

Abstract
Misfolded amyloid beta peptide (Aβ) is a pathological hallmark of Alzheimer’s disease (AD), a neurodegenerative illness characterized by cognitive deficits and neuronal loss. Transgenic mouse models of Aβ over-production indicate that Aβ-induced cognitive deficits occur in the absence of overt neuronal death, suggesting that while extensive neuronal death may be associated with later stages of the human disease, subtle physiological changes may underlie initial cognitive deficits. Therefore, identifying signaling elements involved in those Aβ-induced cognitive impairments that occur prior to loss of neurons may reveal new potential pharmacological targets. Here we report that the enzymatic activity of calcineurin, a key protein phosphatase involved in phosphorylation-dependent kinase activity crucial for synaptic plasticity and memory function, is up-regulated in the CNS of the Tg2576 animal model for Aβ over production. Furthermore, acute treatment of Tg2576 mice with the calcineurin inhibitor FK506 (10 mg/kg ip) improves memory function. These results indicate that calcineurin may mediate some of the cognitive effects of excess Aβ such that inhibition of calcineurin shall be further explored as a potential treatment to reverse cognitive impairments in AD.
Keywords: Amyloid beta, calcineurin, Tg2576 mice, fear conditioning, behavior, FK506, SY5Y cells
Introduction
Alzheimer’s disease (AD) is a terminal neurodegenerative illness characterized by cognitive deficits and extensive neuronal loss in the CNS (Selkoe and Schenk, 2003). Hallmark features of AD are the presence of intracellular neurofibrillary tangles formed by hyper-phosphorylated tau and the presence of high levels of the 40- and 42-amino acid long amyloid beta (Aβ) peptide, which is generated through β- and γ-secretase cleavage of the larger amyloid precursor protein (APP). At elevated levels, Aβ peptides are prone to aggregation and eventually become a major component of the characteristic insoluble amyloid plaques that litter the extracellular space of the AD brain. While there is a general consensus that the presence of excess Aβ is perhaps the most fundamental neurotoxic event in AD (Van Gassen and Annaert, 2003), several lines of evidence indicate that oligomeric, soluble forms of Aβ, rather than amyloid plaques, initiate the cognitive deficits characteristic of the disease (Cleary et al., 2005a; Glabe, 2006; Lesne et al., 2006). For example, transgenic mouse models for AD in which Aβ is over produced and accumulates in the CNS, develop memory impairments long before plaques are detected and in the absence of significant neuronal loss (Irizarry et al., 1997; Takeuchi et al., 2000; Dineley et al., 2002; Westerman et al.). Therefore, some of the cognitive impairments in AD may not be associated with extensive neuronal death; rather, they may be the result of more subtle and reversible changes induced by soluble Aβ. Identifying molecular elements mediating these actions of Aβ may reveal significant pharmacological targets to reverse some of the cognitive impairments in AD.
One of the intracellular signaling elements that has recently attracted attention as a potential modulator of both memory function and cell degeneration is the Ca++/Calmodulin-dependent protein phosphatase 2B (calcineurin, CaN), the most abundant phosphatase in the CNS (Mansuy, 2003). Up-regulation of CaN in aged rats negatively correlates with cognitive performance (Foster et al., 2001) and down-regulation of CaN by an autoinhibitory peptide improves memory in rodents (Malleret et al., 2001). In Amyloid Precursor Protein/Presenilin-1 (APP/PS1) doubly transgenic mice, CaN has been shown to be upregulated in astrocytes where CaN triggers inflammatory responses typical of AD (Norris et al., 2005). In neurons, CaN activation leads to endocytosis of the NMDA receptor and reduces synaptic currents (Snyder et al., 2005). Furthermore, CaN can promote cell death by dephosphorylation/activation of pro-apoptotic members of the Bcl-2 protein family (Wang et al., 1999; Springer et al., 2000). More significantly, inhibition of CaN protects neuronal cells from Aβ-induced cell death (Agostinho and Oliveira, 2003) and in hippocampal slices abolishes perturbations of LTP that are promoted by Aβ (Chen et al., 2002). Thus, there is evidence suggesting that CaN may be involved in both the cognitive and neurodegenerative effects of Aβ. However, direct in vivo evidence of such a relationship between Aβ and CaN has yet to be acquired. Here, we report that CaN is up-regulated in the CNS of Tg2576 mice and that its pharmacological inhibition reverses Aβ-dependent associative learning and memory impairment in these mice.
