

Abstract
The transcription regulatory protein Sp3 shares more than 90% sequence homology with Sp1 in the DNA-binding domain and they bind to the same cognate DNA-element. However, the transcriptional activities of these two Sp-family factors are not equivalent. While Sp1 functions strictly as a transcriptional activator, Sp3 has been shown to be transcriptionally inactive for promoters containing multiple Sp-binding sites. In the present study, we show that the DNA-binding property of Sp3 is promoter dependent and is different from Sp1. The 116 kDa Sp3 polypeptide binds as a monomer to a single Sp-binding site but readily forms slower migrating complexes with adjacent Sp-binding sites. The slower migrating Sp3–DNA complexes are significantly more stable than monomeric Sp3–DNA complexes or multimeric Sp1–DNA complexes. As a consequence, Sp3 can efficiently compete with Sp1 for binding to regions containing multiple Sp sites. The transcription regulatory function of Sp3 is also significantly different from Sp1. Unlike Sp1, Sp3 does not synergistically activate transcription of promoters containing multiple Sp-binding sites. Therefore, although Sp3 is a transcription activator, Sp3 reduces Sp1-dependent transcription of promoters containing adjacent Sp-binding sites by competing with Sp1 for promoter occupancy and thereby blocking the synergistic transactivation function of Sp1. Taken together, this study provides a possible mechanism of the promoter-specific transcription repression function of Sp3.
INTRODUCTION
Sp1 is one of the most well characterized transcription factors, and it plays a major role in the expression of numerous cellular as well as viral genes (reviewed in 1,2). Sp1 binds to GC-rich promoter elements and stimulates transcription of promoters containing these consensus elements (reviewed in 1,2). Three other Sp-family transcription factors, namely Sp2, Sp3 and Sp4 have been cloned (3,4). Among them, the Sp3 mRNA is expressed in all mammalian cells that express Sp1 (3,4). Studies from several laboratories have shown that Sp3 functions as both transcriptional activator and repressor (3,5–16). Sp3 represses Sp1-activated transcription of the SV40 early promoter (3,5,12), dhfr promoter (5), ornithine decarboxylase (8) and HIV-1 LTR (10). In contrast, Sp3 stimulates transcription of many promoters including PDGF-B (9), thymidine kinase (5), p21 (12), and human α2 (I) collagen gene (16). These results show that Sp3 is not functionally equivalent to Sp1. Consistent with these observations, a comparison of the Sp1 and Sp3 knockout phenotypes shows that Sp1 and Sp3 have distinct functions in vivo and Sp3 function is crucial for survival of mice (17).
Although Sp3 and Sp1 differ in function, they share extensive structural similarities. For example, Sp3 shares 90% sequence homology with Sp1 in the zinc-finger DNA-binding domain, and both proteins bind to the GC- or GT-rich DNA sequences with similar specificity (2–4). Sp3 has long glutamine-rich regions, which share 35% identity with the transactivation domains of Sp1 (1–4,7). Studies with chimeric GAL4-Sp3 have shown that the glutamine-rich regions function as activator domains of Sp3 (6,12,14). Sp1 has a C-terminal domain D involved in multimerization and synergistic transactivation functions; a similar D domain is not found in Sp3 (3,6). Furthermore, two independent studies described a unique inhibitory domain in Sp3, located between the second glutamine-rich region and the zinc-finger DNA-binding motif (6,12). Mutations within the inhibitory domain change Sp3 to a strong transactivator of promoters containing multiple Sp-binding sites (6,12).
Strikingly, the transcriptional repression function of Sp3 is dependent upon the number of Sp-binding sites of the promoters. Several studies have shown that Sp3 is a transactivator for promoters containing a single Sp-binding site (5,6,13,14). However, Sp3 preferentially inhibits Sp1-activated transcription of promoters containing multiple Sp-binding sites (5,6,11). Drosophila cells are devoid of endogenous Sp1 or Sp3 proteins (1,3). There are contradictory reports about Sp3-dependent transcription in Drosophila cells. Dennig et al. (6) reported that Sp3 could not transactivate promoters containing multiple Sp-binding sites in Drosophila cells (6). However, other reports suggested that Sp3 functions as a transactivator to promoters containing multiple Sp-binding sites in Drosophila cells (15).
In this study, we compare the DNA-binding properties of purified cellular Sp3 with Sp1. The results reveal that the 116 kDa Sp3 polypeptide binds differently to promoters containing single or adjacent Sp-binding sites. Promoters containing multiple adjacent Sp-sites form significantly more stable Sp3–DNA complexes than those with single Sp-binding sites. Moreover, Sp3–DNA complexes on adjacent sites are also more stable than similar Sp1–DNA complexes and as a consequence Sp3 efficiently displaces Sp1 from preformed Sp1–DNA complexes from such sites. Previous studies have shown that formation of multimeric complexes coincides with the synergistic transactivation of Sp1; however, in sharp contrast, Sp3 showed no synergistic transactivation to promoters containing either single or multiple Sp-binding sites. The results presented in this study provide evidence for the promoter-specific DNA binding activity of Sp3, which is linked to its differential transcription regulatory activities.
MATERIALS AND METHODS
Cells, extracts and antibodies
Spinner cultures of mouse L cells were grown in minimum essential medium (S-MEM, GIBCO-BRL) supplemented with 5% calf serum. Extracts from different cell lines were prepared by a previously described procedure (18). The Sp1 antibody (against residues 520–538 of Sp1 protein) and the Sp3 antibody (against residues 676–695 of Sp3 protein) used in this study were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Purification of Sp3 from mouse L cell extracts
Heparin-agarose and DEAE-Sepharose chromatography. Whole cell extracts of mouse L cell were fractionated by heparin-agarose chromatography with a buffer series containing successively 0.1, 0.25 and 0.6 M KCl, as previously described (18). Column fractions were analyzed by both western blot assay and gel mobility shift assays. The fractions eluted with 0.6 M KCl containing buffer after a thorough wash with 0.25 M KCl containing buffer were found to contain most of the Sp1 and Sp3 proteins. The 0.6 M KCl eluate was dialyzed against the buffer A containing 25 mM Tris–HCl (pH 7.5), 0.1 M KCl, 1 mM MgCl2, 0.1 mM EDTA, 0.2 mM DTT, 0.1% NP40, 0.2 mM phenylmethylsulfonyl fluoride and 10% glycerol. The dialyzed proteins were applied to a DEAE-Sepharose column equilibrated with buffer A and the proteins were eluted with a KCl step gradient. The Sp1 flowed through the DEAE column with 0.1 M KCl containing buffer and the Sp3 was eluted from the column with 0.2 M KCl containing buffer.
