

Abstract
Several γ-herpesviruses encode proteins related to the mammalian cyclins, regulatory subunits of cyclin-dependent kinases (cdks) essential for cell cycle progression. We report a 2.5 Å crystal structure of a full-length oncogenic viral cyclin from γ-herpesvirus 68 complexed with cdk2. The viral cyclin binds cdk2 with an orientation different from cyclin A and makes several novel interactions at the interface, yet it activates cdk2 by triggering conformational changes similar to cyclin A. Sequences within the viral cyclin N-terminus lock part of the cdk2 T-loop within the core of the complex. These sequences and others are conserved amongst the viral and cellular D-type cyclins, suggesting that this structure has wider implications for other cyclin–cdk complexes. The observed resistance of this viral cyclin–cdk complex to inhibition by the p27Kip cdk inhibitor is explained by sequence and conformational variation in the cyclin rendering the p27Kip-binding site on the cyclin subunit non-functional.
Keywords: cyclin/cyclin-dependent kinase/herpesvirus/p27Kip/structure
Introduction
Progression through the mammalian cell cycle is controlled by distinct cyclin-dependent protein kinases whose catalytic activities are regulated by a variety of mechanisms (Morgan, 1995; Pines, 1999). These serine/threonine kinases consist of two subunits: a cyclin protein and a cyclin-dependent kinase (cdk) (Morgan, 1997). Members of the cyclin family bind to and activate specific cdk isozymes and their relative abundance varies during the cell cycle, leading to the periodic activation and inactivation of different cdks (Morgan, 1997). Cdk phosphorylation and levels of cdk inhibitors (CKIs) also affect cdk catalytic activity (Pines, 1999). One well studied cell cycle transition in mammalian cells is the mitogen-dependent entry into S-phase (DNA synthesis), which requires the coordinated activation of cyclin D–cdk4, cyclin D–cdk6 and cyclin E–cdk2 complexes (Sherr, 1994, 1995). Deregulation of the cyclin D-dependent kinases and their downstream targets has frequently been associated with many forms of cancer, underlining the importance of this pathway (Palmero and Peters, 1996; Sherr, 1996). In addition to their catalytic role as protein kinases, specific cyclin–cdk complexes have also been proposed to have non-catalytic roles. The cyclin D–cdk4 and cyclin D–cdk6 holoenzymes phosphorylate the retinoblastoma protein pRb but also sequester the cdk inhibitors p21Cip/WAF and p27Kip, which otherwise block cyclin A–cdk2 and cyclin E–cdk2 kinase activity necessary for S-phase entry (reviewed in Sherr and Roberts, 1999).
Cyclin homologues have been identified within the genomes of several γ-herpesviridae associated with neoplastic disorders, these include herpesvirus saimiri (HSV) (Albrecht et al., 1992; Jung et al., 1994), Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV8) (Chang et al., 1996; Russo et al., 1996a) and murine γ-herpesvirus 68 (Virgin et al., 1997). Herein these viral cyclins will be referred to as V-, K- and M-cyclins, respectively. The viral cyclins are most closely related to the mammalian D-type cyclins, and K- and V-cyclin have been shown to form active complexes with cdk6 (Jung et al., 1994; Chang et al., 1996; Godden-Kent et al., 1997; Li et al., 1997). Cdk6 bound by K-cyclin exhibits potent activity towards an extended range of substrates including histone H1 and p27Kip (Godden-Kent et al., 1997; Ellis et al., 1999; Mann et al., 1999). In addition, the K-cyclin–cdk6 and V-cyclin–cdk6 complexes are resistant to the p16Inka and p21Cip/WAF/p27Kip CKIs and thereby overcome constraints to cell cycle progression mediated by their inhibitors (Swanton et al., 1997). The ability of these viral cyclins to deregulate the cell cycle may provide a cellular environment conducive to viral replication, although their precise role in oncogenicity is not clear (reviewed in Laman et al., 2000).
Structural analyses of a fragment of cyclin A bound to cdk2 have revealed the molecular basis for the partial and full catalytic activation of cdk2 (Jeffrey et al., 1995; Russo et al., 1996b). Cyclin A associates with its catalytic partner cdk2 through a conserved region of ∼150 amino acids known as the cyclin box (Brown et al., 1995; Jeffrey et al., 1995). In the presence of cyclin A, two regions of particular importance, the PSTAIRE helix and the T-loop bearing the activating phosphorylation site at Thr160, undergo a dramatic conformational change to influence ATP coordination and the proper formation of a substrate-binding cleft (Jeffrey et al., 1995; Russo et al., 1996b). Further studies on a ternary complex of a p27Kip peptide bound to cdk2 and a cyclin A fragment defined two independent p27Kip-binding sites, one within cyclin A and a second within the cdk2 ATP-binding pocket (Russo et al., 1996c). p27Kip directly inhibits cyclin A–cdk2 by mimicking the adenosine moiety of ATP and making similar contacts to cdk residues within the ATP-binding site (Russo et al., 1996c). The structures of two further cyclins, cyclin H and V-cyclin, have been determined in their unbound form, demonstrating that all three cyclins have similar three-dimensional folds despite their limited sequence similarity (Brown et al., 1995; Kim et al., 1996; Andersen et al., 1997; Schulze-Gahmen et al., 1999). As part of a study to understand the biochemical and structural properties of viral cyclins, we have determined the structure of the cyclin from γ-herpesvirus 68 bound to cdk2. We describe novel features of the viral cyclin–cdk2 interaction when compared with the structure of cyclin A–cdk2 and discuss the functional implications for cdk recognition and regulation.
Results
We have focused on the cyclin from γ-herpesvirus 68 as a model cyclin to understand the interaction between virus-derived cyclins and the mammalian cdk enzymes. We first determined which cdk subunit was bound by M-cyclin. As shown by co-immunoprecipitation experiments using FLAG-tagged M-cyclin, we demonstrated that M-cyclin binds to cdk2 in vivo rather than the canonical D-type cdk-binding partners cdk4 and cdk6 (Figure 1A). However, the closely related viral cyclin, K-cyclin, has been shown to associate with and activate cdk2, cdk4 and cdk6 (Figure 1A) (Godden-Kent et al., 1997; Li et al., 1997; Mann et al., 1999).
