

Abstract
The 90 kDa ribosomal S6 kinase-2 (RSK2) is a growth factor-stimulated protein kinase with two kinase domains. The C-terminal kinase of RSK2 is activated by ERK-type MAP kinases, leading to autophosphorylation of RSK2 at Ser386 in a hydrophobic motif. The N-terminal kinase is activated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) through phosphorylation of Ser227, and phosphorylates the substrates of RSK. Here, we identify Ser386 in the hydrophobic motif of RSK2 as a phosphorylation-dependent docking site and activator of PDK1. Treatment of cells with growth factor induced recruitment of PDK1 to the Ser386-phosphorylated hydrophobic motif and phosphorylation of RSK2 at Ser227. A RSK2-S386K mutant showed no interaction with PDK1 or phosphorylation at Ser227. Interaction with Ser386-phosphorylated RSK2 induced autophosphorylation of PDK1. Addition of a synthetic phosphoSer386 peptide (RSK2373–396) increased PDK1 activity 6-fold in vitro. Finally, mutants of RSK2 and MSK1, a RSK-related kinase, with increased affinity for PDK1, were constitutively active in vivo and phosphorylated histone H3. Our results suggest a novel regulatory mechanism based on phosphoserine-mediated recruitment of PDK1 to RSK2, leading to coordinated phosphorylation and activation of PDK1 and RSK2.
Keywords: ERK/growth factor/MSK1/PDK1/RSK2
Introduction
The 90 kDa ribosomal S6 kinases (RSK1–4) are a family of broadly expressed serine/threonine kinases that are activated by extracellular signal-regulated protein kinase (ERK1 and ERK2) in response to many growth factors, peptide hormones and neurotransmitters (reviewed in Frödin and Gammeltoft, 1999; Nebreda and Gavin, 1999). RSK is an important effector of ERK in the regulation of cell division and survival. In Xenopus laevis oocytes, RSK regulates G2–M phase progression in meiosis I through phosphorylation of the p34cdc2-inhibitory kinase Myt1 (Palmer et al., 1998). During meiosis II, RSK induces metaphase arrest to prevent cell division in the unfertilized egg (Bhatt and Ferrell, 1999; Gross et al., 1999). In cerebellar granule neurons, RSK phosphorylates and inactivates the pro-apoptotic protein BAD (Bonni et al., 1999). RSK has been proposed to phosphorylate and activate transcription factors like estrogen receptor-α (Joel et al., 1998) and CREB (Xing et al., 1996), and to bind to and modulate the function of CBP and p300, which are transcriptional co-activators (Nakajima et al., 1996). Moreover, RSK2 phosphorylates histone H3 at Ser10, which may cause chromatin remodeling and facilitate gene transcription (Sassone-Corsi et al., 1999). Inactivating mutations in the RSK2 gene are responsible for human Coffin–Lowry syndrome, which is characterized by severe mental retardation and progressive skeletal deformations (Trivier et al., 1996). Mutations in RSK4 may also lead to mental retardation (Yntema et al., 1999).
Recently, two mitogen- and stress-activated protein kinases (MSK) were discovered that are homologous to RSK (Deak et al., 1998; Pierrat et al., 1998). MSK is activated by ERK as well as by p38 MAP kinase in response to growth factors and various cellular stress stimuli. MSK appears to be a major growth factor-activated CREB kinase (Deak et al., 1998; Pierrat et al., 1998) and has been associated with phosphorylation of histone H3 and HMG-14 (Thomson et al., 1999), suggesting a role of MSK in transcriptional regulation.
RSK is composed of two kinase domains connected by an ∼100-amino-acid linker region (Figure 1). The N-terminal kinase (NTK) phosphorylates the substrates of RSK and its activity is regulated by the C-terminal kinase (CTK) and the linker. Activation of RSK requires phosphorylation at four sites (Dalby et al., 1998). As a probable sequence of events, ERK phosphorylates Ser369 in the linker (using mouse RSK2 numbering) and Thr577 in the activation loop of the CTK, leading to its activation (Fisher and Blenis, 1996; Dalby et al., 1998). The CTK then phosphorylates Ser386 in the linker (Vik and Ryder, 1997). Finally, the NTK requires phosphorylation at Ser227 in the activation loop, which is catalyzed by 3-phosphoinositide-dependent protein kinase-1 (PDK1) (Jensen et al., 1999; Richards et al., 1999; Williams et al., 2000). While phosphorylation at Ser227 directly activates the NTK (Jensen et al., 1999), it is not known how phosphorylation at Ser386 modulates the activity of the NTK.

Fig. 1. Structure and regulatory phosphorylation sites of RSK2. RSK is composed of two kinase domains connected by a regulatory linker region. The C-terminal tail contains a docking site for ERK (Gavin and Nebreda, 1999; Smith et al., 1999). The locations of five phosphorylation sites and the kinases that phosphorylate these sites are shown. Phosphorylation at Ser227, Ser369, Ser386 and Thr577 regulates kinase activity, whereas the role of Thr365 phosphorylation is unclear (mouse RSK2 numbering). Amino acid sequences show the PDK1 consensus phosphorylation motif and the hydrophobic motif with conserved residues in bold. The alignment illustrates that both motifs are present in many growth factor-activated kinases, including RSK, MSK, p70 S6K, PKBα, PKCδ, SGK and PRK2. Serines/threonines that require phosphorylation for kinase activation are shown in red. Sequences are human, except RSK1 (rat), RSK2 and PKCδ (both mouse).
PDK1 plays a central role in the activation of several growth factor-induced protein kinases (reviewed in Belham et al., 1999), which include protein kinase B (PKB) (Alessi et al., 1997a; Stephens et al., 1998), p70 S6 kinase (Alessi et al., 1997b; Pullen et al., 1998), several PKC isotypes (Dutil et al., 1998; Le Good et al., 1998) and serum and glucocorticoid-induced kinase (SGK) (Kobayashi and Cohen, 1999; Park et al., 1999). These kinases are all phosphorylated by PDK1 at a serine or threonine in the activation loop analogous to Ser227 in RSK2 (Figure 1). In addition, activation of these kinases requires phosphorylation at a serine or threonine residue located C-terminally to the kinase domain analogous to Ser386 in RSK. This phosphorylation site is situated in a characteristic hydrophobic motif (Figure 1), first recognized in p70 S6 kinase (Pearson et al., 1995).
