, J=7.0 Hz), 1.19 (3H, t, J=7.0 Hz), 2.18 (3H, d, 4J=0.9 Hz), 2.46 (3H, s) 4.41 (2H, q, J=7.0 Hz), 4.42 (2H, q, J=7.0 Hz), 7.28 (1H, br), 8.32 (1H, s), 12.06 (1H, br s) ppm; 13C NMR d: 12.2, 12.4, 12.5, 12.8, 43.2, 43.8, 108.1, 111.3, 120.1, 129.5, 146.2, 150.4, 159.6, 162.6, 179.1 ppm; 13C NMR ((CD3)2SO) d: 11.5, 12.2, 12.28, 12.33, 42.3, 49.9, 105.3, 110.8, 119.9, 130.3, 149.07, 149.08, 158.7, 161.7, 178.3 ppm.
Anal. Calcd for C15H19N3O2S (305.4): C, 58.99; H, 6.27; N, 13.76.
Found: C, 59.05; H, 6.10; N, 13.78.
4.5. Condensation of ortho-Tolualdehyde with Thiobarbituric Acid
A mixture of 3.0 mmol TBA or diethyl TBA, 3.5 mmol of freshly purified o-tolualdehyde, 4 mL of acetic acid, and 0.4 mL of acetic anhydride was heated under N2 at gentle reflux for 2 h. When TBA is used, after cooling, the reaction mixture was diluted with anh. Et2O (3 mL), and the product was separated by filtration. When diethyl TBA is used, the diethyl TBA product was extracted with CHCl3 after diluting with H2O, and purified by radial chromatography (eluent CH2Cl2:CH3OH = 100:0.25) and recrystallization from ethyl acetate-hexane.
4.5.1. 5-(2-Methylphenyl)methylidene-4,6-dioxo-2-thioxo-(1H,3H,5H)-pyrimidine (16a)
This yellow conjugate was isolated in 89% yield. It had mp 255–257°C (lit.29 mp 200°C); 1H NMR ((CD3)2SO) δ: 2.29 (3H, s), 7.19 (1H, m), 7.26 (1H, m), 7.35 (1H, m), 7.64 (1H, d, J=6.9 Hz), 8.41 (1H, s), 12.25 (1H, s), 12.44 (1H, s) ppm; 13C NMR ((CD3)2SO) δ: 19.7, 120.4, 124.8, 129.6, 130.2, 130.8, 132.9, 138.0, 154.4, 159.0, 161.3, 178.8 ppm.
4.5.2. 5-(2-Methylphenyl)methylidene-1,3-diethyl-4,6-dioxo-2-thioxo-(1H,3H,5H)-pyrimidine (16b)
This yellow conjugate was isolated in 57% yield. It had mp 77–79°C and 1H NMR δ: 1.26 (3H, t, J=7.0Hz), 1.34 (3H, t, J=7.0 Hz), 2.39 (3H, s), 4.48 (2H, q, J=7.0 Hz), 4.57 (2H, q, J=7.0 Hz), 7.24 (1H, m), 7.26 (1H, m), 7.38 (1H, m), 7.69 (1H, d, J=7.6 Hz), 8.76 (1H, s) ppm; 13C NMR δ: 12.38, 12.39, 20.3, 43.5, 44.0, 119.3, 125.2, 130.0, 130.1, 131.7, 132.9, 138.7, 157.9, 159.5, 160.3, 179.0 ppm.
