

Surgical bypass via autologous vein remains an evidence-based treatment of choice for selected patients with infra-inguinal lower extremity or coronary occlusive disease. However, contemporary data shows that almost 40% of lower extremity vein bypass grafts develop occlusive lesions or fail within a year(1), and almost half of cardiac bypass patients will lose (> or = 75% stenosis) a vein graft within a year(2). Since many technical avenues for improved patency have been exhausted, the future of enhancing the durability of these reconstructions lies in a better knowledge of and interventions based on the biology of the vein graft wall.
This article will briefly review vein graft failure research to date, and then focus on the pro-inflammatory cytokine TNF-α and the early vein graft. Finally, the current status of the field will be outlined in the context of cytokine based research, and challenges and opportunities for the future discussed. Certainly a multitude of biochemicals (growth factors, cell cycle regulators, etc.) have been linked to mechanisms of vein graft failure, and the following is in no means comprehensive.
Evolution of Current Vein Graft Concepts
Vein grafts undergo a defined sequence of anatomic adaptations after placement, though not all favor long-term patency. The principal cause of failure is traditionally cited as development of neointimal hyperplasia which leads to an obliterative stenosis(3-7). Early work in the vein graft research field focused on mechanical factors(3-5;8). Like arteries, vein graft wall structure adapts to the hemodynamic environment(9;10), though there may be some subtle wall differences(5). Intimal hyperplasia has been noted to occur at vein graft areas of low flow(3), probably areas of low wall shear stress(4;8). Conversely, high flow appears to have protective effects(6;11), in association with decreased wall inflammation(12). The early 1990's also brought a recognition of the importance of circumferential wall tension on the adapting vein graft (5;13).
The 1990's saw increased recognition in vascular biology of the interplay between the inflammatory and cardiovascular systems. The arterial wall response to injury was associated with early inflammatory events including monocyte and T cell adhesion to vascular endothelial cells(14-16). Platelet activation and mural thrombus formation were also implicated in this cascade, as well as cytokine and growth factor elaboration, all leading to subsequent vascular remodeling(17) through cellular migration, proliferation, and matrix deposition(14-16;18;19). In the mid- 1990's, these paradigms began to transfer to the vein graft arena(20). Works specifically examined the role of inflammatory mediators in vein graft failure(10;21-24). For instance, the macrophage was identified as a pivotal cellular mediator of vein graft neointimal hyperplasia, with macrophage depletion suppressesing this process(22).
However, despite incremental progress over these decades, specific cause/effect links between hemodynamic factors → inflammatory biochemical mediators → cellular effectors → vein wall adaptations remain lacking. Thus, not surprisingly, few therapeutic agents to improve vein graft durability have been identified. Anti-platelet and anticoagulant approaches show only a modest benefit under specific circumstances (25;26). Recent trials testing edifoligide (an oligonucleotide decoy that binds to and inhibits E2F transcription factors) failed to yield substantial clinical benefit(1;2).
Pro-inflammatory Cytokines and Vein Graft Failure
Pro-inflammatory cytokines (e.g. TNF-α and IL-1β) were implicated in vein graft intimal hyperplasia almost a decade ago(10;23), though the initiating factors for their expression and the biologic implications of these inflammatory mediators in vein wall adaptations remained largely unknown. Expanding knowledge in cytokine-driven inflammatory pathways in other organ systems has led to effective methods for treating pathologies such as rheumatoid arthritis and inflammatory bowel disease(27-35), and several anti-inflammatory cytokine based pharmacologic compounds have emerged(36-38). While anti-inflammatory cytokine therapies have received recent attention as a means to abrogate primary arterial occlusive disease (39), cytokine manipulation strategies remain relatively unexplored with regards to vein graft failure.
TNF-α is a pleiotropic pro-inflammatory cytokine(40;41). Its expression is controlled at the level of both gene transcription and translation, and it can be synthesized by several cell lines relevant to vascular biology, including macrophages, T-cells, endothelial cells, fibroblasts, and smooth muscle cells(42;43). This potent pro-inflammatory cytokine is initially synthesized and processed to a transmembrane form(44). TNF-α-converting enzyme (TACE), a member of the matrix metalloproteinase superfamily, releases TNF-α from the cell surface(45;46), and as a homotrimer, the soluble TNF-α elicits responses in distal target cells(43). Since its description over three decades ago, several related ligands have been described and are grouped in a TNF superfamily of genes(47).
The tissue response to TNF-α is mediated through two distinct receptors, p55 (type 1 TNF receptor) and p75 (type 2 TNF receptor)(40;48). These receptors belong to a large TNF receptor superfamily which also includes NGFR, CD95, and Apo2(47;49;50). Most cell types co-express both TNF receptors, though expression of the two receptors appears to be differentially regulated and show tissue-specific prevalence. More importantly, the two receptors differ markedly in their intracellular structure and signaling pathways(40;43). The majority of the pro-inflammatory responses classically attributed to TNF-α appear to be mediated by p55 signaling. Studies have shown that administration of TNF-α muteins with specificity for the p55 receptor are pro-inflammatory and shock inducing, whereas p75 muteins lack any pro-inflammatory properties(51;52).
The theory for a pivotal role for TNF-α in vein graft neointima formation and the related pathologic process of atherogenesis is founded on in vitro cell culture studies, pathologic observations, and a limited number of in vivo studies. In cell culture, TNF-α augments expression of intercellular adhesion molecules in human vascular endothelial cells(53) and vascular smooth muscle cells(54), thus increasing the possibility of interactions between mononuclear cells, endothelial cells, and smooth muscle cells in neointimal lesions and atherosclerotic plaques. Additionally, TNF-α induces prostanoid synthesis, corticosteroids, and other cytokines(41), and stimulates smooth muscle cell migration(55) and proliferation after vascular injury(42). Via receptors, TNF-α signaling activates caspases leading to apoptosis, MAP kinases and NF-kappaB(40)—intracellular mediators linked to numerous fundamental vascular processes.