The mechanisms that determine the specificity of PDK1 action towards its multiple kinase substrates are only partly characterized. Activation of PKB by PDK1 requires co-localization at the plasma membrane, mediated by 3-phosphoinositide binding of the pleckstrin homology (PH) domain of the two kinases (Andjelkovic et al., 1997; Anderson et al., 1998; Currie et al., 1999). In addition, 3-phosphoinositides convert PKB into a good substrate for PDK1, possibly through a conformational change (Alessi et al., 1997a; Stokoe et al., 1997). Phosphorylation of p70 S6 kinase and SGK by PDK1 depends on phosphorylation events occurring elsewhere in these kinases, including the hydrophobic motif (Alessi et al., 1997b; Dennis et al., 1998; Pullen et al., 1998; Kobayashi and Cohen, 1999). The catalytic activity of PDK1 is not known to be regulated. Thus, the apparently constitutive activity of PDK1 was not increased by growth factor treatment of cells or by interaction of 3-phosphoinositides with the PH domain of PDK1 (Alessi et al., 1997a,b; Pullen et al., 1998).
In this study, we have investigated the interaction and regulation of RSK2 and PDK1. We report a mechanism for activation of PDK1 and specificity of PDK1 action that is based on phosphoserine recognition. Thus, growth factor-induced phosphorylation at Ser386 in the hydrophobic motif of RSK2 creates a binding site that recruits PDK1 and stimulates its catalytic activity towards Ser227 in RSK2.
Results
Ser386 in the hydrophobic motif of RSK2 is required for phosphorylation of Ser227 and activation of the NTK domain
To study the role of the linker in regulation of RSK2, we generated a series of mutants composed of the NTK domain with increasing portions of the linker region. The mutants were expressed in COS7 cells and their kinase activity was determined and compared with full-length RSK2. The deletion mutants RSK21–360, RSK21–373 and RSK21–380 were nearly inactive in cells (Figure 2). In contrast, RSK21–389 showed 2.5-fold higher kinase activity than full-length RSK2 from unstimulated cells, thus behaving as a constitutively active RSK2 mutant. Residues 381–389 include the hydrophobic motif and Ser386, which is phosphorylated by the CTK. Finally, RSK21–417 and RSK21–422 possessed roughly the same activity as RSK21–389 when corrected for their lower expression (Figure 2). RSK21–360, RSK21–373 and RSK21–380 were not inactive due to misfolding, since they could be activated to the same level as RSK21–389 by co-expression with PDK1 (data not shown). In conclusion, the hydrophobic motif is capable of activating the isolated NTK domain of RSK2 in vivo.
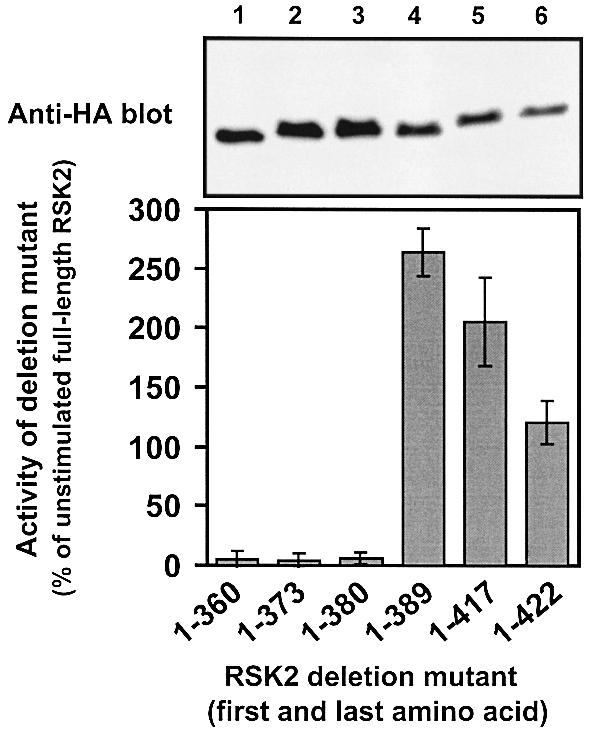
Fig. 2. Kinase activity of RSK2 deletion mutants in vivo. COS7 cells were transfected with plasmids expressing HA epitope-tagged full-length RSK2 or deletion mutants. After 48 h and a final 3 h serum starvation period, cells were lysed and RSK was precipitated with Ab to the HA epitope tag and subjected to kinase assay (lower panel). Data are expressed as a percentage of full-length RSK2 from unstimulated cells and are the mean ± SD of three independent experiments performed in duplicate. The activities of RSK21–389 and unstimulated RSK2 were different (p<0.001) when compared by non-paired t-test. After the kinase assay, RSK was subjected to SDS–PAGE and immunoblotting with Ab to the HA epitope tag (upper panel).
We investigated the phosphorylation of RSK21–389 using phospho-specific antibodies (Abs) to the four phosphorylation sites present in this mutant: Ser227, Thr365, Ser369 and Ser386. Surprisingly, RSK21–389 was heavily phosphorylated at Ser386, even though the mutant contains no CTK domain (Figure 3B, lanes 3 and 4). Apparently, phosphorylation of Ser386 is required for the activity of RSK21–389, since the mutant RSK21–389S386A showed greatly reduced kinase activity (Figure 3A, lanes 5 and 6). Exposure of the cells to epidermal growth factor (EGF) did not increase phosphorylation at Ser386 or kinase activity of RSK21–389, in contrast to full-length RSK2 (Figure 3A and B). Interestingly, the PDK1 site, Ser227, was also highly phosphorylated in RSK21–389, but only marginally phosphorylated in RSK21–389S386A and not phosphorylated in RSK21–380 (Figure 3C). Phosphorylation at Ser227 was strongly induced by EGF in full-length RSK2, but not in the mutants (Figure 3C). Finally, none of the RSK2 mutants showed any phosphorylation at the two ERK phosphorylation sites in the linker (Thr365 and Ser369) under basal or EGF-stimulated conditions. In contrast, full-length RSK2 showed EGF-induced phosphorylation at these sites (Figure 3D and E). All constructs were present at roughly similar levels in the precipitates, as shown by immunoblotting for their hemagglutinin (HA) epitope tag (Figure 3F). These data suggest that RSK21–389 is active in cells because it is constitutively phosphorylated at Ser386 and Ser227. Moreover, the two phosphorylation sites exhibit a hierarchical relationship since phosphorylation at Ser386 is required for the phosphorylation of Ser227.
