


Abstract
One of the key components of proteomics initiatives is the production of high affinity ligands or probes that specifically recognize protein targets in assays that detect and capture proteins of interest. Particularly versatile probes with tremendous potential for use as affinity molecules are aptamers. Aptamers are short single-stranded DNA or RNA sequences that are selected in vitro based on affinity for a target molecule. Aptamers offer advantages over traditional antibody-based affinity molecules in their ease of production, regeneration and stability, largely due to the chemical properties of nucleic acids versus amino acids. We describe an improved in vitro selection protocol that relies on magnetic separations for DNA aptamer production that is relatively easy and scalable without the need for expensive robotics. We demonstrate the ability of aptamers that recognize thyroid transcription factor 1 (TTF1) to bind their target protein with high affinity and specificity, and detail their uses in a number of assays. The TTF1 aptamers were characterized using surface plasmon resonance, and shown to be useful for enzyme-linked assays, western blots and affinity purification.
INTRODUCTION
The future success of proteomics depends on its ability to follow in the footsteps of genomics, where the development of new technologies generated an abundance of sequence data enabling researchers to probe problems that relate to the entire nucleic acid component of the cell. For the promise of proteomics to be realized, new tools are needed that will enable large-scale investigations of protein structure, function and interactions. Significant progress has been made in proteomic technology development in many areas (1), including high-throughput gene cloning (2), protein production (3,4), mass spectrometry (5), two-dimensional PAGE (6) and microfluidics, to allow large-scale proteomics to proceed. One important set of tools that has been improved with moderate success are affinity reagents that function as antibodies to serve as protein probes. Affinity molecules that specifically bind proteins of interest can detect bound proteins in a protein microarray, or capture protein complexes for functional identification (7). Often these molecules can alter biological activity due to their binding and inhibit critical interactions by sterically blocking access to active sites and interaction surfaces, and thus present an opportunity to serve as functional probes as well as therapeutics. Traditionally, antibodies have satisfied the demand for such ligands; however, as recombinant protein production gains throughput and pharmaceutical target repertoires expand, the ability to efficiently generate antibodies quickly falls short.
Several alternatives to antibodies have been investigated, such as single chain antibodies (scFv) (8), peptides displayed on protein domain scaffolded surfaces (9), peptides and peptoids (synthetic peptides) (10). Each of these alternatives has drawbacks that limit their uses, such as problems of stability in varying conditions (ionic strength, temperature and pH) and of low affinity, making some antibody alternatives ineffective for detecting proteins under many conditions (1).
The idea of using single-stranded nucleic acids (aptamers) as affinity molecules for proteins, first described in 1990 (11–13), has shown modest progress. The concept is based on the ability of short (20–80mer) sequences to fold, in the presence of a target, into unique three-dimensional structures that bind the target with high affinity and specificity. Aptamers are generated by a process that combines combinatorial chemistry with in vitro evolution, known as SELEX (systematic evolution of ligands by exponential enrichment). Following the incubation of a protein with a library of DNA or RNA sequences, typically 1014 molecules in complexity, protein–DNA complexes are isolated, the DNA is amplified and the process is repeated until the sample is enriched with sequences that display high affinity for the protein of interest. Since the selection pressure for this in vitro method of evolution is high affinity for the target, aptamers with low nanomolar affinities are often obtained. Aptamers offer advantages over protein-based affinity reagents due to the nature of nucleic acids, which provides for increased stability, ease of regeneration (PCR or oligonucleotide synthesis) and simple modification for detection and immobilization.
Although SELEX appears to be technically very simple, small alterations to a protocol can have a large impact on the success of generating aptamers. Perhaps this explains why 13 years since its first citation in the literature, only approximately 40 protein-detecting aptamer sequences have been published, and very few have been well characterized. Although high-throughput methods for aptamer production have been published, most require expensive robotics and have not produced aptamers against large numbers of diverse targets (14). Of course, in order for aptamer production to be truly high throughput, a supply of purified proteins must also be available as targets.
In this report, we describe an improved protocol for DNA aptamer production that is relatively easy and scalable without the need for expensive robotics. In addition, we fully demonstrate the abilities of our aptamers to bind their target protein with high affinity and specificity, and detail their uses in a number of assays. As a target, we use thyroid transcription factor 1 (TTF1), a well characterized member of the NK homeodomain transcription factors (15,16). TTF1 is expressed in the developing thyroid, lung and brain of vertebrates, and several effector genes have been identified in thyroid and lung tissues (17). The DNA recognition site of TTF1 differs from that of other homeodomain-containing proteins, attributed to the NK-type homeodomain (18). Following 15 rounds of selection, we characterized the affinity and specificity of several aptamers, and describe their uses in assays for the capture and identification of proteins, such as western blots, enzyme-linked assays and affinity purification.
MATERIALS AND METHODS
Cloning, protein expression and purification
TTF1 was cloned from Ciona intestinalis 16 h embryos following RNA isolation (Trizol reagent, Gibco) and first strand synthesis (Superscript First Strand Synthesis System for RTPCR, Gibco) by PCR using the following gene-specific primers that contained 5′ ligation-independent cloning (LIC) (Novagen) compatible ends: forward 5′-GGTATTGAGGGT CGCTCAGTTAGCCCAAAGCATTCG-3′; reverse 5′-AGA GGAGAGTTAGAGCCTTATCGGTAAACACTGTACAG GATCG-3′. LIC was performed as previously described to insert the coding sequence of C.intestinalis TTF1 into the vector pNHis, which adds a His6 tag to the N-terminus of the encoded protein (4).
