

Abstract
Members of the small Maf family of transcription factors play important roles in hematopoiesis. Using transgenic assays, we discovered a tissue-specific enhancer 3′ to the mafK gene. This enhancer directs mafK transcription in hematopoietic as well as in developing cardiac muscle cells, and was thus designated the hematopoietic and cardiac enhancer of mafK (HCEK). Only two of four GATA consensus motifs identified within HCEK contributed to enhancer activity, and both of these sites were required for both cardiac and hematopoietic transcriptional activation. The expression profile of MafK significantly overlapped that of GATA-1 in hematopoietic cells and of GATA-4/-6 in cardiac tissues. Each of these GATA factors bound with high specificity to both of the critical GATA sites in HCEK. Hence, the mafK gene is regulated by different GATA proteins in the hematopoietic and cardiac compartments through the same two GATA-binding sites in HCEK. These data provide the first in vivo demonstration that distinct members of a related transcription factor family activate the tissue-specific expression of a single target gene using the same cis-regulatory element.
Keywords: enhancer/GATA factor/MafK/transgenic mouse
Introduction
The maf oncogene was identified originally as the transduced oncogenic component of the transforming avian retrovirus, AS42 (Kawai et al., 1992). The product of the maf proto-oncogene and its relatives (the Maf family proteins) share a conserved basic region and amphipathic helix (bZip) motif that mediate DNA binding and dimer formation to the Maf recognition element (MARE; Kataoka et al., 1994b). Large Maf proteins, c-Maf, MafB, L-Maf and NRL, have an acidic domain to enable transcriptional activation (Nishizawa et al., 1989; Swaroop et al., 1992; Kataoka et al., 1994a; Ogino and Yasuda, 1998), while the small Maf proteins, MafF, MafG and MafK, do not (Fujiwara et al., 1993; Kataoka et al., 1995).
The small Maf proteins were first shown to function as a subunit of NF-E2, an erythroid/myeloid transcription factor (Andrews et al., 1993b; Igarashi et al., 1994). The NF-E2-binding site closely resembles a MARE, and is an essential cis-regulatory element for erythroid-specific transcription (Reitman and Felsenfeld, 1988; Mignotte et al., 1989). The large subunit of NF-E2 is called p45 (Andrews et al., 1993a), and several other proteins bearing similarity to p45 were isolated subsequently. These proteins, referred to as the cap’n’collar (CNC) family, are comprised of Nrf1, ECH/Nrf2, Nrf3, Bach1 and Bach2 (Chan et al., 1993; Caterina et al., 1994; Luna et al., 1994; Moi et al., 1994; Itoh et al., 1995; Oyake et al., 1996; Kobayashi et al., 1999). Since the CNC proteins do not bind to MARE sites efficiently as monomers or homodimers, they require small Maf proteins as obligatory partners to promote site-specific DNA binding (Andrews et al., 1993b; Igarashi et al., 1994).
The small Maf proteins are widely expressed during embryogenesis. Preliminary observations thereby led to the notion that the small Maf factors were ubiquitous (Andrews et al., 1993b; Fujiwara et al., 1993). Subsequent studies showed that the level of MafK expression changed in parallel with hematopoietic differentiation in the murine fetal liver (Motohashi et al., 1996). Furthermore, detailed examination of day 7.0 (E7.0) lacZ ‘knock-in’ embryos showed that MafK mRNA was highly expressed in extra-embryonic tissue, while MafG mRNA was expressed much more abundantly in the embryo proper (Shavit et al., 1998). These and other studies led to the contrary conclusion that the small maf genes are regulated in a tissue-specific manner, and therefore implied that they might play important, distinct roles during development. Gene targeting revealed that mafG-null mutant mice had decreased platelet numbers and neurological defects (Shavit et al., 1998), whereas mafK- and mafF-null mutant mice were viable, fertile and had normal longevity (Kotkow and Orkin, 1996; Shavit et al., 1998; Onodera et al., 1999). Very recently, we found that the phenotypes displayed in the mafG mutant mouse were exacerbated in compound null mutant mafG::mafK animals (Onodera et al., 2000). This led to the conclusion that MafK is an important contributor to the pool of MARE-binding proteins in vivo during both hematopoiesis and neurogenesis.
With this new found insight that the tissue-specific regulation of mafK might significantly impact our understanding of small Maf function in vivo, we initiated further studies to determine the modes of transcriptional regulation for mafK. We previously showed that the mafK gene contains two alternative promoters/first exons: the distal IM exon is used primarily in mesenchymal and hematopoietic tissues, whereas the gene-proximal IN exon is activated for neural expression (Motohashi et al., 1996). We localized regulatory sequences for mesenchymal and neuronal expression of the mafK gene to the two promoters (Motohashi et al., 1996, 1998), but the element(s) controlling hematopoietic expression were not found.
To document the contributions of MafK to hematopoiesis, we sought to resolve the position of any mafK hematopoietic regulatory sequences within recombinant P1 bacteriophage. P1 recombinants typically bear more genomic information than an average cosmid, but less than a typical bacterial artificial chromosome (BAC) or yeast artificial chromosome (YAC). After modification of subclones isolated from the P1 recombinants to generate reporter genes, we examined their expression in vivo. We report here the identification and characterization of an enhancer lying 3′ to the mafK gene that is coincident with an erythroid DNase I-hypersensitive (HS) site. Importantly, the enhancer possesses the ability to direct the expression of mafK in both hematopoietic and cardiac tissues in developing transgenic animals. Deletion analysis of the enhancer region showed that a 400 bp core of HCEK (the hematopoietic and cardiac enhancer of mafK) is sufficient for its activity, and that the hematopoietic and cardiac enhancer activities are inseparable. Through site-specific mutagenesis of HCEK, two GATA sites (of four) were found to be essential for enhancer activity in both hematopoietic and cardiac tissues. We found that GATA-1, -2, -4 and -6 all bind to the two essential GATA sites in HCEK, but since no single GATA factor is expressed in the same cardiac and hematopoietic cells, we conclude that mafK is a target gene regulated by distinct GATA factors in the two different cell lineages. By analysis of GATA-1-deficient embryonic stem (ES) cells subjected to in vitro differentiation, we further conclude that GATA-1 is an important activator for HCEK in hematopoietic cells. These data constitute the first direct evidence that the expression of a target gene (mafK) is regulated by different members of the same transcription factor family in distinct cell lineages through a common cis-regulatory element.
Results
Erythroid-specific DNase I HS sites in the mafK locus
Our first goal was to identify hematopoietic cell-specific regulatory region(s) in the mafK locus. We therefore screened a murine P1 recombinant library to isolate large contiguous fragments of genomic DNA containing the locus. The three isolated P1 recombinants contained extensive overlap with one another (Figure 1A), and conformed to the mafK gene structure determined from bacteriophage λ clones (Motohashi et al., 1996).