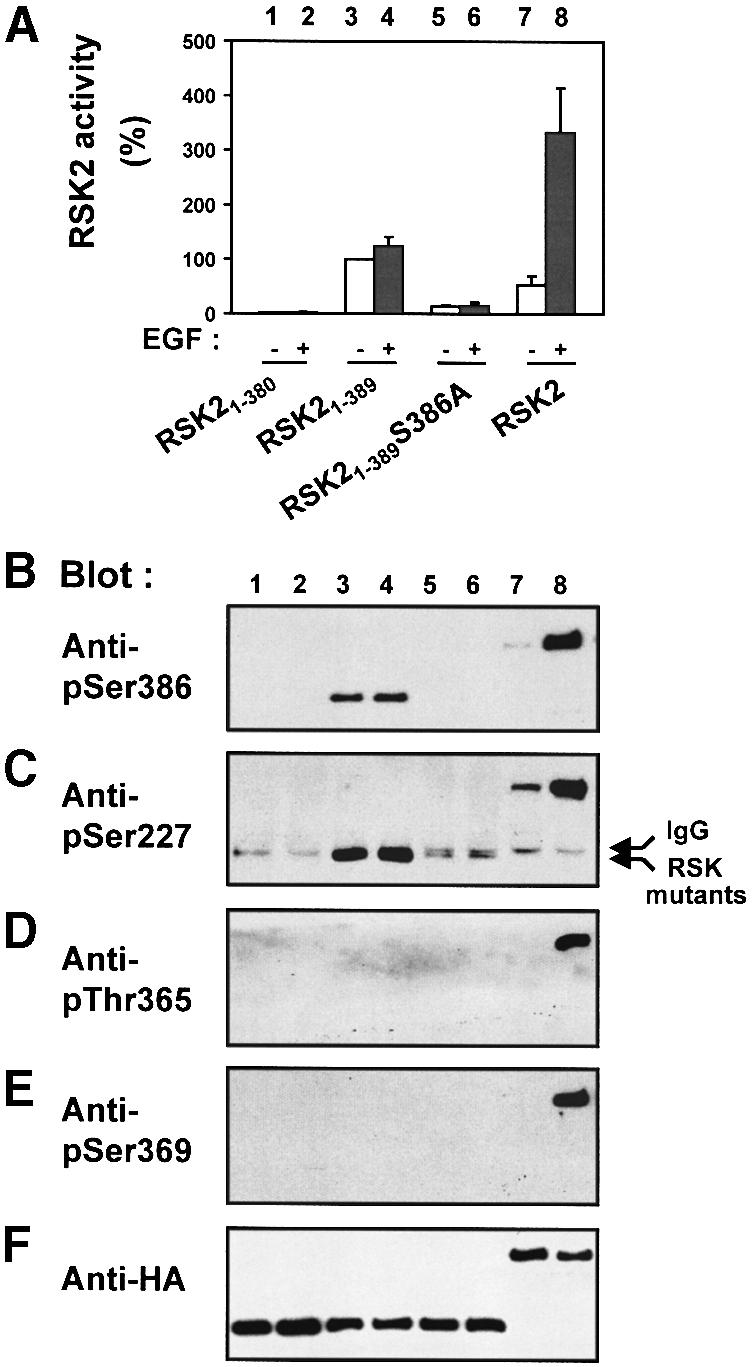
Fig. 3. Role of the hydrophobic motif in regulation of phosphorylation sites in the N-terminal kinase of RSK2. COS7 cells were transfected with plasmids expressing HA epitope-tagged wild-type RSK2 or mutant RSK2. After 48 h and a final 3 h serum starvation period, cells were exposed, or not, to 20 nM EGF for 20 min and lysed. Thereafter, RSK was precipitated from the cell lysates with Ab to the HA epitope. (A) The kinase activity of RSK was determined and expressed as a percentage of basal RSK21–389 activity. Data are the mean ± SD of four independent experiments performed in duplicate. The data of the following bars were different when compared by non-paired t-test: 3 versus 5 (p <0.001); 3 versus 7 (p <0.05). After the kinase assay, aliquots of the precipitated RSKs were subjected to SDS–PAGE and immunoblotting with Abs that bind RSK2 when phosphorylated at Ser386 (B), Ser227 (C), Thr365 (D) or Ser369 (E), or with Ab to the HA tag (F).
A hierarchy between phosphorylation at Ser386 and Ser227 was also observed in full-length RSK2. Thus, the mutant RSK2-S386K showed no phosphorylation at Ser227 in unstimulated or EGF-treated cells (Figure 4B) and was inactive (Figure 4A). In contrast, phosphorylation at the two ERK phosphorylation sites, Thr365 and Ser369, occurred normally in RSK2-S386K in response to EGF (Figure 4C and D). Moreover, mutation of the two ERK sites did not affect phosphorylation at Ser227 or Ser386 (data not shown). The failure of RSK2-S386K to undergo phosphorylation at Ser227 provides an explanation for the observation that Ser386 is required for the activation of RSK1 (Dalby et al., 1998) and RSK2 (Jensen et al., 1999).