After screening several expression conditions, TTF1 was found to be most highly expressed in Rosetta (DE3) pLysS cells (Novagen) induced with 1.0 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 37°C for 4 h. One liter of culture grown under these conditions was harvested by centrifugation at 5000 g for 10 min at 4°C. The pellet was resuspended in 30 ml of resuspension buffer [50 mM Na2HPO4, 300 mM NaCl, 20 mM imidazole, 0.1% Triton X-100, 5 mM 2-mercaptoethanol at pH 7.0 with 1 mM PMSF Plus (Roche), and 1× protease inhibitor cocktail (Sigma)]. The cells were then lysed with the Emulsiflex-C5 (Avestin) homogenizer at 15 000 p.s.i. The lysate was centrifuged at 15 000 g for 25 min at 4°C to remove insoluble material. Chelating Sepharose High Performance resin (Amersham) was charged with 0.1 M NiSO4 and washed with 10 column volumes of sterile water. The cleared lysate was incubated with 300 µl of 50% slurry of Ni2+ charged resin and bound in batch for 20 min with constant rotation then loaded onto an empty polypropylene column (Qiagen) and allowed to drain by gravity flow. The 150 µl column was then washed with 50 column volumes of resuspension buffer containing 20% glycerol and the bound proteins were eluted with 0.5 column volumes of elution buffer (1 M imidazole, 50 mM Na2HPO4 pH 7.0, 150 mM NaCl, 0.1% Triton X-100, 20% glycerol and 5 mM 2-mercaptoethanol). The concentration and purity were determined using 4 µl in a Protein 200 Lab-chip kit (Caliper) run on an Agilent 2100 Bioanalyzer.
In vitro selection of aptamers
A degenerate oligonucleotide library was synthesized at 1 µmol scale and HPLC purified (Operon). This material was diluted to 0.1 nmol/µl in 10 mM Tris pH 8, and stored at –20°C. This library, referred to as ‘LICSelexApt’, is composed of 40 random nucleotides flanked by sequences suitable for LIC: 5′-ggtattgagggtcgcatc-40N-gatggctctaactctcctct-3′. Primers that anneal to the 5′ and 3′ sequences flanking the degenerate region of LICSelexApt that were used during the selection and cloning were: ‘LICSelexF’, 5′-ggtattgagggtcgcatc-3′; ‘LICSelexR’, 5′-agaggagagttagagccatc-3′; in biotinylated and non-biotinylated forms (HPLC purified, Operon).
Protein-bound Ni-NTA magnetic beads were prepared by first equilibrating 150 µl of a 5% slurry (∼45 µg capacity) of Ni-NTA magnetic beads (Qiagen) into PBS-T (50 mM K2HPO4 pH 7.5, 150 mM NaCl, 0.05% Tween-20). The equilibrated beads were resuspended in 1250 µl of PBS-T, and 25 µl of 2 mg/ml purified TTF1 was added (a 1:50 dilution to lower the imidazole concentration) and mixed with rotation for 30 min at 4°C. The bead-bound TTF1 was then washed three times with 1 ml of PBS-T, diluted to 0.25 µg/µl (5 pmol/µl of 50 kDa TTF1) with PBS-T and stored at 4°C.
In the initial round of selection, the ‘LICSelexApt’ library was incubated with the bead-bound TTF1 using a 10-fold molar excess of ssDNA in a volume that gave a 10 nM TTF1 concentration. One nanomole of ‘LICSelexApt’ was diluted into 100 µl of PBS-T in a PCR tube, heated to 95°C for 2 min and immediately cooled at 4°C. This material was added to 10 ml of PBS-T containing 1 µg/ml bovine serum albumin (BSA), 0.1 µg/ml dIdC. Bead-bound TTF1 (100 pmol) was then added to this mixture and incubated with rotation for 30 min at room temperature. The tubes were then applied to a magnet (Dexter), the supernatant removed, and the beads were washed three times with 1 ml of PBS-T, mixing by inversion for each wash step. The proteins and bound aptamers were eluted from the Ni-NTA magnetic beads with 10 µl of 20 mM Tris pH 7.5, 500 mM imidazole, and transferred to PCR tubes. The 100 µl PCRs contained 1.25 U of Pfx polymerase (Invitrogen), 1 µM primers ‘LICSelexF’ and biotinylated ‘LICSelexR’, 0.1 mM dNTPs, 0.5 mM MgSO4 and 0.1× enhancer solution. Amplification conditions were 2 min at 95°C; 15 cycles of 30 s at 95°C; 30 s at 56°C; 30 s at 68°C; 2 min at 68°C. This protocol produced 2–5 µg of the correct size product as determined using a DNA 500 lab-chip (Caliper) on an Agilent 2100 Bioanalyzer. After the amplification step, 90 µl of the PCR product and 23 µl of 5 M NaCl were mixed with 1 mg of M-280 streptavidin magnetic beads (Dynal) for 10 min at room temperature, then washed with 3 × 1 ml of PBS-T. Single-stranded aptamers (non-biotinylated strand) were separated from the immobilized complementary strand using a 5 min incubation of 50 µl of fresh 100 mM NaOH. The tubes were applied to a magnet and the ssDNA was removed and diluted into 1 ml of PBS-T, containing 10 µl of 100 mM monobasic phosphate buffer to adjust the pH to 7.5. Finally, the material was heated to 95°C for 2 min then immediately placed at 4°C until the next round of SELEX.