Materials and Methods
Materials
FK506 (Prograf, tacrolimus injection, Fujisawa Healthcare Inc.) was purchased from the UTMB hospital pharmacy. Amyloid beta 1–42 for cell culture experiments was purchased from Sigma Chemicals. All other reagents were molecular biology purity grade and were purchased from Sigma Chemicals unless otherwise noted.
Animals
Animals were bred in the UTMB animal care facility and maintained under IACUC standards. Dams were C6/SJL F1 females purchased from Jackson Labs (Bar Harbour ME), and sires were Tg2576 males from our colony. Females were housed 3–5 per cage and males 1–4 per cage. According to IACUC-approved protocols, animals were sacrificed by decapitation and the brain removed from the skull. Brains were blocked using a mouse brain matrix and the hippocampus (HIPP), lateral cortex (LCTX), posterior cortex (PCTX), cerebellum (CB), basal forebrain/septum (BFS) and striatum (STR) rapidly dissected and stored frozen at −80ºC until further analysis was performed.
Fear Conditioning
Six hrs before the training session of the behavioral test, half of the mice were treated with FK506. The injection solution was diluted to 2 mg/ml with sterile 0.9% saline and administered at 10 mg/kg i.p. Age and sex matched animals were injected with a comparable volume of 0.9% saline. There were 10 animals per treatment group. The standard protocol consisted of a training phase when the mice were placed in a particular environment (a training chamber with particular lighting, geometry, odor that constitutes the context conditioned stimulus, CS) and allowed to explore for 3 min. An auditory CS (80dB white noise) was then presented for 30 sec and one footshock (0.8 mA, 2 sec duration; the unconditioned stimulus, US) delivered during the last 2 sec of the auditory CS. A second presentation of the auditory CS and the US was delivered at the 5 min mark and the animals then left in the cage for another 2 min. Twenty-four hours later, the mice were returned to the same training chamber and the context test for fear learning performed. The amount of freezing the mice exhibited during 5 min in the training chamber was measured. One hour later the cued test was performed in a completely novel context. The animals were placed in the testing chamber and freezing was measured for 3 min before the auditory CS was re-presented and freezing quantified over the next 3 min.
Cell cultures
Stock cultures of human neuroblastoma SY5Y cells (ATCC) were maintained in 75cm2 tissue culture flasks in 12mL DMEM/F12 culture medium (Invitrogen) supplemented with 5% (v/v) heat-inactivated donor horse serum (Invitrogen, Carlsbad, CA), 5% heat-inactivated fetal bovine serum (Gemini) and 1% antibiotic mixture penicillin, streptomycin (Cellgro) in a humidified cell incubator at 37°C under a 5% CO2 atmosphere. Five mL of medium were replaced every other day for maintenance of cell viability.
Calcineurin and PP1+PP2A assay
Calcineurin and PP-1+PP-2A combined enzymatic activity in brain tissue and cell extracts was measured using a commercial colorimetric kit (Calcineurin Cellular Activity Assay Kit, catalog No. 207007, Calbiochem) according to the manufacturer’s instructions.
Aβ1–42 assay
Presence of human Aβ1–42 in CNS tissue extracts was determined using a commercially available enzyme-linked immunosorbant assay (ELISA) according to the manufacturer’s instructions (BioSource).
Statistical analysis
Statistical analysis of behavior data was accomplished with either one-way or two-way ANOVA (Prism, GraphPad software), followed by Bonferroni post hoc multiple comparisons tests. A value of p<0.05 was considered significant. For paired analyses, Students’ t-test was performed. Where appropriate (unequal variance amongst groups), Bonferroni correction for four-way comparisons was done by reducing the p value for significance to 0.0125.
Results
In order to determine whether Aβ may directly and specifically induce CaN in neuronal type cells, we first measured the enzymatic activity of CaN and the combined activity of PP-1 and PP-2A (two other phosphatases that are Ca++/calmodulin-independent) in human SY5Y neuroblastoma cells treated for 24 hr with increasing concentrations of human Aβ1–42 (Figure 1). There was a significant dose-response increase of CaN activity in SY5Y cells treated with Aβ1–42, whereas the combined PP-1/PP2A enzymatic activity was not affected.
Figure 1.

CaN (TOP) and PP-1/PP-2A (BOTTOM) enzymatic activity in cytosolic extracts from human SY5Y neuroblastoma cells treated for 24 hr with increasing concentrations of human Aβ1–42. Each point represents the average ± S.D. of three independent replicated experiments. *: p<0.05 vs. control (two-tailed Student’s t test).