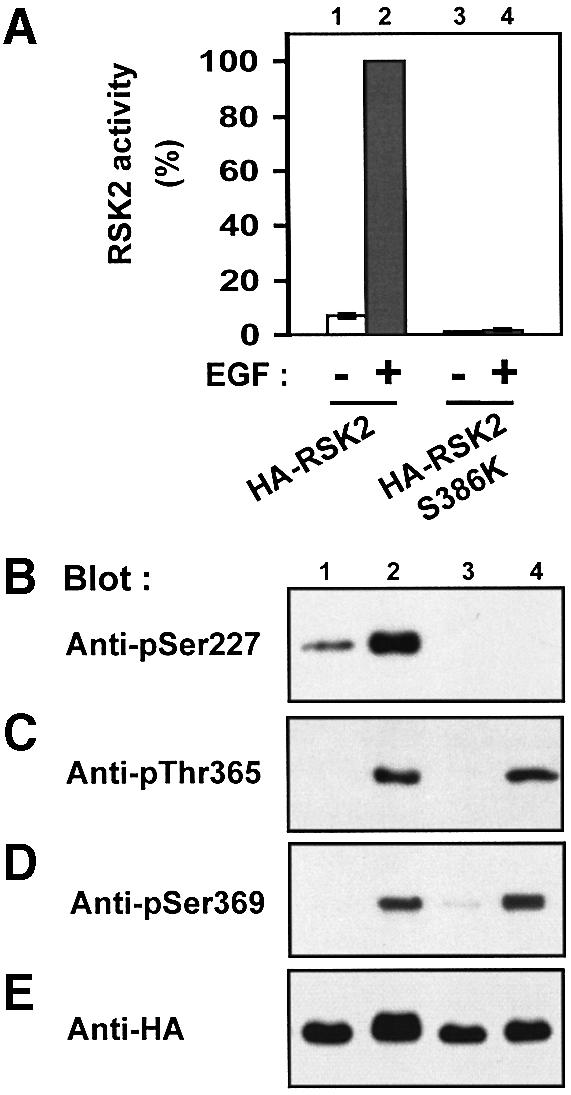
Fig. 4. Ser386 is required for phosphorylation of Ser227 in RSK2. COS7 cells were transfected with plasmids expressing HA-tagged RSK2 or RSK2-S386K. After 48 h and a final 3 h serum starvation period, cells were exposed, or not, to 20 nM EGF for 20 min and lysed. (A) RSK was precipitated from the cell lysates with Ab to the HA tag, its kinase activity was determined and expressed as a percentage of EGF-stimulated wild-type RSK2 activity. Data are the mean ± SD of three independent experiments performed in duplicate. After the kinase assay, aliquots of the precipitated RSKs were subjected to SDS–PAGE and immunoblotting with Abs that bind RSK2 when phosphorylated at Ser227 (B), Thr365 (C), Ser369 (D) or with Ab to the HA epitope tag (E).
The hydrophobic motif of RSK2 is a phosphorylation-dependent docking site for PDK1
Since RSK2-S386K was not phosphorylated at the PDK1 site, we investigated whether this mutant was incapable of interacting with PDK1 as compared with wild-type RSK2. Myc epitope-tagged PDK1 was co-expressed with RSK2 or RSK2-S386K in COS7 cells, followed by precipitation of RSK2 with Ab to the HA tag and immunoblotting for co-precipitated PDK1. Cells were not exposed to EGF prior to lysis, since co-expression with PDK1 induces substantial phosphorylation of RSK2 at Ser386 by a direct or indirect mechanism (Jensen et al., 1999). As shown in Figure 5A, RSK2 precipitated significantly more PDK1 than did RSK2-S386K. Control immunoblots showed that the precipitates contained equal amounts of RSK2 and RSK2-S386K (Figure 5B), and that PDK1 was expressed at similar levels in the two co-transfection conditions (Figure 5C). PDK1 was also found to precipitate with RSK1 and RSK3 from lysates of co-transfected cells (data not shown).
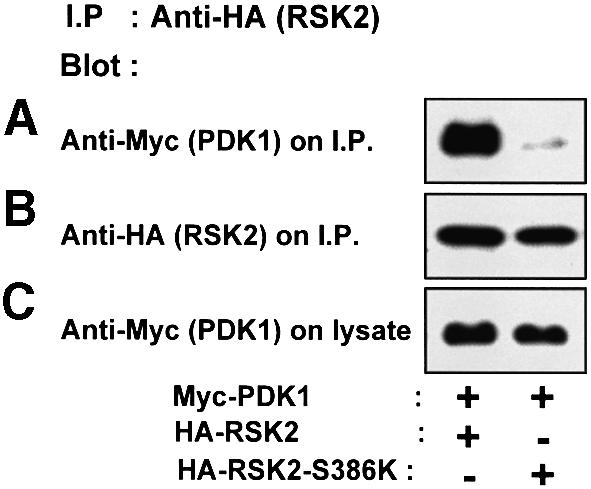
Fig. 5. Ser386 is required for RSK2 to interact with PDK1. COS7 cells were co-transfected with plasmids expressing myc-PDK1 and HA-RSK2 or HA-RSK2-S386K. After 48 h and a final 3 h serum starvation period, the cells were lysed and cell lysates were subjected to immunoprecipitation with Ab to the HA tag. Aliquots of the precipitates were subjected to SDS–PAGE and immunoblotting with Ab to the myc epitope tag on PDK1 (A) or to the HA epitope tag on RSK2 (B). Equal expression of PDK1 in the cells was verified by subjecting pre-immunoprecipitation lysates to immunoblotting for the myc tag (C). The experiments were performed three times with similar results.
In order to identify the binding site for PDK1 in RSK, we assayed the ability of linker deletion mutants of RSK2 to precipitate co-expressed myc-PDK1. As shown in Figure 6A, PDK1 co-precipitated with RSK21–389, but much less so with RSK21–380 or with RSK21–389S386A. This suggests that the hydrophobic motif constitutes the major binding site for PDK1 in RSK2 and that the motif must be phosphorylated at Ser386 to bind PDK1 efficiently. In agreement with this conclusion, the omission of phosphatase inhibitors in the precipitation buffer led to nearly complete dephosphorylation of Ser386 and to disruption of the complex of RSK21–389 and PDK1 (Figure 6B). In an attempt to mimic the negative charge of a phosphate group, we mutated Ser386 in RSK21–389 to aspartic acid. RSK21–389S386D showed less complex formation with PDK1 than RSK21–389, indicating that Asp at this position only partially mimicks a phosphate group. Importantly, however, the association of RSK21–389 S386D with PDK1 was hardly affected by omission of phosphatase inhibitors during immunoprecipitation (Figure 6B). This suggests that the dephosphorylation that disrupts the complex of PDK1 and RSK21–389 primarily occurs at phosphorylated Ser386.