Fig. 1. Isolation of P1 phage clones and DNase I HS site mapping in the murine mafK locus in MEL and NIH 3T3 cells. (A) Three independent P1 phage clones were isolated. Closed triangles marked N and open triangles marked S indicate NotI and SfiI sites residing in the phage arms, respectively. Mouse genomic DNA containing the mafK locus is shown beneath the three P1 phage clones. (B) The mouse mafK locus is shown, and exons are depicted as open boxes. IM and IN indicate the relative positions of the two alternative first exons. Probes used for DNase I HS site analysis are indicated by closed boxes marked a–d. The lines beneath the closed boxes depict the length and direction of genomic fragments that hybridized with the probes in the absence of DNase I. The positions of the DNase I HS sites are indicated by vertical arrowheads marked A–I. Closed and open arrowheads indicate DNase I HS sites in MEL cells and in NIH 3T3 cells, respectively. (C) DNase I HS site analysis of MEL cells (left four panels) and NIH 3T3 cells (right two panels). The genomic DNA was purified from the nuclei treated with increasing amounts of DNase I (0–0.01 U/µl). Restriction enzymes and probes for Southern blot analysis are indicated at the bottom of each autoradiograph, and size markers (kbp) are shown on the left. The bands marked with asterisks indicate parental fragments. The appearance of fragments marked by lettered arrows indicates the existence of HS sites of the same letter as shown in (B). N, NotI; S, SfiI; n.d., not determined.
Since DNase I HS sites often coincide with gene-regulatory domains, we first examined the mafK locus within the P1 recombinants by DNase I HS analysis. To this end, we identified four independent single copy probes within the locus (shown in Figure 1B, in lower case letters a, b, c and d). Eight DNase I HS sites were identified in nuclei isolated from mouse erythroleukemia (MEL) cells (Figure 1C). These sites were classified into four arbitrary groups, shown diagrammatically in Figure 1B: group 1 (5′ to exon IM; site A), group 2 (around the IM promoter; site C), group 3 (the intergenic region between IM and IN; sites D, E and F) and group 4 (3′ to mafK; sites G, H and I). No other HS sites were detected in the 16 kbp interval between site A and the IM promoter. The IM promoter region, containing site C, was examined previously in transgenic assays and, as stated before, did not direct hematopoietic expression of mafK (Motohashi et al., 1996). We also compared the pattern in MEL (erythroid) cells with the pattern reflected in NIH 3T3 (fibroblast) cells (Figure 1B, open arrowheads, and C). One HS site within group 3 (E) and all three HS sites within group 4 (G–I) were MEL cell specific. We therefore focused on these unique erythroid HS sites as candidate hematopoietic regulatory elements of mafK.
Identification of putative mafK hematopoietic regulatory elements
To delineate the regulatory requirements for mafK expression in hematopoietic cells, we used in vivo transgenic assays. For this purpose, the bacterial lacZ gene was ligated to genomic DNA fragments from the mafK locus, and these were then injected into fertilized ova. We first examined transgenic mice bearing either erythroid HS site E (IMIN-lacZ) or HS sites G–I (IMIN-lacZ-HA; Figure 2). Stable lines bearing each of the transgenes were established and then analyzed in the F1 and F2 generations.

Fig. 2. Transgenic constructs. The mouse mafK locus is depicted at the top. The mafK locus bearing the lacZ knock-in is shown beneath the wild-type locus. Transgenes were constructed by ligating various genomic fragments from the mafK locus to the lacZ gene (Materials and methods). The DNase I HS sites in MEL cells are indicated by vertical arrowheads and correspond to those shown in Figure 1B. A, AscI; B, BspHI; H, HindIII; K, KpnI; S, StuI; X, XbaI.
For these initial studies, β-galactosidase (β-gal) enzymatic activity was examined in situ at a single developmental time point (E12.5) for all transgenic lines (Table I). We found reproducible β-gal expression in only half of the lines. This was probably due to the position of integration effects, where expression of transgenes is detected only at permissive genomic integration sites. In support of this hypothesis, the β-gal expression level was uncorrelated with integrated transgene copy number (Table I). To exclude possible false-positive or -negative staining of the reporter gene, we examined at least three independent transgenic lines (as well as founders; below) for each construct, and the consistency of the expression patterns for the positive animals was always confirmed in more than three independent lines. The observations for two of the independent transgenic lines for each construct are reported in Table II. As the control in these studies, we compared the transgene expression patterns with the expression profile of animals bearing lacZ targeted into the mafK genomic locus (lacZ knock-in; Figures 2 and 3; Shavit et al., 1998). In this line, β-gal activity was detected in E6.5 extra-embryonic ectoderm (Figure 3A), E12.5 cardiac cells (Figure 3B) and E12.5 fetal liver hematopoietic cells (Figure 3C). These observations indicated that the genomic sequences replaced by the lacZ gene (from exon II to exon III of mafK; Figure 2) were not necessary for mafK regulation in hematopoietic or cardiac tissues.
Table I. Expression of β-gal in the neural tube, heart and liver of transgenic lines at E12.5.
| Construct | Transgenicline | Transgene copynumber | β-gal expression |
||
|---|---|---|---|---|---|
| Neural tubea | Heartb | Liverb | |||
| IMIN-lacZ | 880 | 5 | + | – | – |
| 886 | 5 | + | – | – | |
| 885 | 17 | + | – | – | |
| 884 | 7 | – | – | – | |
| 889 | 2 | – | – | – | |
| IMIN-lacZ-HA | 359 | 3 | + | + | + |
| 221 | 2 | + | + | + | |
| 980 | 12 | NDc | + | + | |
| 218 | 15 | – | – | – | |
| IM-lacZ-SB | 28 | 70 | – | + | + |
| 40 | 30 | – | + | + | |
| 25 | 40 | – | + | + | |
| 29 | 6 | – | – | + | |
| 15 | 25 | – | – | – | |
| 24 | 5 | – | – | – | |
| 41 | 70 | – | – | – | |
| 43 | 100 | ectopicd | |||
aβ-gal expression was observed in the floor plate of the neural tube.
bThe criteria for positive β-gal expression are described in the legend to Figure 4.
cND, not determined.
dEctopic expression.
Table II. β-Gal expression in the mafK lacZ knock-in mouse and in transgenic lines.
| Embryonic day | Tissue | lacZ knock-in | IMIN-lacZ |
IMIN-lacZ-HA |
IM-lacZ-SB |
|||
|---|---|---|---|---|---|---|---|---|
| 880 | 886 | 359 | 221 | 28 | 40 | |||
| 6.5 | extra-embryonic | |||||||
| ectoderm | + | + | + | + | + | – | – | |
| mesoderm | NDb | ND | ND | ND | ND | + | + | |
| 9.5 | hearta | + | – | – | + | + | + | + |
| blood cellsc | + | – | – | + | + | + | + | |
| 12.5 | hearta | + | – | – | + | + | + | + |
| livera | + | – | – | + | + | + | + | |
aThe criteria for positive β-gal expression are described in the legend to Figure 4.
bND, not determined.
cβ-Gal expression was observed in blood cells in yolk sac and cardiac cavity.