Few studies have extended these in vitro cell culture and pathologic studies into the more complex in vivo vascular setting. Pathologically, TNF-α co-localizes to areas of occlusive lesions in human arteries(56) and arterialized vein grafts(23). In a rabbit heterotopic cardiac transplantation model, in vivo blockade of TNF-α by way of TNF soluble receptor suppressed the acute development of neointima formation by selectively reducing the vascular inflammatory reaction and accumulation of fibronectin(57). Nonetheless, the mechanisms of transplant atherosclerosis may be quite different from those of vein graft neointimal hyperplasia. Another set of in vivo experiments demonstrated that exogenous TNF-α causes coronary arteriosclerosis-like cellular changes in a porcine model(58).
Recent Research
For the last decade, our group has probed the role of cytokines in vascular biology. Initially, we were interested in the role of TNF-α in low shear stress induced arterial intimal hyperplasia. Using murine models and molecular approaches, we documented induction of TNF-α by acute lowering of arterial wall shear stress(59). We have also probed the role of cytokine signaling in the arterial wall response to high shear stress. It had been shown that TNF-α co-localizes to areas of active positive remodeling in response to increased wall shear stress(60). Working with collaborators, we completed experiments utilizing a novel murine model of arteriogenesis, a clinically relevant form of outward arterial remodeling in response to increased wall shear stress. The results showed that TNF-α positively modulates arteriogenesis, probably signaling via the p55 receptor(61). Recent experiments demonstrate that this process is blocked with the administration of TNF-α inhibitors(62).
Based on the above findings linking vascular wall adaptations to changes in hemodynamic environment via pro- and anti-inflammatory cytokine signaling, we hypothesized similar mechanisms in the vein graft. The vein graft is essentially an extreme example of acute hemodynamic change, coupled with a local injury response, leading to morphologic adaptations within the vascular wall. We developed and validated a bilateral jugular vein into carotid artery vein graft model with clinically relevant differential hemodynamic environments(6;63-65). In this model, unilateral reduction in carotid artery (and thus vein graft) flow is accomplished via placement of 8-0 silk suture ligatures to completely occlude the internal carotid and three of the four primary branches of the external carotid artery. Distal branch ligation results in an immediate 90% flow reduction (p<0.001) in the vein graft on the ligated side and 36% flow augmentation (p=0.01) in the contralateral vein graft. The vein grafts develop physiologically relevant levels of wall shear, and neointimal hyperplasia volume that is inversely proportional to wall shear.
To initiate studies into molecular mediators of these vein graft adaptations, quantitative real-time two-step polymerase chain reaction (RT-PCR) was performed for TNF -α, IL-1β and IL-10 on the paired high and low wall shear vein grafts in this rabbit model longitudinally. The results revealed several shear and time dependent cytokine expression signatures (Figure 1)(66;67). TNF-α induction was maximal at day one and gradually decreased over time, but was persistently elevated even four-weeks later (p<0.001). Low shear (associated with increased neointimal hyperplasia) resulted in significantly higher TNF-α mRNA expression (p=0.03). TNF-α was induced 198 and 110 fold in low and high shear vein grafts respectively by the first post-arterialization day. This elevation gradually decreased over time but was persistently elevated from baseline even four weeks later (p<0.001). Over the course of the study, low shear resulted in significantly higher TNF -α mRNA (p<0.003) vein graft wall expression.
Figure 1.
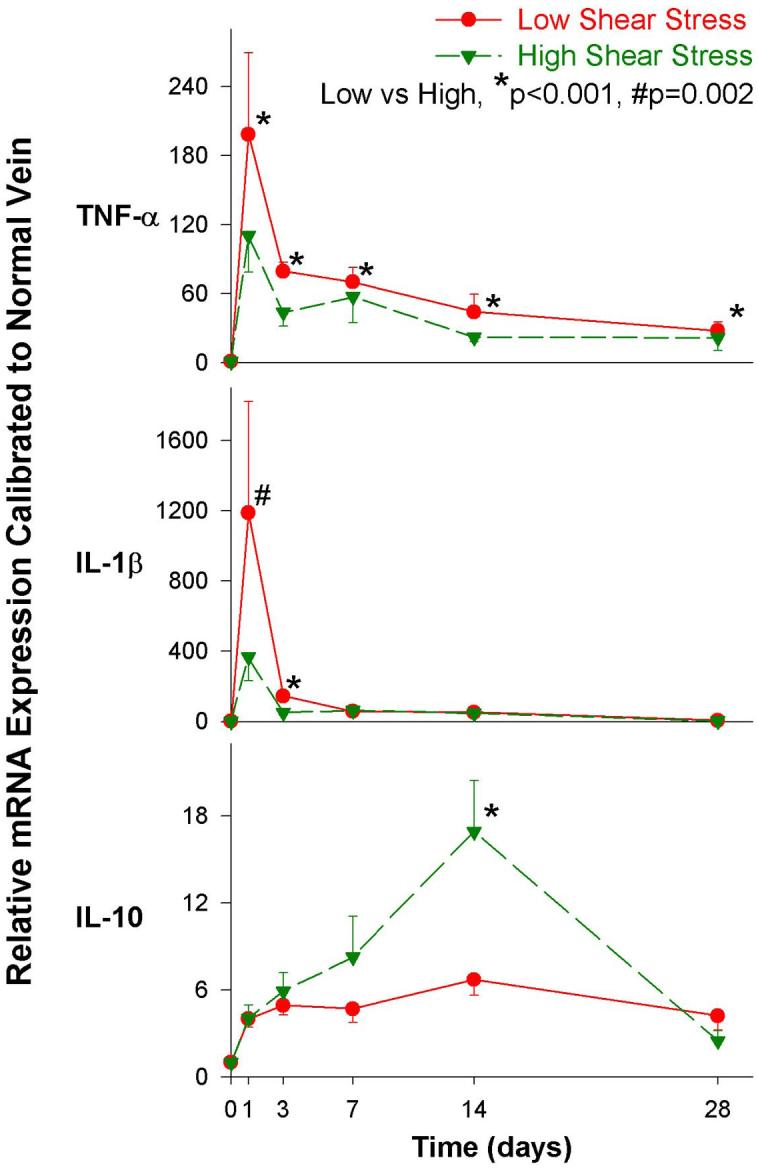
Time course of cytokine mRNA expression in a rabbit bilateral vein graft model with differential shear. Data is combined from two prior J Vasc Surg reports(66;67).