We then measured CaN enzymatic activity and human Aβ1–42 in the hippocampus, posterior cortex and cerebellum of 5 month-old Tg2576 mice, an age at which Aβ levels are elevated in the CNS but not yet deposited into insoluble plaques (Hsiao et al., 1996; Kawarabayashi et al., 2001), and determined the effect thereupon of the CaN inhibitor FK506 (Figure 2a,b). CaN activity was approximately 30 percent greater in Tg2576 mice compared to wild type littermates, paralleling the presence of measurable transgene-derived human Aβ1–42 in all three areas assayed. A single injection of FK506 (10 mg/kg ip) reduced CaN activity in brain regions of Tg2576 mice to basal levels as measured 6 hr after the treatment; however this treatment regimen did not affect CaN activity in wild type mice. Furthermore, Aβ1–42 levels in the Tg2576 mice were not significantly altered by FK506.
Figure 2.

The hippocampus, posterior cortex and cerebellum of Tg2576 mice have measurable Aβ1–42 and display increased CaN activity, which is reversed by treatment with FK506. a) CaN (PP-2B) enzymatic activity assay and b) human Aβ1–42 ELISA in the hippocampus (HIPP), posterior cortex (PCTX) and cerebellum (CB) of 5 month old Tg2576 mice treated with FK506 (10 mg/kg ip) 6 hr before sacrifice. Bars represent the average ± S.D. of five animals per group. *p<0.001 vs. vehicle-treated wild type (WT) mice (two-tailed Student’s t test with Bonferroni correction for multiple comparison). n.d.: none detected (below the ELISA detection limit).
Next, we tested the hypothesis that such CaN inhibition would alleviate the associative learning deficit exhibited by 5 month old Tg2576 mice (Corcoran et al., 2002; Dineley et al., 2002; Comery et al., 2005). Tg2576 mice and their wild type littermates were subjected to 2-pair training in the rodent fear conditioning paradigm. Six hrs before the training session, the mice received a single injection of FK506 (10 mg/kg ip) or vehicle. This dose and time point for FK506 treatment was chosen based on the results shown in Figure 2 and published evidence demonstrating that FK506 reaches maximum CNS concentrations ~5 hrs after i.p. injection in rodents and this level is maintained for at least 24 hr (Butcher et al., 1997; Fukudo et al., 2005). During training (Figure 3a), each group of animals did not freeze significantly to the novel context prior to the first CS-US pairing. Analysis of percent freezing per 30 second epoch after the first CS-US pairing showed that all groups of animals froze significantly more than prior to the first CS-US pairing; however freezing during any of the four epochs following the first CS-US did not differ among groups. FK-506 had no effect on freezing following the first or second CS-US pairing. Twenty-four hr after the training session, mice were tested for contextual and cued fear learning. Wild type mice receiving vehicle or FK-506 froze to a similar extent in the contextual test for fear learning (Figure 3b,c). As expected, vehicle-treated Tg2576 froze significantly less than wild type littermates in the contextual test as measured by either total freezing (Figure 3b) or when each one-minute epoch was separately analyzed (Figure 3c). On the contrary, Tg2576 mice receiving FK506 prior to the training session showed no significant difference vs. wild type mice in freezing behavior in the contextual test and froze significantly more than Tg2576 mice receiving vehicle. On the other hand, all groups of animals froze to a similar extent in the cued test, regardless of genotype or treatment received (Figure 3d). Two-way ANOVA did not reveal a time by genotype interaction. However, during representation of the CS in a novel context, each genotypic group of animals froze significantly more than prior to the onset of the CS. There was no genotype effect in that none of the groups froze significantly different from each other before or after the onset of the CS.
Figure 3.

FK506 reverses contextual fear conditioning deficit in 5 month old Tg2576. a, During training, all groups of animals froze significantly more than prior to the first CS-US pairing; freezing during any of the four epochs following CS-US #1 did not differ among groups. FK-506 had no effect on freezing during training. *p<0.0001 one-way ANOVA F(18,5)=15.92. b, Tg2576 contextual fear conditioning deficit is rescued with FK506. *p<0.0001, one-way ANOVA F(3,16)=32.86. c, percent freezing per one-minute epoch in the contextual test. *p<0.0001, one-way ANOVA F(3,40)=3.102 and =4.065 for epochs 2 and 4, respectively). d, Tg2576 are unimpaired in the cued test for fear learning; FK506 has no effect on performance. *p<0.0001; two-way ANOVA F(15, 3, 5)=1.52, 0.18, 7.03 for interaction, genotype, and time, respectively. All data expressed as average percent freezing +/− SEM.