Fig. 3. Expression patterns of the lacZ knock-in and transgenic mouse lines. The ‘lacZ knock-in’ mouse (A, B and C) and IMIN-lacZ (line 880, D, E and F), IMIN-lacZ-HA (line 359, G, H and I) and IM-lacZ-SB (line 28, J, K and L) transgenic mouse embryos are shown. Embryos at E6.5 were processed for whole-mount staining (A, D, G and J), while older embryos were sectioned for detection of β-gal activity. Blue staining for β-gal activity was detected strongly in extra-embryonic tissue of the lacZ knock-in embryo (A) and IMIN-lacZ (D) and IMIN-lacZ-HA (G) transgenic embryos, whereas IM-lacZ-SB embryos showed blue staining only in the mesodermal region (indicated by an arrow in J). Heart and hematopoietic cells are stained strongly in E12.5 embryos of the lacZ knock-in (B and C), IMIN-lacZ-HA (H and I) and IM-lacZ-SB (K and L) transgenic embryos, but not in IMIN-lacZ embryos (E and F). Arrowheads in (A), (D) and (G) indicate the boundary between extra-embryonic tissue and the embryo proper. ex, extra-embryonic region; e, embryonic region. The scale bar corresponds to 180 µm (B, E, H and K) and 30 µm (C, F, I and L).
3′-flanking sequences are necessary for mafK hematopoietic and cardiac expression in vivo
To localize the region(s) conferring mafK hematopoietic expression, we first examined transgenic lines bearing IMIN-lacZ (Figure 2), an 8 kbp fragment containing exons IM and IN and the intergenic region between them. All group 3 HS sites (D–F) were included in this construct, and five transgenic lines were examined (Table I). While β-gal activity was observed in the extra-embryonic ectoderm at E6.5 (Figure 3D), no activity was detected in E9.5 hematopoietic cells (Table II) or in E12.5 liver or heart (Figure 3E and F). These expression profiles were in clear contrast to lacZ knock-in mouse, and indicated that the 8 kbp genomic fragment in IMIN-lacZ does not contain the cis-elements required for mafK transcription in the fetal heart or in hematopoietic cells.
We then examined group 4 HS sites (Figure 1B, sites G–I) with transgenic mouse lines bearing IMIN-lacZ-HA. In addition to IMIN-lacZ, this construct includes a 6.6 kbp genomic fragment located 3′ to exon III (Figure 2). As anticipated from the HS site mapping analysis, strong β-gal activity was observed in primitive blood cells in the E9.5 cardiac cavity (Table II) and in definitive hematopoietic cells in the E12.5 fetal liver (Figure 3I). Concomitantly, β-gal activity was also observed in E9.5 (Table II) and E12.5 heart (Figure 3H). These data showed that a 6.6 kbp fragment 3′ to the mafK gene, including erythroid HS sites G, H and I, is necessary for hematopoietic (both primitive and definitive) and cardiac tissue-specific mafK regulation. We further reduced the 6.6 kbp 3′-flanking region to a 1.6 kbp fragment (IMIN-lacZ-SB; Figure 2), which retained all three group 4 HS sites (G–I). Three out of four IMIN-lacZ-SB F0 embryos (see below) showed β-gal activity in the fetal liver and heart (Figure 4A), indicating that this 1.6 kbp fragment contained the sequences for hematopoietic mafK regulation. Furthermore, a small segment containing only the exon IM promoter was able to collaborate with the 1.6 kbp SB 3′ fragment (IM-lacZ-SB) to reflect the identical β-gal expression pattern in E12.5 definitive hematopoietic cells (fetal liver; Figure 3L) and E12.5 heart (Figure 3K) as well as E9.5 primitive blood cells (data not shown). Hence, the 1.6 kbp 3′ fragment plus the IM promoter are sufficient for tissue-specific regulation of the mafK gene in vivo; neither exon IN nor the group 3 HS sites appear to contribute to hematopoietic or cardiac expression of mafK.

Fig. 4. Founder analysis of mafK 3′ regulatory sequences. All embryos were analyzed at E12.5, and liver and heart were examined for β-gal expression. (A) The structure of IMIN-lacZ-SB is shown on the top, which is a parent construct for IMIN-lacZ-SX, IMIN-lacZ-DB and IMIN-lacZ-XB. The truncated genomic fragments used to generate IMIN-lacZ-SB, IMIN-lacZ-SX, IMIN-lacZ-DB and IMIN-lacZ-XB are shown on the right. The incidence of β-gal-expressing embryos is shown to the left of the scheme of each construct. (B) The structure of IM-lacZ-XB is shown on the top, which is the parent construct for IM-lacZ-XBst, IM-lacZ-XApa and IM-lacZ-ApaB. The truncated genomic fragments used to generate IM-lacZ-XB, IM-lacZ-XBst, IM-lacZ-XApa and IM-lacZ-ApaB are shown on the right. The incidence of β-gal-expressing embryos is shown to the left of the scheme of each construct. The incidence is expressed as the number of β-gal-positive embryos/the number of transgene-positive embryos. The criteria for positive β-gal expression are blue staining in >20% of hematopoietic cells for the liver and either partial or complete staining of cardiac muscle cells for the heart. The constructs that retained enhancer activity are marked with +, and those without activity are marked with –. Apa, ApaI; B, BspHI; Bst, Bst1107I; D, DraI; K, KpnI; S, StuI; X, XbaI.
To refine the position of regulatory elements responsible for tissue-specific expression of the mafK gene, we tested several further constructs bearing truncations in the 3′ region (IMIN-lacZ-SX, -DB and -XB; Figure 4A). To localize the cis-elements of interest more rapidly, we examined transgenic founders (F0 assay). Whereas embryos bearing HS sites H and I (IMIN-lacZ-DB) showed specific β-gal expression in both the fetal liver and heart (3/7 and 3/7, respectively), those bearing HS sites G and H (IMIN-lacZ-SX) did not (0/9). Finally, we prepared IMIN-lacZ-XB that possessed only HS site I. Mouse embryos bearing this transgene again showed β-gal expression in the fetal liver and heart (4/16 and 5/16). We also showed that the XB fragment and IM promoter (IM-lacZ-XB) are sufficient for the tissue-specific regulation (Figure 4B). These results demonstrated that the regulatory element(s) controlling hematopoietic and cardiac-specific expression of the mafK gene resides in the XB fragment, corresponding to DNase I HS site I. Accordingly, the XB fragment was given the preliminary designation hematopoietic and cardiac enhancer of mafK (HCEK).
We dissected the HCEK element by founder assays. The 0.6 kbp XB fragment was subdivided further into three components (Figure 4B). mafK gene-distal 200 and 400 bp fragments were removed to generate IM-lacZ-XBst and IM-lacZ-XApa, respectively, while the gene-proximal 200 bp were deleted to generate construct IM-lacZ-ApaB (Figure 4B). At low frequency, transgenic embryos bearing IM-lacZ-XBst had β-gal activity in the fetal liver and heart (5/18 and 3/18, respectively). In contrast, neither IM-lacZ-ApaB nor IM-lacZ-XApa displayed reproducible β-gal staining. Thus, HCEK activity requires sequence elements in the interval between the XbaI and Bst1107I sites (Figure 4B).