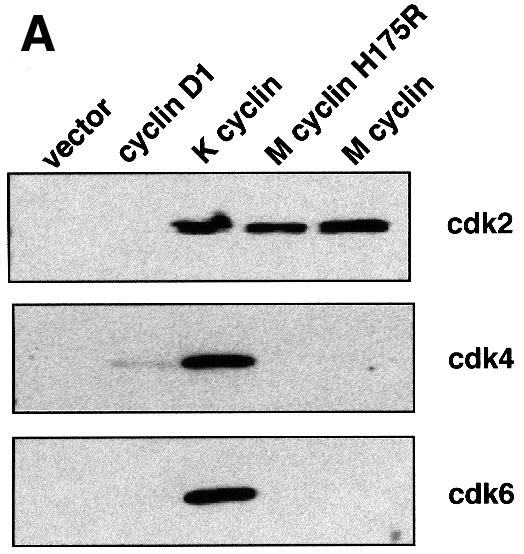

Fig. 1. (A) Wild-type and H175R M-cyclin associate with cdk2 but not with cdk4 or cdk6 in vivo, whilst K-cyclin associates with cdk2, cdk4 and cdk6. (B) The left panel shows wild-type and H175R M-cyclin complexes with cdk2 immunoprecipitated through the cyclin component and assayed against pRb. In vitro kinase assays were performed in the presence of increasing amounts of p27Kip and show the reduced sensitivity of wild-type and mutant M-cyclin towards this inhibitor compared with cyclin A. The lower panel shows a bar graph of the decrease in kinase activity due to increased inhibitor concentrations. 32P counts incorporated into pRb were quantitated on a phosphoimager and normalized to the amount of total kinase activity present when no inhibitor was used.
One of the striking features of V-cyclin–cdk6 and K-cyclin–cdk6 complexes is their ability to evade inhibition by p21Cip/WAF and p27Kip. This is due to the lack of stable interaction between these cyclin–cdk complexes and CKIs. We assessed the degree of resistance of M-cyclin–cdk2 complexes to inhibition by p27Kip. As shown in Figure 1B, M-cyclin–cdk2 was at least an order of magnitude less sensitive to inhibition by p27Kip than cyclin A–cdk2. We also characterized an H175 to R175 mutant of M-cyclin (denoted as H175R in Figure 1 and elsewhere in the text as M-cyclin) and showed that it has identical functional properties to wild-type M-cyclin (Figure 1B). Since this mutant had an improved solubility profile (G.L.Card, N.Jones and N.Q.McDonald, unpublished data), we concentrated our structural analysis on the H175R M-cyclin bound to cdk2.
M-cyclin–cdk2 complexes were prepared using an Escherichia coli host to produce M-cyclin and a recombinant baculovirus to produce cdk2 in Sf9 insect cells. The two purified proteins were mixed in solution and gel filtered prior to concentration and crystallization. In vitro kinase assays using histone H1 as substrate indicated that the M-cyclin–cdk2 complex was partially active in the absence of cdk2 phosphorylation at T160k2 (Thr160 of cdk2, the superscript letter identifies the protein to which the residue belongs, i.e. A for cyclin A, M for M-cyclin, p27 for p27Kip and k2 for cdk2) (data not shown). The same is also true for cyclin A–cdk2 (Russo et al., 1996b). We observed an ∼50-fold increase in kinase activity using T160k2-phosphorylated cdk2 and M-cyclin (data not shown). Crystallization screens identified conditions that gave monoclinic crystals belonging to space group P21 with cell dimensions 87.6 × 73.5 × 107.7 Å, β = 102.2° and two M-cyclin–cdk2 complexes per asymmetric unit. These crystals grew as thin plates with dimensions rarely greater than 5 µm and required intense synchrotron radiation to collect high resolution data from a single crystal. The M-cyclin–cdk2 structure was determined by molecular replacement and refined at 2.5 Å to an R-factor of 23.5% (Rfree of 28.5%) with excellent geometry (Table I). There were no appreciable differences between the structures of either complex, and the root-mean-square deviation (r.m.s.d.) was 0.719 Å for 529 Cα atoms. One of the two complexes (complex A) within the asymmetric unit had better defined electron density and correspondingly lower temperature factors, and only this complex is discussed. For complex A, residues 6–252 of M-cyclin are ordered, as are all of the residues in cdk2 with the exception of 10 and 11.
Table I. Data processing and refinement statistics.
| X-ray source | ESRF, ID13 Microfocus beamline |
| Space group | P21 |
| Unit cell | 87.6 × 73.5 × 107.7 Å, β = 102.2° |
| Resolution | 30–2.5 Å |
| Total observations | 140 317 |
| Unique reflections | 45 057 |
| Intensity I/σIa | 5.7 (1.6) |
| Completenessa | 97.1% (95.6%) |
| Rsyma | 10.0% (37.9%) |
| Rfree (%) | 28.5 |
| R-factor (%) | 23.5 |
| Non-hydrogen protein atoms | 8505 |
| Solvent molecules | 202 |
| Mean B-factor | |
| protein atoms (Å2) | 37.46 |
| solvent molecules (Å2) | 32.71 |
| R.m.s.d. from ideal stereochemistry | |
| bond lengths (Å) | 0.0088 |
| bond angles (°) | 1.41 |
Rsym is defined as Σ│I – <I>|/ΣI, where I is the observed intensity and <I> is the average intensity from multiple measurements. R-factor is defined as Σ││Fp – |Fc||/ΣFp, where |Fp| is the protein structure factor amplitude and |Fc| is the calculated structure factor amplitude.
aHighest resolution shell in parentheses.
Overall structure of the M-cyclin–cdk2 complex
The bi-lobal structure of cdk2 comprises an ATP-binding site and catalytic centre located between an N-terminal (N-lobe) and C-terminal lobe (C-lobe). M-cyclin binds cdk2 by interacting with hydrophobic sequences from the PSTAIRE region (residues 38–56) and strands β4/β5 of the N-lobe (Figure 2). It also makes important contacts with the T-loop in the C-lobe (Figure 2C). The cdk2-binding site on M-cyclin includes sequences within the cyclin box (residues 44–145) and the N-terminus (Figure 2). There are several unique features to the M-cyclin–cdk2 structure; M-cyclin binds cdk2 with a relative disposition different from that of cyclin A such that structurally equivalent residues make different contacts with cdk2 with few interactions identical to the cyclin A–cdk2 interface (Figure 2C; Table II). The flexibility of the cdk2 N-lobe allows for an optimized packing with M-cyclin and ensures that M-cyclin is still able to partially activate cdk2 by promoting the realignment of the PSTAIRE helix similarly to cyclin A.