For additional rounds of selection, the amount of protein was reduced to 50 pmol (rounds 2–10), and subsequently 25 pmol (rounds 11–15) in a binding volume of 1 ml, and the incubation time was reduced to 10 min. After round 2, the PCR cycle number was reduced to 10 cycles because of the amplification of products of incorrect size. More than 15 cycles of amplification often led to the production of larger fragments, later identified as concatamers. In order to remove aptamers that bind to the Ni-NTA magnetic beads, counter-selection was performed after rounds 3, 6, 9 and 12. A 20 µl aliquot of a 5% slurry of Ni-NTA magnetic beads was added to the 1 ml of ssDNA in PBS-T and incubated for 10 min with rotation, then applied to a magnet and the supernatant removed for the next round.
After round 15, the material was amplified by PCR with ‘LICSelexF’ and ‘LICSelexR’ primers, and the products were purified with MinElute (Qiagen), LIC-cloned into pET30XaLIC vector (Novagen), and transformed into NovaBlue Escherichia coli (Novagen). Thirty-two colonies were picked for each sample, and the plasmids purified by 96-well mini-prep (Qiagen). The plasmids were sequenced using a T7 promoter primer in the Big-dye Terminator kit and run on an ABI 3730. Sequences were aligned using ClustalX v.1.81 (19). Pattern analysis was performed using Consensus (20).
Aptamer-enzyme linked assay
To measure the binding of aptamers to proteins immobilized on microtiter plates, 500 ng of purified TTF1 or purified HOX4 fragment was bound to wells of an Ni-NTA HisSorb plate (Qiagen) in 200 µl of PBS-T for 2 h at room temperature. The wells were then washed three times with 200 µl of PBS-T. Biotinylated aptamers (Operon) were diluted to 1 ng/µl in 200 µl of PBS-T, heated to 95°C and then cooled quickly to 4°C. A 200 µl aliquot of aptamer was incubated with proteins in the HisSorb plate overnight at 4°C on a plate vortex, shaking gently. The wells were washed four times with 200 µl of PBS-T for 5 min each on a plate vortex. Streptavidin–horseradish peroxidase (HRP; Molecular Probes) was diluted 1:10 000 into PBS-T and a 200 µl aliquot was incubated with the proteins and bound aptamers in the HisSorb plate for 30 min at room temperature. The wells were washed again as described above, then 150 µl of Turbo-TMB (Pierce) was added to each well and incubated for 20 min at room temperature in the dark. The reactions were stopped with the addition of 150 µl of 1 M H2SO4 and the protein-bound aptamer–streptavidin complex was quantified by determining the absorbance at 450 nm using a SpectraMax Plus (Molecular Devices).
BIAcore surface plasmon resonance
The affinity of the aptamers for their protein targets was measured using surface plasmon resonance (SPR) with a BIAcore X instrument. Biotinylated aptamer (Operon) was diluted to 0.5 ng/µl in HBS-P (10 mM HEPES pH 7.5, 150 mM NaCl, 0.05% Tween-20), heated to 95°C, and rapidly cooled at 4°C before use. Approximately 100 RU of biotinylated aptamer (ligand) was immobilized to one flow cell of a streptavidin-coated sensor chip. Purified TTF1 protein was diluted into HBS-P to give a series of concentrations of TTF1 protein (3, 12, 31, 62, 125 and 250 nM or 2.5, 5, 10, 20, 40 and 100 nM) that were injected over the surface for 2 min at a flow rate of 20 µl/min (to minimize mass transfer limitations). Bulk shift and non-specific interactions with the streptavidin were subtracted using the response from a reference flow cell. After measuring the off rates for 2 min for each analyte injection, complete regeneration of the surface was achieved with two 30 s injections of 0.05% SDS at 50 µl/min. The affinity, as described by the equilibrium dissociation constant (KD), was determined globally by fitting to the kinetic simultaneous ka/kd model, assuming Langmuir (1:1) binding. The steady-state affinity was determined from curve fitting to a plot of the Req values, derived from sensorgrams fitted locally, against the concentrations.
Protein blot analysis with aptamers
Protein samples were prepared for SDS–PAGE by boiling in Laemmli sample buffer and then resolved on denaturing 4–20% polyacrylamide gels using the mini-Protean 3 system (Bio-Rad). The proteins were either stained with Gelcode Blue (Pierce) or transferred to PVDF (Schleicher and Schuell). The PVDF membranes were blocked overnight at 4°C with 5% BSA in PBS-T, and then probed with biotinylated TTF1 aptamer ‘A’ diluted to 1 µg/ml in 5 ml of PBS-T for 2 h at room temperature with rotation. The blots were washed three times for 5 min with 10 ml of PBS-T and then probed with streptavidin–HRP diluted 1:10 000 in PBS-T. The blots were washed three times for 5 min before chemiluminescence detection using pico-west substrate (Pierce). The blots were imaged using a Fluor-S Multi-Imager (Bio-Rad).