HCEK is an embryonic stage-oriented enhancer
Detailed developmental analysis of HCEK activity was examined using IM-lacZ-SB transgenic lines. Mesodermal expression of the lacZ gene began at gastrulation (Figures 3J and 5A–D) and continued in the primitive blood cells in the E9.5 yolk sac (Figure 5E). Cardiac expression of β-gal began at E8.0 in the cardiac primordium (Figure 5D). By E12.5, β-gal expression was observed in the atrium (data not shown) and left and right ventricles. Both the interventricular septum and trabeculated myocardium stained strongly (Figure 3K), whereas staining was not observed in the endocardial cushions (data not shown). β-gal activity in definitive hematopoietic cells was observed in the fetal liver (Figure 3L) and in the adult spleen (Figure 5F), albeit that the latter was weaker than the former. Whereas megakaryocytes in the fetal liver stained strongly (Figure 3L), megakaryocytes from the adult spleen did not (Figure 5F). Cardiac expression of lacZ also weakened in the adult, and β-gal expression was restricted to the endocardium (Figure 5G). Thus, HCEK appears to exert its most potent activity during the embryonic, rather than adult, stages.

Fig. 5. Developmental expression profile of IM-lacZ-SB. Transgenic mice bearing IM-lacZ-SB (line 40 for A, B, C1 and C2; line 28 for D, E, F and G) were examined at gastrula (A, B, C1 and C2; C1 a frontal view, C2 a lateral view), E8.5 (D), E9.5 (E) and adult stages (F and G). Blue staining was first detected in emerging mesodermal cells during gastrulation (A is earliest, B is in the middle and C1, C2 are the latest) and continued in the blood islands (arrowheads in D) and in blood cells in yolk sac vessels (E). β-gal expression started in heart primordium at E8.5 (arrow in D). In adult mice, blood cells in the spleen are stained (F). In the adult heart, only the endocardium is stained, but myocardial staining is not observed (G). The scale bar corresponds to 100 µm (F) or 200 µm (G).
Two GATA sites are required for HCEK activity
To identify potentially critical transcription factor-binding sites that might be responsible for HCEK activity in hematopoietic and/or cardiac tissues, we determined the nucleotide sequence of HCEK (Figure 6). We identified consensus sequences to which numerous transcription factors involved in hematopoietic and cardiac gene expression can bind (underlined). For example, there are four putative GATA factor-binding sites in HCEK, and the second proximal site is a tandem repeat of TATC. Since GATA-1 and -2 are known to be critical regulators of hematopoietic cell differentiation (Tsai et al., 1994; Takahashi et al., 1997), and since GATA-4 and -6 may both be essential for heart development (Charron and Nemer, 1999), the GATA sites were good candidates for HCEK activity in either or both hematopoietic and cardiac cells.

Fig. 6. Nucleotide sequence of HCEK. Potential transcription factor-binding sites are underlined. Restriction sites are highlighted by inverted triangles. Four GATA sites are found in the region between XbaI and Bst1107I. One of them is composed of tandem GATA motifs.
To examine whether each site within HCEK was functional, we mutated the four GATA sites individually within the HCEK element of construct IM-lacZ-XB, and the activity of the reporter was then tested by F0 analysis at E12.5 (Figure 7). F0 embryos bearing IM-lacZ-XB/Gm1, in which the most gene-proximal GATA site was mutated, showed no β-gal expression in the fetal liver or heart (0/11 and 0/11, respectively), indicating that this GATA site is indispensable for HCEK activity. Similarly, F0 embryos bearing IM-lacZ-XB/Gm2, in which the second tandem GATA site was mutated, showed no β-gal expression in the fetal liver or heart (0/11 and 0/11, respectively). In contrast, F0 embryos bearing either IM-lacZ-XB/Gm3 or IM-lacZ-XB/Gm4, in which the two more distal GATA sites were mutated, showed β-gal expression similar to the parental control (data not shown). These experiments demonstrated that the two gene-proximal GATA sites are essential for HCEK activity in both hematopoietic and cardiac cells, while the distal two GATA sites are less important.

Fig. 7. Mutation of GATA sites eliminates HCEK activity in transgenic mice. Left: each of the four GATA sites was mutated within IM-lacZ-XB. Mutated GATA sequences (Gm1–Gm4) are shown under the wild-type sequences. Middle: the incidence of β-gal-expressing embryos bearing each construct with a mutated GATA site is shown in the same way as described in the legend to Figure 4. Right: the structures of IM-lacZ-XB and its derivatives are depicted. Each mutated GATA site was marked with a small x within the XB region. The constructs that retain enhancer activity are marked with +, and those without activity are marked with –. For abbreviations, see legend to Figure 4.
Hematopoietic and cardiac GATA factors bind to the HCEK GATA sites
To test whether tissue-restricted GATA factors were capable of binding to the two indispensable GATA sites (above), we carried out electrophoretic mobility shift assay (EMSA). We examined binding to two oligonucleotide probes corresponding to the most gene-proximal site and to the vital tandem GATA motif. Recombinant GATA factors were prepared by transfecting expression plasmids containing the mouse GATA-1, -2, -4 and -6 cDNAs into cultured 293T cells (Materials and methods). At 24 h post-transfection, nuclear extracts were prepared. Their expression in the transfected cells was confirmed by immunoblot analysis using antibodies specific for each GATA factor (data not shown).
In EMSA with probe A (the most gene-proximal GATA site), all four GATA factors tested (GATA-1, -2, -4 and -6) bound to the probe (Figure 8A, lanes 3, 8, 13 and 18). This binding was competed efficiently by the addition of excess unlabeled probe (Figure 8A, lanes 4, 9, 14 and 19), but not with an unlabeled GATA mutant probe (A/mt; Figure 8A, lanes 5, 10, 15 and 20). These results showed that GATA factor binding depends on the integrity of the GATA sequence. Similarly, all four GATA factors bound to the tandem GATA motifs in probe B (Figure 8B, lanes 3, 10, 17 and 24). This binding was also eliminated by the addition of excess unlabeled probe B (Figure 8B, lanes 4, 11, 18 and 25) and, as before, competition was not observed when unlabeled mutant probe B/mt1 was added (Figure 8B, lanes 5, 12, 19 and 26). Incomplete competition was observed with probes B/mt2 and mt3, in which only one of the two GATA motifs in the tandem GATA site was mutated (Figure 8B, lanes 6, 13, 20 and 27; or lanes 7, 14, 21 and 28, respectively). Thus, both sites contribute to stable GATA factor binding in the tandem GATA motif.