Acknowledgments
We thank the U.S. National Institutes of Health (HD 17779) for generous support of this research. Dr. Stefan E. Boiadjiev is on leave from the Institute of Organic Chemistry, Sofia, Bulgaria.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.(a) Falk H. The Chemistry of Linear Oligopyrroles and Bile Pigments. Springer; Wien: 1989. [Google Scholar]; (b) Boiadjiev SE, Lightner DA. Org Prep Proc Int. 2006;38:347–399. [Google Scholar]
- 2.McDonagh AF. The Porphyrins. In: Dolphin D, editor. Bile Pigments: Bilatrienes and 5,15-Biladienes, Chapter 6. VI. Academic Press; New York: 1979. pp. 293–491. [Google Scholar]
- 3.(a) Fischer H, Hess R. Hoppe-Seyler’s Z physiol Chem. 1931;194:193–228. [Google Scholar]; (b) Fischer H, Adler E. Hoppe-Seyler’s Z physiol Chem. 1931;200:209–231. [Google Scholar]; (c) Siedel W, Fischer H. Hoppe-Seyler’s Z physiol Chem. 1933;214:145–172. [Google Scholar]; (d) Fischer H, Reinecke H. Hoppe-Seyler’s Z physiol Chem. 1939;258:9–15. [Google Scholar]
- 4.Manitto P, Monti D. J Chem Soc Chem Commun. 1980:178–180. [Google Scholar]
- 5.(a) Xie M, Lightner DA. Spectr Lett. 1992;25:1057–1066. [Google Scholar]; (b) Lindner I, Knipp B, Braslavsky SE, Gärtner W, Schaffner K. Angew Chem Int Ed. 1998;37:1843–1846. [Google Scholar]
- 6.Sawamoto D, Inomata K. Chem Lett. 2001:588–589. [Google Scholar]
- 7.Nogales DF, Ma JS, Lightner DA. Tetrahedron. 1993;49:2361–2372. [Google Scholar]
- 8.(a) Falk H, Grubmayr K. Synthesis. 1977:614–615. [Google Scholar]; (b) Shrout DP, Puzicha G, Lightner DA. Synthesis. 1992:328–332. [Google Scholar]; (c) Trull FR, Franklin RW, Lightner DA. J Heterocyclic Chem. 1987;24:1573–1579. [Google Scholar]
- 9.Falk H, Grubmayr K, Höllbacher G, Hofer O, Leodolter A, Neufingerl F, Ribó JM. Monatsh Chem. 1977;108:1113–1130. [Google Scholar]
- 10.(a) Mautner HG, Clayton EM. J Am Chem Soc. 1959;81:6270–6273. [Google Scholar]; (b) Briggs AG, Sawbridge JE, Tickle P, Wilson JM. J Chem Soc (B) 1969:802–805. [Google Scholar]; (c) Biggs AI. J Chem Soc. 1956:2485–2488. [Google Scholar]; (d) Eigen M, Ilgenfritz G, Kruse W. Chem Ber. 1965;98:1623–1638. [Google Scholar]
- 11.Fischer H, Adler E. Hoppe-Seyler’s Z physiol Chem. 1932;210:139–167. [Google Scholar]
- 12.Huggins MT, Lightner DA. Tetrahedron. 2000;56:1797–1810. [Google Scholar]
- 13.(a) Boiadjiev SE, Anstine DT, Lightner DA. J Am Chem Soc. 1995;117:8727–8736. [Google Scholar]; (b) Chen Q, Lightner DA. J Org Chem. 1998;63:2665–2675. doi: 10.1021/jo972227r. [DOI] [PubMed] [Google Scholar]; (c) Huggins MT, Lightner DA. J Org Chem. 2000;65:6001–6008. doi: 10.1021/jo000393k. [DOI] [PubMed] [Google Scholar]
- 14.Trull FR, Ma JS, Landen GL, Lightner DA. Isr J Chem. 1983;23:211–218. and references therein. [Google Scholar]
- 15.a) Cullen DL, Black PS, Meyer EF, Jr, Lightner DA, Quistad GB, Pak CS. Tetrahedron. 1977;33:477–483. [Google Scholar]; (b) Cullen DL, Pèpe G, Meyer EF, Jr, Falk H, Grubmayr K. J Chem Soc Perkin Trans II. 1979:999–1004. [Google Scholar]; (c) Hori A, Mangani S, Pèpe G, Meyer EF, Jr, Cullen DL, Falk H, Grubmayr K. J Chem Soc Perkin Trans II. 1981:1525–1528. [Google Scholar]; (d) Huggins MT, Lightner DA. Monatsh Chem. 2001;132:203–221. [Google Scholar]; (e) Tipton AK, Lightner DA. Monatsh Chem. 1999;130:425–440. [Google Scholar]