While the general expression pattern of IL-1β expression mirrored that of TNF -α, several notable differences exist. IL-1β was induced a striking 1188- and 366- fold in low and high shear vein grafts respectively at day 1, but the return toward baseline was more rapid. Flow impacted IL-1β expression overall (p<0.001), though the differential was greatest at the 1 (p=0.002) and 3 (p<0.0001) day time points. Consistent with the theory that pro-inflammatory cytokine mechanisms drive downstream vein graft adaptations, these early quantitative TNF-α and IL-1β mRNA level changes were temporally distinct from the time course of later morphologic and cellular changes in the vein graft wall. High pro-inflammatory cytokine levels (as in the low flow setting) correlated positively with greater intimal hyperplasia. Via immunohistochemistry, TNF-α and IL-1β protein localize to the vein intima in the first three days after graft placement.
Finally, vein graft arterialization more slowly and modestly induced IL-10 mRNA expression. Overall this occurred independent of shear (p=0.152), though there was a statistically significant higher expression for high shear grafts at the 14-day time point (p<0.001). IL-10 is an immunosuppressive and anti-inflammatory cytokine produced by T-cells, B-cells, natural killer cells, and monocyte/macrophage cell lines(12;68). It has been shown to suppress the production of numerous inflammatory cytokines, including TNF-α(41). Conversely, TNF-α is a principal inducer of IL-10 biosynthesis(69). This acts in a negative feedback loop to suppress TNF-α production and processing.
IL-10 is believed to exert its anti-inflammatory effects on the vascular system through inhibition of leukocyte-endothelial cell interactions and inhibition of pro-inflammatory cytokine and chemokine production(12;68). In support of the hypothesis that the anti-inflammatory cytokine IL-10 downregulates vein graft neointimal hyperplasia, researchers have demonstrated an effect of IL-10 on vascular smooth muscle cell proliferation. Physiologic doses of IL-10 inhibited TNF-α and bFGF-stimulated DNA synthesis and cell proliferation(70), suggesting that endogenous IL-10 not only suppresses pro-inflammatory cytokine expression, but also may antagonize pathologic vascular remodeling induced by cytokines such as TNF-α(70).
These results(66;67) in the context of the medical literature (10;23;71-75) led our group to formulate the general hypothesis outlined in Figure 2. As an initial step to test this hypothesis, we utilized a pharmacologic approach to abrogate TNF-α signaling in the early vein graft of our validated rabbit model(66). Animals received pegylated soluble TNF-α Type I receptor (PEG sTNF-RI; Amgen) or vehicle via either short or long-term dosing. PEG sTNF-RI is a 20 kd molecule containing a homodimer of human p55 covalently linked to a polyethylene glycerol backbone(76). Molecular modification of these pegylated receptors through deletion of 1.4 intracellular domains reduces immunogenicity while having no impact on ligand binding(77). Due to a conserved sequence homology, the compound has been demonstrated to abrogate the adaptive immune response across a range of species, including rabbits(76;78). After 14-28 days, grafts were analyzed. PEG-sTNF-R1 was found in high concentrations in the serum, and localized to neointimal hyperplasia microscopically. Both high and low flow vein grafts from treated animals demonstrated similar volumes of neointimal hyperplasia compared to controls. PEG-sTNF-R1 had minimal impact on vascular wall cell turnover, as reflected by TUNEL and anti-Ki-67 assays(66).
Figure 2.

Hypothetical mechanisms by which pro- and anti-inflammatory cytokines may interplay with wall shear to modulate vein graft wall adaptations.
Thus, while placement of a vein into the arterial circulation acutely upregulates TNF-α(23;66;75) (whose expression level correlates with the degree of subsequent neointimal hyperplasia), pharmacologic interruption of this signaling pathway has no significant impact on neointimal hyperplasia or smooth muscle cell proliferation/apoptosis(66). These data suggest that early vein graft adaptations can proceed via TNF-α independent mechanisms. Recent work by other investigators, however, supports a differing conclusion. Using p55 receptor knockout animals, functional TNF-α inhibition has been shown to attenuate vein graft neointimal hyperplasia(71). Further investigation is required to elucidate these apparently contrasting observations.
Interesting comparisons can be drawn with observed challenges in application of anti-inflammatory approaches in other pathologies. Early anti-TNF-α clinical trials in acute inflammatory processes, such as sepsis, have had disappointing results(79-82) and may be attributed to an over-simplistic view of these disease processes and TNF-α mediated cytotoxicity(41). A similar situation arose in the setting of anti-TNF-α trials for heart failure(83). These experiences may have lessons for work with the vein graft. We have recently completed microarray analyses of vein graft wall in both mice and rabbits (both high and low flow)(84). The results reveal a large number of gene perturbations across multiple families of mediators. These results show that the overwhelming determinant of the wall's transcriptome is the temporal relationship to the operative graft placement—that is, the trauma of the operation itself, rather than the details (neointimal volume, etc.) of the wall adaption. Thus, it may be naive to believe that abrogation of a single mediator would have substantial lasting impact on the final morphology of the wall. Perhaps strategies that block central signaling molecules that control numerous genes for various cytokines and adhesion molecules will be effective (e.g. NF-kappaB)(85), though targets such as TNF-α seem to meet this criteria.
Future Considerations and Directions
To date several lessons have become apparent, and some considerations for future progress are summarized:
Single- vs Multi-agent Strategies Narrowly focused molecular targets hold the appeal of limited unwanted side effects. However, in view of the large number of mediators implicated in the vein graft wall adaptation, pertubation of several pathways may be necessary to consistently achieve substantial and durable effect. For example, multimodality approaches stand as a mainstay of anti-neoplastic therapies. The multitude of processes involved in vein graft failure support use of such strategies, yet the rationale and safety of each component must be confirmed, and substantial investigative work will be required to define the composition of this “synergistic” cocktail.