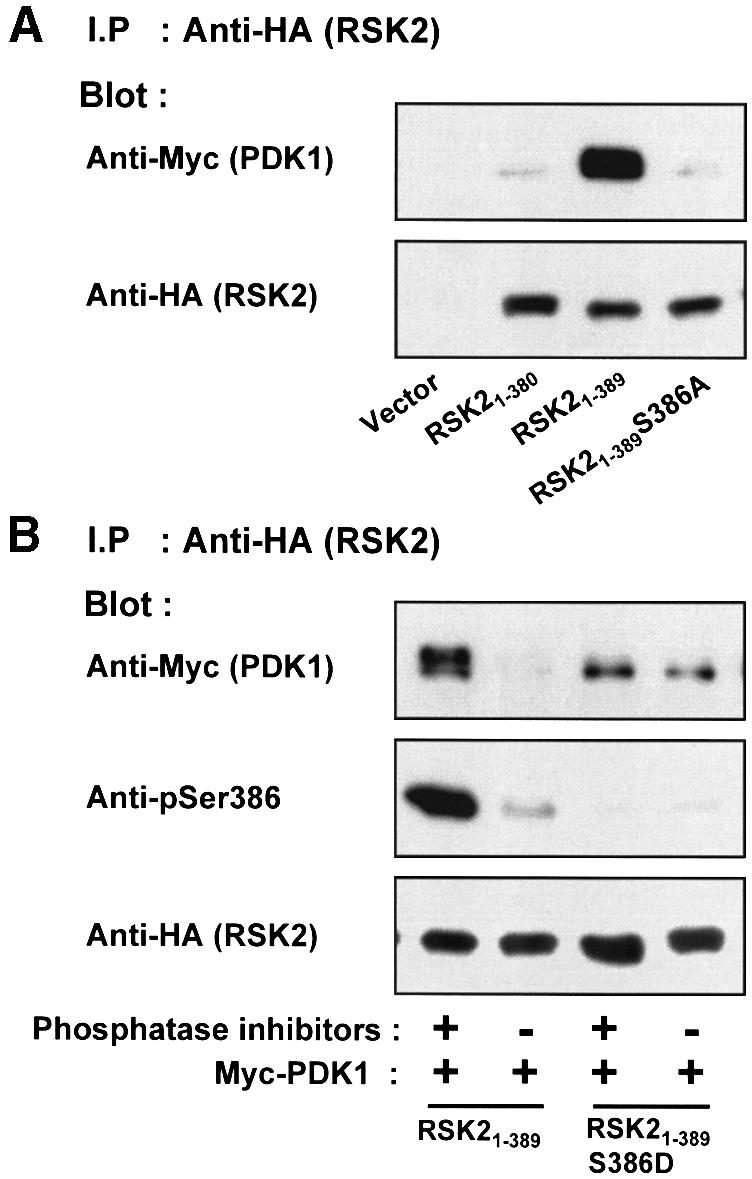
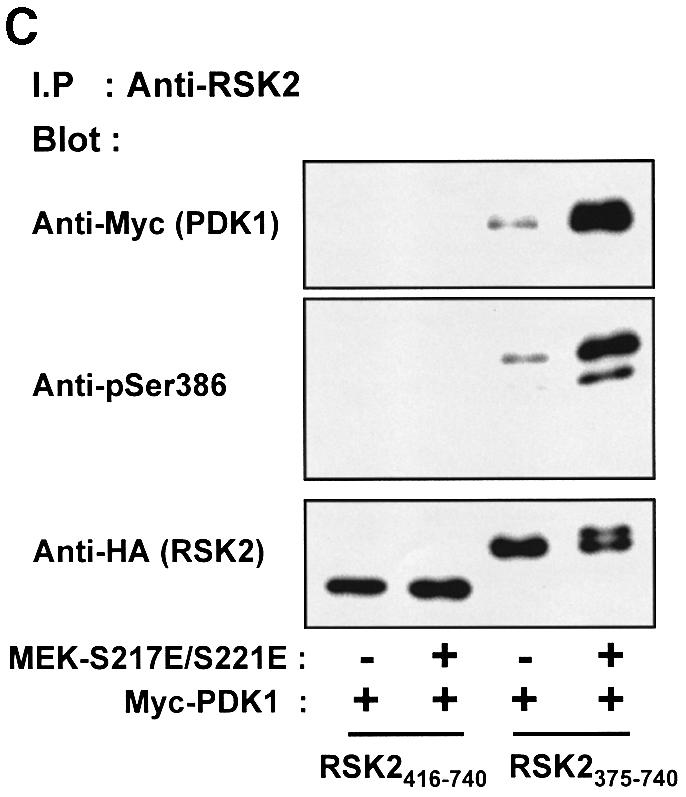
Fig. 6. PDK1 interacts with RSK2 via the Ser386-phosphorylated hydrophobic motif. COS7 cells were transfected with plasmids expressing myc-PDK1, HA-RSK2, HA-RSK2 mutant, MEK-S217/221E or with empty vector (–), as indicated in each panel. After 48 h and a final 3 h serum starvation period, the cells were lysed. (A) Cell lysates were subjected to immunoprecipitation with Ab to the HA tag, whereafter aliquots of the precipitates were subjected to SDS–PAGE and immunoblotting with Ab to the myc tag in PDK1 or to the HA tag in RSK2. (B) Cell lysates were subjected to immunoprecipitation with Ab to the HA tag in lysis buffer with or without phosphatase inhibitors (orthovanadate, calyculin, sodium flouride). Aliquots of the precipitates were subjected to SDS–PAGE and immunoblotting with Ab to the myc tag in PDK1, to phosphoSer386 in RSK2 or to the HA tag in RSK2. (C) Cell lysates were subjected to immunoprecipitation with Ab to the CTK domain of RSK2. Aliquots of the precipitates were subjected to SDS–PAGE and immunoblotting with Ab to the myc tag in PDK1, to phosphoSer386 in RSK2 or to the HA tag in RSK2. The experiments were performed three times with similar results.
It could be argued that the hydrophobic motif is not the actual PDK1 binding site, but that the motif induces a conformational change in RSK2 that allows binding of PDK1 to a distinct site in the NTK domain. We therefore generated truncated mutants of RSK2 composed of the isolated CTK domain and linker sequence with (RSK2375–740) or without the hydrophobic motif (RSK2416–740) and assayed their ability to co-precipitate PDK1. As shown in Figure 6C, PDK1 only formed a complex with the CTK mutant that includes the hydrophobic motif. Moreover, larger amounts of PDK1 were co-precipitated with RSK2375–740 if the hydrophobic motif was phosphorylated at Ser386, achieved through co-expression with constitutively active MEK-S217E/S221E (Figure 6C). In this experiment, MEK-S217E/S221E presumably activates endogenous ERK, which in turn activates the CTK domain, resulting in autophosphorylation at Ser386.