A separate set of Tg2576 and wild type mice were treated with FK506 or vehicle as above and subjected to the fear conditioning and testing. Immediately after completing the testing phase, these mice were sacrificed and the hippocampus (HIPP), lateral cortex (LCTX), posterior cortex (PCTX), striatum (STR), basal forebrain/septum (BFS) and cerebellum (CB) were rapidly dissected and stored frozen until further use. CaN activity and combined PP-1/PP-2A activity assays were performed on freshly homogenized tissue (Figure 4). We found that CaN activity was significantly increased in HIPP, PCTX and CB of Tg2576 mice as compared to wild type littermate mice. CaN activity also showed an upward trend in the LCTX, however this did not reach statistical significance. On the other hand, BFS samples from Tg2576 mice exhibited decreased CaN activity. Treatment with FK506 restored CaN activity in the affected areas of Tg2576 CNS to levels comparable to wild type mice; in several instances, these values were significantly different from vehicle-treated Tg2576. On the other hand, the combined PP-1/PP-2A enzymatic activity (Figure 4b) was affected neither by the genetic make-up of the mice nor by FK506 treatment in all of the CNS areas assayed, with the exception of the HIPP where it followed the same pattern as CaN, being significantly elevated in Tg2576 mice and restored to basal values by the FK506 treatment.
Figure 4.
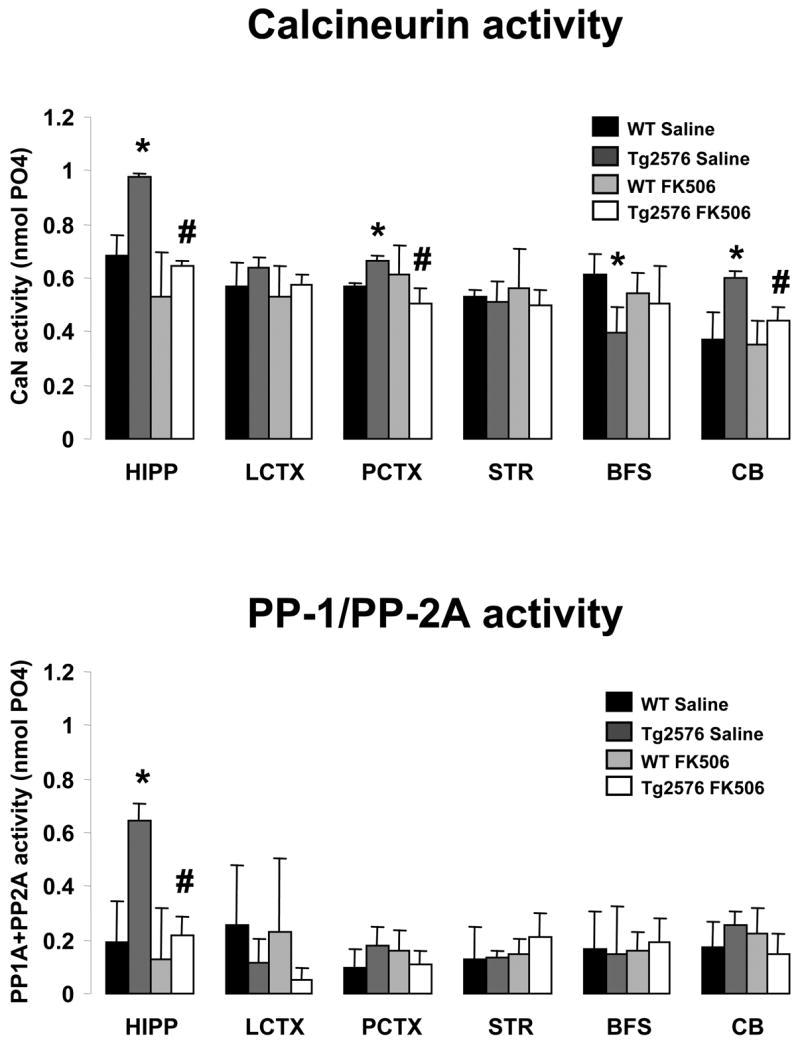
FK506 reverses CaN hyper-activity in several brain regions of Tg2576. Calcineurin (TOP) and combined PP-1/PP-2A enzymatic activity (BOTTOM) in various brain areas of Tg2576 mice and wild type littermate control mice (WT) treated with the calcineurin inhibitor FK506 (10 mg/kg ip) or vehicle. Treatment was administered 6 hr before training. Mice were sacrificed immediately after being subjected to contextual and cued fear conditioning memory tests, approximately 24 hr after training.