Understanding the Interplay of Systems (e.g. Inflammatory, Thrombotic), including Genetic Factors Large amounts of information (e.g. biologic and genetic) can now be rapidly acquired and analyzed via high throughput experimental and statistical techniques. Vascular biologist must embrace contemporary information management and modeling approaches to understand the interplay of these factors in vein graft failure.
Broadening Focus to the Entire Conduit Wall Vein graft researchers must broaden their observations to the behavior of the entire conduit wall, not just the neointima. Adventitial events leading to fibrosis probably contribute substantially to vein graft failure(86).
Consideration of the Injured Host Patient Simple harvest dramatically changes the vein wall cellular phenotype(87). Furthermore, surgical trauma globally impacts the phenotype of pivotal cells such as the leukocyte(88;89), and these effects may all biologically modulate local processes such as vein graft wall adaptations.
Delivery Strategies—local vs systemic While vein grafts offer the unique situation of ability to treat the conduit wall(1;85;90), more systemic approaches may be needed in view of the extra-wall mediators (e.g. circulating cells) that participate in occlusive adaptations(91).
Optimization of Trial Design to Insure Gain of New Knowledge Regardless of Outcome Animal models of vein graft disease hold substantial biologic relevance limitations, and clinical trials are expensive. While certainly breaking new ground, as designed, the PREVENT Trials(1;2) failed to generate substantial new biologic insights despite substantial work and fiscal investments. Thoughtful addition of mechanistic endpoints when feasible will insure some progress independent of human trial outcome.
Translation into other Arterial Occlusive Adaptations (Primary Atherosclerosis, Angioplasty Restenosis, etc) Emerging endovascular approaches bring into question the relevance of vein graft research. However, contemporary evidence based guidelines confirm that a substantial portion of our aging population will require vein conduits for arterial revascularization. Additionally, basic biologic mechanisms delineated in the vein graft field hold a strong likelihood of relevance for other vascular responses to injury.
Summary
Understanding the cytokine mediated molecular mechanisms of vein graft arterialization may suggest clinical interventions that will alter the conduit's natural history. The field appears especially ripe for transfer of knowledge and therapeutic approaches that have evolved in the arterial system, and inflammatory mediated processes such as inflammatory bowel disease and arthritis. However, more robust research approaches such as broadening of the scope beyond focus on single mediators and neointimal hyperplasia will be necessary to reach translatable strategies to prolong human vein graft durability.
Acknowledgment
The author gratefully acknowledges the editorial assistance of Dr. Scott A. Berceli and Dr. Zhihua Jiang.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- 1.Conte MS, Bandyk DF, Clowes AW, Moneta GL, Seely L, Lorenz TJ, et al. Results of PREVENT III: a multicenter, randomized trial of edifoligide for the prevention of vein graft failure in lower extremity bypass surgery. J Vasc Surg. 2006;43(4):742–751. doi: 10.1016/j.jvs.2005.12.058. [DOI] [PubMed] [Google Scholar]
- 2.Alexander JH, Hafley G, Harrington RA, Peterson ED, Ferguson TB, Jr., Lorenz TJ, et al. Efficacy and safety of edifoligide, an E2F transcription factor decoy, for prevention of vein graft failure following coronary artery bypass graft surgery: PREVENT IV: a randomized controlled trial. JAMA. 2005;294(19):2446–2454. doi: 10.1001/jama.294.19.2446. [DOI] [PubMed] [Google Scholar]
- 3.Berguer R, Higgins RF, Reddy DJ. Intimal hyperplasia. An experimental study. Arch Surg. 1980;115(3):332–335. doi: 10.1001/archsurg.1980.01380030078019. [DOI] [PubMed] [Google Scholar]
- 4.Dobrin PB, Littooy FN, Endean ED. Mechanical factors predisposing to intimal hyperplasia and medial thickening in autogenous vein grafts. Surgery. 1989;105(3):393–400. [PubMed] [Google Scholar]
- 5.Galt SW, Zwolak RM, Wagner RJ, Gilbertson JJ. Differential response of arteries and vein grafts to blood flow reduction. J Vasc Surg. 1993;17(3):563–570. [PubMed] [Google Scholar]
- 6.Jiang Z, Wu L, Miller BL, Goldman DR, Fernandez CM, Abouhamze ZS, et al. A novel vein graft model: adaptation to differential flow environments. Am J Physiol Heart Circ Physiol. 2004;286(1):H240–H245. doi: 10.1152/ajpheart.00760.2003. [DOI] [PubMed] [Google Scholar]
- 7.Meyerson SL, Skelly CL, Curi MA, Shakur UM, Vosicky JE, Glagov S, et al. The effects of extremely low shear stress on cellular proliferation and neointimal thickening in the failing bypass graft. J Vasc Surg. 2001;34(1):90–97. doi: 10.1067/mva.2001.114819. [DOI] [PubMed] [Google Scholar]
- 8.Morinaga K, Okadome K, Kuroki M, Miyazaki T, Muto Y, Inokuchi K. Effect of wall shear stress on intimal thickening of arterially transplanted autogenous veins in dogs. J Vasc Surg. 1985;2(3):430–433. [PubMed] [Google Scholar]