Fig. 2. (A) Structure of the M-cyclin–cdk2 complex. Cdk2 is shown in blue except for the PSTAIRE region (residues 38–56) in red and the T-loop (residues 146–166) in yellow. M-cyclin is shown in gold. (B) An equivalent view following rotation by 90° about the horizontal axis. The T-loop forms two antiparallel β-strands, which are sandwiched between the PSTAIRE helix and the N-terminal region of M-cyclin. This figure and Figure 3A were prepared using PREPI (S.Islam and M.J.Sternberg, unpublished). (C) Highly schematic view of the two cyclin–cdk interfaces illustrating key structural elements and selected interface residues. The left hand panel is M-cyclin–cdk2, the right hand panel is cyclin A–cdk2. Colours for the indicated secondary structures follow (A) and Figure 3A.
Table II. Side chain hydrogen bonds at the cyclin–cdk2 interface.
| Cdk2 | M-cyclin | Cdk2 | Cyclin A |
|---|---|---|---|
| t39 | K289 | ||
| e40 | K288 | ||
| e42 | K104 | e42 | K266 |
| v44 | E295 | ||
| v44 | K104 | v44 | K266 |
| S46 | k266 | ||
| R50 | k104 | R50 | f267 |
| R50 | a107 | ||
| S53 | d145 | ||
| K56 | D305 | ||
| E57 | Y185 | ||
| H71 | D138 | H71 | H296 |
| R122 | d11 | R122 | a307 |
| R122 | Y155 | R122 | E269 |
| R150 | D11 | R150 | e269 |
| R150 | i270 | ||
| v156 | N173 | ||
| y180 | N173 | ||
| S181 D11 | |||
| N272 | e174 | ||
| S276 | D177 | ||
| a277 | Y178 | ||
| K278 | N16 | K278 | D181 |
| k278 | Y178 |
Hydrogen bonds were defined by CONTACT (CCP4, 1994). Side chain–side chain hydrogen bonds are indicated by upper case and side chain–main chain by lower case, with conserved bonds indicated in bold. A distance criterion of 3.2 Å was used. The major hydrophobic contacts are shown in Figure 4B and are described in the text.
Basis for the resistance of M-cyclin–cdk2 to the p27Kip cdk inhibitor
Structural studies with a p27Kip-derived peptide complexed to cyclin A–cdk2 have confirmed that two distinct binding sites for p27Kip exist within the cyclin A–cdk2 complex (Russo et al., 1996c). One is located in the cyclin subunit and a second in the ATP-binding cleft of the cdk subunit. Additional contacts are made with the cdk2 N-lobe as p27Kip bridges these two independent sites (Russo et al., 1996c). To establish why viral cyclins such as M- and K-cyclin form active complexes that are resistant to the cdk inhibitor p27Kip, we examined equivalent regions on cyclin A and M-cyclin that define the p27Kip-binding site. As we predicted previously, the acidic residues E220A and D283A, which form salt bridges with R30p27 and K25p27, are replaced by uncharged side chains in M-cyclin, suggesting that electrostatic repulsion prevents p27Kip interaction (Swanton et al., 1999). In addition, the presence of basic or polar residues S93M and K135M in place of L255A and L297A replaces two critical buried interface residues within the p27–cyclin A complex and would also be expected to disrupt the interaction between p27Kip and viral cyclins.
Unexpectedly, we found that two structural elements of M-cyclin also impede p27Kip interaction. The main chain position of the H4–H5 loop leads to a steric clash with the p27Kip peptide conformation observed bound to cyclin A (Figure 3C). Also, one of two helices found only in M-cyclin, helix HC (residues 247–250), lies close to the cyclin A-binding site for p27Kip (Figure 3C). This positions residue F248M within 2.1–2.2 Å of R30p27 and L32p27 side chains. It is also possible that the different disposition of the viral cyclin to cdk2 compared with cyclin A may further preclude interaction between p27Kip and viral cyclin by disrupting a continuous complementary surface bridging across from the cyclin to cdk2 (Russo et al., 1996c).

Fig. 3. (A) Relative position of M-cyclin (gold) and cyclin A (purple) following superposition through their respective cdk2 subunits. The helices of each cyclin are rendered as cylinders emphasizing the structural differences close to the M-cyclin N- and C-termini. These regions have important implications for interaction with the cdk2 T-loop and the basis for p27Kip resistance as described in the text. Helices of M-cyclin referred to in the text are labelled. (B) Sequence alignment of three viral cyclins (accession codes: mhu91858, cgh2_hsvsa and ksu79416) with the D-type cyclins (accession codes: hscycd1, hscycd2 and hscycd3) and cyclin A3 (accession code: hscyclina). M-cyclin, V-cyclin and cyclin A3 were aligned based on structural superposition and analysis using SAP (Taylor and Orengo, 1989). Helices in M-cyclin are indicated by boxes above the sequences and named following the convention used by Schulze-Gahmen et al. (1999). The blue outline box highlights the cyclin box. The helices unique to M-cyclin are shown as blue boxes. Every tenth residue in the M-cyclin is marked with a dot. Conserved residues amongst mammalian cyclins are highlighted in green, whereas those specific to the viral and D-type cyclins are shaded black in reverse contrast or grey for R106M (see text). M-cyclin (above sequences) and cyclin A (below sequences) residues contributing to the cdk interface are indicated by a number representing the amount of surface area of the side chain buried in the cyclin–cdk2 interface. 0 represents 1–10 Å2, up to 9, which indicates that >91 Å2 of the side chain is buried in the interface. (C) Stereo view of the p27Kip-binding site of cyclin A (purple) and the equivalent region of M-cyclin (gold) following superposition through their respective cdk2 subunits. p27Kip is shown in pink. Some residues are omitted from the figure for clarity.
Structural comparison of a viral and non-viral cyclin bound to cdk2
Superposition of the cdk2 subunit from the cyclin A–cdk2 structure onto M-cyclin–cdk2 indicates no gross changes in cdk2 conformation (excepting the T-loop) with an r.m.s.d. of 1.209 Å for 234 Cα atoms of cdk2 (from a total of 284). As mentioned previously, the superposition reveals that the M-cyclin is translated by ∼4 Å towards the cdk2 N-lobe, relative to cyclin A (Figure 3A). This brings the M-cyclin N-terminus into the cdk2 interface and ensures that sequences from M-cyclin make different contacts with cdk2 than equivalent residues in cyclin A (Figure 4B; Table II). The M-cyclin–cdk2 interface has a smaller total buried surface area than that found for cyclin A–cdk2 (2269 Å2 compared with 3002 Å2). M-cyclin contributes 1146 Å2 to the complex (cyclin A contributes 1679 Å2), of which just under a third is due to the N-terminus. This is smaller than cyclin A as fewer side chains from helix HN1 participate in the M-cyclin–cdk2 interface (Figure 3B).