Bars represent the average ± S.D. of 6–8 animals per group. * and # p<0.0125 vs. WT or Tg2576, respectively (two-tailed Student’s t-test with Bonferroni correction for multiple comparison).
CaN activity was also evaluated in mice that received FK-506 or vehicle but were not subjected to behavioral analysis. Hippocampal samples collected 24 hours following FK-506 injection showed comparable CaN activity to that shown in Figure 4 indicating that fear conditioning testing does not influence CaN activity or the effectiveness of FK-506 (data not shown).
Discussion
The results presented here show that there is a significant up-regulation of CaN enzymatic activity in hippocampus, cortex and cerebellum of 5 month old Tg2576 APP transgenic mice. Up-regulation of CaN activity is coincident with the appearance of Aβ1–42 in these CNS areas and impaired performance in a hippocampus-dependent associative learning task, the contextual fear conditioning test. This cognitive phenotype is reversed following a single treatment with the CaN inhibitor FK506 six hours prior to the training session. Furthermore, direct treatment of human SY5Y neuroblastoma cells with increasing concentrations of Aβ1–42 produced a dose-dependent increase in CaN phosphatase activity, while leaving PP-1 and PP-2A activities unaffected. These in vitro results support the idea that high levels of Aβ in the CNS of 5 months old Tg2576 mice leads to increased CaN activity and that such excess activity, particularly in the hippocampus, is directly involved in mediating impairments in the contextual fear learning and memory test in these mice. Therefore we conclude that increased CaN activity may be an important component of the events leading to impaired contextual fear conditioning in Tg2576 mice.
FK-506 treatment reduced CaN levels in the CNS of Tg2576 mice to values comparable to control mice for the training session and CaN activity remained “normalized” through the testing sessions 24 hours later as indicated by CaN activity measurements performed on brain tissue harvested either 6 hours post injection or immediately following the fear learning tests (Figure 4). Normalization of CaN activity in Tg2576 resulted in reversal of the contextual fear conditioning deficit in these mice at 5 months of age. The increased freezing exhibited by FK506-treated Tg2576 mice was unlikely due to a non-specific effect of the drug since it had no effect on freezing behavior in WT littermate controls.
Our protocol for the dosage and timing of FK506 treatment resulted in normalized CaN activity in the CNS of Tg2576 mice rather than total blockade. This finding is supported by previous studies in which complete abolishment of CaN expression in anterior cortex of mice worsened cognitive performance (Zeng et al., 2001), whereas partial reduction of CaN activity through antisense knock-down enhanced contextual fear learning in rats (Ikegami and Inokuchi, 2000). Collectively these findings suggest that complete loss of CaN activity may be as detrimental as excessive CaN activity.
FK506 treatment lowered CaN activity in several brain regions of Tg2576 mice but left CaN in WT mice unaffected. This suggests that, under the FK506 dose and regimen utilized in this study, regulation of CaN by FK506 in WT CNS is more resilient than in Tg2576; indeed, the observed aberrant activation of CaN in this mouse model supports this notion. Again, decreased CaN activity through antisense knock-down can enhance contextual fear learning in rats (Ikegami and Inokuchi, 2000). The lack of an effect of our FK506 regimen on CaN activity in WT mice is consistent with their behavioral performance in the contextual fear conditioning test: WT mice given 10 mg/kg FK506 exhibited neither enhanced fear learning nor reduced CaN activity.
Five month old Tg2576 mice have Aβ levels that are ~7-fold higher than normal but is not yet associated with plaque deposits (Hsiao et al., 1996; Kawarabayashi et al., 2001). As demonstrated by ours and others’ studies, Tg2576 mice at this age exhibit associative learning and memory impairments in the contextual fear conditioning test (Corcoran et al., 2002; Dineley et al., 2002; Comery et al., 2005). Several lines of evidence support the notion that early cognitive deficits in AD mouse models are triggered by oligomeric assemblies of Aβ rather than plaque-associated Aβ (Cleary et al., 2005b; Selkoe, 2005; Lesne et al., 2006). Recently, it has been demonstrated that purified oligomers (dimers and trimers as well as a 56 kDa dodecamer aggregate) of in vitro- and in vivo-produced Aβ can disrupt cognitive function (Cleary et al., 2005b; Lesne et al., 2006). The emergence of the 56kDa species correlates with the onset of spatial memory impairment in these mice and can induce the same cognitive deficits in wild type rodents (Lesne et al., 2006). While several oligomeric species are evident in Tg2576 brain at different ages, the 56kDa dodecamer is absent in 5 month old Tg2576 brain but is present from 6 months of age onward. Collectively, these findings indicate that aberrant CaN activity observed here is related to the presence of soluble oligomeric forms of Aβ rather than being the result of insoluble Aβ aggregates. Our results further suggest that the associative memory deficits in 5 months Tg2576 mice are triggered by Aβ assemblies that are distinct from the 56kDa species reported to initiate spatial learning and memory deficits in 6 months old Tg2576 mice.