- 9.Davies MG, Klyachkin ML, Dalen H, Svendsen E, Hagen PO. Regression of intimal hyperplasia with restoration of endothelium-dependent relaxing factor-mediated relaxation in experimental vein grafts. Surgery. 1993;114(2):258–270. [PubMed] [Google Scholar]
- 10.Sterpetti AV, Cucina A, Lepidi S, Randone B, Corvino V, D'Angelo LS, et al. Formation of myointimal hyperplasia and cytokine production in experimental vein grafts. Surgery. 1998;123(4):461–469. [PubMed] [Google Scholar]
- 11.Paszkowiak JJ, Dardik A. Arterial wall shear stress: observations from the bench to the bedside. Vasc Endovascular Surg. 2003;37(1):47–57. doi: 10.1177/153857440303700107. [DOI] [PubMed] [Google Scholar]
- 12.Tedgui A, Mallat Z. Anti-inflammatory mechanisms in the vascular wall. Circ Res. 2001;88(9):877–887. doi: 10.1161/hh0901.090440. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz LB, O'Donohoe MK, Purut CM, Mikat EM, Hagen PO, McCann RL. Myointimal thickening in experimental vein grafts is dependent on wall tension. J Vasc Surg. 1992;15(1):176–186. doi: 10.1067/mva.1992.33805. [DOI] [PubMed] [Google Scholar]
- 14.Schwartz SM, deBlois D, O'Brien ER. The intima. Soil for atherosclerosis and restenosis. Circ Res. 1995;77(3):445–465. doi: 10.1161/01.res.77.3.445. [DOI] [PubMed] [Google Scholar]
- 15.Gimbrone-MA J. Atherogenesis: current concepts. Monogr Pathol. 1995;37:1–11. [PubMed] [Google Scholar]
- 16.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362(6423):801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 17.Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med. 1994;330(20):1431–1438. doi: 10.1056/NEJM199405193302008. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz SM, Reidy MA, de BD. Factors important in arterial narrowing. J Hypertens Suppl. 1996;14(5):S71–S81. [PubMed] [Google Scholar]
- 19.Ross R. Cell biology of atherosclerosis. Annu Rev Physiol. 1995;57:791–804. doi: 10.1146/annurev.ph.57.030195.004043. [DOI] [PubMed] [Google Scholar]
- 20.Motwani JG, Topol EJ. Aortocoronary saphenous vein graft disease: pathogenesis, predisposition, and prevention. Circulation. 1998;97(9):916–931. doi: 10.1161/01.cir.97.9.916. [DOI] [PubMed] [Google Scholar]
- 21.Hoch JR, Stark VK, Hullett DA, Turnipseed WD. Vein graft intimal hyperplasia: leukocytes and cytokine gene expression. Surgery. 1994;116(2):463–470. [PubMed] [Google Scholar]
- 22.Hoch JR, Stark VK, van Rooijen N, Kim JL, Nutt MP, Warner TF. Macrophage depletion alters vein graft intimal hyperplasia. Surgery. 1999;126(2):428–437. [PubMed] [Google Scholar]
- 23.Faries PL, Marin ML, Veith FJ, Ramirez JA, Suggs WD, Parsons RE, et al. Immunolocalization and temporal distribution of cytokine expression during the development of vein graft intimal hyperplasia in an experimental model. J Vasc Surg. 1996;24(3):463–471. doi: 10.1016/s0741-5214(96)70203-3. [DOI] [PubMed] [Google Scholar]
- 24.Ryomoto M, Wolff RA, Tomas JJ, Miyamoto T, Hoch JR. 17beta-estradiol attenuates intimal hyperplasia and macrophage accumulation with a reduction in monocyte chemoattractant protein 1 expression in a vein graft model. J Vasc Surg. 2002;36(3):613–621. doi: 10.1067/mva.2002.125845. [DOI] [PubMed] [Google Scholar]
- 25.Becquemin JP. Effect of ticlopidine on the long-term patency of saphenous-vein bypass grafts in the legs. Etude de la Ticlopidine apres Pontage Femoro-Poplite and the Association Universitaire de Recherche en Chirurgie. N Engl J Med. 1997;337(24):1726–1731. doi: 10.1056/NEJM199712113372404. [DOI] [PubMed] [Google Scholar]
- 26.Sarac TP, Huber TS, Back MR, Ozaki CK, Carlton LM, Flynn TC, et al. Warfarin improves the outcome of infrainguinal vein bypass grafting at high risk for failure. J Vasc Surg. 1998;28(3):446–457. doi: 10.1016/s0741-5214(98)70130-2. [DOI] [PubMed] [Google Scholar]
- 27.Bresnihan B, Cobby M. Clinical and radiological effects of anakinra in patients with rheumatoid arthritis. Rheumatology (Oxford) 2003;42(Suppl 2):ii22–8. doi: 10.1093/rheumatology/keg329. ii22-ii28. [DOI] [PubMed] [Google Scholar]
- 28.Lovell DJ, Giannini EH, Reiff A, Cawkwell GD, Silverman ED, Nocton JJ, et al. Etanercept in children with polyarticular juvenile rheumatoid arthritis. Pediatric Rheumatology Collaborative Study Group. N Engl J Med. 2000 Mar 16;342(11):763–9. doi: 10.1056/NEJM200003163421103. 342(11):763-769. [DOI] [PubMed] [Google Scholar]
- 29.Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein [see comments] N Engl J Med. 1997;337(3):141–147. doi: 10.1056/NEJM199707173370301. [DOI] [PubMed] [Google Scholar]
- 30.Srivastava MD. Immunomodulatory effects of etanercept (TNFR:Fc) and its use in a patient with Crohn's disease. Res Commun Mol Pathol Pharmacol. 2001;109(12):125–141. [PubMed] [Google Scholar]
- 31.Taylor PC. Anti-tumor necrosis factor therapies. Curr Opin Rheumatol. 2001;13(3):164–169. doi: 10.1097/00002281-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 32.Scott DL, Kingsley GH. Tumor necrosis factor inhibitors for rheumatoid arthritis. N Engl J Med. 2006;355(7):704–712. doi: 10.1056/NEJMct055183. [DOI] [PubMed] [Google Scholar]