Aptamer affinity purification
Aptamers immobilized to magnetic beads were utilized for native protein purification. A 10 µg aliquot of biotinylated TTF1 aptamer ‘A’ was diluted into 200 µl of PBS-T in a PCR tube and heated to 95°C for 2 min, then immediately placed at 4°C for 5 min. This material was added to 2 mg of M-280 streptavidin magnetic beads (Dynal), and 50 µl of 5 M NaCl was added, and then mixed for 30 min with rotation at room temperature. In order to determine the level of non-specific binding to the M-280 beads, we performed the purification with biotin bound, instead of aptamer. The beads were washed twice with 1 ml of PBS-T before the purification. A 100 µl aliquot of cleared lysate from the protein purification described above was spiked with 10 µg of partially purified target protein and then diluted 1:3 with PBS-T. The protein was eluted with DNase treatment using 12 µl of PBS-T containing 50 mM NaCl, 5 mM MgCl2 and 60 U of Benzonase (Novagen). Several binding and elution schemes were tested: (i) 10 min binding at 4°C and 2 h nuclease treatment at 4°C; (ii) 30 min binding at 4°C and 15 min nuclease treatment at room temperature; and (iii) 5 min binding and 5 min nuclease treatment at room temperature. For each set of conditions, the beads were washed four times with 1 ml of PBS-T containing 600 mM NaCl, and then washed twice with 1 ml of PBS-T containing 50 mM NaCl to adjust the ionic strength for optimal nuclease acitivity. Protein that remained after nuclease treatment was removed from the aptamer beads with 12 µl of 0.05% SDS. The samples were analyzed by SDS–PAGE on a 4–20% gel that was stained with GelCode blue (Pierce).
RESULTS
Enrichment of aptamers to TTF1
We have optimized an in vitro ssDNA selection protocol that utilizes His6-tagged protein targets bound to Ni-NTA magnetic beads to screen for high affinity binding aptamers from a library of approximately 6 × 1014 sequences. We used Ni-NTA magnetic beads to provide a universal support for His6-tagged proteins as well as to facilitate the rapid partitioning of protein–aptamer complexes from unselected sequence pools. During the course of optimizing our protocol, we had observed enrichment of sequences that were not unique to a particular protein, and therefore incorporated counter-selection steps against Ni-NTA magnetic beads to prevent enrichment of aptamers that recognize the beads only. The number of PCR cycles was also optimized to avoid overamplification, which is evidenced by mis-annealed products. The stringency of the selection was controlled by adjusting the target protein concentrations, the incubation times and the washes. In order to assess our protocol, the enrichment of aptamers that bind TTF1 was monitored after 5, 10 and 15 rounds of selection. Multiple sequence alignments using ClustalX revealed that no enrichment was evident after five rounds of selection; however, after 10 rounds of selection, several groups of sequences were modestly enriched (two or three of 32, not shown). Satisfactory enrichment was accomplished with 15 cycles of selection (Fig. 1). Five groups of identical sequences were identified after the 15 rounds of selection, including one aptamer ‘A’ that represented 30% of the total evaluated. Therefore, our selection conditions using magnetic beads were sufficiently stringent for successful enrichment in 15 rounds. The aptamers that were generated (Fig. 1) did not have affinity towards the Ni-NTA magnetic beads (not shown). Interestingly, there were two aptamers ‘A’ and ‘C’ that contained a consensus sequence (Fig. 1), although this consensus may not necessarily be responsible for the affinity towards TTF1 since all the aptamer sequences obtained displayed affinity towards the protein (not shown). In addition, several G repeats were found in each group (20). The TTF1 dsDNA binding consensus sequence [5′ T(C/T)AAGTG 3′] is not contained in any of the aptamer sequences. In addition to the aptamers described in Figure 1, there were three sequences that were each represented as singletons (not shown).
Figure 1.

Enrichment of aptamers to TTF1 from 15 rounds of selection. The 40 base variable region of each aptamer sequence is shown (5′–3′). The number of times that each sequence was obtained from a total of 30 isolates is displayed on the right. A consensus sequence found in two of the aptamers is underlined.
Determination of the specificity of a TTF1 aptamer
We used an enzyme-linked assay in order to prioritize the aptamers from the TTF1 selection for further characterization (not shown). This assay provided a rapid assessment of the relative binding capabilities of many aptamers from a particular selection experiment. The results suggested that TTF1 aptamer ‘A’ may have the highest affinity for TTF1 (later confirmed by kinetic studies) and thus was chosen for further characterization. In addition, the enzyme-linked assay was used to provide information regarding cross-reactivity (Fig. 2). Employing a colorimetric detection system (Turbo-TMB + sulfuric acid) for peroxidase activity conjugated to streptavidin, we observed a significant (100×) signal over background, and the data for triplicate samples ranged from 0.006 ± 0.0002 to 0.63 ± 0.05 absorbance units. In order to determine if the TTF1 aptamer ‘A’ would recognize another homeodomain family member, the enzyme-linked assay was used to show that the TTF1 aptamer ‘A’ does not cross-react with the homeodomain of HOX4 (Fig. 2), nor did the aptamer bind BSA. In addition, an aptamer that was selected for HOX4 binding (not described here) did not cross-react with the TTF1 protein.
Figure 2.

Determination of specificity of aptamers using an enzyme-linked assay. Combinations of TTF1, HOX4 and BSA protein and either a TTF1 aptamer ‘A’ or a HOX4 aptamer (indicated by plus signs below the graph) were evaluated for their binding activity and cross-reactivity. The data are from triplicate samples.
