

Abstract
In melanocytes and melanoma cells, cAMP activates extracellular signal-regulated kinases (ERKs) and MEK-1 by an unknown mechanism. We demonstrate that B-Raf is activated by cAMP in melanocytes. A dominant-negative mutant of B-Raf, but not of Raf-1, blocked the cAMP-induced activation of ERK, indicating that B-Raf is the MEK-1 upstream regulator mediating this cAMP effect. Studies using Clostridium sordelii lethal toxin and Clostridium difficile toxin B have suggested that Rap-1 or Ras might transduce cAMP action. We show that Ras, but not Rap-1, is activated cell-specifically and mediates the cAMP-dependent activation of ERKs, while Rap-1 is not involved in this process in melanocytes. Our results suggest a novel, cell-specific mechanism involving Ras small GTPase and B-Raf kinase as mediators of ERK activation by cAMP. Also, in melanocytes, Ras or ERK activation by cAMP is not mediated through protein kinase A activation. Neither the Ras exchange factor, Son of sevenless (SOS), nor the cAMP-responsive Rap-1 exchange factor, Epac, participate in the cAMP-dependent activation of Ras. These findings suggest the existence of a melanocyte-specific Ras exchange factor directly regulated by cAMP.
Keywords: B-Raf/cAMP/ERKs/melanocytes/Rap-1
Introduction
Melanocytes are neural crest-derived skin cells specialized in the synthesis of melanin pigments. This process is regulated by a physical stimulus such as ultraviolet (UV) A/B radiation or hormonal stimuli evoked by pro-opiomelanocortin hormones like α-melanocyte stimulating hormone (α-MSH). α-MSH, by binding to the melanocortin 1 receptor (MC1-R) on melanocytes and up-regulating the cAMP pathway, induces melanocyte and melanoma cell differentiation (Levine et al., 1991). This process is characterized by increased melanin synthesis (Hearing and Jimenez, 1989; Hearing and Tsukamoto, 1991) and dendrite outgrowth crucial for pigment delivery within the epidermis (Buscà et al., 1996, 1998). In previous studies undertaken to characterize the molecular events triggered by cAMP during melanocyte differentiation, we have shown that the elevation of intracellular cAMP provokes the activation of the mitogen-activated protein kinase (MAP kinase, or MAPK) ERK1 in B16 mouse melanoma cells (Englaro et al., 1995).
ERK1 and ERK2 are serine/threonine protein kinases activated upon dual phosphorylation by the MAPK kinase (MAPKK) MEK (reviewed in Robinson and Cobb, 1997), which in its turn is phosphorylated and therefore activated by the MAPKK kinases of the Raf family (Moodie et al., 1993). Raf kinases are activated after their interaction with the GTP-bound form of p21Ras (reviewed in Morrison and Cutler, 1997), and the levels of Ras–GTP are mainly controlled by Ras-specific guanine nucleotide exchange factors such as Son of sevenless (SOS) or R-Ras–GRF. When activated, ERKs translocate to the nucleus where they regulate gene expression, leading to cell proliferation or differentiation depending on the cell type. Interestingly, the activation of ERKs by cAMP has been reported in a limited number of cell systems, including B16 melanoma cells and PC12 phaeochromocytoma cells (Frodin et al., 1994; Young et al., 1994; Englaro et al., 1995) which are also derived from the neural crest. Several other studies have tried to determine the mechanism of ERK activation by cAMP in PC12 cells. Vossler et al. (1997) have shown that agents that increase the intracellular cAMP content, through protein kinase A (PKA) and the sequential activation of Rap-1 small GTPase and B-Raf kinase, activate ERK in a Ras-independent manner.
The Raf family of kinases is composed of the ubiquitously expressed Raf-1 and by A-Raf and B-Raf. B-Raf, which is highly present in neural cells and tissues (Marx et al., 1988; Eychene et al., 1992; Barnier et al., 1995), lies downstream of Ras and upstream of MEK during nerve growth factor (NGF)-induced signal transduction in PC12 cells. B-Raf appears to be the major MAPKK activated in these cells after stimulation by NGF (Troppmair et al., 1992; Jaiswal et al., 1994; Lange-Carter and Johnson, 1994; Traverse and Cohen, 1994). It is also the major MEK activator in bovine brain (Catling et al., 1994).
Rap-1 is a member of the Ras superfamily of small GTP-binding proteins. It has an effector domain almost identical to that of Ras, indicating that both small GTPases might share common downstream effectors. Rap-1 can be activated by several second messengers like calcium and diacylglycerol (McLeod et al., 1998; Zwartkruis et al., 1998), and by cAMP, since Rap-1 is phosphorylated by PKA (Lerosey et al., 1991) and this seems to participate to the Rap-1 GTP loading (Altschuler et al., 1995). The physiological role of Rap-1 remains to be elucidated. It has been suggested that Rap-1 could antagonize Ras-dependent signalling (Cook et al., 1993; Hu et al., 1997, 1999), but the recent work of Zwartkruis et al. (1998) does not support this notion. Recent studies have reported the cloning of different nucleotide exchange factors for Rap-1 [Epac (also named cAMP-GEFI) and cAMP-GEFII) directly activated by cAMP (De Rooij et al., 1998; Kawasaki et al., 1998). This finding has revealed that, in addition to a PKA-dependent regulation of Rap-1, the activity of the small GTPase can be controlled by cAMP in a PKA-independent manner.
In B16 melanoma cells, we have previously shown that cAMP activates MEK but fails to activate Raf-1. In an attempt to identify the molecular events connecting cAMP to ERK activation in melanocytes, we examined the involvement of Rap-1 and B-Raf in the cAMP-induced activation of ERKs in human melanocytes and in B16 mouse melanoma cells. Our results demonstrate that cAMP can activate Rap-1 and B-Raf kinase, but only B-Raf mediates the cAMP effects on ERK activation. cAMP activated the Ras small GTP-binding protein by a mechanism that involves neither PKA nor the exchange factors SOS or Epac. Finally, the activation of ERK by cAMP was blocked by a dominant-negative mutant of Ras, demonstrating that Ras mediates the action of cAMP. Our results reveal novel mechanism of MAPK pathway activation by cAMP involving Ras and B-Raf as signalling molecules.
Results
cAMP activates MAPKs and B-Raf in melanocytes
We have previously shown that, in B16 mouse melanoma cells, agents that increase cAMP concentrations activate the ERK1 MAPK (Englaro et al., 1995). Using specific antibodies against the phosphorylated, and thus activated, forms of ERKs or MEK-1, we show here that cAMP-elevating agents, isobutylmethylxanthine (IBMX) plus forskolin or α-MSH, rapidly (within 5–10 min) activate ERK1, ERK2 and MEK (Figure 1A and B). ERK activation by cAMP also occurred in normal human melanocytes, which constitute a more physiological system (Figure 1A). In contrast, cAMP-elevating agents did not activate ERKs in NIH 3T3 mouse fibroblasts (Figure 1A). 12-O-tetradecanoyl phorbol-13-acetate (TPA) was used in all cases as a positive control, since it strongly activates ERKs. Thus, MAPK activation by cAMP seems to be a tissue-specific process, which takes place in melanocyte cells.

Fig. 1. cAMP activates ERKs, MEK and B-Raf in melanocyte. Lysates from non-stimulated cells (NS), cells stimulated for 10 min with the cAMP-elevating agents, IBMX plus forskolin (I+F) or α-MSH (α-M) or with the protein kinase C activator, TPA (T) were subjected to western blot analysis with an anti-phospho-ERK antibody (A) or with an anti-phospho-MEK1 antibody (B). A polyclonal antibody to ERK2 (1/2000) (A) or MEK (1/1000) (B) was used as a control of protein loading. (C) B16 cells were treated as above or pre-incubated for 20 min with cAMP-elevating agents and then stimulated for 10 min with TPA (I+F+T). B-Raf or Raf-1 was then immunoprecipitated and their kinase activities were assayed using the His–MEK fusion protein as substrate. Samples were resolved by 10% SDS–PAGE and analysed by autoradiography.
In an attempt to discover the precise mechanisms leading to MEK and ERK activation by cAMP in melanocyte cells, we measured Raf-1 and B-Raf activities upon cAMP treatment of B16 melanoma cells. Raf-1 was not activated by cAMP in B16 cells, as has been described previously (Englaro et al., 1995). TPA strongly activated Raf-1, but the pretreatment of cells with cAMP-elevating agents markedly impaired the TPA-induced Raf-1 activity (Figure 1C). In contrast, B-Raf immunokinase assays showed that the cAMP-elevating agents (IBMX plus forskolin, or α-MSH), as well as TPA, used as a positive control, significantly activated the kinase (Figure 1C). These results suggest that B-Raf, but not Raf-1, mediates the cAMP-stimulation of MEK and ERKs in B16 cells.
B-Raf is the MAPKKK that transduces the cAMP activation of ERKs in B16 melanoma cells
To verify this hypothesis, haemagglutinin-tagged ERK1 (HA–ERK1) was cotransfected with epitope-tagged dominant-negative mutants of B-Raf or Raf-1 or with constitutive active mutants of the kinases. Then, after treatment with cAMP-elevating agents and immunoprecipitation of the tagged ERK1, we evaluated ERK activation by western blot experiments using an antibody specific to the phosphorylated (active) ERKs. ERK1 phosphorylation was greatly increased by the overexpression of constitutive active mutants of both B-Raf and Raf-1 (Figure 2A). As expected, cAMP-elevating agents activated HA–ERK1 in B16 cells. This activation was blocked by the dominant-negative mutant of B-Raf but not by the dominant-negative mutant of Raf-1, indicating that the cAMP activation of ERKs follows the B-Raf ‘sub-pathway’.