- 16.Kaplan D, Navon G. J Chem Soc Perkin Trans I. 1981:1374–1383. [Google Scholar]
- 17.Dolphin D, Harris RLN, Huppatz JL, Johnson AW, Kay IT. J Chem Soc (C) 1966:30–40. [Google Scholar]
- 18.Hinman RL, Theodoropulos S. J Org Chem. 1963;28:3052–3058. [Google Scholar]
- 19.Bijev AT, Prodanova PP. Bulg Chem Commun. 2004;36:125–130. [Google Scholar]
- 20.a) Ahluwalia VK, Aggarwal R. Proc Indian Nat Sci Acad. 1996;62(Part A):369–413. [Google Scholar]; (b) Kidwai M, Mohan R, Rastogi S. Synth Commun. 2003;33:3747–3759. [Google Scholar]
- 21.(a) El-Hashash M, Mahmoud M, El-Ficky H. Rev Roumaine Chem. 1979;24:1191–1202. [Google Scholar]; (b) Osman AN, El-Gendy AA, Kandeel MM, Ahmed EM, Hussein MMM. Bull Fac Pharm (Cairo Univ) 2003;41:59–68. (Chem. Abst. 143, 286367) [Google Scholar]; (c) D’yachkov AI, Ivin BA, Smorygo NA, Sochilin EG. Zhur Org Khim. 1976;12:1115–1122. [Google Scholar]
- 22.(a) Dox AW, Plaisance GP. J Am Chem Soc. 1916;38:2156–2164. [Google Scholar]; (b) Dox AW, Plaisance GP. J Am Chem Soc. 1916;38:2164–2166. [Google Scholar]; (c) Van Order RB, Lindwall HG. J Org Chem. 1945;10:128–133. doi: 10.1021/jo01181a001. [DOI] [PubMed] [Google Scholar]
- 23.(a) Bohanon TM, Caruso PL, Denzinger S, Fink R, Möbius D, Paulus W, Preece JA, Ringsdorf H, Schollmeyer D. Langmuir. 1999;15:174–184. [Google Scholar]; (b) Weißenfels M, Pulst M, Greif D, Werner T. Zeitschrift für Chemie. 1990;30:19–20. [Google Scholar]; c) Sha CK, Tsou CP. J Org Chem. 1990;55:2446–2450. [Google Scholar]
- 24.(a) Ivin BA, D’yachkov AI, Sochilin EG. Zhur Org Khim. 1977;13:121–127. [Google Scholar]; (b) Ivin BA, D’yachkov AI, Vishnyakov IM, Smorygo NA, Sochilin EG. Zhur Org Khim. 1975;11:1337–1342. [Google Scholar]; (c) Ivin BA, D’yachkov AI, Vishnyakov IM, Sochilin EG. Zhur Org Khim. 1975;11:1550–1554. [Google Scholar]; (d) D’yachkov AI, Ivin BA, Vishnyakov IM, Sochilin EG. Zhur Org Khim. 1975;11:2182–2187. [Google Scholar]
- 25.(a) Fischer H, Zeile K. Justus Liebig’s Ann Chem. 1930;483:251–271. [Google Scholar]; (b) Fischer H, Höfelmann H. Justus Liebig’s Ann Chem. 1938;533:216–230. [Google Scholar]; (c) Paine JB, III, Woodward RB, Dolphin D. J Org Chem. 1976;41:2826–2835. [Google Scholar]; (d) Woodward RB, Ayer WA, Beaton JM, Bickelhaupt F, Bonnett R, Buchschacher P, Closs GL, Dutler H, Hannah J, Hauck FP, Ito S, Langemann A, Le Goff E, Leimgruber W, Lwowski W, Sauer J, Valenta Z, Volz H. Tetrahedron. 1990;46:7599–7659. [Google Scholar]
- 26.(a) Falk H, Schlederer T. Monatsh Chem. 1981;112:501–510. [Google Scholar]; (b) Bobál P, Lightner DA. Synthesis. 2000:1835–1838. [Google Scholar]
- 27.(a) Clezy PS, Fookes CJR, Liepa AJ. Aust J Chem. 1972;25:1979–1900. [Google Scholar]; (b) Gossauer A, Hirsch W. Justus Liebig’s Ann Chem. 1974:1496–1513. [Google Scholar]; (c) Gossauer A, Hinze RP. J Org Chem. 1978;43:283–285. [Google Scholar]
- 28.Pihlaja K, Seilo M. Acta Chem Scand. 1968;22:3053–3062. [Google Scholar]
- 29.Ramana DV, Viswanadham SK. Indian J Chem. 1988;27B:613–616. [Google Scholar]