Sp-affinity chromatography. The 0.2 M KCl eluates from the DEAE-Sepharose column were further purified by DNA-affinity chromatography. The sequence specific Sp-DNA affinity column was prepared by covalently linking ligated double stranded synthetic oligonucleotides 5′GATCTGGGTGGGGC3′ containing the high-affinity Sp-binding sequence to the CNBr-activated Sepharose 4B following a previously described method (19). The DEAE-Sepharose 0.2 M KCl eluate was first mixed with non-specific competitor DNA such as sonicated salmon sperm DNA and poly (dI). Poly (dC) incubated at 4°C for 20 min was allowed to bind to the affinity resin with Buffer A containing 0.1 M KCl. After extensive washing with the same buffer, the Sp3 was eluted from the column with step gradients of 0.15, 0.2, 0.25, 0.3, 0.4 and 0.6 M KCl. The eluted proteins were analyzed by gel mobility shift assays and western blot assays. Sp3 DNA-binding activity was eluted from the affinity column with 0.4 M KCl containing buffer. Sp1 was purified from the mouse L cell extracts by sequential chromatography using previously described methods (2,19).
Electrophoresis mobility shift assays
Mobility shift assays were performed as previously described (20,21) with the following modifications. Different amounts of proteins and antibodies (as indicated in the figure legends) were mixed with 0.1–0.5 ng of 32P-labeled DNA probe and 1 µg of poly (dA-dT) in 20 µl reaction buffer and incubated at room temperature for 20 min. An aliquot of the reaction mixtures (7.5 µl) was analyzed by gel retardation assay using 4% native polyacrylamide gel containing 0.25× TBE and 0.1% NP-40. Oligonucleotides 5′GATCTGGGTGGGGC3′ containing high-affinity Sp-binding sequence (1) were subcloned into the BamHI site of the pBluescript II SK+ plasmid. The DNA fragment was excised with XbaI and HindIII digestion, labeled with Klenow and [32P]dATP, and was used as a GT-box Sp-probe in the gel retardation assays. The DNA fragments excised from BCAT-1 and BCAT-2 plasmids following digestion with EcoR1 and HindIII were used as one Sp-site and two-site Sp-binding DNA probes. The DNA-fragment excised from the pCAT-promoter vector (Promega) plasmid following digestion with HindIII and BglII was used as a probe of SV40 early promoter containing six Sp-binding sites. For competition experiments, a 100-fold molar excess of unlabeled probe DNA was added to the reaction mixture before adding the proteins. The wild-type (5′GGGGCGGGG3′) and mutant Sp1-oligos (5′GGTTCGGGC3′) from Santa Cruz Biotechnology, Inc. were used as competitors.
Dissociation rate measurements
The dissociation rates of Sp1– or Sp3–DNA complexes were analyzed following a previously described procedure (22). Briefly, Sp1– or Sp3–DNA complexes were allowed to form for 15 min at room temperature under standard conditions, and then a large excess (100-fold) of unlabeled probe DNA was added. The samples were removed at designated times and immediately loaded onto a 4% native polyacrylamide gel that was already running. The binding of purified Sp3 to different DNA probes was calculated following densitometric scanning of the autoradiograms and was expressed as percentage binding. The time 0 was set at 100. The averages plus the standard deviations for three independent experiments were shown.
Western blot assay and transient transfection assays
Western blot assay and transient transfection assays were performed using previously described procedures (21). Drosophila SL2 cells were transfected by the calcium phosphate co-precipitation method (21). The CAT activity of the reporter construct and the transactivator was divided by the CAT activity of the reporter construct alone to determine the fold activation. All transfections were repeated at least three times and the mean fold activation was calculated. The results include standard error of the mean.
RESULTS
An 116 kDa polypeptide co-purifies with the Sp3 DNA-binding activity
The biochemical and DNA-binding properties of Sp1 have been studied extensively using purified protein from mammalian cell extracts (reviewed in 1). However, similar analyses of Sp3 have not been reported. Sp1 is extensively modified by glycosylation and phosphorylation, which affect its function (19,23,24). Therefore, to study the biochemical properties of Sp3, we sought to purify Sp3 from mouse L cell extracts. Figure 1A illustrates the purification scheme. Briefly, the soluble proteins obtained by high salt extraction of mouse L cells were separated by sequential column chromatography and the column fractions were analyzed by both gel mobility shift assay and western blot assay to detect separation of the Sp1 and Sp3 proteins (described in Materials and Methods).
Figure 1.

Purification of cellular Sp3. A 116 kDa polypeptide co-purifies with Sp3–DNA binding activity in mouse L cell extracts. (A) Schematic diagram of the purification scheme of Sp3 from mouse L cell extract. (B) The 0.2 M KCl eluate from the DEAE-Sepharose column of mouse L cell extract was subjected to DNA-affinity chromatography as described in Materials and Methods. A 1 µl aliquot of the indicated KCl eluate of the DNA-affinity column was analyzed for DNA-binding activity by gel retardation assay. In lanes 8, 9 and 10, the reaction mixtures were assayed in the presence of a 100-fold molar excess of unlabeled oligonucleotides containing either GT-box sequence (5′TGGGTGGGGC3′, lane 8) or GC-box sequence (5′GGGGCGGGG3′, lane 9) or mutant GC-box sequence (5′GGTTCGGGGC3′, lane 10). (C) Two hundred microliters of 0.4 M KCl eluates of the Sp3-affinity column were analyzed by western blot assay using either the Sp1 or Sp3 antibody. (D) Two hundred microliter aliquots of the 0.4 M KCl eluates of the Sp-affinity columns for Sp1 and Sp3 were analyzed by SDS–PAGE (7.5% acrylamide) and visualized by silver staining. Migrations of the molecular-weight marker proteins are shown for each gel.