Fig. 2. In melanocytes, B-Raf mediates the cAMP-induced activation of ERK. (A) B16 cells were co-transfected with HA-tagged ERK1 together with an empty vector (vector), with dominant-negative mutants of B-Raf (N-B-Raf) or Raf-1 (N-Raf-1), or with constitutively active forms of B-Raf (B-Raf-CAAX) or Raf-1 (Raf-1-CAAX) tagged with the HA epitope. Cells were then treated or not (NS) for 10 min with the cAMP-elevating agents, IBMX plus forskolin (I+F). Lysates were then immunoprecipitated with an anti-HA antibody and subjected to western blot analysis using an anti-phopho ERK antibody (upper panel). A western blot of the same lysates was carried out using an anti-tag antibody (1/1000 dilution) as a control of the transfected protein expression (lower panel). (B) CHO cells co-transfected with HA–ERK1 and N-Raf-1 or an empty vector (vector), were left untreated (NS) or treated with TPA (T) for 10 min. HA–ERK activation was monitored as above (upper panel). A western blot of the cell lysates was performed using an anti-tag antibody as a control for transfected protein expression (lower panel).
The Raf-1 dominant-negative function was verified in Chinese hamster ovary (CHO) cells, in which Raf-1 is the predominantly expressed MAPKKK (not shown). As expected, N-Raf-1 blocked the TPA-activated ERK activity (Figure 2B). The level of the transfected protein expression was verified by western blot analysis using the anti-tag antibody (Figure 2A and B, lower panels).
Rap-1 and Ras are putative upstream B-Raf activators mediating the cAMP stimulation of ERKs
As an approach to identifying the upstream activators of B-Raf likely to mediate cAMP stimulation of ERKs, we investigated the role of small GTP-binding proteins of the Ras family by using Clostridium sordelii lethal toxin (LT) or Clostridium difficile toxin B. LT acts specifically by inhibiting the small GTPases Ras, Rap-1 and Rac (Just et al., 1996; Popoff et al., 1996) and toxin B specifically inhibits the Rho family of small GTPases Rho, Rac and CDC42 (Just et al., 1995; Chaves-Olarte et al., 1996). In our hands, low concentrations of LT (70 ng/ml) completely inhibited the cAMP-induced activation of MAPK (Figure 3), suggesting that Ras, Rap-1 or Rac mediated the action of cAMP. On the other hand, toxin B did not affect the cAMP-induced ERK activation (Figure 3), indicating that the small GTPases of the Rho family, including Rac, do not play a significant role in this process in melanocytes.
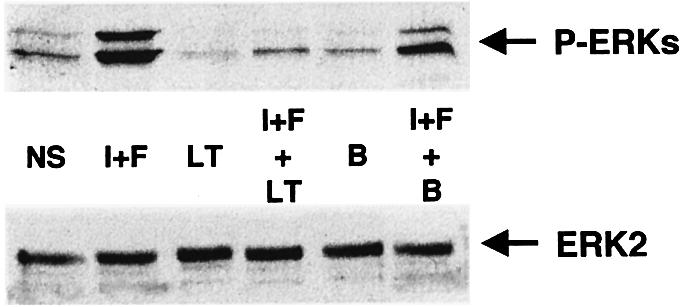
Fig. 3. Rap-1 and Ras as putative mediators of cAMP signalling. B16 cells treated or not for 4 h with C.sordelii lethal toxin (LT) or C.difficile toxin B (B) were exposed or not (NS) to cAMP elevating agents IBMX plus forskolin (I+F). Cells lysates were then subjected to western blot analysis using an antibody to activated ERKs (1/5000) (upper panel). The nitrocellulose membrane was stripped and probed with an anti-ERK2 antibody as a control for protein loading (lower panel).
We have previously reported that the inhibition of Rho-family proteins by toxin B induced disruption of stress fibres, leading to characteristic morphological alterations in B16 cells (Buscà et al., 1998). Toxin B action was therefore verified by examining cells by phase-contrast microscopy (not shown). Taken together, these results suggest that Ras and Rap-1 may mediate the activation of MAPK by cAMP in B16 cells.
cAMP activates Ras and Rap-1
Next, pull-down experiments were carried out to measure the effect of cAMP on Ras and Rap-1 activities. To detect activated Ras, we used a glutathione S-transferase (GST) fusion protein containing the B-Raf Ras-binding domain (B-Raf RBD); to detect activated Rap-1, we used a Ral–GDS RBD (RGF–RBD) fusion protein (Herrmann et al., 1996).
Cells were treated with IBMX and forskolin or with α-MSH to increase cellular cAMP levels, and cell lysates were incubated with each GST fusion protein coupled to glutathione–Sepharose beads. The bound proteins were analysed by western blotting to detect Ras or Rap-1. Rap-1 binding to the RGF–RBD was increased upon treatment with cAMP-elevating agents in both B16 melanoma cells and in NIH 3T3 cells, indicating that Rap-1 is activated by cAMP in these cell lines (Figure 4A). In B16 cells exposed to cAMP-elevating agents, Ras binding to the B-Raf RBD was strongly stimulated, so cAMP can also activate Ras (Figure 4B). We also detected significant Ras activation by cAMP in normal human melanocytes, thus giving a more physiological relevance to our observation (Figure 4B). Conversely, in NIH 3T3 cells, cAMP did not activate Ras while platelet-derived growth factor (PDGF), used as a positive control, clearly increased the binding of Ras to B-Raf RBD (Figure 4B).

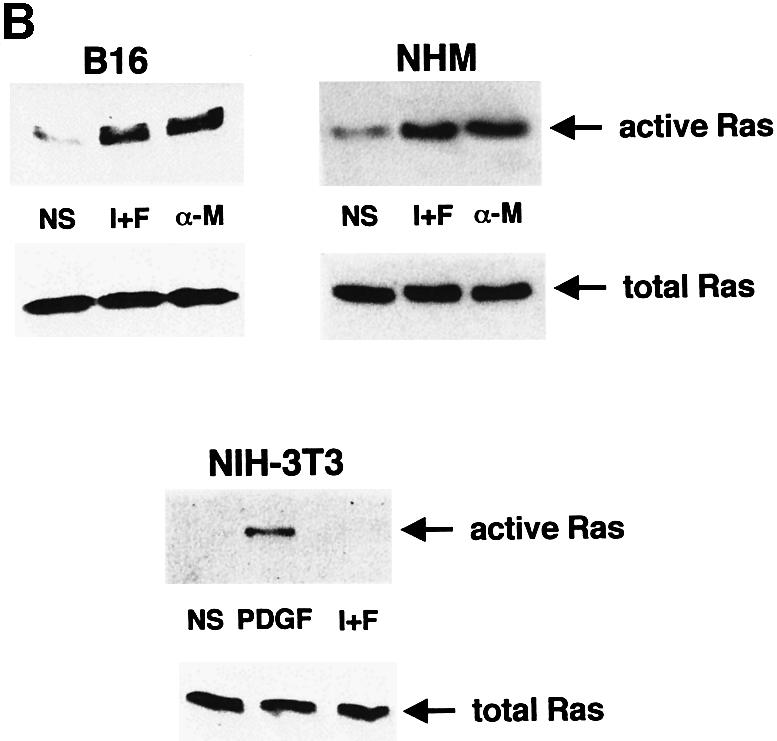
Fig. 4. cAMP activates Ras and Rap-1 in melanocytes. (A) B16 melanoma cells and NIH 3T3 fibroblasts were treated with IBMX plus forskolin (I+F) or with α-MSH (α-M) for the indicated times. Cells were lysed and subjected to pull-down experiments using the Ral–GDS RBD GST fusion to detect activated Rap-1. Precipitated proteins were subjected to western blotting using a polyclonal antibody to Rap-1 (1/500 dilution) (upper panels). Thirty micrograms of each protein lysate were subjected to direct western blotting using the anti-Rap-1 antibody to detect the total Rap-1 content in the cell (lower panels). (B) B16 cells, normal human melanocytes or NIH 3T3 fibroblasts were treated as described in (A). NIH 3T3 fibroblasts were also stimulated for 5 min with PDGF as a positive control. Pull-down experiments were performed using a B-Raf RBD GST fusion protein to detect activated Ras. Precipitated proteins were subjected to western blotting using an anti-Ras monoclonal antibody at a 1/100 dilution (upper panels). Extracts of each condition were also analysed for total Ras expression (lower panels).
Taken together, these results show that Rap-1 is activated by cAMP in both B16 melanoma cells and NIH 3T3 cells. However, Ras, which is specifically activated by cAMP in melanocytes but not in NIH 3T3 fibroblasts, fulfils the conditions required to transduce the cAMP-induced stimulation of ERK in a cell-specific manner.
Ras but not Rap-1 mediates the cAMP-activation of ERKs in B16 melanoma cells
To analyse directly whether Rap-1 or Ras mediated the cAMP-induced stimulation of MAPK in B16 melanoma cells, we investigated ERK activation by cAMP in cells expressing dominant-negative (HA–Rap-1-N17 or HA–Ras-N17) or constitutive active mutants (HA–Rap-1-V12 or HA–Rap-1-E63, or HA–Ras-V12) of both small GTPases (Figure 5). Cells cotransfected with these constructs and the HA-tagged ERK1 were treated or not with cAMP-elevating agents and the HA–ERK1 was immunoprecipitated with an anti-HA antibody. The activated ERK was then detected by western blotting using an antibody against the phosphorylated forms of ERK. As shown in Figure 5, overexpression of the constitutively active Ras greatly increased ERK activation whereas overexpression of two constitutively active forms of Rap-1 (Rap-1-V12 and Rap-1-E63) did not affect the activation state of the kinase. Only the dominant-negative mutant of Ras, and not that of Rap-1, completely blocked the cAMP-induced ERK activation. The level of expression of the transfected protein expression was verified by western blotting with an anti-HA antibody (Figure 5A, lower panels). Pull-down experiments after overexpression of Rap-1-V12 and Rap-1-E63 confirmed that these mutants were constitutively active (Figure 5B).