- 33.Sandborn WJ. New concepts in anti-tumor necrosis factor therapy for inflammatory bowel disease. Rev Gastroenterol Disord. 2005;5(1):10–18. [PubMed] [Google Scholar]
- 34.Narula SK, Cutler D, Grint P. Immunomodulation of Crohn's disease by interleukin-10. Agents Actions Suppl. 1998;49:57–65. doi: 10.1007/978-3-0348-8857-8_9. 57-65. [DOI] [PubMed] [Google Scholar]
- 35.Rogler G, Andus T. Cytokines in inflammatory bowel disease. World J Surg. 1998;22(4):382–389. doi: 10.1007/s002689900401. [DOI] [PubMed] [Google Scholar]
- 36.Furst DE, Breedveld FC, Kalden JR, Smolen JS, Burmester GR, Bijlsma JW, et al. Updated consensus statement on biological agents, specifically tumour necrosis factor {alpha} (TNF{alpha}) blocking agents and interleukin-1 receptor antagonist (IL-1ra), for the treatment of rheumatic diseases, 2005. Ann Rheum Dis. 2005;64(Suppl 4):iv2–14. doi: 10.1136/ard.2005.044941. iv2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Anti-TNF-alpha therapies: the next generation. Nat Rev Drug Discov. 2003;2(9):736–746. doi: 10.1038/nrd1175. [DOI] [PubMed] [Google Scholar]
- 38.Economides AN, Carpenter LR, Rudge JS, Wong V, Koehler-Stec EM, Hartnett C, et al. Cytokine traps: multi-component, high-affinity blockers of cytokine action. Nat Med. 2003;9(1):47–52. doi: 10.1038/nm811. [DOI] [PubMed] [Google Scholar]
- 39.Terkeltaub RA. IL-10: An “Immunologic Scalpel” for atherosclerosis? Arterioscler Thromb Vasc Biol. 1999;19(12):2823–2825. doi: 10.1161/01.atv.19.12.2823. [DOI] [PubMed] [Google Scholar]
- 40.Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11(9):372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 41.Ksontini R, MacKay SL, Moldawer LL. Revisiting the role of tumor necrosis factor alpha and the response to surgical injury and inflammation. Arch Surg. 1998;133(5):558–567. doi: 10.1001/archsurg.133.5.558. [DOI] [PubMed] [Google Scholar]
- 42.Tanaka H, Sukhova G, Schwartz D, Libby P. Proliferating arterial smooth muscle cells after balloon injury express TNF-alpha but not interleukin-1 or basic fibroblast growth factor. Arterioscler Thromb Vasc Biol. 1996;16(1):12–18. doi: 10.1161/01.atv.16.1.12. [DOI] [PubMed] [Google Scholar]
- 43.Bluethmann H. Physiological, Immunological, and Pathological Functions of Tumor Necrosis Factor (TNF) Revealed by TNF Receptor -Deficient Mice. In: Durum SK, Muegge K, editors. Cytokine Knockouts. Humana Press; Totowa, New Jersy: 1998. pp. 69–87. [Google Scholar]
- 44.Kriegler M, Perez C, DeFay K, Albert I, Lu SD. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell. 1988;53(1):45–53. doi: 10.1016/0092-8674(88)90486-2. [DOI] [PubMed] [Google Scholar]
- 45.Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. 1997;385(6618):733–736. doi: 10.1038/385733a0. [published erratum appears in Nature 1997 Apr 17;386(6626):738] [DOI] [PubMed] [Google Scholar]
- 46.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor- alpha from cells. Nature. 1997;385(6618):729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 47.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104(4):487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 48.Tartaglia LA, Weber RF, Figari IS, Reynolds C, Palladino MAJ, Goeddel DV. The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc Natl Acad Sci U S A. 1991;88(20):9292–9296. doi: 10.1073/pnas.88.20.9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tartaglia LA, Rothe M, Hu YF, Goeddel DV. Tumor necrosis factor's cytotoxic activity is signaled by the p55 TNF receptor. Cell. 1993;73(2):213–216. doi: 10.1016/0092-8674(93)90222-c. [DOI] [PubMed] [Google Scholar]
- 50.Tartaglia LA, Goeddel DV. Two TNF receptors. Immunol Today. 1992;13(5):151–153. doi: 10.1016/0167-5699(92)90116-O. [DOI] [PubMed] [Google Scholar]
- 51.Van Zee KO, Stackpole SA, Montegut WJ, Rogy MA, Calvano SE, Hsu KC, et al. A human tumor necrosis (TNF) alpha mutant that binds exclusively to the p55 TNF receptor produces toxicity in the baboon. Journal of Experimental Medicine. 1994;179:1185–1191. doi: 10.1084/jem.179.4.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Welborn MB, Van Zee K, Edwards PD, Pruitt JH, Kaibara A, Vauthey JN, et al. A human tumor necrosis factor p75 receptor antagonist stimulates in vitro T cell proliferation but does not produce inflammation or shock in the baboon. Journal of Experimental Medicine. 1996;184:165–171. doi: 10.1084/jem.184.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wertheimer SJ, Myers CL, Wallace RW, Parks TP. Intercellular adhesion molecule-1 gene expression in human endothelial cells. Differential regulation by tumor necrosis factor-alpha and phorbol myristate acetate. J Biol Chem. 1992;267(17):12030–12035. [PubMed] [Google Scholar]
- 54.Couffinhal T, Duplaa C, Labat L, Lamaziere JM, Moreau C, Printseva O, et al. Tumor necrosis factor-alpha stimulates ICAM-1 expression in human vascular smooth muscle cells. Arterioscler Thromb. 1993;13(3):407–414. doi: 10.1161/01.atv.13.3.407. [DOI] [PubMed] [Google Scholar]