To relate these findings to the activation mechanism of RSK, we investigated whether stimulation of cells with EGF resulted in increased complex formation between PDK1 and full-length RSK2. In this experiment, we used a kinase-deficient PDK1 mutant (PDK1-KD), since co-expression with wild-type PDK1 induces phosphorylation at Ser386 in full-length RSK2 (Jensen et al., 1999). As shown in Figure 7A, EGF treatment increased the amount of PDK1-KD in the RSK2 precipitate (concomitantly with induction of Ser386 phosphorylation; data not shown). In contrast, EGF did not stimulate complex formation between RSK2-S386K and PDK1-KD. Control immunoblots showed that precipitates from untreated and EGF-stimulated cells contained equal amounts of RSK2 or RSK2-S386K (Figure 7B) and that PDK1-KD was present at similar levels in the various cell lysates (Figure 7C).
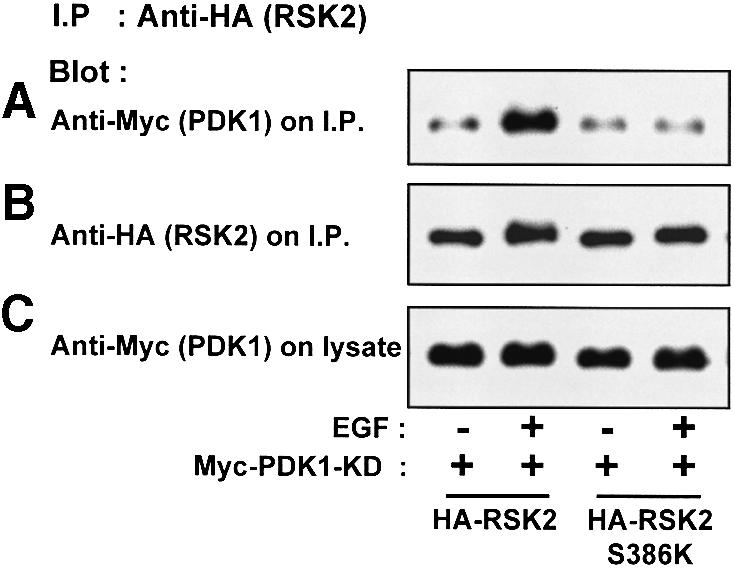
Fig. 7. Recruitment of PDK1 to RSK2 in growth factor-treated cells. COS7 cells were co-transfected with plasmids expressing kinase-deficient (KD) myc-PDK1 and HA-RSK2 or HA-RSK2-S386K, as indicated. After 48 h and a final 3 h serum starvation period, cells were exposed, or not, to 20 nM EGF for 25 min and then lysed. The cell lysates were subjected to immunoprecipitation with Ab to the HA tag. Aliquots of the precipitates were subjected to SDS–PAGE and immunoblotting with Ab to the myc tag in PDK1 (A) or to the HA tag in RSK2 (B). Equal expression of PDK1 in the cells was verified by subjecting pre-immunoprecipitation lysates to immunoblotting for the myc tag (C). The experiments were performed three times with similar results.
In conclusion, our findings strongly suggest that the hydrophobic motif of RSK2 is a phosphoSer386-dependent docking site for PDK1. A model can be proposed in which signal input from the ERK pathway induces phosphorylation of the hydrophobic motif of RSK2, thereby creating a binding site that recruits PDK1 for phosphorylation of Ser227 and activation of the NTK domain.
Binding of PDK1 to the Ser386-phosphorylated hydrophobic motif stimulates its autophosphorylation and catalytic activity
In the experiments described above, we noticed that PDK1 exhibits an upward shift in electrophoretic mobility when co-expressed with RSK2 or RSK21–389, which is indicative of phosphorylation of PDK1. Extended electrophoresis of co-expression lysates revealed that PDK1 migrates as up to four discrete bands when co-expressed with RSK2, RSK21–389, kinase-deficient RSK21–389 and RSK2375–740 (co-expressed with active MEK), which all contain phosphorylated Ser386 (Figure 8A). In contrast, PDK1 migrated as one band when co-expressed with RSK2 mutants that did not contain phosphorylated Ser386, i.e. RSK2-S386K, RSK2375–740 (without co-expression of active MEK), RSK2416–740 and RSK21–389S386A. Kinase-deficient PDK1 showed no mobility shift when co-expressed with RSK2 or RSK21–389 (Figure 8A). These data suggest that PDK1 undergoes autophosphorylation at several sites when it interacts with the Ser386-phosphorylated hydrophobic motif of RSK2. To investigate whether autophosphorylation of PDK1 occurs in an intra- or intermolecular manner, COS7 cells were co-transfected with RSK21–389, PDK1-KD and the truncated mutant PDK151–556. As shown in Figure 8B, RSK21–389 induced autophosphorylation of PDK151–556, but did not promote PDK151–556 to phosphorylate PDK1-KD. This suggests that PDK1 undergoes intramolecular autophosphorylation following binding to the hydrophobic motif of RSK2.