We found increased CaN activity levels in several CNS areas of 5 months old Tg2576, some of which are affected by Aβ deposits in older Tg2576 mice (e.g., the hippocampus and cortex) and some of which are not (e.g., the cerebellum) (Gordon et al., 2002). In the same CNS areas we also found significant levels of Aβ1–42, thus suggesting that the mechanisms that lead to Aβ deposition into plaques and CaN activation are unrelated in the CNS of these mice.
Notably, we found elevated CaN activity in parietal cortex samples, which in our dissections include the amygdala -- the brain region crucial for fear learning and memory (Maren et al., 1996). However, Tg2576 mice exhibit a selective impairment in the contextual fear conditioning test whereas, as previously demonstrated, they are not impaired in cued fear conditioning (Dineley et al., 2002). The lack of impairment in cued fear conditioning in spite of an apparent enhancement of CaN activity in amygdala may reflect the fact that the CS in the cued portion of the paradigm is much more salient than the contextual CS. This is reflected by the greater amount of freezing on behalf of all animal groups during the cued test. Alternatively, our results may indicate that proper CaN regulation in hippocampus is more crucially involved in the modulation of contextual memory in comparison to the amygdala. In fact, increased CaN expression and activity in the amygdala has been established as a key signal in the extinction of fear memory rather than during the establishment or retrieval of fear memory (Lin et al., 2003a; Lin et al., 2003b). Likewise, while CaN is aberrantly activated in the cerebellum of Tg2576 mice, these animals show no apparent motor coordination or cerebellum-dependent motor learning abnormalities as measured by the rotating rod test (Dineley et al., 2002). This suggests that either the amount of CaN dysregulation in Tg2576 cerebellum is insufficient to induce a phenotype as measured by rotating rod, or CaN does not play a significant role in motor coordination or motor learning in this mouse model.
With the exception of the hippocampus, we did not observe any changes in the combined PP-1/PP-2A enzymatic activity due to the presence of the transgene or FK506 treatment. In contrast to CaN, PP-1 and PP-2A are independent of Ca++/Calmodulin for their enzymatic activity (Mansuy, 2003). This observation, along with our current result of a selective induction of CaN but not PP-1/PP-2A in S5Y5 cells by Aβ1–42 (Figure 1), suggests that an Aβ-induced increase in intracellular Ca++ may be responsible for CaN activation. While previous evidence from us and others has demonstrated that Aβ induces Ca++ influx through α7 nAChRs in neurons and leads to CaN activation (Dineley et al., 2001; Dougherty et al., 2003; Snyder et al., 2005), the exact pathway linking Aβ and increased CaN activity in Tg2576 CNS remains to be established. An additional consideration is that the coupling mechanisms between increased intracellular Ca++ and CaN activation in different brain regions is likely though distinct mechanisms. Indeed, we found that in the basal forebrain/septum region CaN activity was reduced in untreated Tg2675 brain as compared to the increase observed in the hippocampus, which receives rich cholinergic innervation from the basal forebrain. This suggests that although α7 nAChRs are present both pre- and post-synaptically, the mechanisms by which they might couple to CaN activation in post-synaptic hippocampal neurons is different than in pre-synaptic basal forebrain neurons, possibly through differential compartmentalization of α7 nAChRs, Ca++ and CaN.