The L-cell extracts were first fractionated by heparin agarose chromatography as described in Materials and Methods. Both Sp1 and Sp3 proteins co-purified in the column and were detected in the 0.6 M KCl eluate of the heparin-agarose column (data not shown). The 0.6 M KCl eluate of the heparin-agarose column was further fractionated by DEAE-Sepharose chromatography. The majority of the Sp3 DNA-binding activity was eluted from the DEAE-Sepharose column with 0.2 M KCl containing buffer (data not shown). Sp1 eluted from the DEAE-Sepharose column by 0.1 M KCl containing buffer, and thus, a DEAE-Sepharose column can effectively separate Sp3 from Sp1. To further purify Sp3, the 0.2 M KCl eluate of the DEAE-Sepharose column was fractionated by DNA-affinity chromatography. The Sp-DNA affinity column was prepared with tandemly ligated oligonucleotides containing Sp-recognition (GT-box) sequences, as described in Materials and Methods. After extensive washing, the proteins were eluted from the affinity column with a KCl step gradient. As shown in Figure 1B, the Sp3–DNA binding activity was specifically bound to the column and was eluted from the column with 0.4 M KCl containing buffer (Fig. 1B, lane 6). The Sp3 DNA-binding activity in the affinity eluate was specifically competed by both GT-box and GC-box containing oligonucleotides (Fig. 1B, lanes 8 and 9), but not by oligonucleotides containing a mutant Sp-binding site (Fig. 1B, lane 10). Analysis of the Sp3-affinity column eluate by western blot analysis showed that it contained a 116 kDa polypeptide that was specifically recognized by the Sp3 antibody, but not by Sp1 antibody (Fig. 1C). Moreover, analysis of the fraction by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and silver staining revealed a major band corresponding to a 116 kDa polypeptide (Fig. 1D). Taken together, these results are consistent with the notion that the 116 kDa polypeptide represents the major Sp3 DNA binding activity in mouse L cells. Besides the 116 kDa Sp3 polypeptide, all cell extracts contained 66 kDa doublet Sp3 polypeptides (Fig. 5A), which are two isoforms of Sp3 formed via internal translational initiation (15,25). The 66 kDa Sp3 polypeptides co-purified with the 116 kDa Sp3 polypeptide during the heparin agarose chromatography but were separated during the DEAE-Sepharose chromatography and were not purified further.
Figure 5.

Sp3 does not form heterodimers with Sp1. (A) The Sp3 antibody does not co-immunoprecipitate Sp1 or vice versa. Aliquots of the heparin agarose fraction containing most of the Sp-like activity (200 µg) were separately immunoprecipitated with Sp1 antibody or Sp3 antibody. The immunoprecipitates were eluted with SDS sample buffer and subjected to western blot analysis. The Sp1 and Sp3 immunoprecipitates were probed with Sp1 antibody (Left panel, lanes 1–2) or Sp3 antibody (right panel, lanes 3–4). (B) Gel Shift assay. A low concentration of purified Sp1 (1.2 ng) was incubated with the 32P-labeled BCAT-1 probe to generate a predominantly monomeric complex of Sp1 (lane1). Sp1 antibody (1 µl) was added to the reaction mixture to separate the Sp1 and Sp3 complexes (Lanes 2–6). In lanes 3–6, increasing levels of purified Sp3 protein (1.2, 2.4, 6.0 and 12 ng) were added, and Sp3 formed a complex with the probe that migrates differently from the Sp1/Sp1-Ab complex. About a 50% reduction in the level of Sp1/Sp1-Ab complex was observed following the addition of 12 ng of Sp3 (lane 6).
Sp3, unlike Sp1, fails to generate oligomeric complexes on promoters containing a single Sp-site
In this study, we compared the DNA-binding activities of Sp3 with Sp1. We analyzed the DNA-binding properties of the purified Sp3 by gel mobility shift assay using different 32P-labeled DNA probes containing either one Sp-binding site (BCAT-1 probe), two adjacent Sp-binding sites (BCAT-2 probe) or multiple Sp-binding sites (SV40 early promoter fragment). On a single-site probe, purified Sp3 formed one predominant complex, which probably results from interaction of a single Sp3 molecule with DNA (Fig. 2A, panel 1). Even in the presence of a large excess of Sp3-protein, only one major Sp3–DNA complex was formed on a single-site probe, suggesting that Sp3 does not form significant homo-oligomers on a probe containing a single Sp-binding site. In contrast, consistent with previously published results, Sp1 readily formed homo-oligomeric complexes on a single site probe (Fig. 2B). Sp1 has been shown to bind as oligomers with promoters containing a single binding site (20). Our analysis indicates that Sp3 did not form such oligomeric complexes on a single site probe. However, when a probe containing two adjacent Sp-binding sites (BCAT-2) was used, Sp3 readily formed a slower migrating complex corresponding to simultaneous occupation of both the DNA-binding sites by Sp3 (Fig. 2A, panel 2). Even at a very low concentration of Sp3-protein, the slower migrating Sp3-complexes were formed on the two-site probe (Fig. 2A, lane 1, panel 2). In a parallel assay, Sp1 readily formed homo-oligomeric complexes with the probe containing two Sp-binding sites (20 and Fig. 2B). Antibody supershift assays in both panel A and panel B show specificities of the Sp1 and Sp3 complexes (Fig. 2).
Figure 2.