Fig. 4. (A) Surface representation of M-cyclin (gold, left) and cyclin A (purple, right) bound to their respective cdk2 partners (cyan Cα worm). The cdk2 PSTAIRE region is shown in red and the T-loop in yellow for both complexes as in Figure 2. The green surface of M-cyclin and cyclin A indicates the hydrophobic patch, which packs against the PSTAIRE helix. The shift in position of M-cyclin relative to cyclin A can be seen as well as the different contacts to the PSTAIRE helix. (B) Close-up of the hydrophobic patch of M-cyclin–cdk2 and cyclin A–cdk2 revealing differences in their interface contacts. (C) Stereo view of a conserved cyclin–cdk2 interaction centred on the salt bridge between K104M and E133M (top view) equivalent to K266A and E295A (bottom view) as described in the text. Certain residues in this region are omitted for clarity.
The major hydrophobic contacts in the M-cyclin–cdk2 interface centre on the PSTAIRE helix and strands β4 and β5 of the cdk2 N-lobe (Figure 4A and B). A critical residue in this region is W142M, which packs against H71k2, V69k2 and L76k2 within a hairpin turn between strands β4 and β5 at the tip of the cdk2 N-lobe (Figure 4B). This region has much lower atomic temperature factors than the equivalent region of cyclin A-bound cdk2. The Cα atoms of the equivalent residues W142M and F304A are ∼3.5 Å apart but, in contrast to W142M, the side chain of F304A makes van der Waals contact with I52k2 of the PSTAIRE helix (Figure 4B). M-cyclin residues I101M, V105M, L137M and A144M define a shallow hydrophobic cavity, which packs against residue I49k2 of the PSTAIRE helix (Figure 4A and B). An equivalent cavity in cyclin A is located closer to the C-lobe of cdk2 (Figure 4A). Residues I49k2 and L54k2 adopt different rotamers to cyclin A-bound cdk2, and the PSTAIRE helix is tilted at the N-terminal end optimizing packing interactions with M-cyclin (Figure 4A). In the M-cyclin–cdk2 structure, I49k2 fills part of the space occupied by F304A whilst L54k2 tucks into the core of cdk2 towards the T-loop (Figure 4B). Also R50k2 makes two hydrogen bonds to main chain carbonyl groups of K104M and A107M, reminiscent of the hydrogen bonds it makes to cyclin A in phosphorylated cdk2 (Russo et al., 1996b).
Residues 6–16 of the M-cyclin tuck under F152k2 of the T-loop, tethering this region at the centre of the cyclin–cdk interface. Notably, D11M hydrogen-bonds with S181k2 and forms a salt bridge to R150k2 as discussed later. L14M occupies a small pocket below R122k2. These sequences form an important part of the cyclin–cdk interface and also contribute to a small hydrophobic core within M-cyclin, packing against several aromatic side chains from the cyclin box (F9M and L15M contact C154M, Y155M, H158M and Y167M).
Of the few contacts to cdk2 conserved between both cyclin A and M-cyclin, the majority cluster either side of the PSTAIRE helix. The salt bridge formed by K104M and E133M (equivalent to K266A and E295A) is of particular importance as it also makes three hydrogen bonds with cdk2, identical to cyclin A: one to the main chain carbonyl group of E42k2, and one to both the amide and carbonyl groups of V44k2 (Table II). This is surprising in view of the different position of the two cyclins, but can be explained by a compensating shift in residues 42–51 of cdk2, which include the N-terminal end of the PSTAIRE helix and its preceding loop (Figure 4C). The position of the PSTAIRE helix in the M-cyclin–cdk2 structure still permits E51k2 to salt-bridge with the functionally important K33k2 (2.7 Å Oε1 of E51A to Nζ K33A), analogous to the situation with cyclin A (Jeffrey et al., 1995). L130M and L137M (equivalent to L292A and L299A) flank either side of the K104M–E133M salt bridge, the latter being part of the cavity contacting the PSTAIRE helix described earlier. Close to the C-terminal end of the PSTAIRE helix, V143k2 packs against the aliphatic portion of K104M, as well as side chains from R26M and D145M, which form an ion pair and also make van der Waal contacts to F152k2. Also, D19M (equivalent to D181A) makes a favourable charge interaction with K278k2 but is too distant to form the salt bridge seen for cyclin A–cdk2. Four ordered water molecules are also found at the cyclin–cdk interface, of which the most significant solvent-mediated contact is that between the Oε1 atom of E133M and carbonyl oxygen of V44k2, identical to that seen in cyclin A (Oε1 atom of E295A) (Figure 4C).
T-loop conformation in the M-cyclin–cdk2 complex
The conformation of the T-loop of cdk2 is highly sensitive to phosphorylation of T160k2 by cdk-activating protein kinases and to the presence of a bound cyclin subunit (Jeffrey et al., 1995; Russo et al., 1996b). Both M-cyclin–cdk2 complexes within the asymmetric unit have an identical T-loop conformation and this differs appreciably from previously reported structures. T-loop residues 148–157 form two antiparallel β-strands as part of a three-stranded β-sheet directly below the PSTAIRE helix (Figures 2 and 5B). This β-sheet platform contributes several hydrophobic side chains, which pack against the PSTAIRE helix, notably A149k2, A151k2 and V156k2. In contrast, other sequences in this region, including R150k2, F152k2 and V154k2, are involved intimately in the M-cyclin–cdk2 interface (Figures 4B and 5B). In particular, R150k2 forms a salt bridge with D11M from the N-terminus of M-cyclin and adopts a quite different position to that found in cyclin A-activated cdk2 (Figure 5A and B). R150k2 is one of three arginine side chains that coordinate phospho-T160k2 in cyclin A-bound cdk2 (Figure 5B) (Russo et al., 1996b). The other two arginines in close proximity, R50k2 and R126k2, have a very similar conformation to that seen in a cyclin A–phosphorylated cdk2 complex (Russo et al., 1996b).

Fig. 5. (A) Stereo view of the electron density at the interface of the cdk2 T-loop and the N-terminus of M-cyclin in the last stage of refinement. The (2|Fo| – |Fc|) Fourier synthesis was calculated at 2.5 Å and the map contoured at 1.2σ. T-loop residues are labelled in yellow, and M-cyclin N-terminal residues in gold. (B) Close-up view of the T-loop of cdk2 (yellow) in complex with M-cyclin (top) or cyclin A (bottom). Coordinates of a substrate peptide are also shown derived from the cyclin A–phosphorylated cdk2 complex (Brown et al., 1999). Superposition of the substrate peptide onto M-cyclin–cdk2 shows a number of steric clashes in the region of the T-loop.