We did find increased levels of both CaN and combined PP-1/PP-2A enzymatic activity in the hippocampus of Tg2576 mice. CaN is particularly abundant in the hippocampus, and high levels of CaN activity were further elevated in Tg2576 mice. PP-1 can be activated through CaN-mediated dephosphorylation (Mansuy and Shenolikar, 2006). FK506 reduced both CaN and PP-1/PP-2A activities in the hippocampus of Tg2576 mice suggesting that increased hippocampal PP-1/PP-2A activity is a direct consequence of elevated CaN activity. It is thus possible that direct inhibition of PP-1/PP-2A in the hippocampus (e.g. through the use of okadaic acid) may bypass CaN and provide partial rescue of fear conditioning memory task in Tg2576 mice. Our current data, however, allows us only to speculate on the possible effects of PP-1/PP-2A inhibitors on fear conditioning memory in Tg2576 mice and more specific studies are needed to directly address this question. Nonetheless, the hippocampus is an early and particularly vulnerable target in AD and in APP transgenic animals, elevated Aβ perturbs several forms of hippocampus-dependent cognitive function including contextual learning and memory (Dineley et al., 2002; Westerman et al., 2002). Our observation of increased CaN and PP-1/PP-2A activity in this brain region that, when normalized with a selective CaN inhibitor, suggests that the hippocampus represents an important anatomical locale where excess Aβ impinges upon these factors to produce memory deficits.
We observed an amelioration of associative memory deficits after a single injection of the CaN inhibitor FK506 in Tg2576 mice. The same treatment did not affect Aβ1–42 levels in the CNS of these mice (Figure 2). This indicates that the molecular mechanisms leading to memory deficits in Tg2576 mice at this age are reversible, independently of continued high levels of Aβ. This observation supports the idea that memory impairments in AD are initially the result of reversible functional changes in molecules important for synaptic plasticity, learning and memory induced by increasing levels of Aβ in the CNS. Indeed, it has been reported that in Aβ can disturb synaptic plasticity and LTP (both essential components of memory function) through down regulation of NMDA receptors at the cell surface (Snyder et al., 2005), a process requiring the α7 AChR and CaN. It is therefore tempting to argue that the memory-improving action of FK506 in Tg2576 mice may result from rescuing the CaN-mediated NMDA receptor decrease induced by Aβ in hippocampal and cortical neurons. While the present results allow us only to speculate about the possible mechanism of action of FK506 in the memory-deficient Tg2576 mouse model, ongoing studies in our laboratory are currently aimed at answering this important question.
In conclusion, our data indicate that CaN is part of the machinery mediating the detrimental effects on memory of Aβ and thus encourage further consideration of CaN inhibition as a pharmacological strategy to ameliorate cognitive deficits in AD. However, while we observed that a single injection with FK506 improved fear conditioned memory function in Tg2576 mice, prolonged treatment of humans with FK506 has been reported to be associated with significant side effects, most notably nephrotoxicity (Olyaei et al., 2001; Baran et al., 2004; Staatz and Tett, 2004). Therefore, before considering clinically attractive the idea of CaN inhibitors for treating AD, future studies will have to determine whether lower, less nephrotoxic doses of FK506 administered for longer periods of time still provide behavioral benefits or whether novel behaviorally-active drugs that selectively target only CaN in the CNS could eventually be designed.
Acknowledgments
This work was supported by the John Sealy Memorial Endowment for Biomedical Research to KTD and 1R21NS053986 from the NIH/National Institute of Neurological Diseases and Stroke to GT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agostinho P, Oliveira CR. Involvement of calcineurin in the neurotoxic effects induced by amyloid-beta and prion peptides. Eur J Neurosci. 2003;17:1189–1196. doi: 10.1046/j.1460-9568.2003.02546.x. [DOI] [PubMed] [Google Scholar]
- Baran DA, Galin ID, Gass AL. Calcineurin inhibitor-associated early renal insufficiency in cardiac transplant recipients: risk factors and strategies for prevention and treatment. Am J Cardiovasc Drugs. 2004;4:21–29. doi: 10.2165/00129784-200404010-00003. [DOI] [PubMed] [Google Scholar]