Sp3 does not form oligomeric complexes on promoters containing Sp-binding sites. (A) Complex formation of Sp3 with 32P-labeled DNA probes containing either one (BCAT-1) or two (BCAT-2) Sp-binding sites. Increasing amounts of affinity purified Sp3 (1.2, 2.4, 5.9 and 18 ng protein) were added to the reaction mixtures (20 µl) containing 0.2 ng of BCAT-1 (lanes 1–4) or BCAT-2 probes (lanes 5–9). After 20 min of incubation at room temperature, 7.5 µl of the reaction mixture was separated on a native polyacrylamide (4%) gel as described in Materials and Methods. The reaction mixtures in lanes 10–12 contained 10 ng of Sp3. The reaction mixture in lane 10 has no antibody whereas the reaction mixture in lane 11 received 0.1 µg of Sp3-antibody and reaction mixture in lane 12 received 0.1 µg of Sp1-antibody. (B) Complex formation of affinity purified Sp1 with 32P-labeled DNA probes containing either one (BCAT-1) or two (BCAT-2) Sp-binding sites. Increasing amounts of Sp1 (0.8, 1.6, 3.2 and 7 ng protein) were added to the reaction mixtures (20 µl) containing 0.2 ng of BCAT-1 or BCAT-2 probes. After 20 min incubation at room temperature, 7.5 µl of the reaction mixture was separated on a native polyacrylamide (4%) gel as described in Materials and Methods. The reaction mixtures in lanes 9–11 contained 5 ng of Sp1. The reaction mixture in lane 9 has no antibody whereas the reaction mixture in lane 10 received 0.1 µg of Sp1 antibody and the reaction mixture in lane 11 received 0.1 µg of Sp3 antibody.
Sp3–DNA complexes on a two-site probe are more stable than monomeric Sp3–DNA complex or oligomeric Sp1–DNA complexes
To investigate whether the slower migrating Sp3–DNA complexes on a two-site probe are more stable than a monomeric Sp3–DNA complex, we compared the off-rates of the single-site and two-site Sp3–DNA complexes. The preformed Sp3–DNA complexes were challenged with an excess of unlabeled oligonucleotides for various time periods. As shown in Figure 3, a significant difference in the off-rates between the two types of Sp3 complexes was readily detected. In the case of the monomeric Sp3–DNA complex, there was almost complete displacement of Sp3 from the single-site interaction within 30 s of incubation with the unlabeled competitor (Fig. 3A). Quantitative analysis revealed that more than 90% of the initial Sp3–DNA complexes were displaced by 0.5 min of chase with cold DNA. In contrast, only partial displacement of Sp3 from two-site probe was observed even after 3 min of incubation (Fig. 3B). Quantitative analysis showed that only 50% of the Sp3–DNA complexes were displaced after 2 min of chase and 5% of the initial Sp3–DNA complexes were detected even after 10 min of chase (Fig. 3D). A large number of cellular promoters carry tandem Sp-binding sites, including the SV40 early promoter that contains six closely spaced Sp-binding sites. We analyzed the pattern and stability of Sp3–DNA complexes on the SV40 early promoter. Purified Sp3 formed multiple complexes of different mobilities with the SV40 promoter fragment (Fig. 3C). Upon incubation with an excess of unlabeled competitor DNA, only 50% of the slower migrating Sp3–DNA complexes were dissociated from the promoter after 5 min of chase (Fig. 3C). These results further confirm the notion that the stability of Sp3-binding to the promoter depends on the number of adjacent Sp-binding sites on the promoter.
Figure 3.

Sp3–DNA complexes on two-Sp site probe are more stable than Sp3–DNA complex on one Sp-site probe. (A and B) Purified Sp3 (12 ng) was pre-bound with radiolabeled probe (1 ng) for 15 min at room temperature in a 30 µl reaction mixture. At time zero, a 100-fold molar excess of unlabeled probe DNA (100 ng) was added to the reaction, and 6 µl aliquots of the reaction mixture were taken at the indicated time intervals and were loaded onto a running polyacrylamide gel. (A) Dissociation of the Sp3–DNA complexes from the probe containing one Sp-binding site; (B) Dissociation of Sp3–DNA complexes from the probe containing two Sp-binding sites. The slower migrating Sp3–DNA complexes on the two Sp-site probe have significantly slower off-rates. Since the electrophoresis was continuous during the chase experiments, the probes and the shifted complexes run progressively higher on the gel with increasing time. (C) Purified Sp3 (12 ng protein) was pre-incubated for 15 min at room temperature with a 32P-labeled DNA fragment (0.5 ng) of the SV40 early promoter. After addition of a 100-fold molar excess of the unlabeled DNA at time zero, aliquots of reaction mixture at the indicated times were loaded on a running polyacrylamide gel. The numbers on the top of the lanes represent percent Sp3 DNA complex at different time of chase. (D) Quantitation of the Sp3–DNA complexes with one Sp-site, and two Sp-site containing probes. The Sp3–DNA complexes formed with one Sp-site and two Sp-site containing probes were expressed as a percentage following densitometric scanning of autoradiograms represented in (A) and (B). Bars represent the average plus standard deviations for at least three independent experiments.
Under similar conditions, Sp1 was completely dissociated from interactions with either one- or two-site probes within 2 min of incubation with the unlabeled competitor (Fig. 4). Comparison of the dissociation rates of the Sp1 complexes on a single or two-site probe revealed no major difference in the stabilities of the monomeric versus oligomeric Sp1–DNA complexes (data not shown). Taken together, these results show that although Sp1 readily formed oligomers on both single-site and multiple-site probes, the oligomeric Sp1–DNA complexes are significantly less stable than Sp3–DNA complexes on a two-site probe.
Figure 4.

Comparison of the stabilities of the Sp1–DNA complexes with probes containing one or two Sp-binding sites. (A and B) Identical experiments to that described in Figure 3 were performed with affinity purified Sp1 protein (5 ng) and 32P-labeled BCAT-1 (1 ng) and BCAT-2 probe (1 ng). Two differently migrating Sp1–DNA complexes were visible, representing monomeric and multimeric Sp1–DNA complexes.