- 55.Jovinge S, Hultgardh-Nilsson A, Regnstrom J, Nilsson J. Tumor necrosis factor-alpha activates smooth muscle cell migration in culture and is expressed in the balloon-injured rat aorta. Arterioscler Thromb Vasc Biol. 1997;17(3):490–497. doi: 10.1161/01.atv.17.3.490. [DOI] [PubMed] [Google Scholar]
- 56.Rus HG, Niculescu F, Vlaicu R. Tumor necrosis factor-alpha in human arterial wall with atherosclerosis. Atherosclerosis. 1991;89(23):247–254. doi: 10.1016/0021-9150(91)90066-c. [DOI] [PubMed] [Google Scholar]
- 57.Clausell N, Molossi S, Sett S, Rabinovitch M. In vivo blockade of tumor necrosis factor-alpha in cholesterol-fed rabbits after cardiac transplant inhibits acute coronary artery neointimal formation. Circulation. 1994;89(6):2768–2779. doi: 10.1161/01.cir.89.6.2768. [DOI] [PubMed] [Google Scholar]
- 58.Fukumoto Y, Shimokawa H, Ito A, Kadokami T, Yonemitsu Y, Aikawa M, et al. Inflammatory cytokines cause coronary arteriosclerosis-like changes and alterations in the smooth-muscle phenotypes in pigs. J Cardiovasc Pharmacol. 1997;29(2):222–231. doi: 10.1097/00005344-199702000-00011. [DOI] [PubMed] [Google Scholar]
- 59.Rectenwald JE, Moldawer LL, Huber TS, Seeger JM, Ozaki CK. Direct evidence for cytokine involvement in neointimal hyperplasia. Circulation. 2000;102(14):1697–1702. doi: 10.1161/01.cir.102.14.1697. [DOI] [PubMed] [Google Scholar]
- 60.Arras M, Ito WD, Scholz D, Winkler B, Schaper J, Schaper W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J Clin Invest. 1998;101(1):40–50. doi: 10.1172/JCI119877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hoefer IE, van Royen N, Rectenwald JE, Bray EJ, Abouhamze Z, Moldawer LL, et al. Direct evidence for tumor necrosis factor-alpha signaling in arteriogenesis. Circulation. 2002;105(14):1639–1641. doi: 10.1161/01.cir.0000014987.32865.8e. [DOI] [PubMed] [Google Scholar]
- 62.Grundmann S, Hoefer I, Ulusans S, van Royen N, Schirmer SH, Ozaki CK, et al. Anti-tumor necrosis factor-{alpha} therapies attenuate adaptive arteriogenesis in the rabbit. Am J Physiol Heart Circ Physiol. 2005;289(4):H1497–H1505. doi: 10.1152/ajpheart.00959.2004. [DOI] [PubMed] [Google Scholar]
- 63.Fernandez CM, Goldman DR, Jiang Z, Ozaki CK, Tran-Son-Tay R, Berceli SA. Impact of shear stress on early vein graft remodeling: a biomechanical analysis. Ann Biomed Eng. 2004;32(11):1484–1493. doi: 10.1114/b:abme.0000049033.65547.cf. [DOI] [PubMed] [Google Scholar]
- 64.Berceli SA, Jiang Z, Klingman NV, Schultz GS, Ozaki CK. Early differential MMP-2 and -9 dynamics during flow-induced arterial and vein graft adaptations. J Surg Res. 2006;134(2):327–334. doi: 10.1016/j.jss.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 65.Berceli SA, Jiang Z, Klingman NV, Pfahnl CL, Abouhamze ZS, Frase CD, et al. Differential expression and activity of matrix metalloproteinases during flow-modulated vein graft remodeling. J Vasc Surg. 2004;39(5):1084–1090. doi: 10.1016/j.jvs.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 66.Jiang Z, Shukla A, Miller BL, Espino DR, Tao M, Berceli SA, et al. TNF- and the early vein graft. J Vasc Surg. doi: 10.1016/j.jvs.2006.08.049. In press. [DOI] [PubMed] [Google Scholar]
- 67.Jiang Z, Berceli SA, Pfahnl CL, Wu L, Goldman D, Tao M, et al. Wall shear modulation of cytokines in early vein grafts. J Vasc Surg. 2004;40(2):345–350. doi: 10.1016/j.jvs.2004.03.048. [DOI] [PubMed] [Google Scholar]
- 68.Scumpia PO, Moldawer LL. Biology of interleukin-10 and its regulatory roles in sepsis syndromes. Crit Care Med. 2005;33(12 Suppl):S468–S471. doi: 10.1097/01.ccm.0000186268.53799.67. [DOI] [PubMed] [Google Scholar]
- 69.Wang P, Wu P, Siegel MI, Egan RW, Billah MM. IL-10 inhibits transcription of cytokine genes in human peripheral blood mononuclear cells. J Immunol. 1994;153(2):811–816. [PubMed] [Google Scholar]
- 70.Selzman CH, McIntyre RC, Jr, Shames BD, Whitehill TA, Banerjee A, Harken AH. Interleukin-10 inhibits human vascular smooth muscle proliferation. J Mol Cell Cardiol. 1998;30(4):889–896. doi: 10.1006/jmcc.1998.0642. [DOI] [PubMed] [Google Scholar]
- 71.Zhang L, Peppel K, Brian L, Chien L, Freedman NJ. Vein graft neointimal hyperplasia is exacerbated by tumor necrosis factor receptor-1 signaling in graft-intrinsic cells. Arterioscler Thromb Vasc Biol. 2004;24(12):2277–2283. doi: 10.1161/01.ATV.0000147766.68987.0d. [DOI] [PubMed] [Google Scholar]
- 72.Zimmerman MA, Reznikov LL, Raeburn CD, Selzman CH. Interleukin-10 attenuates the response to vascular injury. J Surg Res. 2004;121(2):206–213. doi: 10.1016/j.jss.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 73.Zimmerman MA, Reznikov LL, Sorensen AC, Selzman CH. Relative contribution of the TNF-alpha receptors to murine intimal hyperplasia. Am J Physiol Regul Integr Comp Physiol. 2003;284(5):R1213–R1218. doi: 10.1152/ajpregu.00434.2002. [DOI] [PubMed] [Google Scholar]