The conformation of T-loop residues 157–165, including T160k2, also deviates from that seen in previous structures. Comparison with a cyclin A–phosphorylated cdk2 bound to a substrate peptide indicates that the T-loop lies directly along the path of a substrate peptide with several steric clashes and may contribute to the partial activity exhibited by the M-cyclin–cdk2 complex. Similarly to the T-loop of cyclin A-bound unphosphorylated cdk2, V164k2 does not adopt an αL conformation critical for substrate binding (Brown et al., 1999).
M-cyclin–cdk2 as a model for other cyclin–cdk complexes
In order to establish whether the M-cyclin–cdk2 complex would provide a good model for other viral and D-type cyclin–cdk complexes, we first compared the structure of M-cyclin with those of V-cyclin and cyclin A (Brown et al., 1995; Jeffrey et al., 1995; Schulze-Gahmen et al., 1999). The r.m.s.d. is 1.44 Å for 164 Cα atoms between M-cyclin and cyclin A, and 1.79 Å for 173 Cα atoms between M-cyclin and V-cyclin. Superposition of the three cyclins reveals several structural elements unique to M-cyclin. These include helices HN and HN2, the H4–H5 loop and helix HC (Figure 3B). Helices HN and HC were not observed in the V-cyclin structure due either to flexibility or to the intermediate resolution of the structure (Schulze-Gahmen et al., 1999), whereas sequences in V-cyclin and cyclin A equivalent to helix HN2 adopt a loop conformation. The H4–H5 loop is more extended in M-cyclin than in other cyclins and has the sequence G121M, G122M and A123M, which overlaps the p27Kip-binding site on cyclin A, as described earlier.
The majority of sequences conserved between the M-cyclin and V-cyclin are located within their buried cores, and these proteins clearly demonstrate a closer structural relationship with one another than with cyclin A (Figure 3B). This is consistent with the sequence similarity of viral cyclins and cellular cyclins in the cyclin box (the second helical cyclin domain is far more divergent in sequence than the cyclin box and is therefore an unreliable guide). The cyclin box of M-cyclin shares 36% identity to K-cyclin and 30% to V-cyclin and cyclin D3, compared with 23% identity to cyclin A. Several residues on the surface of M-cyclin and V-cyclin are also strictly conserved in all viral cyclin and D-type cyclin sequences (Figure 3B). These include D11M, W142M, D151M and R106M (conserved as a basic side chain; R106M forms a salt bridge with D151M), all of which contribute to the M-cyclin–cdk2 interface. This ‘fingerprint’ is distinct from the nine residues that are conserved amongst the majority of mammalian cyclin sequences, six of which are buried within the cyclin hydrophobic core (R49M, W55M, L72M, D78M, Q92M and L140M) whereas the other three contribute to the cdk interface (K104M, P110M and E133M). We therefore propose that the presence of the four conserved surface residues in viral and D-type cyclins is consistent with these cyclins engaging their cdk partners in a manner similar to M-cyclin.
Discussion
To understand the basis of viral cyclin interaction with mammalian cdk enzymes, we have focused our biochemical and structural efforts on M-cyclin, the viral cyclin from murine γ-herpesvirus 68 (Virgin et al., 1997). Although the in vivo oncogenic effects of M-cyclin expression have been established in transgenic mice (van Dyk et al., 1999), its molecular properties remain uncharacterized. We first examined M-cyclin’s ability to associate with and activate different cdks and showed that its preferred cdk partner is cdk2, similar to cyclins A and E. M-cyclin differs from other viral cyclins in its restricted cdk preference. For example, K-cyclin is more promiscuous and can activate cdk2, cdk4 and cdk6 (Godden-Kent et al., 1997; Mann et al., 1999). We also demonstrated that M-cyclin is able to reduce the sensitivity of cdk2 to inhibition by p27Kip by at least 10-fold, analogous to the ability of K-cyclin to block inhibition of cdk6 by p27Kip (Swanton et al., 1997).
We then determined the crystal structure of a partially active M-cyclin–cdk2 complex at 2.5 Å resolution by molecular replacement. This structure provides the first crystallographic evidence that significant differences exist in the way two different cyclins can bind to and activate the same cdk subunit. This has important implications for other cyclin–cdk complexes. M-cyclin and cyclin A bind to cdk2 and activate it through repositioning the same cdk structural elements. Remarkably, however, the different relative disposition of cyclin and cdk in each complex ensures that very few interface contacts are conserved identically. The exception is the K104M/E133M sequence which contacts carbonyl and amide groups of E42k2 and V44k2, a ‘hot spot’ binding epitope at the cyclin–cdk interface. That such differences might exist was suggested from a model for V-cyclin–cdk6 derived from cyclin A–cdk2, which indicated several bad contacts close to the PSTAIRE helix and elsewhere (Schulze-Gahmen et al., 1999).
The different cyclin disposition in M-cyclin–cdk2 is indicative of the plasticity of the cyclin–cdk interface. Despite a translation of 4 Å relative to cyclin A, M-cyclin packs against the cdk2 N-lobe using primarily hydrophobic interactions close to W142M and the PSTAIRE helix. The conformational flexibility of cdk2 within the N-lobe allows for optimization of interactions with M-cyclin close to W142M by readjusting the position of the hairpin turn between strands β4 and β5. Although the PSTAIRE helix orientation differs slightly from that found in cyclin A–cdk2, it still suffices to preserve the interaction of E51k2 with K33k2 required for cdk2 catalytic activation. A further consequence of the position of M-cyclin bound to cdk2 is that almost half of the cdk2 T-loop is buried within the interface pincered between the N-terminus of M-cyclin and the PSTAIRE helix (Figure 2C). The presence of the N-terminus of M-cyclin within the interface with cdk2 was unexpected, but in preliminary experiments we have confirmed that deletion mutants of M-cyclin lacking these sequences are unable to bind or activate cdk2 in vitro (P.Knowles and N.Q.McDonald, unpublished). Equivalent N-terminal sequences were not observed in the V-cyclin structure, which was determined in the absence of a bound cdk subunit (Schulze-Gahmen et al., 1999). This suggests that binding to the cdk subunit increases the degree of order in these sequences.
One of the remarkable features of viral cyclins is their capacity to evade inhibition by CKIs, suggesting that their expression would result in unrestrained cdk kinase activity in an infected cell. Previous predictions anticipated that sequence differences near the p27Kip-binding site on viral cyclins would render it non-functional (Swanton et al., 1999). From our M-cyclin–cdk2 structure, a more complex molecular explanation emerges for the inability of p27Kip to inhibit active viral cyclin–cdk complexes. Rather than single or multiple sequence changes in the cyclin, it is a combination of sequence and conformational variation, particularly the main chain position of two structural elements (the H4–H5 loop and helix HC), that leads to a markedly reduced affinity for p27Kip. In order to bind M-cyclin–cdk2, p27Kip would have to overcome both unfavourable electrostatic and steric interactions. Several reports indicate that cyclin D–cdk6 complexes are able to engage p27Kip but are not inhibited by p27Kip in vivo (reviewed in Sherr and Roberts, 1999). Whether the different mode of cdk engagement we describe for M-cyclin is used by cyclin D and how this perturbs p27Kip–cdk6 interaction and cdk6 activity are important questions raised by our study.