Sp3 does not form heterodimers with Sp1
Sp1 and Sp3 recognize the same promoter element. To investigate a possible interaction between Sp1 and Sp3, we used both the co-immunoprecipitation assay and the gel mobility shift assay. For the co-immunoprecipitation assay, partially fractionated mouse L cell extracts (0.6 M heparin agarose eluate) containing significant amounts of both Sp1 and Sp3 were immunoprecipitated with either Sp1 or Sp3 antibody. The immunoprecipitates were collected with protein-A Sepharose beads, washed extensively with a buffer containing 0.25 M KCl, and were analyzed by western blot assay using either Sp1 or Sp3 antibody. As shown in Figure 5A, immunoprecipitates obtained with the Sp3 antibody contained the116 kDa polypeptide and a doublet of 66 kDa polypeptides, which were specifically recognized by the Sp3 antibody (Fig. 5A, lane 4). The Sp3 antibody did not co-immunoprecipitate the Sp1 polypeptides. On the other hand, the Sp1 antibody efficiently immunoprecipitated the Sp1 polypeptides (Fig. 5A, lane 1), but did not co-immunoprecipitate the Sp3-polypeptides (Fig. 5A, lane 3). These results suggest that Sp3 and Sp1 do not form a stable complex in solution. Similar co-precipitation assays in our laboratory had previously identified an interaction between Sp1 and p107 (21). These results also show that the Sp1 and Sp3 antibodies do not cross-react.
We also used the gel shift assay to investigate the interaction between Sp1 and Sp3. We preformed the Sp1–DNA complex by incubating purified Sp1 with a promoter fragment containing one Sp-binding site. In order to distinguish the Sp1–DNA complex from the Sp3–DNA complex, Sp1 antibody was also included in the reaction mixture. To investigate the effect of Sp3, an increasing amount of Sp3 was added to the reaction mixture containing preformed Sp1–DNA complex and Sp1 antibody. The reaction was continued for an additional 20 min. A low level of Sp1-protein (1.2 ng) generated a predominantly monomeric complex of Sp1 (Fig. 5B, lane 1), and the Sp1-antibody supershifted the entire Sp1–DNA complex (Fig. 5B, lane 2). Addition of a 2-fold excess of Sp3 (2.4 ng) did not significantly displace Sp1 from the probe. Sp3 generated a new Sp3–DNA complex on the probe, and we did not detect any heteromeric Sp1/Sp3 complex when the two purified proteins were added together (Fig. 5B, lanes 3–6). Significant reduction in the Sp1/Sp1-Ab complex was observed only following the addition of a 5–10-fold excess of Sp3 protein (Fig. 5B, lanes 5 and 6).
Sp3 efficiently displaces Sp1 from a promoter containing adjacent Sp-binding sites
Since Sp3 forms a more stable complex than Sp1 on a promoter containing adjacent Sp-binding sites, we examined the possibility that Sp3 might displace Sp1 efficiently from such a promoter DNA. To investigate this possibility, we preformed the Sp1–DNA complex by incubating purified Sp1 with a promoter fragment containing two Sp-binding sites. In order to distinguish the Sp1–DNA complex from the Sp3–DNA complex, Sp1 antibody was also included in the reaction mixture as described above. Interestingly, when a competition experiment was performed using a probe containing two Sp-binding sites and 1.2 ng Sp1 protein, addition of 1.2 ng of Sp3 protein resulted in a significant loss of the Sp1-specific DNA complex (Fig. 6, lane 3). Furthermore, addition of 2.4 ng of Sp3 (twice the level of Sp1) caused a significant decrease in the Sp1–DNA complex and coincided with the appearance of a distinctly migrating Sp3–DNA complex (Fig. 6, lanes 4). The identity of the newly formed Sp3 complex was confirmed by adding Sp3 antibody to the reaction mixture which efficiently supershifted the complex (Fig. 6, lane 10). The formation of a slower migrating Sp3-complex with promoters containing multiple Sp-binding sites was not restricted to the purified Sp3 protein, Sp3 can also form a slower migrating more stable DNA binding complex with the two-site probe, even in the presence of a large excess of Sp1 protein in different cell extracts (data not shown). Taken together, these results suggest that Sp3 has promoter-specific DNA binding activity and it forms a more stable complex with promoters containing multiple clustered Sp-binding sites.
Figure 6.
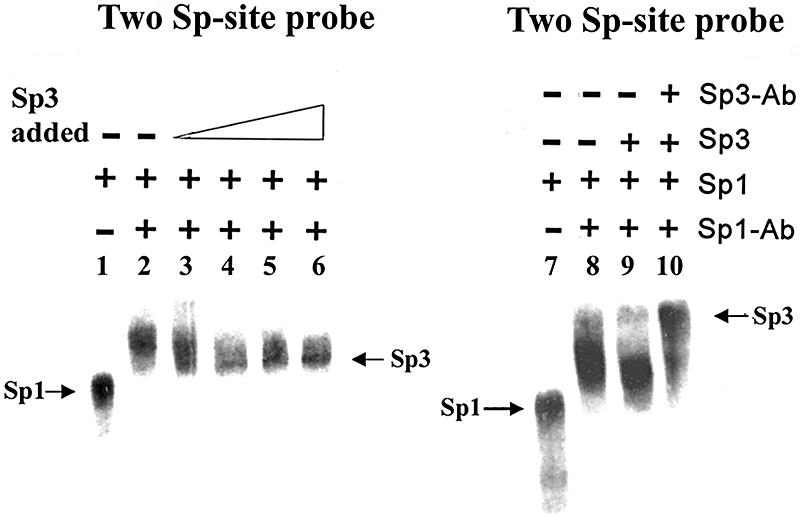
Sp3 displaces Sp1 from promoter containing adjacent Sp-sites. The Sp1–DNA complex was preformed by incubating purified Sp1 (1.2 ng protein) with 32P-labeled BCAT-2 probe (0.5 ng) containing two Sp-binding sites (lane1). To distinguish the Sp1–DNA complex from the Sp3–DNA complex, Sp1 antibody (1 µl) was also added to the reaction mixture (lane 2). Increasing amounts of Sp3 (1.2, 2.4, 6.0 and 12 ng protein) were added to the reaction mixtures (lanes 3–6) containing preformed Sp1–DNA complex and Sp1 antibody and the incubation was continued for an additional 20 min. Addition of Sp3 was followed by a loss of the Sp1–DNA complex and the appearance of a distinctly migrating Sp3–DNA complex (lanes 3–6). To determine the identity of the newly formed complex, 1 µl of Sp3-antibody was added to the reaction mixture containing 1.2 ng Sp1, 2.4 ng Sp3 and 1 µl of Sp1 antibody, and it completely supershifted the complex (lane 10). The reaction mixture in lane 7 contained 1.2 ng Sp1, lane 8 contained 1.2 ng Sp1 and 1 µl Sp1 antibody, and lane 9 contained 1.2 ng Sp1, 2.4 ng Sp3 and 1 µl Sp1 antibody.