Determination of the affinity of aptamers for TTF1
We utilized SPR employing a BIAcore X instrument to measure the affinity of the interaction of TTF1 with aptamers immobilized on a sensor chip. Sensorgrams of a concentration series of TTF1 injected over aptamers ‘A’ or ‘C’ are shown in Fig. 3A and C, respectively. The affinity, as described by the KD, was determined by a global fit using the kinetic simultaneous ka/kd model, assuming Langmuir (1:1) binding. The KD of aptamer ‘A’ for TTF1 was 3.36 × 10–9 M, and the KD of aptamer ‘C’ for TTF1 was 3.25 × 10–8 M. The steady-state affinities of TTF1 for the aptamers, determined from plots of Req values derived from the sensograms in Figure 3A and C fitted locally, correlated well with the simultaneous ka/kd model (Fig. 3B and D). KD values for the remaining aptamer sequences (B, D and E) and the three singletons showed that these aptamers also displayed affinity towards TTF1, ranging from 2.2 × 10–8 to 6.7 × 10–8 M (not shown).
Figure 3.

Determination of the affinities of aptamers for TTF1 using surface plasmon resonance. (A) Sensorgrams of the binding response to aptamer ‘A’ measured for concentrations of 2.5, 5, 10, 20, 40 and 100 nM TTF1 analyte. The KD = 3.36 × 10–9 M as determined from a global fit of the kinetic simultaneous ka/kd model, assuming Langmuir (1:1) binding, and χ2 = 14.1. (B) Plot of the steady-state affinity for ‘A’ using the Req values derived from sensorgrams in (A) fitted locally. The KD = 5.14 × 10–9 M as determined from the steady-state affinity model. (C) Sensorgrams of the binding responses to aptamer ‘C’ measured for concentrations of 3, 12, 31, 62.5, 125 and 250 nM TTF1 analyte. The KD = 3.25 × 10–8 M as determined from a global fit of the kinetic simultaneous ka/kd model, assuming Langmuir (1:1) binding, and χ2 = 10.9. (D) Plot of the steady-state affinity for ‘C’ using the Req values derived from sensorgrams in (C) fitted locally. The KD = 6.56 × 10–8 M as determined from the steady-state affinity model.
Comparison of the specificity of the TTF aptamer to a monoclonal anti-PentaHis antibody using protein blot analysis
The results of the enzyme-linked assay suggested that the TTF1 aptamer ‘A’ exhibited specificity for TTF1. In order to verify the specificity and determine whether the aptamer recognized the denatured form of TTF1, as well as to investigate further the potential uses of the aptamer, we performed a protein blot analysis (Fig. 4). The TTF1 aptamer ‘A’ was indeed able to bind the denatured TTF1 on the blot (Fig. 4C, lanes 3 and 4) and exhibited very little non-specific binding to the proteins in the cleared E.coli lysate or to the purified HOX4 (Fig. 4C, lanes 1 and 2). The performance of the aptamer was similar to the anti-PentaHis antibody in terms of chemiluminescent signal intensity and specificity (Fig. 4B). Note that the bands in lane 4 of Figure 4B and C below the major TTF1 band (marked with an arrow) are degradation products of TTF1 as determined by MALDI mass spectrometry analysis (not shown). Also, there is an ∼20 kDa protein in the E.coli lysate (Fig. 4C, lanes 1 and 3) that is recognized by the streptavidin–HRP secondary probe and not due to cross-reactivity of the aptamer (not shown).
Figure 4.

Comparison of the specificity of TTF1 aptamer ‘A’ to a monoclonal anti-PentaHis antibody in a protein blot analysis. Lane 1 contains cleared lysate from E.coli expressing the HOX4 homeodomain. Lane 2 contains purified HOX4 homeodomain protein (marked with an arrow). Lane 3 contains cleared lysate from E.coli expressing TTF1. Lane 4 contains purified TTF1 protein (marked with an arrow). (A) 4–20% SDS–PAGE stained with GelCode blue. (B) Blot of the material shown in (A) probed with an anti-PentaHis monoclonal antibody. (C) Blot of the material shown in (A) probed with the biotinylated TTF1 aptamer ‘A’. Note that the lower dark bands in lanes 1 and 3 of (C) were detected by the secondary probe, streptavidin–HRP (not shown).
Aptamer affinity purification
We performed aptamer affinity chromatography from a complex mixture of proteins in the soluble fraction of bacterial lysates using biotinylated aptamers on streptavidin magnetic beads (Fig. 5). TTF1 aptamer ‘A’ specifically purified the recombinant TTF1 protein out of the E.coli lysate in a single purification step. Elution of all proteins bound to the aptamer ‘A’ magnetic beads with SDS, which removes all bound proteins from the beads, showed that the purification of TTF1 was highly specific. We then tested generic elution conditions that would be most amenable to high-throughput methods. Elution of the purified TTF1 from the affinity matrix was inefficient with 1 M NaCl (not shown); therefore, we tested elution with a recombinant DNase. The recovery of purified TTF1 with DNase treatment (lanes 4, 6 and 8) was ∼25–50% of the total protein bound to the affinity column as revealed by a subsequent denaturing elution with SDS (lanes 5, 7 and 9). The efficiency of elution with DNase was better when the affinity beads were not saturated with TTF1 protein. This is probably due to the accessibility of the aptamer, which may be protected in conditions of saturating amounts of TTF1 protein. Additional optimization for improved elution yield of specific proteins without denaturing could be investigated on a case by case basis. This work further illustrates the potential utility of aptamers and demonstrates single-step purification from bacterial lysates.