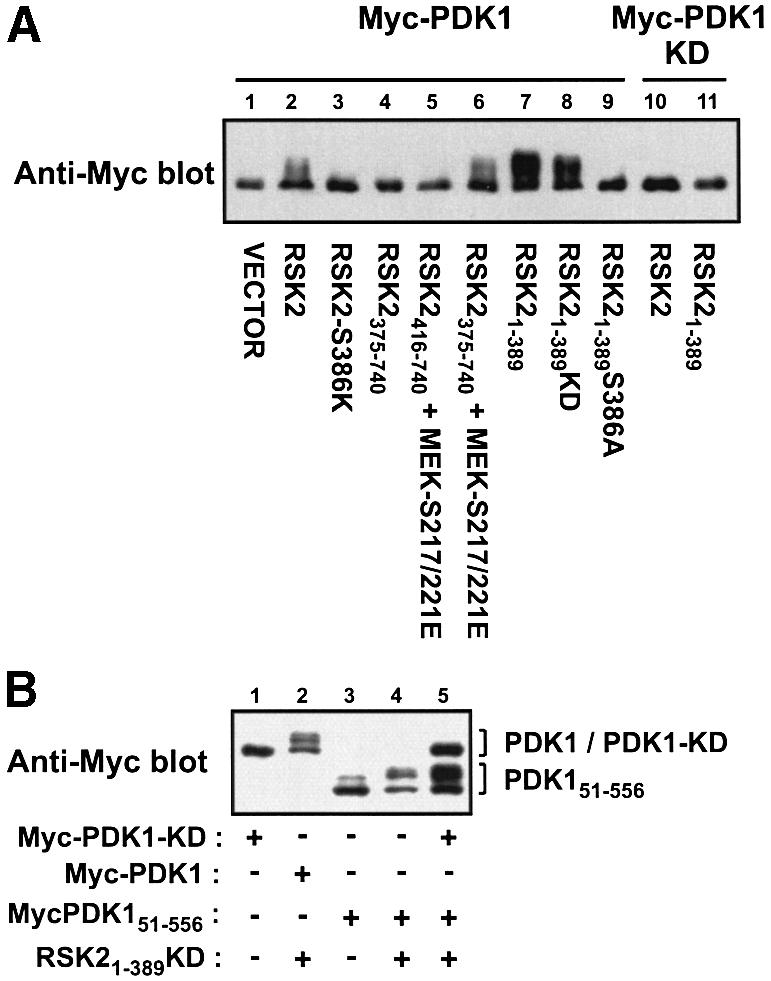
Fig. 8. Autophosphorylation of PDK1 after binding to the Ser386-phosphorylated hydrophobic motif of RSK2 in vivo. (A and B) COS7 cells were co-transfected with plasmids expressing wild-type or mutant forms of myc-PDK1 and HA-RSK2 and with MEK-S217/221E as indicated. KD denotes kinase-deficient mutant. Forty-eight hours after transfection, cells were serum starved for 3 h. Thereafter, cells were lysed in SDS–PAGE sample buffer and subjected to SDS–PAGE and immunoblotting with Ab to the myc tag in PDK1. The experiments were performed three times with similar results.
PDK1 autophosphorylation was induced in vitro by incubation of immunopurified PDK1 with [γ-32P]ATP and pSer386 peptide, a synthetic peptide of RSK2 residues 373–396 containing phosphorylated Ser386 (Figure 9A). In contrast, PDK1 incubated alone, with S6 peptide or with Ser386 peptide (which is non-phosphorylated) showed little autophosphorylation. Electrophoretic mobility shift assay showed that pSer386 peptide induced autophosphorylation of >90% of PDK1 in the in vitro phosphorylation reaction (Figure 9B).
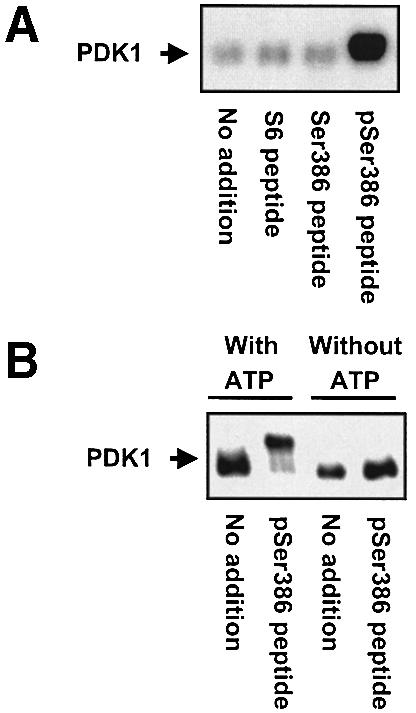
Fig. 9. Autophosphorylation of PDK1 induced by an RSK2 peptide containing phospho-Ser386 in vitro. Immunopurified myc-PDK1 was pre-incubated for 20 min alone (no addition) or with 10 µM S6 peptide or RSK2 peptide, residues 373–396, which was unphosphorylated (Ser386 peptide) or phosphorylated at Ser386 (pSer386 peptide). (A) PDK1 was allowed to autophosphorylate for 20 min in the presence of [γ-32P]ATP, whereafter the reactions were subjected to SDS–PAGE and autoradiography. (B) PDK1 was allowed to autophosphorylate for 35 min in the presence or absence of ATP, whereafter the reactions were subjected to SDS–PAGE and immunoblotting with Ab to the myc tag in PDK1. The experiments were performed twice with similar results.
We next investigated whether the increase in autophosphorylation of PDK1 was associated with catalytic activation of PDK1. Immunopurified PDK1 was subjected to in vitro kinase assay using bacterially expressed, kinase-deficient RSK21–373 as a substrate. Incubation with pSer386 peptide increased the kinase activity of PDK1 6-fold as compared with incubation alone, with S6 peptide or with Ser386 peptide (Figure 10A and B). Phosphorylation of NTK by PDK1 occurred exclusively at Ser227, since the mutant RSK21–360S227E was not phosphorylated by PDK1, in contrast to RSK21–360 (Figure 10C). Activation of PDK1 by pSer386 peptide was also observed when using S6 peptide as a substrate (data not shown), although S6 peptide is a rather poor substrate for PDK1, since it does not contain a PDK1 consensus phosphorylation sequence.