Fig. 5. Ras but not Rap-1 mediates the cAMP activation of MAPK in B16 cells. (A) An empty vector (vector), the dominant-negative forms of Rap-1 or Ras (Rap-1-N17, Ras-N17) and the constitutively active versions of Rap-1 or Ras (Rap-1-V12, Rap-1-E-63 or Ras-V12), all tagged with the HA epitope, were co-expressed in B16 cells with HA-tagged ERK1. Forty-eight hours after transfection, cells were treated or not (NS) with cAMP-elevating agents IBMX plus forskolin (I+F). ERK1 activation was detected after immunoprecipitation of HA–ERK1 and western blotting using an antibody against activated ERKs (upper panel). A western blot of the same lysates was carried out using an anti-tag antibody as a control for transfected protein expression (lower panel). (B) B16 melanoma cells were transfected with an empty vector (vector) or with the constitutively active mutants of Rap-1 (Rap-1-V12 or Rap-1-E63). Cells were then lysed and subjected to pull-down experiments using the Ral–GDS RBD GST fusion to detect activated Rap-1. Precipitated proteins were subjected to western blotting using a polyclonal antibody against Rap-1 (1/500 dilution).
Taken together, these results demonstrate that Ras mediates the activation of ERK by cAMP in B16 melanoma cells, whereas Rap-1 does not seem to play a significant role in the activation of the MAPK cascade in this cell system.
cAMP activation of Ras/ERK is PKA-independent
Most of the cellular effects of cAMP have been attributed to the activation of the cAMP-dependent protein kinase (PKA). To determine whether the activation of Ras by cAMP in melanocyte cells was PKA-dependent, we incubated B16 cells with H89, a potent and selective inhibitor of PKA. Figure 6 shows that H89, which inhibits PKA activity in B16 cells at a concentration of 3 µM (Figure 6C) did not inhibit the cAMP activation of Ras (Figure 6A) or the cAMP-dependent phosphorylation of ERK (Figure 6B). Overexpression of the catalytic subunit of PKA did not affect ERK activation either (Figure 6D). Control experiments to investigate the function of the transfected PKA catalytic domain were performed by cotransfecting this vector with a plasmid encoding the PKA-responsive cAMP-responsive element (CRE) sequence cloned upstream of the luciferase reporter gene. The PKA catalytic subunit strongly stimulated luciferase activity (not shown). These findings demonstrate that cAMP activates the MAPK pathway in a PKA-independent way.
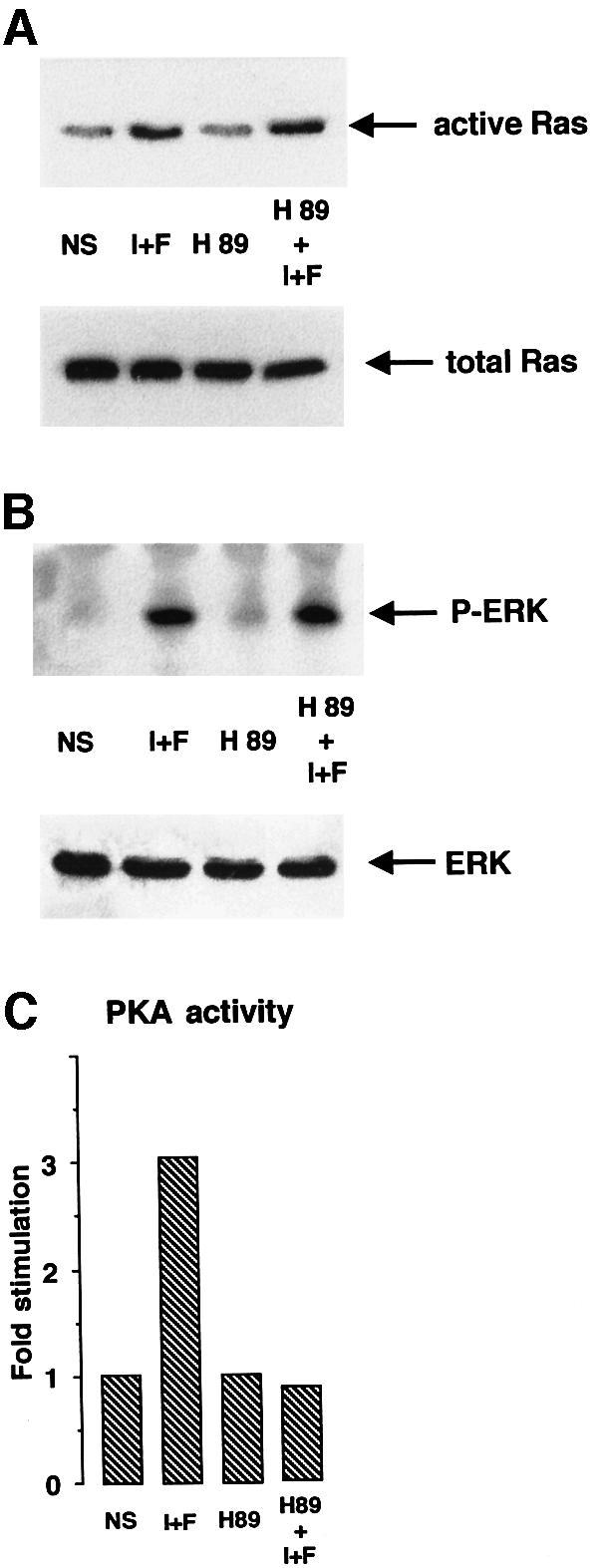
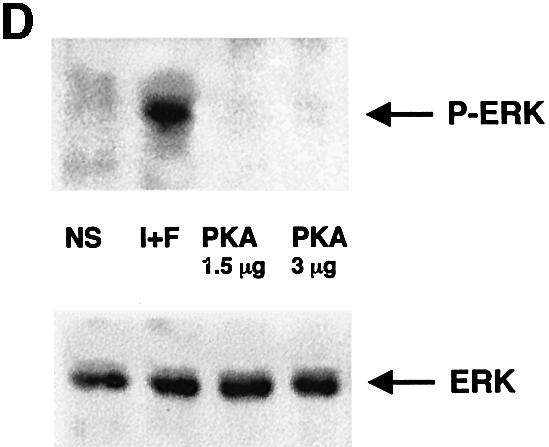
Fig. 6. Activation of the Ras-MAPK cascade by cAMP is PKA independent. (A) B16 cells stimulated or not (NS) with IBMX plus forskolin (I+F) were pretreated for 20 min with the PKA inhibitor H89 (3 µM) (H89). Cell lysates were subjected to pull-down experiments using the B-Raf RBD fusion protein, followed by a western blot analysis to detect activated Ras (upper panel). A control for Ras expression in each condition is shown (lower panel). (B) After transfection with HA–ERK1, B16 cells were pre-incubated with the PKA inhibitor (H89) and then stimulated or not (NS) with IBMX plus forskolin (I+F). Next, cell lysates were immunoprecipitated with an anti-HA antibody and subjected to western blotting using an antibody against phospho-ERKs (upper panel). A western blot of the same lysates was carried out using an anti-HA antibody as a control for HA–ERK expression in each condition (lower panel). (C) B16 cells stimulated or not (NS) with cAMP elevating agents (I+F) were pretreated for 20 min with 3 µM H89 (H89), then PKA activity was measured as described in Materials and methods. (D) B16 cells were cotransfected with HA–ERK1 and two different amounts of a plasmid encoding the catalytic subunit of PKA. Next, cell lysates were immunoprecipitated with an anti-HA antibody and subjected to western blotting using an antibody against the phosphorylated forms of ERKs (upper panel). A western blot of the same lysates was carried out using an anti-HA antibody as a control for HA–ERK1 expression in each condition (lower panel).
SOS and Epac do not participate in the cAMP-dependent activation of the MAPK cascade in melanocytes
Next, we investigated the involvement of SOS, the classical guanine nucleotide exchange factor for Ras, in the cAMP-activation of Ras. We blocked the SOS-dependent regulation of Ras by using a dominant-negative mutant of SOS (ΔSOS) which lacks amino acids 618–1036 corresponding to the guanine nucleotide exchange domain of the molecule. B16 melanoma cells were cotransfected with the HA-tagged ERK1 plasmid and with an empty vector or the dominant-negative version of the factor (ΔSOS). Next, cells were treated or not with cAMP-elevating agents and ERK phosphorylation was examined. cAMP-elevating agents activated ERK in cells transfected with an empty vector. The overexpression of ΔSOS did not impair this activation, so SOS does not constitute the Ras upstream molecule regulated by cAMP (Figure 7A). The levels of expression of the transfected protein were verified by western blotting with anti-HA and anti-SOS antibodies (Figure 7A, lower panels). We confirmed the dominant-negative function of ΔSOS as this construct inhibited the insulin activation of ERK in CHO cells stably expressing the insulin receptor (Figure 7B).