Figure 5.
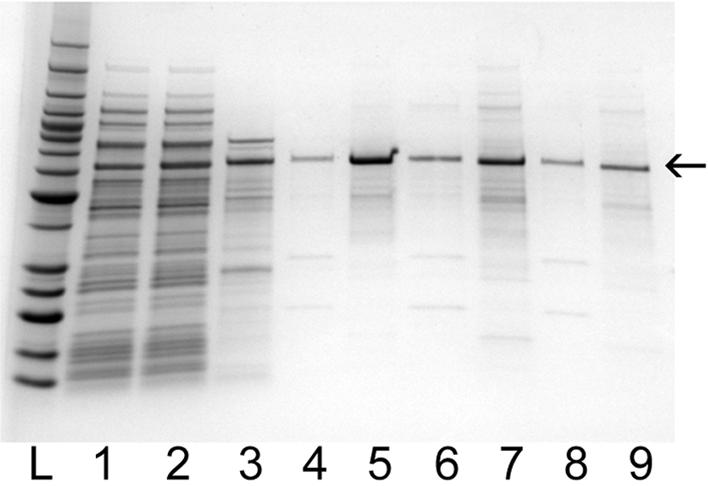
SDS–PAGE analysis of aptamer affinity purification of TTF1 protein from E.coli lysates using biotinylated TTF1 aptamer ‘A’ immobilized on streptavidin magnetic beads. The gel is 4–20% polyacrylamide stained with GelCode blue. Lane 1 contains cleared lysate from E.coli expressing the protein of interest, and lane 2 contains the cleared lysate spiked with Ni-NTA-purified TTF1 protein (lane 3). Material in lane 4 was from 10 min binding and 2 h elution at 4°C. Material in lane 6 was from 30 min binding and 15 min elution at room temperature. Material in lane 8 was from 5 min binding and 5 min elution at room temperature. After each elution with Benzonase, any remaining protein was removed from the aptamer with 0.1% SDS (lanes 5, 7 and 9).
DISCUSSION
We have developed an improved and straightforward protocol for DNA aptamer production and have characterized aptamers that recognize TTF1, a member of the NK homeodomain transcription factors (15). The use of aptamers as protein affinity reagents offers advantages over the use of antibodies. Nucleic acids are easily synthesized or amplified by PCR, therefore a vast supply of consistent quality is available. Also, nucleic acids can easily be modified to incorporate tags, such as biotin or fluorescent molecules, for detection and/or immobilization. Additionally, aptamers are smaller (<25 kDa) and more stable than antibodies. Moreover, unlike the requirement for milligram quantities of protein or peptide for antibody production, only microgram quantities of protein or peptide are required for aptamer SELEX. These properties, coupled to the present technology available for DNA microarrays, make aptamers very suitable for use in protein microarrays as a ligand, or for detecting proteins bound to a chip surface (21).
Despite these advantages, aptamers have rarely been selected for general use since the technology was developed 13 years ago. Approximately 40 unique aptamers against proteins or peptides have been described in more than 300 references in the literature. This lack of widespread use may be attributed to challenges in adapting existing protocols to particular targets and a general lack of fine details in existing methods. Many variations in aptamer production protocols have been described in which the method of protein target partitioning seems to vary the most. Unbound molecules have been removed from target proteins via: (i) filtration on a membrane (13); (ii) column chromatography, in which the targets are bound to a matrix, such as Sepharose, using a covalent linkage or an affinity tag (22); and (iii) binding of the protein to the wells of a microtiter plate (23).
The novelty of the protocol that we have described is the use of Ni-NTA magnetic beads for the immobilization of His6-tagged protein targets during selection. His6 tags are widely used in recombinant protein production, for example our group has described an efficient protein production pipeline for high-throughput generation of His6-tagged proteins in E.coli. (4). The use of a tag for immobilization promotes the proper orientation of proteins uniformly on a bead surface, and simultaneously provides a purification step, thereby reducing the chances of selection toward contaminants. Paramagnetic beads are an optimal solid support for parallel processing of both proteins and nucleic acids. Very small amounts of magnetic beads with proteins bound can be rapidly partitioned from unselected material, stringently washed and subsequently eluted. Others have claimed that filtration is necessary to partition the bound aptamers from unselected molecules in order to obtain sufficient stringency (14). However, in our protocol, the washes were sufficiently stringent using magnetic beads. This is an advantage because partitioning by filtration is a cumbersome process for multiple targets but magnetic bead separations are easily accomplished in parallel, manually or automated. Additionally, we have taken advantage of the highly specific and strong streptavidin–biotin interaction for several applications: (i) generating single-stranded material from biotinylated PCR products after amplification at each round of selection using streptavidin magnetic beads; (ii) detecting biotinylated aptamers in enzyme-linked assays and western blot analysis; (iii) immobilizing biotinylated aptamers to streptavidin beads for purification of protein target; and (iv) immobilizing biotinylated aptamers to streptavidin sensor chips in BIAcore measurements.