Fig. 8. GATA factors bind to critical GATA sites in HCEK. Nuclear extracts were incubated with 32P-labeled oligonucleotides containing GATA sites (probes A and B). (A) Probe A was incubated alone (lanes 1, 6, 11 and 16) or with nuclear extract prepared from mock- (lanes 2, 7, 12 and 17), GATA-1- (lanes 3–5), GATA-2- (lanes 8–10), GATA-4 (lanes 13–15) or GATA-6-transfected cells (lanes 18–20; Materials and methods). Addition of a 400-fold molar excess of identical unlabeled oligonucleotides decreased the intensity of the bands (lanes 4, 8, 14 and 19), while addition of mutated oligonucleotide probe A/mt did not cause substantial changes (lanes 5, 10, 15 and 20). Mutated GATA sequences are shown under the wild-type sequences. (B) Probe B was incubated alone (lanes 1, 8, 15 and 22), with nuclear extracts from untransfected cells (lanes 2, 9, 16 and 23) or with nuclear extract prepared from GATA-1- (lanes 3–7), GATA-2- (lanes 10–14), GATA-4- (lanes 17–21) or GATA-6-transfected cells (lanes 24–28). Addition of a 400-fold molar excess of unlabeled oligonucleotide diminished the intensity of the shifted bands (lanes 4, 11, 18 and 25), while addition of probe B/mt1 (with doubly mutated GATA motifs) did not cause any significant change (lanes 5, 12, 19 and 26). Moderate competition was observed with oligonucleotides in which half of the tandem GATA site was mutated (B/mt2: lanes 6, 13, 20 and 27 and B/mt3: lanes 7, 14, 21 and 28). Arrows indicate the specific shifted bands with each GATA factor. In the case of GATA-1, although two shifted bands were observed, only the upper band is super-shifted by addition of an anti-GATA-1 antibody (data not shown). M, mock transfectant.
The results of the transgenic analyses and EMSA, taken together, show that in different tissues distinct GATA factors bind to the same GATA motifs within HCEK. Thus, the GATA factors contribute differentially to cell lineage-specific transcription of mafK in hematopoietic and cardiac cells through a common cis-element.
mafK transcription in hematopoietic cells is activated by GATA-1
To test the functional relationship between GATA activity and MafK expression during hematopoiesis, we examined MafK mRNA levels in GATA-1 promoter mutant ‘knock-down’ ES cells (Takahashi et al., 1997). Since mice carrying this germline mutant allele die by E12.5, we used in vitro differentiation of OP-9/ES cells for this analysis (Suwabe et al., 1998). This analysis would allow us to examine the regulatory effects of GATA-1 and GATA-2 on mafK expression during both primitive and definitive hematopoiesis. GATA-1 knock-down ES cells have been shown to express 20-fold lower levels of GATA-1, and concurrent 10-fold (or more) higher than normal levels of GATA-2 (both in comparison with wild-type, differentiated ES cells), and therefore we could test directly whether GATA-1, GATA-2 or both were capable of activating hematopoietic mafK transcription.
At days 8 and 14 of ES cell culture, we collected non-adherent cells (representing the primitive or definitive hematopoietic lineages, respectively; Suwabe et al., 1998). We found that mafK mRNA accumulation was severely diminished in both primitive and definitive hematopoietic cells when compared with wild-type ES cells (Figure 9). Since hematopoietic cells generated from the knock-down mutant expressed TER119 antigen and β-globin (Suwabe et al., 1998), they had differentiated to the stage where mafK is normally actively transcribed (Igarashi et al., 1995). Therefore, although GATA-1 and GATA-2 are both reported as strong transcriptional activators (Yamamoto et al., 1990), we conclude that the HCEK is activated by GATA-1, but not effectively by GATA-2, in hematopoietic cells.
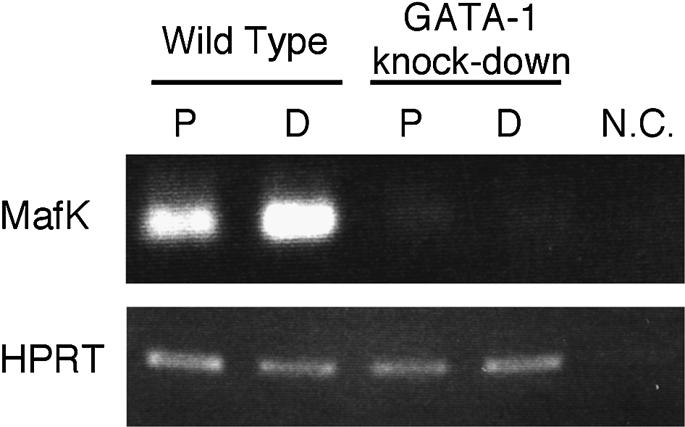
Fig. 9. Inhibited mafK expression in GATA-1 knock-down hematopoietic cells. Wild-type and GATA-1 knock-down ES cells (Takahashi et al., 1997) were differentiated into primitive (P) and definitive (D) hematopoietic cells using in vitro differentiation (Materials and methods). The expression of MafK mRNA was examined by RT–PCR. MafK expression was reduced markedly in both primitive and definitive hematopoietic cells generated from GATA-1 knock-down ES cells in comparison with the levels in wild-type ES cells. HPRT was used as a normalizing control. N.C., negative control (water).
Discussion
This study identified an enhancer 3′ to the mafK gene that is capable of directing its hematopoietic and cardiac cell-specific expression. Of four GATA sites in the core region of the enhancer, two were found to be critical for both hematopoietic and cardiac expression of the gene. Since GATA-1/-2 and GATA-4/-6 regulate gene expression in hematopoietic and cardiac tissues, respectively, these data suggest that the observed activities are based on the binding of at least two distinct GATA factors to the HCEK. These results thus indicate that the mafK gene is under stringent GATA factor control in at least two different tissues, suggesting that the GATA factor–MafK epistatic relationship arose in evolution before the distinct tissues did.
HCEK activity was first detected in the extra-embryonic mesoderm at the gastrulation stage, and the activity continued in primitive hematopoietic cells of the yolk sac and in definitive hematopoietic cells of the fetal liver. HCEK was also very active in the developing heart, in which reporter gene expression was first observed in the cardiac primordium at E8.0. Interestingly, both tissues differentiate from the same germ layer, the splanchnic mesoderm. HCEK became active soon after the commitment of splanchnic mesoderm to the hematopoietic and cardiac lineages. These results suggest that the control over expression of the mafK gene mediated by HCEK may be an important developmental event regulating the timing of novel gene activation in tissues derived from the splanchnic mesoderm.
Although β-gal activity was strong during the embryonic stage, only weak signals were observed in adult tissues. Two explanations are possible for this observation. The first explanation is that artificial loci, in which the transgenes are integrated randomly, might be susceptible to chromatin modifications, resulting in position effect variegation or inactivation of the loci in the adult stage. The second explanation is that the environment of the transcription factor is altered in the adult. This may explain the different HCEK activity observed in both cardiac and hematopoietic cells in the two distinct developmental stages. We believe the latter possibility is more likely, since mafK gene regulation has been shown to be highly complex (below).