The T-loop conformation observed in M-cyclin–cdk2 differs considerably from that seen in other cdk2 structures (Jeffrey et al., 1995; Russo et al., 1996b; Brown et al., 1999). For example, F152k2 of the T-loop is buried within the complex interface and the viral cyclin D11M chelates R150k2, tethering this portion of the T-loop. We anticipate that it is unlikely that residues 146–157 change conformation upon T160k2 phosphorylation. However, residues 158–166 could reorganize about phosphorylated T160k2 as in cyclin A-bound cdk2 such that V164k2 adopts an αL conformation to form the P+1 pocket specific for the proline residue in cdk target sites (Russo et al., 1996b; Brown et al., 1999). Since the viral cyclin–cdk complex is partially active, we infer that the C-terminal part of the T-loop must exhibit some flexibility in solution.
Viral cyclins are known to alter the substrate specificity of their cdk partners (Godden-Kent et al., 1997; Ellis et al., 1999; Mann et al., 1999) and are able to elicit a higher level of kinase activity than D-type cyclin–cdk6 complexes. Both of these properties may reflect the conformation of the phosphorylated T-loop bound to viral cyclin, and this will be clarified by structures of ternary complexes of substrate–viral cyclin–cdk. We cannot at present address whether the viral cyclin influences substrate selection directly through the close proximity of its N-terminus to the T-loop or indirectly by altering the T-loop conformation to promote interaction with appropriate substrates. We note that sequences within the D-type cyclin N-terminus have previously been implicated in recruitment of cdk substrates such as pRb via the LXCXE motif, which is absent from the viral cyclins (Kato et al., 1993).
Whether the M-cyclin–cdk2 complex reveals a novel virus-specific configuration or one that is more widely shared by other cyclins must await further viral cyclin–cdk crystal structures. Given the highly divergent nature of the cyclin sequences, we note that there would need to be considerable selective pressure to retain a particular residue because it plays either a key structural role or a functional role such as contributing to cdk binding. The conservation of sequences D11M, R106M, W142M and D151M in the viral and D-type cyclins argues that cyclins with these residues bind their cdk partners in a similar manner to M-cyclin. Indeed, models of V-cyclin–cdk6 complex derived from M-cyclinH175R–cdk2 appear to be consistent with this proposal (data not shown) but are unable to explain why M-cyclin does not bind cdk4 or cdk6. Further experiments guided by the structure described here are required in order to establish that M-cyclin provides an appropriate model for other cyclin–cdk complexes.
Materials and methods
Plasmids and antibodies
The cyclin A construct, a gift from Dr Jonathan Pines, has the human cyclin A gene with a c-myc tag at its N-terminus transcribed from a cytomegalovirus (CMV) promoter. The following plasmids have been described previously: pcDNA3-cyclin D1 and pcDNA3-K cyclin (Mann et al., 1999). The γHV-68 cyclin clone, a gift from Dr William Burns (Wisconsin Medical School), was PCR subcloned into pRSETA (Invitrogen) using oligonucleotides that eliminated the EcoRI site at nucleotide 13 while conserving the amino acid sequence. In a manner identical to the cyclins described above, γHV-68 cyclin was N-terminally tagged by FLAG epitopes introduced by ligating a double-stranded oligonucleotide into the unique NcoI site in the pRSETA construct. A PCR mutation H175 to R gave a mutant M-cyclin, which was characterized in parallel with the wild-type viral cyclin.
For expression in mammalian cells, the tagged cyclin was subcloned into pcDNA3 via BamHI–EcoRI (Invitrogen). These constructs were a generous gift from C.Swanton. The co-immunoprecipitation experiments used the following antibodies: anti-cdk2 SC-163; anti-cdk4 SC-260; anti-cdk6 SC-177 (Santa Cruz Biochemicals); anti-FLAG (Sigma, M2); and anti-cyclin A (rabbit polyclonal, a gift from Jonathan Pines).
Cell culture, lysis and immunoprecipitation
The culture and transfection of U2-OS cells have been described previously (Mann and Jones, 1996). Cells were split the day before being transiently transfected with 10 µg of mammalian expression constructs encoding cyclins. At 48 h post-transfection, cells were harvested by scraping cells into 50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 10% glycerol, 1 mM dithiothreitol (DTT), 0.1% Tween-20, 10 mM β-glycerophosphate, 0.1 mM phenylmethylsulfonyl fluoride (PMSF). Cell lysates were subject to immunoprecipitation for 2 h at 4°C with anti-FLAG antibodies conjugated to agarose (M2 Affimatrix; Kodak) or with anti-human cyclin A antibodies.
Immunoblotting and in vitro kinase assay
Immunoprecipitates from three parallel transient transfections were pooled and split into five aliquots. One aliquot was used for immunoblotting. Immunoprecipitates were washed extensively, separated by SDS–PAGE and transferred to Protran (Schleicher and Schuell). Membranes were incubated with antibodies against cdk subunits (anti-cdk2 SC-163, anti-cdk4 SC-260 and anti-cdk6 SC-177; Santa Cruz Biochemicals) and visualized by ECL (Amersham Pharmacia Biotech). The other four aliquots were used in kinase assays. Immunoprecipitates were resuspended in 30 µl of 50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM DTT, 2.5 mM EGTA, 10 mM β-glycerophosphate, 50 µM ATP, 10 µCi of [γ-32P]ATP (6000 Ci/mmol) and 5 µg of purified GST–Rb (C-terminus) as substrate with either 0, 0.1, 1 or 10 µg of purified p27. Reactions were incubated for 30 min at 30°C and then loaded on a 10% SDS–polyacrylamide gel. Gels were dried and analysed by autoradiography and quantitated by phosphoimaging.