- 74.Zimmerman MA, Selzman CH, Reznikov LL, Miller SA, Raeburn CD, Emmick J, et al. Lack of TNF-alpha attenuates intimal hyperplasia after mouse carotid artery injury. Am J Physiol Regul Integr Comp Physiol. 2002;283(2):R505–R512. doi: 10.1152/ajpregu.00033.2002. [DOI] [PubMed] [Google Scholar]
- 75.Sharony R, Pintucci G, Saunders PC, Grossi EA, Baumann FG, Galloway AC, et al. Matrix metalloproteinase expression in vein grafts: role of inflammatory mediators and extracellular signal-regulated kinases-1 and -2. Am J Physiol Heart Circ Physiol. 2006;290(4):H1651–H1659. doi: 10.1152/ajpheart.00530.2005. [DOI] [PubMed] [Google Scholar]
- 76.Edwards CK, III, Martin SW, Seely J, Kinstler O, Buckel S, Bendele AM, et al. Design of PEGylated soluble tumor necrosis factor receptor type I (PEG sTNF-RI) for chronic inflammatory diseases. Adv Drug Deliv Rev. 2003;55(10):1315–1336. doi: 10.1016/s0169-409x(03)00112-1. [DOI] [PubMed] [Google Scholar]
- 77.Rosenberg JJ, Martin SW, Seely JE, Kinstler O, Gaines GC, Fukuzuka K, et al. Development of a novel, nonimmunogenic, soluble human TNF receptor type I (sTNFR-I) construct in the baboon. J Appl Physiol. 2001;91(5):2213–2223. doi: 10.1152/jappl.2001.91.5.2213. [DOI] [PubMed] [Google Scholar]
- 78.Porat R, Paddock HN, Schwaitzberg SD, Connolly RJ, Wilkens T, Dasch JR, et al. Glycosylated recombinant human tumor necrosis factor binding protein-1 reduces mortality, shock, and production of tumor necrosis factor in rabbit Escherichia coli sepsis. Crit Care Med. 1995;23(6):1080–1089. doi: 10.1097/00003246-199506000-00014. [DOI] [PubMed] [Google Scholar]
- 79.Fisher CJJ, Agosti JM, Opal SM, Lowry SF, Balk RA, Sadoff JC, et al. Treatment of Septic Shock with the Tumor Necrosis Factor Receptor:fc Fusion Protein. N Engl J Med. 1996;334(26):1697–1702. doi: 10.1056/NEJM199606273342603. [DOI] [PubMed] [Google Scholar]
- 80.Abraham E, Glauser MP, Butler T, Garbino J, Gelmont D, Laterre PF, et al. p55 Tumor necrosis factor receptor fusion protein in the treatment of patients with severe sepsis and septic shock. A randomized controlled multicenter trial. Ro 45-2081 Study Group. JAMA. 1997;277(19):1531–1538. [PubMed] [Google Scholar]
- 81.Cohen J, Carlet J. INTERSEPT: an international, multicenter, placebo-controlled trial of monoclonal antibody to human tumor necrosis factor-alpha in patients with sepsis. International Sepsis Trial Study Group. Crit Care Med. 1996;24(9):1431–1440. doi: 10.1097/00003246-199609000-00002. [DOI] [PubMed] [Google Scholar]
- 82.Reinhart K, Wiegand-Lohnert C, Grimminger F, Kaul M, Withington S, Treacher D, et al. Assessment of the safety and efficacy of the monoclonal anti-tumor necrosis factor antibody-fragment, MAK 195F, in patients with sepsis and septic shock: a multicenter, randomized, placebo-controlled, dose- ranging study [see comments] Crit Care Med. 1996;24(5):733–742. doi: 10.1097/00003246-199605000-00003. [published erratum appears in Crit Care Med 1996 Sep;24(9):1608] [DOI] [PubMed] [Google Scholar]
- 83.Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. 2003;107(25):3133–3140. doi: 10.1161/01.CIR.0000077913.60364.D2. [DOI] [PubMed] [Google Scholar]
- 84.Transcriptional analyses of early shear stress induced vein graft adaptations. Presented at Vascular 2006. 2006 Jun 2; [Google Scholar]
- 85.Miyake T, Aoki M, Shiraya S, Tanemoto K, Ogihara T, Kaneda Y, et al. Inhibitory effects of NFkappaB decoy oligodeoxynucleotides on neointimal hyperplasia in a rabbit vein graft model. J Mol Cell Cardiol. 2006;41(3):431–440. doi: 10.1016/j.yjmcc.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 86.Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, et al. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ Res. 2001;89(12):1111–1121. doi: 10.1161/hh2401.100844. [DOI] [PubMed] [Google Scholar]
- 87.Hinokiyama K, Valen G, Tokuno S, Vedin JB, Vaage J. Vein graft harvesting induces inflammation and impairs vessel reactivity. Ann Thorac Surg. 2006;82(4):1458–1464. doi: 10.1016/j.athoracsur.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 88.Laudanski K, Miller-Graziano C, Xiao W, Mindrinos MN, Richards DR, De A, et al. Cell-specific expression and pathway analyses reveal alterations in trauma-related human T cell and monocyte pathways. Proc Natl Acad Sci U S A. 2006;103(42):15564–15569. doi: 10.1073/pnas.0607028103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Biberthaler P, Bogner V, Baker HV, Lopez MC, Neth P, Kanz KG, et al. Genome-wide monocytic mRNA expression in polytrauma patients for identification of clinical outcome. Shock. 2005;24(1):11–19. doi: 10.1097/01.shk.0000163394.93467.77. [DOI] [PubMed] [Google Scholar]
- 90.Mann MJ, Whittemore AD, Donaldson MC, Belkin M, Conte MS, Polak JF, et al. Ex-vivo gene therapy of human vascular bypass grafts with E2F decoy: the PREVENT single-centre, randomised, controlled trial. Lancet. 1999;354(9189):1493–1498. doi: 10.1016/S0140-6736(99)09405-2. [DOI] [PubMed] [Google Scholar]
- 91.Zhang L, Freedman NJ, Brian L, Peppel K. Graft-extrinsic cells predominate in vein graft arterialization. Arterioscler Thromb Vasc Biol. 2004;24(3):470–476. doi: 10.1161/01.ATV.0000116865.98067.31. [DOI] [PubMed] [Google Scholar]