- Butcher SP, Henshall DC, Teramura Y, Iwasaki K, Sharkey J. Neuroprotective actions of FK506 in experimental stroke: in vivo evidence against an antiexcitotoxic mechanism. J Neurosci. 1997;17:6939–6946. doi: 10.1523/JNEUROSCI.17-18-06939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QS, Wei WZ, Shimahara T, Xie CW. Alzheimer amyloid beta-peptide inhibits the late phase of long-term potentiation through calcineurin-dependent mechanisms in the hippocampal dentate gyrus. Neurobiol Learn Mem. 2002;77:354–371. doi: 10.1006/nlme.2001.4034. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-protein specifically disrupt cognitive function. Nature Neuroscience. 2005a;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005b;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Comery TA, Martone RL, Aschmies S, Atchison KP, Diamantidis G, Gong X, Zhou H, Kreft AF, Pangalos MN, Sonnenberg-Reines J, Jacobsen JS, Marquis KL. Acute gamma-secretase inhibition improves contextual fear conditioning in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2005;25:8898–8902. doi: 10.1523/JNEUROSCI.2693-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran KA, Lu Y, Turner RS, Maren S. Overexpression of hAPPswe impairs rewarded alternation and contextual fear conditioning in a transgenic mouse model of Alzheimer’s disease. Learn Mem. 2002;9:243–252. doi: 10.1101/lm.51002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Xia X, Bui D, Sweatt JD, Zheng H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J Biol Chem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD. Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci. 2001;21:4125–4133. doi: 10.1523/JNEUROSCI.21-12-04125.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JJ, Wu J, Nichols RA. Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci. 2003;23:6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster TC, Sharrow KM, Masse JR, Norris CM, Kumar A. Calcineurin links Ca2+ dysregulation with brain aging. J Neurosci. 2001;21:4066–4073. doi: 10.1523/JNEUROSCI.21-11-04066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukudo M, Yano I, Masuda S, Okuda M, Inui K. Distinct inhibitory effects of tacrolimus and cyclosporin a on calcineurin phosphatase activity. J Pharmacol Exp Ther. 2005;312:816–825. doi: 10.1124/jpet.104.074930. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol Aging. 2006;27:570–575. doi: 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O’Callaghan JP, Morgan D. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol. 2002;173:183–195. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Ikegami S, Inokuchi K. Antisense DNA against calcineurin facilitates memory in contextual fear conditioning by lowering the threshold for hippocampal long-term potentiation induction. Neuroscience. 2000;98:637–646. doi: 10.1016/s0306-4522(00)00161-5. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lin CH, Lee CC, Gean PW. Involvement of a calcineurin cascade in amygdala depotentiation and quenching of fear memory. Mol Pharmacol. 2003a;63:44–52. doi: 10.1124/mol.63.1.44. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Leu TH, Chang WC, Wang ST, Gean PW. Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci. 2003b;23:1574–1579. doi: 10.1523/JNEUROSCI.23-05-01574.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell. 2001;104:675–686. doi: 10.1016/s0092-8674(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Mansuy IM. Calcineurin in memory and bidirectional plasticity. Biochem Biophys Res Commun. 2003;311:1195–1208. doi: 10.1016/j.bbrc.2003.10.046. [DOI] [PubMed] [Google Scholar]
- Mansuy IM, Shenolikar S. Protein serine/threonine phosphatases in neuronal plasticity and disorders of learning and memory. Trends Neurosci. 2006;29:679–686. doi: 10.1016/j.tins.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Maren S, Aharonov G, Fanselow MS. Retrograde abolition of conditional fear after excitotoxic lesions in the basolateral amygdala of rats: absence of a temporal gradient. Behav Neurosci. 1996;110:718–726. doi: 10.1037//0735-7044.110.4.718. [DOI] [PubMed] [Google Scholar]
- Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J Neurosci. 2005;25:4649–4658. doi: 10.1523/JNEUROSCI.0365-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olyaei AJ, de Mattos AM, Bennett WM. Nephrotoxicity of immunosuppressive drugs: new insight and preventive strategies. Curr Opin Crit Care. 2001;7:384–389. doi: 10.1097/00075198-200112000-00003. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Defining molecular targets to prevent Alzheimer disease. Archives of Neurology. 2005;62:192–195. doi: 10.1001/archneur.62.2.192. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Springer JE, Azbill RD, Nottingham SA, Kennedy SE. Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury. J Neurosci. 2000;20:7246–7251. doi: 10.1523/JNEUROSCI.20-19-07246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staatz CE, Tett SE. Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation. Clin Pharmacokinet. 2004;43:623–653. doi: 10.2165/00003088-200443100-00001. [DOI] [PubMed] [Google Scholar]
- Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T. Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol. 2000;157:331–339. doi: 10.1016/s0002-9440(10)64544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gassen G, Annaert W. Amyloid, presenilins, and Alzheimer’s disease. Neuroscientist. 2003;9:117–126. doi: 10.1177/1073858403252227. [DOI] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH. The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2002;22:1858–1867. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Chattarji S, Barbarosie M, Rondi-Reig L, Philpot BD, Miyakawa T, Bear MF, Tonegawa S. Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell. 2001;107:617–629. doi: 10.1016/s0092-8674(01)00585-2. [DOI] [PubMed] [Google Scholar]