Sp3, unlike Sp1 does not show synergistic transactivation of promoters containing adjacent Sp-binding sites
In an attempt to correlate the DNA-binding activity with the transcription activity of Sp3, we compared the transcription activation function of Sp3 with Sp1 using the BCAT-1 and BCAT-2 promoters in Sp-factor negative Drosophila cells. Sp1 and Sp3 were expressed from matched promoter constructs, pPacSp1 and pPacSp3 (15). Drosophila cell extracts transfected with 5 µg each of pPacSp1 and pPacSp3 showed comparable levels of Sp1 and Sp3 protein by western blot assay (Fig. 7D). The results as presented in Figure 7A and B show that Sp3 is a transcriptional activator for both BCAT-1 and BCAT-2 promoters in Drosophila cells. For BCAT-1 promoter containing only one Sp-binding site, 1 µg of Sp3 cDNA transfection led to a 7–8-fold activation of the promoter. Comparison with the Sp1-dependent transactivation revealed that Sp3 is a weaker transactivator than Sp1. Transfection of only 50 ng of Sp1-expression plasmid gave a 13-fold increase in transcription of the BCAT-1 promoter (Fig. 7A). Signifi cantly, Sp3 was found to be a much weaker transactivator compared to Sp1 for the BCAT-2 promoter containing adjacent Sp-binding sites (Fig. 7A and B). Previous studies have demonstrated that Sp1 can synergistically transactivate the BCAT-2 promoter and in agreement with these previous reports, we found that as low as 50 ng of Sp1-expressing plasmid led to more than a 90-fold increase in the BCAT-2 promoter activity (20, Fig. 7A). With the Sp3-expressing plasmid, a dosage-dependent increase in the promoter activity was observed, and transfection of 1 µg of Sp3 expression plasmid led to about a 17-fold activation of BCAT-2 promoter activity. Clearly, Sp3 did not show any synergistic transactivation like Sp1. One hundred nanograms of Sp3 led to a 15-fold activation and 1 µg of Sp3 led to a 17-fold activation of the BCAT-2 promoter (Fig. 7B). Interestingly, in the Sp1/Sp3 co-transfection assays, over-expression of Sp3 blocked the Sp1-dependent transactivation of the BCAT-2 promoter (Fig. 7C). Due to the synergistic transactivation, transfection of 50 ng of Sp1-expression plasmid led to more than a 95-fold activation of the BCAT-2 promoter. However, co-expression of 0.25 µg of Sp3 expression plasmid decreased the BCAT-2 promoter activity to 54-fold and 1 µg of Sp3 expression plasmid reduced the promoter activity further to 16-fold. Interestingly, co-expression of 25 ng of Sp3 expression plasmid slightly increased the BCAT-2 promoter activity (Fig. 7C). Since both Sp1 and Sp3 can independently increase transcription of the BCAT-2 promoter (Fig. 7A and B), the decrease in the Sp1-dependent transcription after over-expression of Sp3 is likely due to replacement of Sp1 by Sp3 at the promoter. These results are consistent with the results of the binding experiments (Figs 3 and 6) showing that Sp3 forms a more stable complex with the Sp-binding sites of the BCAT-2 promoter and a high level of Sp3 can efficiently displace preformed Sp1 complexes from the BCAT-2 promoter.
Figure 7.

Sp3 show no synergistic transactivation of promoters containing multiple Sp-binding sites. (A and B) The indicated reporter plasmids (5 µg) were transfected into Drosophila SL2 cells with various amounts of the Sp1 expression plasmid (pPacSp1) and Sp3 expression plasmid (pPacSp3). The cells were subsequently lysed, assayed for CAT activity and the values represent the fold induction of each reporter plasmid calculated from three independent experiments. (C) The CAT reporter plasmid BCAT-2 (2.5 µg) was transfected into Drosophila SL2 cells along with Sp1 expression plasmid pPacSp1 (50 ng), and varying levels of Sp3 expression plasmid pPacSp3 (25 ng to 1 µg). The cells were subsequently lysed and assayed for CAT activity. The values represent the average of three independent experiments. (D) Comparable expression of Sp3 and Sp1 in transfected Drosophila cells. The pPacSp1 (5 µg) and pPacSp3 (5 µg) were transfected into Drosophila SL2 cells. The cells were subsequently lysed, and 50, 100 and 200 µg of cell extracts were analyzed for the Sp1 and Sp3 protein using western blot assay.