Preparation of the M-cyclin–cdk2 complex
Human cdk2 was produced by a recombinant baculovirus (a gift from Professor D.Morgan, UCSF) used to infect Sf9 insect cells according to published protocols (Rosenblatt et al., 1993). M-cyclin was produced as a GST-tagged fusion protein using the vector pGEX-KG vector (Guan and Dixon, 1991) in a DH5α bacterial host. Each 1 l culture, containing 200 mg of ampicilin, was grown at 37°C until an OD600 nm of 0.7 was reached. The temperature was then lowered to 25°C until an OD600 nm of 1.3 was attained, whereupon it was lowered further to 18°C before overnight induction with 0.025 mM isopropyl-β-d-thiogalactopyranoside (IPTG). The cells were harvested by centrifugation at 5000 g for 15 min at 4°C, before being resuspended in pre-cooled lysis buffer: 20 mM Tris pH 7.9, 100 ml NaCl, 1 mM EDTA, 0.5% Tergitol NP-40, 10 mM DTT, 10 mM leupeptin, 1 mM pepstatin A, 10 mM benzamidine and 1 mM PMSF. Sonication on ice was followed by centrifugation at 25 000 g for 30 min at 4°C, before the supernatant was batch-purified on pre-equilibrated glutathione–Sepharose 4B resin (Pharmacia). Following three washes in lysis buffer and one in PBSA buffer (1.4 M NaCl, 30 mM KCl, 100 mM Na2HPO4, 20 mM NaH2PO4, 10 mM DTT), the resin was incubated overnight at 4°C with 6 U of thrombin (Boehringer Mannheim), leaving M-cyclin in the soluble fraction. The resin was removed using a PD10 column and any excess thrombin inactivated using inhibitors. The M-cyclin was then concentrated using an Amicon stirred cell, before being loaded onto a Superdex 75 FPLC column (Pharmacia), pre-equilibrated in running buffer: 50 mM Tris pH 7.9, 150 mM NaCl, 10 mM DTT. M-cyclin was isolated in an elution volume corresponding to a mol. wt of ∼29 000 Da, consistent with SDS–PAGE. The identity of M-cyclin was confirmed by mass spectroscopy fingerprinting following limited trypsin digest.
Crystallization of the M-cyclin–cdk2 complex
The purified human cdk2 and M-cyclin were mixed in an equimolar ratio before being concentrated in an Amicon stirred cell to 20 mg/ml. The complex was purified further on a Superdex 75 FPLC column (Pharmacia), pre-equilibrated in the above running buffer. The M-cyclin–cdk2 complex eluted at a volume corresponding to a mol. wt of ∼63 000 Da. Crystals of the complex grew by vapour diffusion or micro-batch as thin single plates by mixing equal volumes of the complex at 6 mg/ml with the crystallization buffer; 0.1 M HEPES pH 7.5, 12% propan-2-ol, 5% polyethylene glycol 4000 (PEG 4K) and 10 mM DTT. Crystals regularly took only 4 h to grow at room temperature to dimensions of ∼100 × 100 × 4 µm, with the largest reaching 400 × 500 × 4 µm after 1 day. Crystals were harvested immediately to prevent further nucleation. The crystals tended to grow as multi-clustered plates unsuitable for data collection. This was partially overcome by micro- or streak-seeding clear drops after 6 h.
Data collection and reduction
Owing to the thin nature of these crystals, a high intensity X-ray source was required. We collected a native data set on the micro-focus beam line ID13 at the European Synchrotron Research Facility (ESRF) at Grenoble, France. Data were recorded using a MAR Research CCD detector. Owing to the unique set-up of the station, the X-ray beam was collimated to 30 × 30 µm, allowing several images to be recorded from a single crystal, which was translated when the diffraction limit fell below 4 Å. The data were reduced using the programs DENZO and SCALEPACK (Otwinowski and Minor, 1997). The statistics for the data are shown in Table I. The presence of two M-cyclin–cdk2 complexes per asymmetric unit was established from a self-rotation function and native Patterson synthesis. We note that the M-cyclin–cdk2 complex associates as a non-crystallographic dimer, which is quite different from that observed in three independent crystal forms of cyclin A–cdk2 (Brown et al., 1999). The R175M mutation places the guanidino group between F179M and Y171M, thereby making favourable interactions with the π electron clouds from their respective ring systems. It is still unclear how this improves the solubility of recombinant M-cyclin.
Structural determination and refinement
Initial phases were obtained by molecular replacement after modification of the complex structure of cdk2–cyclin A, PDB ID code 1FIN. The search model for cdk2 was left unaltered, but the cyclin A model was trimmed back to a polyalanine probe model but included side chains from conserved residues identified from sequence alignment (15% of all side chains). The program AMoRE (CCP4, 1994) was used to search for the two molecules expected in the asymmetric unit, and two distinct solutions were obtained. The initial phases were improved further by applying 2-fold non-crystallographic symmetry (NCS) averaging using the programs MAMA and IMP (Jones et al., 1991), and by solvent flattening, histogram matching and phase extension with NCS averaging in the program DM (Cowtan, 1994). Model building was performed using the program O (Jones et al., 1991) and initial refinement carried out using X-PLOR incorporating Rfree validation of the progress of refinement (Brünger, 1996). After four cycles of positional refinement and one cycle of simulated annealing, refinement was continued using CNS (Brünger, 1998). The final R and Rfree values were 23.5 and 28.5%, respectively, with an r.m.s.d. of 0.0088 Å from ideal bond lengths and 1.41° for angles. Eighty-four per cent of residues were within the most favourable region of a Ramachandran plot.
Acknowledgments
Acknowledgements
We thank the staff at the ESRF ID13 microfocus beamline for assistance in data collection, in particular Anastassis Perrakis. We also thank Naomi Clout, Gordon Peters, Charlie Swanton and David Mann for helpful discussions throughout this project. G.L.C. is funded by an EU framework IV grant number BIO4-CT96-0285 to N.Q.M. Co-ordinates and structure factors have been deposited in the Protein Databank.