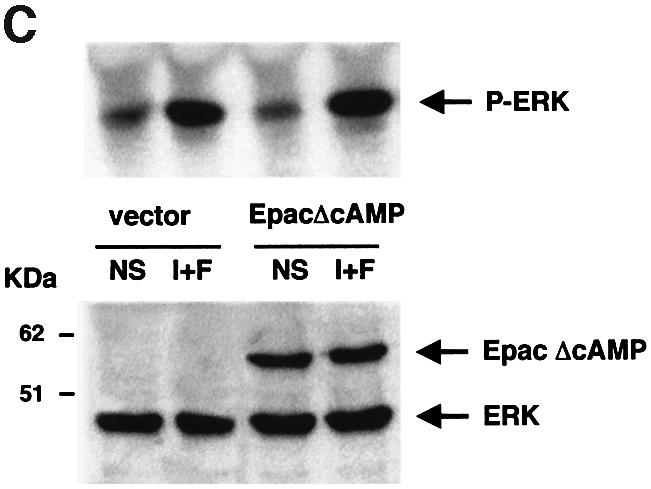
Fig. 7. SOS and Epac do not participate in the cAMP-dependent activation of MAPK in B16 cells. (A) B16 melanoma cells were cotransfected with the HA–ERK1 plasmid together with either an empty vector (vector) or a dominant-negative mutant of SOS (ΔSOS) lacking the nucleotide exchange domain. Cells were treated or not (NS) with IBMX plus forskolin (I+F) and ERK1 phosphorylation was examined as described in Figure 3. A western blot of the same lysates was carried out using anti-HA and anti-SOS antibodies as a control for transfected protein expression (lower panel). (B) CHO cells stably expressing the insulin receptor were cotransfected with the HA–ERK1 and an empty vector (vector) or a vector encoding SOS mutant (ΔSOS). Cells were left untreated (NS) or treated with 0.1 µM insulin (Ins) for 10 min. Cell lysates were immunoprecipitated with an anti-HA antibody and subjected to western blotting with anti-phospho-ERKs (upper panel). A western blot of the same lysates was carried out using an anti-HA antibody as a control for HA–ERK expression. (C) As in (B), except that an activated version of Epac (Epac ΔcAMP) lacking the cAMP-binding domain was used. ERK activation was visualized using an anti-phospho-ERK antibody (upper panel). The lower panel shows the expression of the HA–ERK1 and HA–Epac mutant.
As SOS is not involved in the cAMP-induced cascade in melanoma cells, we assessed the putative involvement of Epac, a recently described exchange factor for Rap-1, directly activated by cAMP. We performed the same experiment described above using a plasmid encoding an Epac protein lacking the cAMP-binding domain (EpacΔcAMP) which represents an activated version of this exchange factor (De Rooij et al., 1998). EpacΔcAMP neither induced ERK activation nor influenced ERK activation by cAMP, showing that Epac is not involved in the cAMP response towards ERK in B16 melanocytes. The level of expression of the transfected protein was verified by western blotting using an anti-HA antibody (Figure 7C, lower panel).
Ras is not involved in the activation of ERK by cAMP in PC12 cells
In PC12 cells, ERKs and B-Raf are activated by cAMP (Vossler et al., 1997). The common embryonic origin of PC12 cells and melanocyte cells has prompted us to investigate whether Ras could be activated by cAMP in PC12 cells. We therefore performed some experiments in this neuronal cell model. We first looked at ERK activation. cAMP-elevating agents, like TPA, strongly activated ERKs, as seen by western blotting with an anti-phospho-ERK antibody (Figure 8A). Next, pull-down experiments were performed to measure Rap-1 and Ras activation in PC12 cells. Rap-1 was activated by cAMP-elevating agents (Figure 8B), as reported by Vossler et al. (1997). In contrast, cAMP did not activate Ras in PC12 cells (Figure 8C). NGF was used as a positive control for Ras activation (Figure 8C).

Fig. 8. Ras is not activated by cAMP and Rap-1 does not mediate the cAMP-dependent ERK activation in PC12 cells. (A) PC12 cells were treated with TPA (T) or IBMX plus forskolin (I+F). Activation of ERKs was assessed using an anti-phospho-ERK antibody (1/5000). (B) PC12 cells treated with IBMX plus forskolin (I+F) were solubilized and subjected to pull-down experiments using a Ral–GDF RBD GST fusion protein to detect active Rap-1. The same lysates were subjected to direct western blotting using an anti-Rap-1 antibody (1/500) to detect the total Rap-1 content in the cell (lower panel). (C) Pull-down experiments using the B-Raf RBD GST fusion protein to detect activated Ras was performed as in (B), using NGF (50 ng/ml) as a positive control (upper panel). The same lysates were also analysed for Ras total expression (lower panels). (D) PC12 cells were cotransfected with HA–ERK1 and an empty vector (vector) or the constitutively active form of Ras (Ras-V12) or the constitutive active mutants of Rap-1 (Rap-1-V12 or Rap-1-E63) all tagged with the HA epitope. Cell extracts were then immunoprecipitated with an anti-HA antibody and immunoblotted using an anti-phospho-ERK antibody (upper panel). The lower panel shows the expression of the transfected proteins analysed by western blotting with an anti-tag antibody.
Finally, we overexpressed HA-tagged constitutively active mutants of Ras, Rap-1 and both Raf isoforms, Raf-1 and B-Raf, together with HA–ERK1. As described before, HA–ERK was immunoprecipitated and a western blot using an antibody to the phosphorylated forms of ERKs was then performed. As expected, the active form of Ras (Ras-V12), as well as both active mutants of Raf-1 (Raf-1-CAAX) and B-Raf (B-Raf-CAAX), activated ERK1 in PC12 cells. Surprisingly, the functional constitutively active mutant of Rap-1, Rap-1-E63, induced no ERK activation in these cells (Figure 8D). The same result was obtained using the other constitutively active mutant of Rap-1, Rap-1-V12 (not shown). The expression levels of the transfected proteins were verified by western blotting using an anti-HA antibody (Figure 8D, lower panels). Identical results were obtained with PC12 cells from three different origins (see Materials and methods), excluding the possibility that our results were not specific to the PC12 cells used in our study.
Taken together, these data indicate that, although Rap-1 is activated by cAMP, it does not transmit the cAMP effects on ERK in PC12 cells. They clearly show that Ras cannot mediate these effects either, since it is not activated by cAMP in this cell system. Thus, the activation of Ras by cAMP appears to be a highly specific process, so far only known to occur in melanocytes.
Discussion
We have investigated the mechanism by which cAMP activates the MAPK(s) ERK(s) in melanocytes and melanoma cells. We found that B-Raf mediates the cAMP-activation of ERK in melanoma cells. We also observed that cAMP activates Rap-1, but does not seem to participate in the regulation of ERK by cAMP. Unexpectedly, we found that p21Ras is activated by cAMP and transduces the cAMP-dependent activation of ERK in melanoma cells. Ras activation occurs through a PKA-independent process that involves neither SOS nor Epac. We detected ERK and Ras activation by cAMP in normal human melanocytes, further strengthening the physiological relevance of the results obtained in the B16 melanoma cell line.
To date, cAMP has been reported to stimulate ERK activity in a very limited number of cell types, including T cells (Saxena et al., 1999), PC12 rat phaeochromocytoma cells (Frodin et al., 1994; Young et al., 1994) and melanocytes (Englaro et al., 1995). In T cells, the effect of cAMP on ERKs is mediated through the inhibition of a protein tyrosine phosphatase (Saxena et al., 1999). In PC12 cells the effect of cAMP has been proposed to involve the phosphorylation and activation by PKA of Rap-1 small GTPase, then Rap-1 up-regulates B-Raf activity which in turn phosphorylates and stimulates MEK, resulting in ERK activation (Vossler et al., 1997). The fact that both PC12 cells and melanocytes derive from the neural crest has prompted us to hypothesize that they share cell-specific signalling mechanisms responsible for this MAPK cascade activation. Indeed, our data and those reported in PC12 cells suggest that B-Raf is a key component in the pathway leading to ERK activation by cAMP.
Next we wished to elucidate the mechanisms by which cAMP regulates B-Raf activity. Experiments using bacterial toxins suggested that Rap-1 and Ras might function upstream of B-Raf. Indeed, cAMP activates Rap-1 in B16 cells but we found that two different functional constitutively active mutants of Rap-1 were not able to induce ERK activation. Furthermore, a dominant-negative mutant of Rap-1 which has been reported to block the function of the endogenous protein (Vossler et al., 1997; York et al., 1998) did not prevent the activation of ERK by cAMP. These observations indicate that Rap-1 does not participate in the molecular events connecting cAMP to ERK in B16 cells.
Although Rap-1 is activated by cAMP in melanocytes, the physiological role of this event remains to be elucidated. Melanocyte differentiation is characterized by increased melanin synthesis and distribution within the epidermis. Melanin is produced and transported within specialized vesicles called melanosomes. The localization of Rap-1 in the vesicular/endocytic compartment (reviewed in Bos, 1998) is compatible with a putative role of Rap-1 in melanosome dynamics. Bourneville’s disease, or tuberous sclerosis complex (TSC), is an autosomal dominant syndrome characterized by the widespread development of hamartomas, mental retardation and a skin pigmentation disorder (Sheth, 1998). The product of the TSC2 gene is tuberin, which has a specific GTPase activity towards Rap-1 (Geist et al., 1996; Wienecke et al., 1997). This observation, indicating that the alteration of Rap-1 function is associated with a pigmentation defect, supports the hypothesis that Rap-1 is involved in melanocyte differentiation.