DISCUSSION
In this study, we compared the transcriptional and the DNA-binding properties of the transcription factor Sp3 with Sp1. Comparison of the DNA-binding activities of Sp3 and Sp1 reveals several interesting differences. First, unlike Sp1, Sp3 binds predominantly as a monomer with promoters containing a single Sp-binding site. However, using the Sp-binding sites of the BCAT-2 promoter (containing two Sp-binding sites of HTLVIII promoter), and SV40 early promoter (six Sp-binding sites), we showed that Sp3 readily forms a slower migrating complex with adjacent Sp-binding sites (Figs 2 and 3). A dosage-dependent DNA-binding assay revealed that even at a very low concentration of Sp3, formation of the multi-site slower migrating Sp3–DNA complex is favored over the faster migrating single-site Sp3–DNA complex (Fig. 2). Most importantly, the results presented in this report show that the slower migrating multi-site Sp3–DNA complexes are significantly more stable than the single-site Sp3–DNA complex (Fig. 3). More than 90% of Sp3 dissociated from the 32P-labeled BCAT-1 promoter fragment containing one Sp-site within 30 s of chase with cold probe, whereas comparable dissociation of Sp3 from the BCAT-2 promoter fragments containing two Sp-binding sites required more than 5 min of chase. The SV40 early promoter contains six Sp-binding sites, and the off-rates of different Sp3 complexes on that promoter varied by at least 10-fold (Fig. 3C). Taken together, these results imply that the stability of the Sp3–DNA complex is promoter-dependent. In contrast, although Sp1 readily binds as multimers to both single and multiple Sp-binding sites, no significant difference was observed between the stabilities of the monomeric and multimeric Sp1–DNA complexes (Fig. 4). Sp1 and Sp3 contain highly conserved, functionally interchangeable DNA-binding domains and the relative affinity or the rates of association of Sp1 and Sp3 to these promoter sequences are comparable (data not shown).
The results presented in this report also revealed major differences between the transactivation properties of Sp1 and Sp3. Previous studies have demonstrated that Sp1 forms homo-oligomeric complexes between multiple Sp-binding sites, which coincide with the synergistic transactivation function of Sp1 (reviewed in 2). Using the BCAT-2 promoter, Pascal and Tjian showed that, besides the transactivation domains of Sp1, the C-terminal D-domain of Sp1 is involved in homo-oligomerization and synergistic transactivation (20). In agreement with these previous studies, results presented in Figure 7 show that only 50 ng of the Sp1 expression plasmid gave a 90–100-fold increase in the BCAT-2 promoter activity in Drosophila cells. However, in sharp contrast, the Sp3 showed no synergistic transactivation of the BCAT-2 promoter (Fig. 7B). A 10-fold increase in the transfection of the Sp3-expression plasmid (from 100 ng to 1 µg) led to only a 3-fold increase in the BCAT-2 promoter activity (Fig. 7B). Like Sp1, Sp3 readily formed slower-migrating complexes with the BCAT-2 promoter fragment (Figs 2 and 3) but that does not result in synergistic transactivation. Sp3 activates transcription of promoters containing both single and multiple Sp-binding sites in Drosophila cells (Fig. 7 and data not shown).
Results presented in Figure 7C show that co-transfection of excess of Sp3-expression plasmid dramatically decreased Sp1-dependent transactivation of the BCAT-2 promoter. In this study, we showed that Sp3 forms a more stable promoter-specific complex and can efficiently displace Sp1 from the BCAT-2 promoter fragment containing two adjacent Sp-binding sites. Thus, these observations now provide a rational explanation for the repression of Sp1-dependent transcription by Sp3. It is conceivable that in promoters containing adjacent Sp-binding sites (like BCAT-2 or SV40 early promoter), a more stable interaction of Sp3 with adjacent Sp-binding sites effectively prevents Sp1 from binding to these sites. Since Sp3 has no synergistic transactivation function like Sp1, preferential binding of Sp3 leads to decreased transcription. Two previous studies have identified a specific repression domain immediately upstream of the Sp3 DNA-binding domain (6,12). From our results, we cannot rule out the involvement of the inhibitory domain of Sp3 in repressing the Sp1-dependent transcription of BCAT-2 promoter. Several scenarios are possible. It is possible that the formation of a stable Sp3–DNA complex involves the unique repression domain of Sp3, and when Sp3 is tethered to the promoter via adjacent Sp-binding sites, it forms transcriptionally less active pre-initiation complexes. It is also possible that formation of slower migrating Sp3–DNA complexes causes structural changes in the protein and exposes the inhibitory domain of Sp3 and thereby prevents formation of a transcriptionally active pre-initiation complex. Two more recent studies show that SUMO-1 modification represses Sp3 transcriptional activation by modulating its sub-nuclear localization (26,27). However, the transcription repression following SUMOlation of Sp3 does not depend on the number of Sp3 binding sites of the promoter. Other earlier studies showed that two low molecular weight Sp3 isoforms mediate transcriptional repression via competition for binding of the Sp-family transcription factors, however that repression does not depend on the number of Sp-binding sites of the promoter (15,25).
The results presented in this communication demonstrate that Sp3 forms complexes of different stability with single versus adjacent Sp-binding sites. Many promoters carry multiple Sp-binding sites either clustered or separate. A prediction from this study would be that Sp3 regulates these promoter activities based on the number and position of the Sp3 binding sites. Sp1 often interacts with a variety of different transcription factors to regulate promoter activity. However, it is not known how these interactions affect the stability of the Sp1–DNA complexes. It will also be interesting to know how the interaction of Sp3 with other promoter-specific factors modulates stability of the Sp3 complex and transcription regulatory activity. The results presented in this report now provide a possible explanation for the selective repression of promoters with multiple Sp-binding sites by Sp3.