Using manual processing (by one person), we were able to complete three rounds of selection per day on eight samples. We found that 15 rounds of selection produced high affinity aptamers. Therefore, our present throughput could be approximately 32 aptamers per month. However, our protocol is amenable to a 96-well approach and could be scaled-up to produce about 384 aptamers per month using manual processing. Others have described an automated aptamer acquisition platform with a throughput of 120/month for eight proteins; however, it requires customized robotics not available to many laboratories (14). Another high-throughput SELEX protocol using 96-well microtiter plates has been described that is compatible with robotics and was tested manually (23). However, that protocol relied on hydrophobic immobilization of proteins on microtiter plates, and the authors concede that the four proteins tested adhered to the wells with varying efficiency, making it difficult to control the amount of protein in each experiment. In addition, use of a hydrophobic immobilization would also result in proteins with variable orientations on the surface, reducing the effective concentration of available active sites. Lastly, since this method is non-specific, it would also result in the immobilization of all proteins in the sample, including protein contaminants that could interfere with the aptamer selection process.
We have illustrated the functional versatility of aptamers in several assays. Enzyme-linked assays provide a means of quickly evaluating a group of aptamers from a selection by measuring their relative affinities, and this kind of triage can be used to prioritize aptamers for more detailed characterization (24). We demonstrate that enzyme-linked assays can also provide information about cross-reactivity. We are able to measure significant signals over very low background by using biotinylated aptamers to detect proteins in a 96-well microtiter plate and a peroxidase-conjugated streptavidin and colorimetric substrate to detect the bound aptamers. Enzyme-linked assays offer advantages over other techniques, such as equilibrium dialysis and electrophoretic mobility shift assays, that are used to evaluate aptamers from a selection. These advantages include the lack of radioisotope usage, increased throughput in a 96-well plate, minimization of waste and ease of precise quantitation of the relative binding affinities.
Using aptamers in a protein blot analysis is another means of characterizing their specificity. We have tested TTF1 aptamers ‘A’ and ‘C’ in chemiluminescent protein blot analysis; however, only the TTF1 aptamer ‘A’ worked in this application, suggesting that, just as some antibodies fail to recognize the denatured form of a protein, some aptamers will recognize epitopes that are absent in the denatured form of the protein. The TTF1 aptamer showed no cross-reactivity to E.coli proteins in a cleared lysate on the blot and was similar in specificity to that observed for the anti-PentaHis–HRP antibody.
The key function of high affinity aptamers in applications such as protein purification, protein profiling chips and diagnostics is to recognize and separate the target protein from a complex mixture of proteins. We have described in this work the first successful application of aptamer affinity chromatography for one-step purification of a protein from the complex mixture of proteins in the soluble fraction of bacterial cell lysates. Although aptamer affinity chromatography has been described and demonstrated for the purification of a protein from conditioned cell culture media, this purification technique has not been previously demonstrated for more complex samples such as cell lysates (25). Detrimental effects from DNase activity in our purification from bacterial lysates were not observed, unlike the problems associated with DNase degradation of aptamers that occurs when purifying targets from serum (25). Importantly, aptamer affinity chromatography provides a means of protein purification of the native form of a protein without relying on affinity tags that may adversely affect protein structure, function or ability to form crystals for structural characterization.
We have recently learned that TTF1 is a highly specific marker for primary lung adenocarcinomas, and antibodies against TTF1 have been recommended to be included in a panel of antibodies for the differential diagnosis between primary and metastatic adenocarcinomas of the lung (26). We have initiated studies in our lab to determine whether or not the aptamers that we have produced against C.intestinalis TTF1 recognize human TTF1; if so, the aptamers described here may become a valuable diagnostic tool for primary lung adenocarcinoma.
Acknowledgments
ACKNOWLEDGEMENTS
The authors are very grateful to Peter Beernink for excellent critical comments on the manuscript. This work was performed under the auspices of the US Department of Energy, Office of Biological and Environmental Research, by the University of California, Lawrence Livermore National Laboratory under contract No. W-7405-ENG-48, Lawrence Berkeley National Laboratory under contract No. DE-AC03-76SF00098, and Los Alamos National Laboratory under contract No. W-7405-ENG-36.