Expression of the mafK gene is detected in many tissues, so initial reports described MafK as a ubiquitous transcription factor (Andrews et al., 1993b; Fujiwara et al., 1993; Igarashi et al., 1995). More recently, we demonstrated that the expression of mafK is finely regulated in distinct tissues by multiple cis-elements within and closely surrounding the gene. The distal IM promoter is utilized mainly in mesenchymal and hematopoietic cells (Motohashi et al., 1996), whereas the proximal IN promoter is utilized for neural expression of the gene (Motohashi et al., 1998). In addition to these two upstream regulatory regions, HCEK was newly identified in the present study. Furthermore, while mesenchymal cell-specific expression was clearly observed in IM-lacZ transgenic mice in intra-embryonic mesoderm at E7.5 and in sclerotome at E9.5 (Motohashi et al., 1996), this expression was weakened when a specific 3′-flanking sequence was added to the test construct (i.e. IM-lacZ-SB; data not shown). These observations suggest the presence of still other unidentified positive elements in the mafK locus, which derepress mafK expression from the negative influence of the 3′-flanking region in specific tissues. Hence, mafK appears to be under the influence of multiple and distinct regulatory elements.
EMSA showed that GATA-1/-2 and GATA-4/-6 can all bind to the critical GATA sites in HCEK. Several lines of evidence strongly suggest that GATA-1 is an important activator for HCEK in hematopoietic cells. First, the increase in MafK mRNA levels during erythroid differentiation closely coincides with the increase in GATA-1 levels (Igarashi et al., 1995). Secondly, a lacZ gene directed by HCEK shows a very similar hematopoietic expression profile to that of G1HE, a GATA-1 gene hematopoietic enhancer (Nishimura et al., 2000). Finally, ES cells bearing a GATA-1 knock-down promoter mutation expressed remarkably lower levels of MafK mRNA than did wild-type ES cells after erythroid differentiation in vitro. We conclude that the reduction in GATA-1 level causes a reduction in mafK transcription, and that mafK might be an immediate downstream target of GATA-1 in hematopoietic differentiation.
As is GATA-1 in hematopoietic cells, GATA-4 and -6 are both believed to be important in cardiac gene regulation. Specifically supporting this hypothesis, gene targeting has shown that GATA-4 is required for heart tube formation (Molkentin et al., 1997). The expression profiles of these two GATA factors overlap with that of MafK in cardiac tissue (Molkentin et al., 1997; Koutsourakis et al., 1999), suggesting that GATA-4 and/or -6 may regulate mafK expression in the heart.
The present analysis identifies a novel mode of gene regulation, whereby one enhancer is regulated by tissue-specific trans-acting factors through the same cis-regulatory element in different tissues. In this case, GATA-1 mainly activates the mafK gene expression in hematopoietic cells, and cardiac factors GATA-4 and -6 regulate the gene in cardiac tissues, with all three proteins binding to the same two GATA sites in HCEK. We speculate that overlapping utilization of the same cis-regulatory elements by lineage-restricted transcription factors in different tissues may be one of the strategies that cells have evolved to produce diversity in the regulation of gene expression during the course of molecular evolution. A key observation here is that the target gene, mafK, is highly expressed in both hematopoietic and cardiac cells (Shavit et al., 1998). This observation suggests the intriguing possibility that, while distinct sets of GATA factors might be used to determine the fate of cells during differentiation, one target molecule, MafK, is commonly used in different sets of cells during differentiation.
In conclusion, we are now one step closer to further elucidation of the regulatory mechanisms that control erythropoiesis, in which various transcription factors contribute to the regulatory hierarchy specifying a unique developmental pathway. We have recently shown that MafK is an important regulator of red cell membrane proteins required for late stages of erythroid cell maturation (Onodera et al., 2000). We have also found recently that forced expression of MafK specifically in erythroid cells results in profound perturbation of erythroid differentiation (H.Motohashi, J.D.Engel and M.Yamamoto, in preparation). Since cell differentiation in metazoans is based on the orderly progression of multiple gene activation steps, elucidation of the control processes that activate erythroid-specific regulatory genes is crucial for a comprehensive understanding of erythroid differentiation. Thus, the present analysis of the mafK gene represents an important step in deciphering how the regulators themselves are regulated.
Materials and methods
Isolation of P1 phage clones of the mouse mafK gene
A P1 phage mouse genomic library from Mus musculus (strain RIII) genomic DNA was screened for clones containing the mafK gene (Genome Systems). Library screening was carried out by PCR. A pair of oligonucleotides, MafK.cod.2u (5′-AACGCCCCTGTTCTTAGCGA-3′) and MafK.cod.2d (5′-GCTCGGCAGATTTGACGATG-3′), were used to amplify a 375 bp fragment located in the third exon of the mafK gene. Three positive clones, designated 25, 26 and 27, were mapped by standard restriction enzyme digestion and Southern blot analysis (Sambrook et al., 1989).
DNase I HS sites analysis
Isolation of nuclei and DNase I treatments were performed as previously described (Gerber et al., 1997). Cultured cells (non-adherent MEL or trypsinized NIH 3T3) were resuspended in ice-cold lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl, 5 mM MgCl2, 0.1% NP-40) and incubated for 10 min. Nuclei were pelleted by centrifugation, treated with various (up to 0.01 U/µl) concentrations of DNase I, and incubated at 37°C for 10 min. Genomic DNA was purified from the nuclease-treated nuclei by phenol–chloroform extraction, and the purified DNA was then digested with restriction enzymes as indicated in the figure legends.
Cell culture
MEL cells (B8 subclone) were cultured in ES medium (Nissui) supplemented with 10% fetal bovine serum (FBS). NIH 3T3 cells and 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL) supplemented with 10% FBS.
Generation of LacZ reporter constructs and transgenic mice
Reporter constructs were generated using restriction enzyme sites present in mafK and lacZ using plasmid pSVβ (Clontech). To generate pIMIN-lacZ, a genomic fragment from the mafK gene [KpnI (at –0.9 kbp) to SmaI (at +7.5 kbp); transcriptional initiation site of IM exon is numbered as +1] and two fragments from pSVβ [lacZ DNA and the poly(A) signal] were inserted into pBluescript SK(+) in the appropriate order. pIMIN-lacZ-HA, pIMIN-lacZ-SB, pIMIN-lacZ-SX, pIMIN-lacZ-DB and pIMIN-lacZ-XB were made by inserting the genomic fragments from the 3′ region of the mafK gene (see Figures 2 and 4A) 3′ to the poly(A) signal sequence in pIMIN-lacZ. IM-lacZ-SB, IM-lacZ-XB, IM-lacZ-XBst, IM-lacZ-XApa and IM-lacZ-ApaB were generated by inserting the genomic fragments from the 3′ region of the mafK gene (see Figures 2 and 4B) 3′ to the poly(A) signal sequence of pIM-lacZ (Motohashi et al., 1996). The GATA sites in region XB were mutated using PCR as previously described (Cormack, 1987). The authenticity of the mutagenesis and deletion was confirmed by DNA sequencing.
The lacZ reporter constructs were purified as described previously, and the purified DNA was microinjected into fertilized oocytes (Motohashi et al., 1996). Transgenic mice were generated using standard procedures (Hogan et al., 1986).