The lack of Rap-1 involvement in cAMP-induced ERK activation in melanoma cells suggests that Ras could be the mediator in this process. This attractive hypothesis was confirmed by pull-down experiments which demonstrated that cAMP activates Ras, as shown by its increased binding to the B-Raf RBD. The fact that Ras activates the ERK pathway is well known (reviewed in Johnson and Vaillancourt, 1994), but our results disclose a novel mechanism in which Ras may act as an early transducer of the cAMP-dependent activation of MAPK in melanocytes. It is well established that Ras–GTP activates both B-Raf and Raf-1. However, we show here that Raf-1 is not activated by cAMP. Furthermore, cAMP inhibits Raf-1 activation by TPA. Raf-1 inhibition by cAMP has already been reported (Wu et al., 1993; Moodie et al., 1994; Vaillancourt et al., 1994) and seems to be mediated by Raf-1 phosphorylation by PKA at Ser43 and Ser621 (Wu et al., 1993; Häfner et al., 1994; Mischak et al., 1996). B-Raf is not inhibited by the up-regulation of the cAMP pathway (Vossler et al., 1997; MacNicol and MacNicol, 1999). This was initially explained by the absence, in B-Raf, of the PKA phosphorylation site homologous to Ser43 in Raf-1. More recently, B-Raf resistance to PKA was explained in a report demonstrating that the N-terminal regulatory domain of B-Raf interfered with the ability of PKA to phosphorylate the kinase and modulate its activity (MacNicol and MacNicol, 1999). Thus, it was tempting to propose that, upon the cAMP-induced Ras activation, the concomitant activation of PKA led to the inhibition of Raf-1, making B-Raf the only MAPKKK involved in the activation of ERK by cAMP. However, this possibility has been ruled out because we have not been able to release the blockade of Raf-1 activity upon cAMP treatment by the use of a PKA inhibitor (not shown). Thus we must conceive another hypothesis that could eventually involve (i) RKIP, a recently described inhibitor of Raf-1 (Yeung et al., 1999), (ii) Akt (PKB), which inhibits Raf-1 and is activated by cAMP (Zimmermann and Moelling, 1999; Scheid and Woodgett, 2000) or (iii) Rap-1 itself, which has been reported to antagonize Ras-dependent Raf-1 activation in some cell systems (Cook et al., 1993; Hu et al., 1997, 1999).
Our data and those of Vossler et al. (1997) reveal that PC12 phaeochromocytoma cells and melanocytes share some specific signal transduction features, at least concerning the activation of ERK, MEK and B-Raf by cAMP. However, our findings clearly demonstrate the existence of different regulatory mechanism since Ras is activated by cAMP in melanocytes but not in PC12 cells. Further, in melanocyte cells, two constitutively active Rap-1 mutants were unable to activate ERK. In our hands, these mutants also failed to activate the MAPK pathway in PC12 cells. Our results are in total agreement with the data of Zwartkruis et al. (1998), demonstrating that, in PC12 cells, Rap-1 activation does not correlate with ERK activation. Thus the activation of B-Raf by Rap-1 may be restricted to a very specific PC12 subclone (the PC12-GR5 variant used by Vossler et al.) or to certain experimental conditions such as upon overexpression of both Rap-1 and B-Raf (Vossler et al., 1997; Okada et al., 1999).
In most cell systems, cAMP effects have been considered to be the result of PKA activation. However, cAMP interacts directly with some ion channels (Zufall et al., 1997; Santoro et al., 1998) and PKA has not been clearly implicated in the cAMP-regulation of certain neural functions (Huang et al., 1995; Liu et al., 1995; Brandon et al., 1997). We have shown that, in B16 cells, the cAMP-dependent activation of Ras, and therefore ERKs, is PKA-independent. When thinking about possible cAMP-dependent regulatory points for Ras, we focused our attention on SOS, the classical guanine nucleotide exchange factor that usually couples tyrosine kinase growth factor receptors to Ras activation (reviewed in Bar-Sagi, 1994; Schlessinger, 1994). Since SOS recruitment to the plasma membrane is sufficient to activate Ras, cAMP, by some unknown mechanism, could target SOS to the membrane, thereby activating Ras. However, the overexpression of a dominant-negative mutant of SOS (ΔSOS) lacking its guanine nucleotide exchange domain (Sakaue et al., 1995), did not block the cAMP-activation of ERK, showing that SOS does not mediate the cAMP action towards the MAPK cascade. R-Ras–GRF is another exchange factor for Ras whose activity is enhanced by increased concentrations of intracellular calcium in neurons (Farnsworth et al., 1995; Buchsbaum et al., 1996; Freshney et al., 1997). cAMP, through PKA phosphorylation of specific types of calcium channels, promotes calcium influx into skeletal muscle and hippocampal neurons (Sculptonearu et al., 1993; Hell et al., 1995; Gray et al., 1997). Taken together, these findings suggest that cAMP, through PKA activation, could increase the intracellular concentration of calcium leading to Ras–GRF and Ras activation in these cell systems. In melanocytes, the fact that cAMP-activation of MAPK is independent of PKA makes the involvement of Ras–GRF in the process very unlikely. Taken together, our data lead us to hypothesize that Ras activation by cAMP in melanocytes could be mediated by an unidentified, cell-specific Ras exchange factor which would be directly activated by cAMP. Two independent groups have recently cloned several exchange factors for Rap-1 containing a cAMP-binding sequence and which are directly activated by cAMP (Epac) (De Rooij et al., 1998) and cAMP-GEFs (Kawasaki et al., 1998). However, these proteins have not been found to function as exchange factors for Ras. Indeed, we confirmed that Epac was not involved in the cascade leading to ERK activation in B16 cells.
Our data have led us to propose a putative model explaining the cAMP-activation of the MAPK pathway in melanocytes (Figure 9). We suggest that this process is based on two regulatory and melanocyte-specific features: (i) a Ras exchange factor directly activated upon cAMP binding, which could account for the cAMP-induced Ras activation; (ii) expression and activation of B-Raf kinase, which is required to permit the downstream activation steps towards MAPK since Raf-1, which is somehow inhibited by cAMP, cannot ensure this function. Future studies will aim to identify melanocyte-specific Ras exchange factors directly regulated by cAMP.

Fig. 9. Model for the cAMP-dependent activation of MAPK in melanocyte cells. α-MSH binds the melanocyte receptor, MC1-R, and activates adenylyl cyclase (AC), leading to an increase in the intracellular cAMP content. cAMP activates Rap-1 through phosphorylation by PKA via cAMP-dependent Rap-1 GEFs (Epac/cAMP Rap-1 GEFs). In melanocytes, cAMP could also directly activate an unidentified cell-specific cAMP-dependent Ras GEF (cAMP Ras GEF) which would activate Ras in response to cAMP. Ras activates Raf-1 and B-Raf, but Raf-1 is inhibited by an unknown mechanism, so activated B-Raf mediates the Ras activation of MEK and ERKs.
Materials and methods
Materials
Reagents. Forskolin, IBMX, TPA, α-MSH, insulin, PDGF, 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), aprotinin, leupeptin, orthovanadate, NaPPi, NaF, bovine serum albumin (BSA), NGF, gelatin, ATP, protein A–Sepharose and protein G–Sepharose were purchased from Sigma. Dulbecco’s modified Eagle medium (DMEM), MCDB medium, Lipofectamine, Optimem, horse serum and streptomycin/penicillin were from Gibco-BRL. Fetal bovine serum (FBS) was from Hyclone laboratories. [γ-32P]ATP and the enhanced chemiluminescence (ECL) detection system were from Amersham Life Sciences. Immobilon-P membranes were from Millipore Corp. MEK–His fusion protein was purchased from Santacruz Biotechnologies. Glutathione–Sepharose beads were from Pharmacia Biotech. The PKA inhibitor H89 was from Calbiochem and the plasmid preparation kit for transfection was from Quiagen. Toxins LT and B were gifts from Dr P.Boquet (U452, INSERM). The FuGene transfection reagent was from Boehringer Mannheim and the Transfast transfection reagent from Promega France.
Antibodies. The polyclonal antibodies against the phosphorylated forms of ERKs and MEK were from Promega France. The specific polyclonal anti-B-Raf IS11 antibody directed against amino acids 396–443, which are conserved in both avian and mammalian species, has been previously described (Eychene et al., 1992; Barnier et al., 1995). Polyclonal antibodies against Raf-1, Rap-1, ERK2 and SOS were from Santacruz Biotechnology. The antibody to Ras was the monoclonal anti-pan-Ras from Calbiochem. The anti-HA antibody was the monoclonal 12CA5 from Babco (Richmond, CA). The anti-mouse and anti-rabbit peroxidase-conjugated antibodies were purchased from Dakopatts.
Plasmids. The pGEX plasmid encoding the Ral–GDS RBD GST fusion protein is described in Herrmann et al. (1996). The plasmid encoding the B-Raf RBD GST fusion protein was obtained by subcloning the Ras-binding domain of B-Raf (amino acids 146–227 of quail B-Raf; Papin et al., 1998) into the BamHI and EcoRI sites of pGEX-2T. The oligomers used for PCR amplification of this fragment were the (+432) 5′-GTCACGGGGATCCCCTAAGTCTCCACA-3′ and (670) 5′-TGGCAGAATTCCCAAGACCTCCAC-3′. The construct was verified by sequencing. The pcDNA3-HA-tagged plasmid containing the ERK1 gene was a gift from Dr Pouyssegur’s laboratory (UMR134, Nice), the B-Raf constitutively active (pcDNA3/HA1-B-Raf-CAAX) was obtained by subcloning the EcoRI–ApaI fragment from pBluescript/B-Raf-CAAX (Papin et al., 1998) in place of the EcoRI–ApaI fragment of pcDNA3/HA1-B-Raf (Papin et al., 1998). The HA-tagged constitutively active form of Raf-1 (pcDNA3/HA1-Raf-1-CAAX) was obtained by subcloning the EcoRV–XbaI 3′ fragment from pRcRSV/HA1-Raf-1-CAAX (Papin et al., 1998) in place of the EcoRV–XbaI 3′ fragment of pcDNA3/HA1 (Papin et al., 1998). The negative mutant of B-Raf (corresponding to the N-terminal region of quail B-Raf) was obtained by subcloning the EcoRI–SalI fragment of pGBT-9N-B-Raf (Papin et al., 1996) into the EcoRI and XhoI sites of pcDNA3. The N-Raf-1-negative form of Raf-1 (also lacking the C-terminal kinase domain, amino acids 330–627) is described in Schaap et al. (1993). PcDNA3/HA1-Ras-V12 was obtained by subcloning the EcoRI fragment of pGBT-9/Ras-V12 (Papin et al., 1996) downstream of the HA1 epitope into the pcDNA3 vector. PcDNA3/HA1-Ras-N17 was obtained by subcloning the SalI–XhoI fragment of pRcRSV/Ras-N17 downstream of the HA1 epitope into the pcDNA3 vector. The plasmid encoding HA-Rap-1-E63 and Rap-1-V12 were kindly provided by Dr J.Gunzburg and plasmids encoding Rap-1-V12 and Rap-1-N17 are described in Vossler et al. (1997) and were tagged with the HA epitope. The plasmid encoding the SOS mutant ΔSOS was obtained from Sakaue et al. (1995). The vector encoding the EpacΔcAMP construct was from De Rooij et al. (1998).