References
- Albrecht J.C., Nicholas,J., Cameron,K.R., Newman,C., Fleckenstein,B. and Honess,R.W. (1992) Herpesvirus saimiri has a gene specifying a homologue of the cellular membrane glycoprotein CD59. Virology, 190, 527–530. [DOI] [PubMed] [Google Scholar]
- Andersen G., Busso,D., Poterszman,A., Hwang,J.R., Wurtz,J.M., Ripp,R., Thierry,J.C., Egly,J.M. and Moras,D. (1997) The structure of cyclin H: common mode of kinase activation and specific features. EMBO J., 16, 958–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N.R., Noble,M.E., Endicott,J.A., Garman,E.F., Wakatsuki,S., Mitchell,E., Rasmussen,B., Hunt,T. and Johnson,L.N. (1995) The crystal structure of cyclin A. Structure, 3, 1235–1247. [DOI] [PubMed] [Google Scholar]
- Brown N.R., Noble,M.E., Endicott,J.A. and Johnson,L.N. (1999) The structural basis for specificity of substrate and recruitment peptides for cyclin-dependent kinases. Nature Cell Biol., 1, 438–443. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. (1996) X-PLOR Reference Manual 3.851. Yale University, New Haven, CT. [Google Scholar]
- Brünger A. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for computational crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Chang Y., Moore,P.S., Talbot,S.J., Boshoff,C.H., Zarkowska,T., Godden,K., Paterson,H., Weiss,R.A. and Mittnacht,S. (1996) Cyclin encoded by KS herpesvirus. Nature, 382, 410. [DOI] [PubMed] [Google Scholar]
- Cowtan K. (1994) DM: an automated procedure for phase improvement by density modification. Joint CCP4/ESF-EACBM newsletter. Protein Crystallogr., 31, 34–38. [Google Scholar]
- Ellis M., Chew,Y.P., Fallis,L., Freddersdorf,S., Boshoff,C., Weiss,R.A., Lu,X. and Mittnacht,S. (1999) Degradation of p27(Kip) cdk inhibitor triggered by Kaposi’s sarcoma virus cyclin–cdk6 complex. EMBO J., 18, 644–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godden-Kent D., Talbot,S.J., Boshoff,C., Chang,Y., Moore,P., Weiss,R.A. and Mittnacht,S. (1997) The cyclin encoded by Kaposi’s sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J. Virol., 71, 4193–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan K. and Dixon,J.E. (1991) Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal. Biochem., 192, 262–267. [DOI] [PubMed] [Google Scholar]
- Jeffrey P.D., Russo,A.A., Polyak,K., Gibbs,E., Hurwitz,J., Massague,J. and Pavletich,N.P. (1995) Mechanism of CDK activation revealed by the structure of a cyclinA–CDK2 complex. Nature, 376, 313–320. [DOI] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Jung J.U., Stager,M. and Desrosiers,R.C. (1994) Virus-encoded cyclin. Mol. Cell. Biol., 14, 7235–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato J., Matsushime,H., Hiebert,S.W., Ewen,M.E. and Sherr,C.J. (1993) Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev., 7, 331–342. [DOI] [PubMed] [Google Scholar]
- Kim K.K., Chamberlin,H.M., Morgan,D.O. and Kim,S.H. (1996) Three-dimensional structure of human cyclin H, a positive regulator of the CDK-activating kinase. Nature Struct. Biol., 3, 849–855. [DOI] [PubMed] [Google Scholar]
- Laman H., Mann,D.J. and Jones,N.C. (2000) Viral-encoded cyclins. Curr. Opin. Genet. Dev., 10, 70–74. [DOI] [PubMed] [Google Scholar]
- Li M., Lee,H., Yoon,D.W., Albrecht,J.C., Fleckenstein,B., Neipel,F. and Jung,J.U. (1997) Kaposi’s sarcoma-associated herpesvirus encodes a functional cyclin. J. Virol., 71, 1984–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann D.J. and Jones,N. (1996) E2f-1 but not E2f-4 can overcome p16-induced G1 cell-cycle arrest. Curr. Biol., 6, 474–483. [DOI] [PubMed] [Google Scholar]
- Mann D.J., Child,E.S., Swanton,C., Laman,H. and Jones,N. (1999) Modulation of p27(Kip1) levels by the cyclin encoded by Kaposi’s sarcoma-associated herpesvirus. EMBO J., 18, 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D.O. (1995) Principles of CDK regulation. Nature, 374, 131–134. [DOI] [PubMed] [Google Scholar]
- Morgan D.O. (1997) Cyclin-dependent kinases: engines, clocks and microprocessors. Annu. Rev. Cell Dev. Biol., 13, 261–291. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Palmero I. and Peters,G. (1996) Perturbation of cell cycle regulators in human cancer. Cancer Surv., 27, 351–367. [PubMed] [Google Scholar]
- Pines J. (1999) Four-dimensional control of the cell cycle. Nature Cell Biol., 1, E73–E79. [DOI] [PubMed] [Google Scholar]
- Rosenblatt J., DeBondt,H., Jancarik,J., Morgan,D.O. and Kim,S.H. (1993) Purification and crystallisation of human cyclin-dependent kinase 2. J. Mol. Biol., 230, 1317–1319. [DOI] [PubMed] [Google Scholar]
- Russo J.J. et al. (1996a) Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl Acad. Sci. USA, 93, 14862–14867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A.A., Jeffrey,P.D. and Pavletich,N.P. (1996b) Structural basis of cyclin-dependent kinase activation by phosphorylation. Nature Struct. Biol., 3, 696–700. [DOI] [PubMed] [Google Scholar]
- Russo A.A., Jeffrey,P.D., Patten,A.K., Massague,J. and Pavletich,N.P. (1996c) Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A–Cdk2 complex. Nature, 382, 325–331. [DOI] [PubMed] [Google Scholar]
- Schulze-Gahmen U., Jung,J.U. and Kim,S.H. (1999) Crystal structure of a viral cyclin, a positive regulator of cyclin-dependent kinase 6. Structure Fold Des., 7, 245–254. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. (1994) G1 phase progression: cycling on cue. Cell, 79, 551–555. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. (1995) D-type cyclins. Trends Biochem. Sci., 20, 187–190. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. (1996) Cancer cell cycles. Science, 274, 1672–1677. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and Roberts,J.M. (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev., 13, 1501–1512. [DOI] [PubMed] [Google Scholar]
- Swanton C., Mann,D.J., Fleckenstein,B., Neipel,F., Peters,G. and Jones,N. (1997) Herpes viral cyclin–Cdk6 complexes evade inhibition by CDK inhibitor proteins. Nature, 390, 184–187. [DOI] [PubMed] [Google Scholar]
- Swanton C., Card,G.L., Mann,D., McDonald,N.Q. and Jones,N. (1999) Overcoming inhibitions: viral cyclin subversion of CKI function. Trends Biochem. Sci., 24, 116–120. [DOI] [PubMed] [Google Scholar]
- Taylor W.R. and Orengo,C.A. (1989) Protein structure alignment. J. Mol. Biol., 208, 1–22. [DOI] [PubMed] [Google Scholar]
- van Dyk L.F., Hess,J.L., Katz,J.D., Jacoby,M., Speck,S.H. and Virgin,H.I. (1999) The murine γ-herpesvirus 68 v-cyclin gene is an oncogene that promotes cell cycle progression in primary lymphocytes. J. Virol., 73, 5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin H.W. IV, Latreille,P., Wamsley,P., Hallsworth,K., Weck,K.E., Dal Canto,A.J. and Speck,S.H. (1997) Complete sequence and genomic analysis of murine γ-herpesvirus 68. J. Virol., 71, 5894–5904. [DOI] [PMC free article] [PubMed] [Google Scholar]