Analysis of transgenic mice and embryos
Mouse embryos were fixed at room temperature for 30 min (E6.5) or 90 min (E12.5) in 1% formaldehyde, 0.2% glutaraldehyde and 0.02% NP-40 in phosphate-buffered saline. 5-bromo-4-chloro-3-indolyl-β-d-galactosidase (X-gal) staining was performed as previously described (Onodera et al., 1997). For sectioned samples, nuclear fast red was used for counterstaining. Genomic DNA was purified from tail or placenta, and integration of the transgenes was verified by PCR using transgene-specific primer sets. Oligonucleotides MafK ex2.53 (5′-ACGATTTTCTGGTGGTTCCG-3′) and LacZ RT2 (5′-GCAACGAAAATCACGTTCTTGTTGG-3′) were used to detect transgenes containing mafK exon II. MafK IM53 (5′-TGTTCTTCGCCGAGTCGGAA-3′) and LacZ RT2 were used to detect transgenes that did not contain mafK exon II.
Preparation of nuclear extracts
Mammalian expression vectors (pEF-BOS) (Mizushima et al., 1990) containing the mouse GATA-1, GATA-2, GATA-4 and GATA-6 cDNAs (DDBJ/EMBL/GenBank accession Nos X15763, AB000096, AF179424 and AF179425, respectively) were transfected into 293T cells. Nuclear extracts were prepared as described (Dignam, 1990).
Electrophoretic mobility shift assay
EMSA was performed as described previously (Yamamoto et al., 1990). Nuclear extracts were incubated on ice with 32P-labeled probe. Sense and antisense oligonucleotides corresponding to nucleotides 81–120 of HCEK (Figure 6) were annealed to generate the wild-type probe A. Sense and antisense oligonucleotides corresponding to nucleotides 130–169 of HCEK were annealed to generate wild-type probe B. The sequences of the GATA mutant probes A/mt and B/mt1, mt2 and mt3 are all shown in Figure 8. For competition, a 400-fold molar excess of unlabeled oligonucleotide was used. The DNA–protein complexes were separated from free probe using a 5% non-denaturing polyacrylamide gel and detected by autoradiography.
In vitro OP-9/ES cells differentiation system
Wild-type and GATA-1 promoter mutant ‘knock-down’ ES cells (Takahashi et al., 1997) were differentiated into primitive and definitive hematopoietic cells as described previously (Suwabe et al., 1998). Day 8 and day 14 non-adherent cells were collected, which represent primitive and definitive hematopoietic cells, respectively (Suwabe et al., 1998). Total RNA was prepared using ISOGEN (Wako). Reverse transcription was performed as previously described using Superscript II (Gibco-BRL) and random hexamer oligonucleotides (Suwabe et al., 1998). The primer set for detection of MafK mRNA was ex1.53 (5′-AAGCGCTTGTGAAAGAGTGC-3′) and ex3.35 (5′-TTAGCTCCCGCACTGACATG-3′). The primer set used for detection of HPRT was described previously (Suwabe et al., 1998). Amounts of the cDNAs were adjusted by dilution to produce equal quantities of the HPRT control amplicon.
Acknowledgments
Acknowledgements
We are grateful to Dr Kim-Chew Lim for critical discussions and advice. We thank Ms N.Kaneko for technical support in histological examination. This work was supported by an NIH grant (R01 CA80088 to J.D.E.), by a Center of Excellence award from the NCI to the Robert H.Lurie Comprehensive Cancer Center, and by grants from the Ministry of Education, Science, Sports and Culture (H.M. and M.Y.), JSPS-RFTF and CREST (M.Y.).
References
- Andrews N.C., Erdjument-Bromage,H., Davidson,M.B., Tempst,P. and Orkin,S.H. (1993a) Erythroid transcription factor NF-E2 is a haematopoietic-specific basic-leucine zipper protein. Nature, 362, 722–728. [DOI] [PubMed] [Google Scholar]
- Andrews N.C., Kotkow,K.J., Ney,P.A., Erdjument-Bromage,H., Tempst,P. and Orkin,S.H. (1993b) The ubiquitous subunit of erythroid transcription factor NF-E2 is a small basic-leucine zipper protein related to the v-maf oncogene. Proc. Natl Acad. Sci. USA, 90, 11488–11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina J.J., Donze,D., Sun,C.W., Ciavatta,D.J. and Towns,T.M. (1994) Cloning and functional characterization of LCR-F1: a bZIP transcription factor that activates erythroid-specific, human globin gene expression. Nucleic Acids Res., 22, 2383–2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J.Y., Han,X.-L. and Kan,Y.W. (1993) Cloning of Nrf1, an NF-E2-related transcription factor, by genetic selection in yeast. Proc. Natl Acad. Sci. USA, 90, 11371–11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron F. and Nemer,M. (1999) GATA transcription factors and cardiac development. Semin. Cell Dev. Biol., 10, 85–91. [DOI] [PubMed] [Google Scholar]
- Cormack B. (1987) Current Protocols in Molecular Biology. Academic Press, New York, NY. [Google Scholar]
- Dignam J.D. (1990) Preparation of extracts from higher eukaryotes. Methods Enzymol., 182, 194–203. [DOI] [PubMed] [Google Scholar]
- Fujiwara K.T., Kataoka,K. and Nishizawa,M. (1993) Two new members of the maf oncogene family, mafK and mafF, encode nuclear b-Zip proteins lacking putative trans-activator domain. Oncogene, 8, 2371–2380. [PubMed] [Google Scholar]
- Gerber A.N., Klesert,T.R., Bergstrom,D.A. and Tapscott,S.J. (1997) Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: a mechanism for lineage determination in myogenesis. Genes Dev., 11, 436–450. [DOI] [PubMed] [Google Scholar]
- Hogan B., Consstantini,F. and Lacy,E. (1986) Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Igarashi K., Kataoka,K., Itoh,K., Hayashi,N., Nishizawa,M. and Yamamoto,M. (1994) Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature, 367, 568–572. [DOI] [PubMed] [Google Scholar]
- Igarashi K., Itoh,K., Motohashi,H., Hayashi,N., Matuzaki,Y., Nakauchi,H., Nishizawa,M. and Yamamoto,M. (1995) Activity and expression of murine small Maf family protein MafK. J. Biol. Chem., 270, 7615–7624. [DOI] [PubMed] [Google Scholar]