Cell culture and specific cell treatments
B16/F10 mouse melanoma cells and NIH 3T3 mouse fibroblasts were cultured in DMEM supplemented with 7% FBS and penicillin/streptomycin [100 IU (50 µg)/ml] in a humidified atmosphere containing 5% CO2 in air at 37°C. Normal human melanocytes were obtained from neonatal foreskins of Caucasian individuals, and grown in MCDB medium according to a modification of the method of Eisinger and Marko (1982) as described previously (Aberdam et al., 1993). Before kinase assays or pull-down experiments, cells were depleted of growth factors for at least 24 h.
When indicated, cells were treated with forskolin (20 µM), IBMX (100 µM), TPA (16 nM), α-MSH (1 µM), PDGF (5 ng/ml) or insulin (0.1 µM) for the times indicated. Cells were incubated with C.sordelii LT at 70 ng/ml for ≥4 h before other treatment or with C.difficile toxin B at 0.5 g/ml for ≥4 h before any treatment. To inhibit PKA activity, cells were treated with H89 at 3 µM for 20 min and then with cAMP-elevating agents for another 10 min.
CHO-IR cells stably expressing the insulin receptor were obtained from Dr Tartare-Deckert (INSERM Unité 145, Nice, France) and cultured in F12 medium supplemented with 10% FBS. PC12 cells from the American Type Culture Collection (CRL-1721), from Dr Schlessinger’s laboratory (Dikic et al., 1994) and from Dr Rogers’ laboratory (Walton et al., 1988) were cultured in DMEM supplemented with 7% horse serum, 7% FBS and 1 mM glutamine. Before transfection, PC12 cells were seeded on dishes coated with collagen.
Western blotting
Cells were grown in six-well dishes, starved of serum for 24 h, treated with agents as indicated and lysed in a buffer containing 1% Triton X-100, 50 mM Tris pH 7.6, 100 mM NaCl, 50 mM NaF, 5 mM EDTA, 1 mM NaPPi, 5 µg/ml leupeptin, 1 mM AEBSF, 100 IU/ml aprotinin, 1 mM NaVO4 and 10 mM p-nitrophenolphosphate. Proteins (30 µg) were separated on 10% sodium dodecylsulfate (SDS)–polyacrylamide gels and transferred to Immobilon-P membranes. Membranes were saturated and proteins were detected with the appropiate primary antibody and a secondary peroxidase-conjugated anti-mouse or anti-rabbit antibody at a dilution of one in 3000. Blots were developed using the enhanced ECL system.
B-Raf and Raf-1 immunokinase assays
B16 cells were grown in 100 mm dishes. After the indicated stimulations, cells were washed and lysed. For B-Raf immunoprecipitation, cells were solubilized in buffer containing 50 mM HEPES pH 7.50, 50 mM NaCl, 50 mM NaF, 10 mM NaPPi, 100 µM sodium orthovanadate, 100 µM AEBSF, 100 IU/ml aprotinin, 1 mg/ml leupeptin and 1% Triton X-100. Extracts were incubated with 2.5 µl of the anti-B-Raf polyclonal antibody (IS11) and the kinase assay was performed as described by Barnier et al. (1995). For Raf-1 kinase assays, cells were lysed in buffer containing 50 mM HEPES pH 7.4, 150 mM NaCl, 10 mM EDTA, 100 mM NaF, 10 mM NaPPi, 2 µM sodium orthovanadate, 100 µM AEBSF, 5 µg/ml leupeptin and 1% Triton X-100. Solubilized proteins were incubated with 10 µl of the anti-Raf-1 polyclonal antibody pre-adsorbed to protein A–Sepharose and the kinase assay was performed as previously described (Englaro et al., 1995) using purified His–MEK-1 as a substrate. Reactions were stopped by addition of Laemmli buffer and analysed by SDS–PAGE (10% acrylamide) and autoradiography.
Cotransfection and HA-immunoprecipitation assays
Since the efficiency of transfection in B16 cells appears to be very low, we tested the action of different effector proteins on ERK activation in these cells as follows. B16 melanoma cells were seeded in six-well dishes and transient transfections were performed the following day using 9.8 µl of Lipofectamine and 3 µg of total plasmid DNA (1.5 µg of pCDNA3 HA–ERK1 and 1.5 µg of plasmids encoding the various mutant cDNAs) in 1 ml final volume of Optimem. Cells were then starved of serum, treated with agents as indicated and lysed in a 1% Triton buffer as described previously. Equal amounts of protein extracts were incubated with 15 µl of the anti-HA monoclonal antibody and protein G–Sepharose was used to collect the antigen–antibody complexes. Immunoprecipitates were washed in 0.1% Triton buffer containing 30 mM HEPES pH 7.4 and 30 mM NaCl, and resolved on 10% SDS–polyacrylamide gels. Gels were transferred to Immobilon-P membranes, probed with a polyclonal antibody against active ERKs and visualized by ECL. Proteins (30 µg) were probed with an anti-tag antibody to detect exogenous ERK1 expression. We used the FuGene reagent (Boehringer Mannheim) to transfect CHO cells, and the Transfast reagent (Promega) to transfect PC12 cells, as described in the manufacturers’ protocols.
Pull-down experiments to detect activated Ras and Rap-1
B16 mouse melanoma cells, NIH 3T3 cells or PC12 cells were grown to 80% confluence and stimulated with agents. Cells were then washed twice with ice-cold phosphate-buffered saline (PBS) and lysed in buffer A containing 50 mM Tris pH 7.5, 15 mM NaCl, 20 mM MgCl2, 5 mM EGTA, 1% Triton X-100, 1% N-octylglucoside, 100 µM AEBSF, 5 µg/ml leupeptin and 1 mM pepstatin A. Lysates were clarified by centrifugation and supernatants were incubated with 30 µg of GST fusion protein precoupled to glutathione–Sepharose beads. To isolate Rap-1–GTP, we used the GST–Ral GDS RBD obtained as described by Herrmann et al. (1996). To detect Ras–GTP, the GST–B-Raf RBD, obtained as described by Herrmann et al. (1995), was used. After incubation for 2 h at 4°C, beads were washed three times in buffer A, resuspended in Laemmli buffer and boiled. Samples were analysed by SDS–PAGE (12.5%) followed by transfer to Immobilon-P membranes. Active Rap-1 and Ras were detected using specific antibodies, as described above.
PKA activity assay
B16 cells were grown in 100 mm dishes, then stimulated as described and lysed in a buffer containing 50 mM HEPES pH 7.6, 150 mM NaCl, 10 mM EDTA, 10 mM NaPPi, 2 mM sodium orthovanadate, 100 mM NaF, 0.5 mM AEBSF, 100 UI/ml aprotinin, 5 µg/ml leupeptin and 1% Triton X-100. The lysates were incubated with phosphorylation buffer containing 10 mM HEPES pH 7.2, 68.5 mM NaCl, 2.7 mM KCl, 0.15 mM KH2PO4, 0.5 mg/ml glucose, 25 mM β-glycerophosphate pH 7.2, 10 mM MgCl2, 1 mM EGTA, 1.85 mM CaCl2, 0.1 mM ATP, 100 µM Kemptide as a substrate peptide and 25 µCi of [γ-32P]ATP per mmol. The phosphorylation reaction was allowed to proceed for 10 min at 37°C and then stopped by spotting on to Whatman P81 papers. The papers were washed twice with 1% (v/v) orthophosphoric acid, rinsed in ethanol and air dried. The radioactivity was determined by Cerenkov counting.
Acknowledgments
Acknowledgements
We thank Dr de Gunzburg for providing vectors encoding Rap-1-N17, Rap-1-V12 and Rap-1-E63, and Dr P.Stork for providing other Rap-1-N17- and Rap-1-V12-encoding plasmids. We thank Dr Schapp for the dominant-negative version of Raf-1 (N-Raf-1), Dr Pouyssegur for HA-tagged ERK1, Dr Tartare-Deckert for the CHO-IR cells and Dr Bos for the EpacΔcAMP construct. We are grateful to Dr Boquet for the C.sordelii LT toxin and the C.difficile B toxin, and Aurore Grima and Christine Ordonez for scanning the autoradiography films. We are grateful to Dr Boulukos and Dr Pouyssegur for critical reading of this manuscript and to Alexandra Charlesworth for her expert editorial help. This work was supported by the Association pour la Recherche sur le Cancer (ARC-grant 5209), La Ligue contre le Cancer and the Centre de Recherche et d’Investigations Epidermiques et Sensorielles (CERIES).