REFERENCES
- 1.Courey A.J. and Tjian,R. (1993) In McKnight,S.L. and Yamamoto,K.R. (eds), Transcriptional Regulation, Book 2. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA, pp. 743–771. [Google Scholar]
- 2.Kadonaga J.T., Jones,K.A. and Tjian,R. (1986) Promoter specific activation of RNA polymerase II transcription by Sp1. Trends Biochem. Sci., 11, 20–23. [Google Scholar]
- 3.Hagen G., Muller,S., Beato,M. and Suske,G. (1994) Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J., 13, 3843–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kingsley C. and Winoto,A. (1992) Cloning of GT box-binding proteins: a novel Sp1 multigene family regulating T-cell receptor gene expression, Mol. Cell. Biol., 12, 4251–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birnbaum M., van Wijnen,A.J., Odgren,P.R., Last,T.J., Suske,G., Stein,G.S. and Stein,J.L. (1995) Sp1 trans-activation of cell cycle regulated promoters is selectively repressed by Sp3. Biochemistry, 34, 16503–16508. [DOI] [PubMed] [Google Scholar]
- 6.Dennig J., Beato,M. and Suske,G. (1996) An inhibitor domain in Sp3 regulates its glutamine-rich activation domains. EMBO J., 15, 5659–5667. [PMC free article] [PubMed] [Google Scholar]
- 7.Hagen G., Muller,S., Beato,M. and Suske,G. (1992) Cloning by recognition site screening of two novel GT box-binding proteins: a family of Sp1 related genes. Nucleic Acids Res., 20, 5519–5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar A.P. and Butler,A.P. (1997) Transcription factor Sp3 antagonizes activation of the ornithine decarboxylase promoter by Sp1. Nucleic Acids Res., 25, 2012–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang Y., Robinson,D.F., Dennig,J., Suske,G. and Fahl,W. (1996) Transcriptional regulation of the SIS/PDGF-B gene in human osteosarcoma cells by the Sp family of transcription factors. J. Biol. Chem., 271, 11792–11797. [DOI] [PubMed] [Google Scholar]
- 10.Majello B., De Luca,P., Hagen,G., Suske,G. and Lania,L. (1994) Different members of the Sp1 multigene family exert opposite transcriptional regulation of the long terminal repeat of HIV-1. Nucleic Acids Res., 22, 4914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majello B., De Luca,P., Suske,G. and Lania,L. (1995) Differential transcriptional regulation of c-myc promoter through the same DNA binding sites targeted by Sp1-like proteins. Oncogene, 10, 1841–1848. [PubMed] [Google Scholar]
- 12.Majello B., De Luca,P. and Lania,L. (1997) Sp3 is a bifunctional transcription regulator with modular independent activation and repression domains. J. Biol. Chem., 272, 4021–4026. [DOI] [PubMed] [Google Scholar]
- 13.Prowse D.M., Bolgan,L., Molnar,A. and Paolo Dotto,G. (1997) Involvement of the Sp3 transcription factor in induction of p21Cip1/WAF1 in keratinocyte differentiation. J. Biol. Chem., 272, 1308–1314. [DOI] [PubMed] [Google Scholar]
- 14.Udvadia A.J., Templeton,D.J. and Horowitz,J.M. (1995) Functional interactions between the retinoblastoma (Rb) protein and Sp-family members: Superactivation by Rb requires amino acids necessary for growth suppression. Proc. Natl Acad. Sci. USA, 92, 3953–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kennett S.B., Udvadia,A.J. and Horowitz,J.M. (1997) Sp3 encodes multiple proteins that differ in their capacity to stimulate or repress transcription. Nucleic Acids Res., 25, 3110–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ihn H. and Trojanowska,M., (1997) Sp3 is a transcriptional activator of the human α2(I) collagen gene. Nucleic Acids Res., 25, 3712–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bouwman P., Gollner,H., Elsasser,H.P., Eckhoff,G., Karis,A., Grosveld,F., Philipsen,S. and Suske,G. (2000). Transcription factor Sp3 is essential for post-natal survival and late tooth development. EMBO J., 19, 655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raychaudhuri P., Rooney,R. and Nevins,J.R. (1989). Identification of an E1A inducible cellular factor that interacts with regulatory sequences within the adenovirus E4 promoter. EMBO J., 6, 4073–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson S.P. and Tjian,R. (1989) Purification and analysis of RNA polymerase II transcription factors by using wheat germ agglutinin affinity chromatography. Proc. Natl Acad. Sci. USA, 86, 1781–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pascal E. and Tjian,R. (1991) Different activation domain of Sp1 govern formation of multimers and mediate transcriptional synergism. Genes Dev., 5, 1646–1656. [DOI] [PubMed] [Google Scholar]
- 21.Datta P.K., Raychaudhuri,P. and Bagchi,S. (1995) Association of p107 with Sp1: Genetically separable regions of p107 are involved in regulation of E2F- and Sp1-dependent transcription. Mol. Cell. Biol., 15, 5444–5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raychaudhuri P., Bagchi,S., Neill,S. and Nevins,J.R. (1990) Activation of the E2F transcription factor in adenovirus infected cells involves E1A dependent stimulation of DNA binding activity and induction of cooperative binding mediated by an E4 gene product. J. Virol., 64, 2707–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson S.P. and Tjian,R. (1988) O-Glycosylation of eukaryotic transcription factors: implications for mechanisms of transcriptional regulation. Cell, 55, 125–133. [DOI] [PubMed] [Google Scholar]
- 24.Black A.R., Jensen,D., Lin,S. and Azizkhan,J.C. (1999) Growth/cell cycle regulation of Sp1 phosphorylation. J. Biol. Chem., 274, 1207–1215. [DOI] [PubMed] [Google Scholar]
- 25.Kennett S.B., Moorefield,K.S. and Horowitz,J.M. (2002) Sp3 represses gene expression via the titration of promoter-specific transcription factors. J. Biol. Chem., 277, 9780–9789. [DOI] [PubMed] [Google Scholar]
- 26.Ross S., Best,J.L., Zon,L.I. and Gill,G. (2002) SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol. Cell, 10, 831–842. [DOI] [PubMed] [Google Scholar]
- 27.Sapetschnig A., Rischitor,G., Braun,H., Doll,A., Schergaut,M., Melchior,F. and Suske,G. (2002) Transcription factor Sp3 is silenced through SUMO modification by PIAS1. EMBO J., 21, 5206–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]