- Itoh K., Igarashi,K., Hayashi,N., Nishizawa,M. and Yamamoto,M. (1995) Cloning and characterization of a novel erythroid-derived CNC family transcription factor heterodimerizing with the small Maf family proteins. Mol. Cell. Biol., 15, 4184–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K., Fujiwara,K.T., Noda,M. and Nishizawa,M. (1994a) MafB, a new Maf family transcription activator that can associate with Maf and Fos but not with Jun. Mol. Cell. Biol., 14, 7581–7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K., Noda,M. and Nishizawa,M. (1994b) Maf nuclear oncoprotein recognizes sequences related to an AP-1 site and forms heterodimers with both Fos and Jun. Mol. Cell. Biol., 14, 700–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K., Igarashi,K., Itoh,K., Fujiwara,K.T., Noda,M., Yamamoto,M. and Nishizawa,M. (1995) Small Maf proteins heterodimerize with Fos and potentially act as competitive repressors of NF-E2 transcription factor. Mol. Cell. Biol., 15, 2180–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai S., Goto,N., Kataoka,K., Saegusa,T., Shinno-Kohno,H. and Nishizawa,M. (1992) Isolation of the avian transforming retrovirus, AS42, carrying the v-maf oncogene and initial characterization of its gene product. Virology, 188, 778–784. [DOI] [PubMed] [Google Scholar]
- Kobayashi A., Ito,E., Toki,T., Kogame,K., Takahashi,S., Igarashi,K., Hayashi,N. and Yamamoto,M. (1999) Molecular cloning and functional characterization of a new cap’n’collar family transcription factor Nrf3. J. Biol. Chem., 274, 6443–6452. [DOI] [PubMed] [Google Scholar]
- Kotkow K.J. and Orkin,S.H. (1996) Complexity of the erythroid transcription factor NF-E2 as revealed by gene targeting of the mouse p18 NF-E2 locus. Proc. Natl Acad. Sci. USA, 93, 3514–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsourakis M., Langeveld,A., Patient,R., Beddington,R. and Grosveld,F. (1999) The transcription factor GATA6 is essential for early extraembryonic development. Development, 126, 723–732. [PubMed] [Google Scholar]
- Luna L., Johnsen,O., Skartlien,A.H., Pedeutour,F., Turc-Carel,C., Prydz,H. and Kolsto,A. (1994) Molecular cloning of a putative novel human bZIP transcription factor on chromosome 17q22. Genomics, 22, 553–562. [DOI] [PubMed] [Google Scholar]
- Mignotte V., Wall,L., deBohr,E., Grosveld,F. and Romeo,P.-H. (1989) Two tissue-specific factors bind the erythroid promoter of human porphobilinogen deaminase gene. Nucleic Acids Res., 17, 37–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima S. and Nagata,S. (1990) pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res., 18, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moi P., Chan,K., Asunis,I., Cao,A. and Kan,Y.W. (1994) Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the β-globin locus control region. Proc. Natl Acad. Sci. USA, 91, 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin J.D., Lin,Q., Duncan,S.A. and Olson,E.N. (1997) Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev., 11, 1061–1072. [DOI] [PubMed] [Google Scholar]
- Motohashi H., Igarashi,K., Onodera,K., Takahashi,S., Ohtani,H., Nakafuku,M., Nishizawa,M., Engel,J.D. and Yamamoto,M. (1996) Mesodermal- vs neuronal-specific expression of MafK is elicited by different promoters. Genes Cells, 1, 223–238. [DOI] [PubMed] [Google Scholar]
- Motohashi H., Ohta,J., Engel,J.D. and Yamamoto,M. (1998) A core region of the mafK gene IN promoter directs neurone-specific transcription in vivo.Genes Cells, 3, 671–684. [DOI] [PubMed] [Google Scholar]
- Nishimura S., Takahashi,S., Kuroha,T., Suwabe,N., Nagasawa,T., Trainor,C. and Yamamoto,M. (2000) A GATA box in the GATA-1 gene hematopoietic enhancer is a critical element in the network of GATA factors and sites that regulate this gene. Mol. Cell. Biol., 20, 713–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa M., Kataoka,K., Goto,N., Fujiwara,K.T. and Kawai,S. (1989) v-maf, a viral oncogene that encodes a leucine zipper motif. Proc. Natl Acad. Sci. USA, 86, 7711–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino H. and Yasuda,K. (1998) Induction of lens differentiation by activation of a bZIP transcription factor, L-Maf. Science, 280, 115–118. [DOI] [PubMed] [Google Scholar]
- Onodera K., Takahashi,S., Nishimura,S., Ohta,J., Motohashi,H., Yomogida,K., Hayashi,N., Engel,J.D. and Yamamoto,M. (1997) GATA-1 transcription is controlled by distinct regulatory mechanisms during primitive and definitive erythropoiesis. Proc. Natl Acad. Sci. USA, 94, 4487–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onodera K., Shavit,J.A., Motohashi,H., Katsuoka,F., Akasaka,J.E., Engel,J.D. and Yamamoto,M. (1999) Characterization of the murine mafF gene. J. Biol. Chem., 274, 21162–21169. [DOI] [PubMed] [Google Scholar]
- Onodera K., Shavit,J.A., Motohashi,H., Yamamoto,M. and Engel,J.D. (2000) Perinatal synthetic lethality and hematopoietic defects in compound mafG::mafK mutant mice. EMBO J., 19, 1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyake T., Itoh,K., Motohashi,H., Hayashi,N., Hoshino,H., Nishizawa,M., Yamamoto,M. and Igarashi,K. (1996) Bach proteins belong to a novel family of BTB-basic leucine zipper transcription factors that interact with MafK and regulate transcription through the NF-E2 site. Mol. Cell. Biol., 16, 6083–6095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitman M. and Felsenfeld,G. (1988) Mutational analysis of the chicken β-globin enhancer reveals two positive-acting domains. Proc. Natl Acad. Sci. USA, 85, 6267–6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Shavit J.A., Motohashi,H., Onodera,K., Akasaka,J., Yamamoto,M. and Engel,J.D. (1998) Impaired megakaryopoiesis and behavioral defects in mafG-null mutant mice. Genes Dev., 12, 2164–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwabe N., Takahashi,S., Nakano,T. and Yamamoto,M. (1998) GATA-1 regulates growth and differentiation of definitive erythroid lineage cells during in vitro ES cell differentiation. Blood, 92, 4108–4118. [PubMed] [Google Scholar]
- Swaroop A., Xu,J., Pawar,H., Jackson,A., Scolnick,C. and Agarwal,N. (1992) A conserved retina-specific gene encodes a basic motif/leucine zipper protein. Proc. Natl Acad. Sci. USA, 89, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S., Onodera,K., Motohashi,H., Suwabe,N., Hayashi,N., Yanai,N., Nabesima,Y. and Yamamoto,M. (1997) Arrest in primitive erythroid cell development caused by promoter-specific disruption of the GATA-1 gene. J. Biol. Chem., 272, 12611–12615. [DOI] [PubMed] [Google Scholar]
- Tsai F.-Y., Keller,G., Kuo,F.C., Weiss,M., Chen,J., Rosenblatt,M., Alt,F.W. and Orkin,S.H. (1994) An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature, 371, 221–226. [DOI] [PubMed] [Google Scholar]
- Yamamoto M., Ko,L.J., Leonard,M.W., Beug,H., Orkin,S.H. and Engel,J.D. (1990) Activity and tissue-specific expression of the transcription factor NF-E1 [GATA] multigene family. Genes Dev., 4, 1650–1662. [DOI] [PubMed] [Google Scholar]