REFERENCES
- 1.Phizicky E., Bastiaens,P.I., Zhu,H., Snyder,M. and Fields,S. (2003) Protein analysis on a proteomic scale. Nature, 422, 208–215. [DOI] [PubMed] [Google Scholar]
- 2.Dieckman L., Gu,M., Stols,L., Donnelly,M.I. and Collart,F.R. (2002) High throughput methods for gene cloning and expression. Protein Expr. Purif., 25, 1–7. [DOI] [PubMed] [Google Scholar]
- 3.Braun P., Hu,Y., Shen,B., Halleck,A., Koundinya,M., Harlow,E. and LaBaer,J. (2002) Proteome-scale purification of human proteins from bacteria. Proc. Natl Acad. Sci. USA, 99, 2654–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doyle S.A., Murphy,M.B., Massi,J.M. and Richardson,P.M. (2002) High-throughput proteomics: a flexible and efficient pipeline for protein production. J. Proteome Res., 1, 531–536. [DOI] [PubMed] [Google Scholar]
- 5.Lipton M.S., Pasa Tolic’,L., Anderson,G.A., Anderson,D.J., Auberry,D.L., Battista,J.R., Daly,M.J., Fredrickson,J., Hixson,K.K., Kostandarithes,H. et al. (2002) Global analysis of the Deinococcus radiodurans proteome by using accurate mass tags. Proc. Natl Acad. Sci. USA, 99, 11049–11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Issaq H.J., Conrads,T.P., Janini,G.M. and Veenstra,T.D. (2002) Methods for fractionation, separation and profiling of proteins and peptides. Electrophoresis, 23, 3048–3061. [DOI] [PubMed] [Google Scholar]
- 7.Templin M.F., Stoll,D., Schrenk,M., Traub,P.C., Vohringer,C.F. and Joos,T.O. (2002) Protein microarray technology. Drug Discov. Today, 7, 815–822. [DOI] [PubMed] [Google Scholar]
- 8.Steinhauer C., Wingren,C., Hager,A.C. and Borrebaeck,C.A. (2002) Single framework recombinant antibody fragments designed for protein chip applications. Biotechniques, Suppl, 38–45. [PubMed] [Google Scholar]
- 9.Li M. (2000) Applications of display technology in protein analysis. Nat. Biotechnol., 18, 1251–1256. [DOI] [PubMed] [Google Scholar]
- 10.Ng S., Goodson,B., Ehrhardt,A., Moos,W.H., Siani,M. and Winter,J. (1999) Combinatorial discovery process yields antimicrobial peptoids. Bioorg. Med. Chem., 7, 1781–1785. [DOI] [PubMed] [Google Scholar]
- 11.Tuerk C. and Gold,L. (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 12.Ellington A.D. and Szostak,J.W. (1990) In vitro selection of RNA molecules that bind specific ligands. Nature, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 13.Ellington A.D. and Szostak,J.W. (1992) Selection in vitro of single-stranded DNA molecules that fold into specific ligand-binding structures. Nature, 355, 850–852. [DOI] [PubMed] [Google Scholar]
- 14.Cox J.C., Rajendran,M., Riedel,T., Davidson,E.A., Sooter,L.J., Bayer,T.S., Schmitz-Brown.M. and Ellington,A.D. (2002) Automated acquisition of aptamer sequences. Comb. Chem. High Throughput Screen, 5, 289–299. [DOI] [PubMed] [Google Scholar]
- 15.Harvey R.P. (1996) NK-2 homeobox genes and heart development. Dev. Biol., 178, 203–216. [DOI] [PubMed] [Google Scholar]
- 16.Ristoratore F., Spagnuolo,A., Aniello,F., Branno,M., Fabbrini,F. and Di Lauro,R. (1999) Expression and functional analysis of Cititf1, an ascidian NK-2 class gene, suggest its role in endoderm development. Development, 126, 5149–5159. [DOI] [PubMed] [Google Scholar]
- 17.Mizuno K., Gonzalez,F.J. and Kimura,S. (1991) Thyroid-specific enhancer-binding protein (T/EBP): cDNA cloning, functional characterization and structural identity with thyroid transcription factor TTF-1. Mol. Cell. Biol., 11, 4927–4933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guazzi S., Price,M., De Felice,M., Damante,G., Mattei,M.G. and Di Lauro,R. (1990) Thyroid nuclear factor 1 (TTF-1) contains a homeodomain and displays a novel DNA binding specificity. EMBO J., 9, 3631–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Higgins D.G. and Sharp,P.M. (1988) CLUSTAL: a package for performing multiple sequence alignment on a microcomputer. Gene, 73, 237–244. [DOI] [PubMed] [Google Scholar]
- 20.Hertz G.Z. and Stormo,G.D. (1995) Identification of consensus patterns in unaligned DNA and protein sequences: a large-deviation statistical basis for penalizing gaps. Proceedings of the Third International Conference on Bioinformatics and Genome Research, pp. 201–216. [Google Scholar]
- 21.Walter G., Bussow,K., Lueking,A. and Glokler,J. (2002) High-throughput protein arrays: prospects for molecular diagnostics. Trends Mol. Med., 8, 250–253. [DOI] [PubMed] [Google Scholar]
- 22.Ylera F., Lurz,R., ErdmannmV.A. and Furste,J.P. (2002) Selection of RNA aptamers to the Alzheimer’s disease amyloid peptide. Biochem. Biophys. Res. Commun., 290, 1583–1588. [DOI] [PubMed] [Google Scholar]
- 23.Drolet D.W., Jenison,R.D., Smith,D.E., Pratt,D. and Hicke,B.J. (1999) A high throughput platform for systematic evolution of ligands by exponential enrichment (SELEX). Comb. Chem. High Throughput Screen, 2, 271–278. [PubMed] [Google Scholar]
- 24.Drolet D.W., Moon-McDermott,L. and Romig,T.S. (1996) An enzyme-linked oligonucleotide assay. Nat. Biotechnol., 14, 1021–1025. [DOI] [PubMed] [Google Scholar]
- 25.Romig T.S., Bell,C. and Drolet,D.W. (1999) Aptamer affinity chromatography: combinatorial chemistry applied to protein purification. J. Chromatogr. B, 731, 275–284. [PubMed] [Google Scholar]
- 26.Reis-Filho J.S., Carrilho,C., Valenti,C., Leitao,D., Ribeiro,C.A., Ribeiro,S.G. and Schmitt,F.C. (2000) Is TTF1 a good immunohistochemical marker to distinguish primary from metastatic lung adenocarcinomas? Pathol. Res. Pract., 196, 835–840. [DOI] [PubMed] [Google Scholar]