References
- Aberdam E., Roméro,C. and Ortonne,J.-P. (1993) Repeated UVB irradiations do not have the same potential to promote stimulation of melanogenesis in cultured normal human melanocytes. J. Cell Sci., 106, 1015–1022. [DOI] [PubMed] [Google Scholar]
- Altschuler D.L., Peterson,S.N., Ostrowski,M.C. and Lapetina,E.G. (1995) Cyclic AMP-dependent activation of Rap1b. J. Biol. Chem., 270, 10373–10376. [DOI] [PubMed] [Google Scholar]
- Bar-Sagi D. (1994) The Sos (Son of sevenless) protein. Trends Endocrinol. Metab., 5, 165–169. [DOI] [PubMed] [Google Scholar]
- Barnier J.V., Papin,C., Eychene,A., Lecoq,O. and Calothy,G. (1995) The mouse B-Raf gene encodes multiple protein isoforms with tissue-specific expression. J. Biol. Chem., 270, 23381–23389. [DOI] [PubMed] [Google Scholar]
- Bos J.L. (1998) All in the family? New insights and questions regarding interconnectivity of Ras, Rap1 and Ral. EMBO J., 17, 6776–6782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon E.P., Idzerda,R.L. and McKnight,G.S. (1997) PKA isoforms, neural pathways, and behaviour: making the connection. Curr. Opin. Neurobiol., 7, 397–403. [DOI] [PubMed] [Google Scholar]
- Buchsbaum R., Telliez,J.B., Goonesekera,S. and Feig,L.A. (1996) The N-terminal pleckstrin, coiled-coil, and IQ domains of the exchange factor Ras-GRF act cooperatively to facilitate activation by calcium. Mol. Cell. Biol., 16, 4888–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscà R., Bertolotto,C., Ortonne,J.-P. and Ballotti,R. (1996) Inhibition of the phosphatidylinositol 3-kinase/p70S6-kinase pathway induces B16 melanoma cell differentiation. J. Biol. Chem., 271, 31824–31830. [DOI] [PubMed] [Google Scholar]
- Buscà R., Bertolotto,C., Abbe,P., Englaro,W., Ishizaki,T., Narumiya,S., Boquet,P., Ortonne,J.-P. and Ballotti,R. (1998) Inhibition of p21Rho is required for cAMP-induced melanoma cell differentiation. Mol. Biol. Cell, 9, 1367–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catling A.D., Reuter,C.W., Cox,M.E., Parsons,S.J. and Weber,M.J. (1994) Partial purification of a mitogen-activated protein kinase kinase activator from bovine brain. Identification as B-Raf or a B-Raf-associated activity. J. Biol. Chem., 269, 30014–30021. [PubMed] [Google Scholar]
- Chaves-Olarte E., Florin,I., Boquet,P., Popoff,M., Von Eichel-Streiber,C. and Thelestam,M. (1996) UDP–glucose deficiency in a mutant cell line protects against glucosyltransferase toxins from Clostridium difficile and Clostridium sordellii. J. Biol. Chem., 271, 6925–6932. [DOI] [PubMed] [Google Scholar]
- Cook S.J., Rubinfeld,B., Albert,I. and Mc Cormick,F. (1993) Rap V12 antagonizes Ras-dependent activation of ERK1 and ERK2 by LPA and EGF in Rat-1 fibroblast. EMBO J., 12, 3475–3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rooij J., Zwartkruis,F.J.T., Verheijen,M.H.G., Cool,R.H., Nijman,S.M.B., Wittinghofer,A. and Bos,J.L. (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature, 396, 474–477. [DOI] [PubMed] [Google Scholar]
- Dikic I., Schlessinger,J. and Lax,I. (1994) PC12 cells overexpressing the insulin receptor undergo insulin-dependent neuronal differentiation. Curr. Biol., 4, 702–708. [DOI] [PubMed] [Google Scholar]
- Eisinger M. and Marko,O. (1982) Selective proliferation of normal human melanocytes in vitro in presence of phorbol ester and cholera toxin. J. Invest. Dermatol., 95, 441–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englaro W., Rezzonico,R., Durand-Clément,M., Lallemand,D., Ortonne,J.-P. and Ballotti,R. (1995) Mitogen-activated protein kinase pathway and AP-1 are activated during cAMP-induced melanogenesis in B-16 melanoma cells. J. Biol. Chem., 270, 24315–24320. [DOI] [PubMed] [Google Scholar]
- Eychene A., Barnier,J.V., Dezelee,P., Marx,M., Laugier,D., Calogeraki,I. and Calothy,G. (1992) Quail neuroretina c-Rmil (B-Raf) proto-oncogene cDNAs encode two proteins of 93.5 and 95 kDa resulting from alternative splicing. Oncogene, 7, 1315–1323. [PubMed] [Google Scholar]
- Farnsworth C.L., Freshney,N.W., Rosen,L.B., Ghosh,A., Greenberg,M.E. and Feig,L.A. (1995) Calcium activation of Ras mediated by neuronal exchange factor Ras–GRF. Nature, 376, 524–527. [DOI] [PubMed] [Google Scholar]
- Freshney N.W., Goonesekera,S.D. and Feig,L.A. (1997) Activation of the exchange factor Ras–GRF by calcium requires an intact Ddl homology domain. FEBS Lett., 407, 111–115. [DOI] [PubMed] [Google Scholar]
- Frodin M., Peraldi,P. and Van Obberghen,E. (1994) Cyclic AMP activates the mitogen-activated protein kinase cascade in PC12 cells. J. Biol. Chem., 269, 6207–6214. [PubMed] [Google Scholar]
- Geist R.T., Reddy,A.J., Zhang,J. and Gutman,D.H. (1996) Expression of the tuberous sclerosis 2 gene product, tuberin, in adult and developing nervous system tissues. Neurobiol. Dis., 3, 111–120. [DOI] [PubMed] [Google Scholar]
- Gray P.C., Tibbs,V.C., Catterall,W.A. and Murphy,B.J. (1997) Identification of a 15-kDa cAMP-dependent protein kinase-anchoring protein associated with skeletal muscle L-type calcium channels. J. Biol. Chem., 272, 6297–6302. [DOI] [PubMed] [Google Scholar]
- Häfner S. et al. (1994) Mechanism of inhibition of Raf-1 by protein kinase A. Mol. Cell. Biol., 14, 6696–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearing V.J. and Jimenez,M. (1989) Analysis of mammalian pigmentation at the molecular level. Pigm. Cell. Res., 2, 75–85. [DOI] [PubMed] [Google Scholar]
- Hearing V.J. and Tsukamoto,K. (1991) Enzymatic control of pigmentation in mammals. FASEB J., 5, 2902–2909. [PubMed] [Google Scholar]
- Hell J.W., Yokoyama,C.T., Breeze,L.J., Chavkin,C. and Catterall,W.A. (1995) Phosphorylation of presynaptic and postsynaptic calcium channels by cAMP-dependent protein kinase in hippocampal neurons. EMBO J., 14, 3036–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann C., Martin,A.G. and Wittinghofer,A. (1995) Quantitative analysis of the complex between p21ras and the Ras-binding domain of the human Raf-1 protein kinase. J. Biol. Chem., 270, 2901–2905. [DOI] [PubMed] [Google Scholar]
- Herrmann C., Horn,G., Spaargaren,M. and Wittinghofer,A. (1996) Differential interaction of the Ras family GTP-binding proteins H-Ras, Rap1A, and R-Ras with the putative effector molecules Raf kinase and Ral-guanine nucleotide exchange factor. J. Biol. Chem., 271, 6794–6800. [DOI] [PubMed] [Google Scholar]
- Hu C.D., Kariya,K.I., Kotani,G., Shirouzu,M., Yokoyama,S. and Kataoka,T. (1997) Coassociation of Rap1A and Ha-Ras with Raf-1 N-terminal region interferes with Ras-dependent activation of Raf-1. J. Biol. Chem., 272, 11702–11705. [DOI] [PubMed] [Google Scholar]
- Hu C.D., Kariya,K.I., Okada,T., Qi,X., Song,C. and Kataoka,T. (1999) Effect of phosphorylation on activities of Rap1A to interact with Raf-1 and to suppress Ras-dependent Raf-1 activation. J. Biol. Chem., 274, 48–51. [DOI] [PubMed] [Google Scholar]
- Huang N.N., Wang,D.J., Heller,E. and Heppel,L.A. (1995) Homologous desensitization of ATP-stimulated mitogenesis: mechanism involves desensitization of arachidonic acid release and cAMP elevation but not the activation of protein kinase A. J. Cell Physiol., 165, 667–675. [DOI] [PubMed] [Google Scholar]
- Jaiswal R.K., Moodie,S.A., Wolfman,A. and Landreth,G.E. (1994) The mitogen-activated protein kinase cascade is activated by B-Raf in response to nerve growth factor through interaction with p21Ras. Mol. Cell. Biol., 10, 6944–6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson G.L. and Vaillancourt,R.R. (1994) Sequential protein kinase reactions controlling cell growth and differentiation. Curr. Opin. Cell Biol., 6, 230–238. [DOI] [PubMed] [Google Scholar]
- Just I., Selzer,J., Wilm,M., Von Eichel-Streiber,C., Mann,M. and Aktories,K. (1995) Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature, 375, 500–503. [DOI] [PubMed] [Google Scholar]
- Just I., Selzer,J., Hofmann,F., Green,G.A. and Aktories,K. (1996) Inactivation of Ras by Clostridium sordellii lethal toxin-catalysed glucosylation. J. Biol. Chem., 271, 10149–10153. [DOI] [PubMed] [Google Scholar]
- Kawasaki H., Springett,G.M., Mochizuki,N., Toki,S., Nakaya,M., Matsuda,M., Housman,D.E. and Graybiel,A.M. (1998) A family of cAMP-binding proteins that directly activate Rap1. Science, 282, 2275–2279. [DOI] [PubMed] [Google Scholar]
- Lange-Carter C.A. and Johnson,G.L. (1994) Ras-dependent growth factor regulation of MEK kinase in PC12 cells. Science, 265, 1458–1461. [DOI] [PubMed] [Google Scholar]
- Lerosey I., Pizon,V., Tavitian,A. and de Gunzburg,J. (1991) The cAMP-dependent protein kinase phosphorylates the Rap1 protein in vitro as well as in intact fibroblasts, but not the closely related Rap2 protein. Biochem. Biophys. Res. Commun., 175, 430–436. [DOI] [PubMed] [Google Scholar]
- Levine N. et al. (1991) Induction of skin tanning by subcutaneous administration of a potent synthetic melanotropin. J. Am. Med. Assoc., 266, 2730–2736. [PubMed] [Google Scholar]
- Liu F.C., Takahashi,H., McKay,R.D. and Graybiel,A.M. (1995) Dopaminergic regulation of transcription factor expression in organotypic cultures of developing striatum. J. Neurosci., 15, 2367–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacNicol M.C. and MacNicol,A.M. (1999) Nerve growth factor-stimulated B-Raf catalytic activity is refractory to inhibition by cAMP-dependent protein kinase. J. Biol. Chem., 274, 13193–13197. [DOI] [PubMed] [Google Scholar]
- Marx M., Eychene,A., Laugier,D., Bechade,C., Crisanti,P., Dezelee,P., Pessac,B. and Calothy,G. (1988) A novel oncogene related to c-mil is transduced in chicken neuroretina cells induced to proliferate by infection with an avian lymphomatosis virus. EMBO J., 11, 3369–3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeod S.J., Ingham,R.J., Bos,J.L., Kurosaki,T. and Gold,M.R. (1998) Activation of Rap-1 GTPase by the B cell antigen receptor. J. Biol. Chem., 273, 29218–29223. [DOI] [PubMed] [Google Scholar]
- Mischak H., Seitz,T., Janosch,P., Eulitz,M., Steen,H., Schellerer,M., Philipp,A. and Kolch,W. (1996) Negative regulation of Raf-1 by phosphorylation of serine 621. Mol. Cell. Biol., 16, 5409–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodie S.A., Willumsen,B.M., Weber,M.J. and Wolfman,A. (1993) Complexes of Ras⋅GTP with Raf-1 and mitogen-activated protein kinase kinase. Science, 260, 1658–1661. [DOI] [PubMed] [Google Scholar]
- Moodie S.A., Paris,M.J., Kolch,W. and Wolfman,A. (1994) Association of MAK1 with p21ras⋅GMPPNP is dependent on B-Raf. Mol. Cell. Biol., 14, 7153–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison D.K. and Cutler,R.E.,Jr (1997) The complexity of Raf-1 regulation. Curr. Opin. Cell Biol., 9, 174–179. [DOI] [PubMed] [Google Scholar]
- Okada T., Hu,C.D., Jin,T.G., Kariya,K.I., Yamawaki-Kataoka,Y. and Kataoka,T. (1999) The strength of interaction at the Raf cysteine-rich domain is a critical determinant of response of Raf to Ras family small GTPases. Mol. Cell. Biol., 19, 6057–6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papin C., Denouel,A., Calothy,G. and Eychène,A. (1996) Identification of signalling proteins interacting with B-Raf in the yeast two-hybrid system. Oncogene, 12, 2213–2221. [PubMed] [Google Scholar]
- Papin C., Denouel-Galy,A., Laugier,D., Calothy,G. and Eychene,A. (1998) Modulation of kinase activity and oncogenic properties by alternative splicing reveals a novel regulatory mechanism for B-Raf. J. Biol. Chem., 273, 24939–24947. [DOI] [PubMed] [Google Scholar]
- Popoff M.R. et al. (1996) Ras, Rap, and Rac small GTP-binding proteins are targets for Clostridium sordellii lethal toxin glucosylation. J. Biol. Chem., 271, 1–8. [DOI] [PubMed] [Google Scholar]
- Robinson M.J. and Cobb,M.H. (1997) Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol., 9, 180–186. [DOI] [PubMed] [Google Scholar]
- Sakaue M., Bowtell,D. and Kasuga,M. (1995) A dominant-negative mutant of mSOS1 inhibits insulin-induced Ras activation and reveals Ras-dependent and -independent insulin signaling pathways. Mol. Cell. Biol., 15, 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B., Liu,D.T., Yao,H., Bartsch,D., Kandel,E.R., Siegelbaum,S.A. and Tibbs,G.R. (1998) Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell, 93, 717–729. [DOI] [PubMed] [Google Scholar]
- Saxena M., Williams,S., Tasken,K. and Mustelin,T. (1999) Crosstalk between cAMP-dependent kinase and MAP kinase through a protein tyrosine phosphatase. Nature Cell Biol., 1, 305–311. [DOI] [PubMed] [Google Scholar]
- Schaap D., Van der Wal,J., Howe,L.R., Marshall,C.J. and Van Blitterswijk,W.J. (1993) A dominant-negative mutant of raf blocks mitogen-activated protein kinase activation by growth factors and oncogenic p21ras. J. Biol. Chem., 268, 20232–20236. [PubMed] [Google Scholar]
- Scheid M.P. and Woodgett,J.R. (2000) Protein kinases: six degrees of separation? Curr. Biol., 10, 191–194. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. (1994) How receptor tyrosine kinases activate Ras. Trends Biochem. Sci., 18, 223–275. [DOI] [PubMed] [Google Scholar]
- Sculptonearu A., Rotman,E., Takahashi,M., Scheuer,T. and Catterall,W.A. (1993) Voltage-dependent potentiation of the activity of cardiac L-type calcium channel alpha 1 subunit due to phosphorylation by cAMP-dependent protein kinase. Proc. Natl Acad. Sci. USA, 90, 10135–10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth P.B. (1998) Tuberous sclerosis complex. In Norlund,J.J., Boissy,R.E., Hearing,V.J., King,R.A. and Ortonne,J.-P. (eds), The Pigmentary System. Oxford University Press, Oxford, UK, pp. 606–610. [Google Scholar]
- Traverse S. and Cohen,P. (1994) Identification of a latent MAP kinase kinase kinase in PC12 cells as B-Raf. FEBS Lett., 350, 13–18. [DOI] [PubMed] [Google Scholar]
- Troppmair J., Bruder,J.T., App,H., Cai,H., Liptak,L., Szeberenyi,J., Cooper,G.M. and Rapp,U.R. (1992) Ras controls coupling of growth factor receptors and protein kinase C in the membrane to Raf-1 and B-Raf protein serine kinases in the cytosol. Oncogene, 9, 1867–1873. [PubMed] [Google Scholar]
- Vaillancourt R., Gardner,A.M. and Johnson,G.L. (1994) B-Raf-dependent regulation of the MEK-1/mitogene-activated protein kinase pathway in PC12 cells and regulation by cyclic AMP. Mol. Cell. Biol., 14, 6522–6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossler M.R., Yao,H., York,R.D., Pan,M.-G., Rim,C.S. and Stork,P.J.S. (1997) cAMP activates MAP kinase and Elk-1 through a B-Raf and Rap1-dependent pathway. Cell, 89, 73–82. [DOI] [PubMed] [Google Scholar]
- Walton K.M., Sandberg,K., Rogers,T.B. and Schnaar,R.L. (1988) Complex ganglioside expression and tetanus toxin binding by PC12 pheochromocytoma cells. J. Biol. Chem., 263, 2055–2063. [PubMed] [Google Scholar]
- Wienecke R., Maizie,J.C.J., Reed,J.A., de Gunzburg,J., Yeung,R.S. and Declue,J.E. (1997) Expression of the TSC2 product tuberin and its target Rap1 in normal human tissues. Am. J. Pathol., 150, 43–50. [PMC free article] [PubMed] [Google Scholar]
- Wu J., Dent,P., Jelinek,T., Wolfman,A., Weber,M.J. and Sturgill,T.W. (1993) Inhibition of the EGF-activated MAPK signaling pathway by adenosine 3′,5′-monophosphate. Science, 262, 1065–1072. [DOI] [PubMed] [Google Scholar]
- Yeung K. et al. (1999) Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature, 401, 173–177. [DOI] [PubMed] [Google Scholar]
- York R.D., Yao,H., Dillon,T., Ellig,C.L., Eckert,S.P., McCleskey,E.W. and Stork,P.S.J. (1998) Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature, 392, 622–626. [DOI] [PubMed] [Google Scholar]
- Young S.W., Dickens,M. and Tavare,J.M. (1994) Differentiation of PC12 cells in response to a cAMP analogue is accompanied by sustained activation of mitogen-activated protein kinase. Comparison with the effects of insulin, growth factors and phorbol esters. FEBS Lett., 338, 212–216. [DOI] [PubMed] [Google Scholar]
- Zimmermann S. and Moelling,K. (1999) Phosphorylation and regulation of Raf by Akt (protein kinase B). Science, 286, 1741–1744. [DOI] [PubMed] [Google Scholar]
- Zufall F., Shepherd,G.M. and Barnstable,C.J. (1997) Cyclic nucleotide gated channels as regulators of CNS development and plasticity. Curr. Opin. Neurobiol., 7, 404–412. [DOI] [PubMed] [Google Scholar]
- Zwartkruis F.J.T., Wolthuis,R.M.F., Nabben,N.M.J.M., Franke,B. and Bos,J.L. (1998) Extracellular signal-regulated activation of Rap1 fails to interfere in Ras effector signalling. EMBO J., 17, 5905–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]