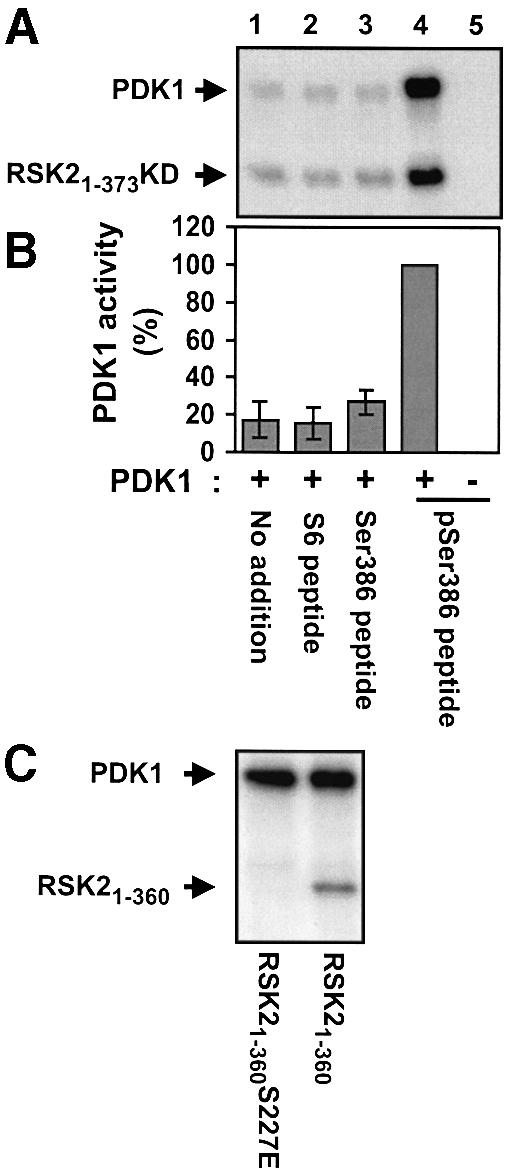
Fig. 10. Activation of PDK1 by a RSK2 peptide containing phosphoSer386 in vitro. Immunopurified myc-PDK1 was pre-incubated for 20 min alone (no addition) or with 10 µM S6 peptide, Ser386 peptide or pSer386 peptide. (A and C) PDK1 was then incubated for 10 min with [γ-32P]ATP and 2 µg of RSK21–373KD or 0.5 µg of RSK21–360 as a substrate, as indicated in each panel, whereafter the reactions were subjected to SDS–PAGE and autoradiography. (B) PDK1 activity in (A) was determined by quantitation of radioactivity incorporated into RSK21–373KD using a PhosphorImager. Data are expressed as a percentage of PDK1 activity in the presence of pSer386 peptide and are the mean ± SD of three independent experiments. The data of the following bars were not different when compared by non-paired t-test: 1 versus 2 or 3 (p >0.2). The data from bars 1 and 4 were different when compared by non-paired t-test (p <0.001).
These data suggest that PDK1 is activated upon interaction with phosphoSer386 in the hydrophobic motif of RSK2, leading to intramolecular autophosphorylation of PDK1 and phosphorylation of RSK2 at Ser227. This represents a novel regulatory mechanism for PDK1, in which PDK1 is activated through interaction with its substrate RSK2 and thus may be indirectly activated by the ERK signaling pathway.
Constitutively active mutants of RSK2 and MSK1 phosphorylate histone H3 in vivo
We used the new insight into the activation mechanism of RSK2 to generate constitutively active kinases of RSK2 and MSK1. RSK21–389 was not phosphorylated at the two ERK phosphorylation sites in the linker (see Figure 3D and E), raising the possibility that the introduction of negatively charged amino acids at these sites would further increase its activity. Indeed, RSK21–389T365E and RSK21–389S369E showed 60–80% higher kinase activity than RSK21–389 (Figure 11A). The increased activity of RSK21–389T365E provides the first evidence for a role of Thr365 phosphorylation in activation of RSK. Mutation of both ERK sites generated a mutant, RSK21–389T365E/S369E, with the same activity as EGF-stimulated wild-type RSK2. Finally, we attempted to activate RSK2 by enhancing the docking of PDK1 to the linker. Recently, the hydrophobic motif of protein kinase C-related kinase-2 (PRK2) was isolated as a PDK1-interacting fragment in a yeast two-hybrid screen (Balendran et al., 1999a). The hydrophobic motif of PRK2 contains the three characteristic aromatic residues, but in addition it contains two Asp residues, one of which is situated at the position analogous to Ser386 in RSK2 (confer Figure 1). Surface plasmon resonance measurements demonstrated a very tight interaction between the PRK2 fragment and PDK1 (Balendran et al., 1999a). We therefore replaced the hydrophobic motif of RSK21–389T365E/S369E with that of PRK2. The resultant chimeric molecule, termed constitutively active (CA)-RSK2, showed a 4-fold increase in its ability to precipitate co-expressed PDK1 compared with RSK21–389 T365E/S369E (data not shown) and was twice as active (Figure 11A), presumably due to efficient recruitment of endogenous PDK1.
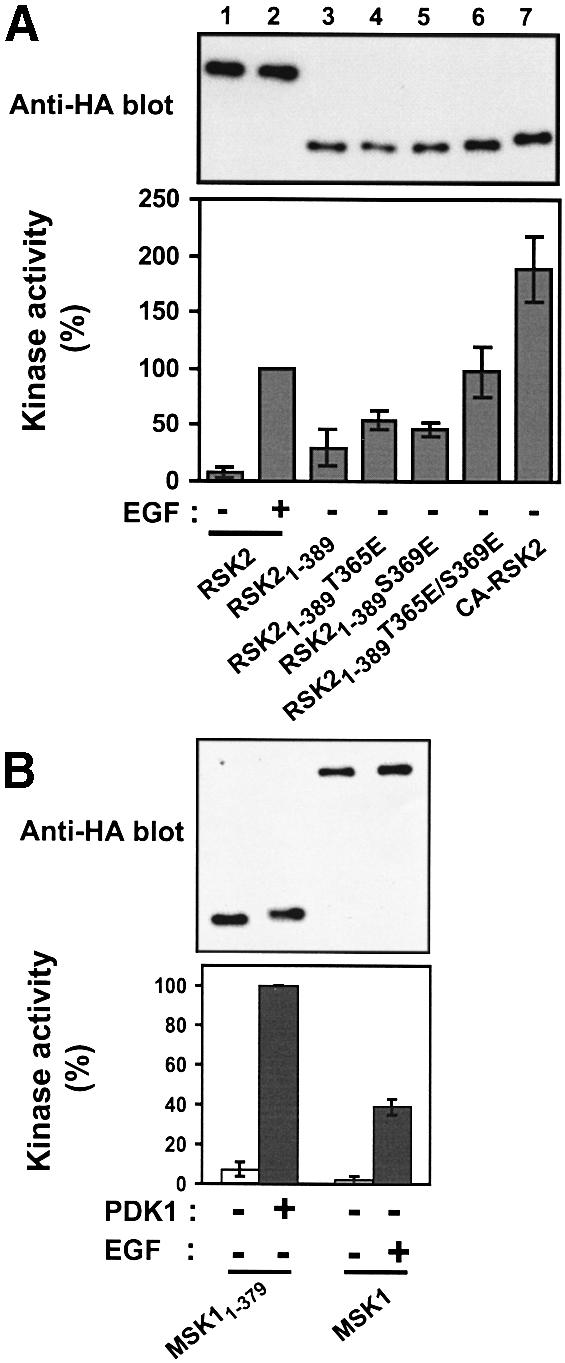
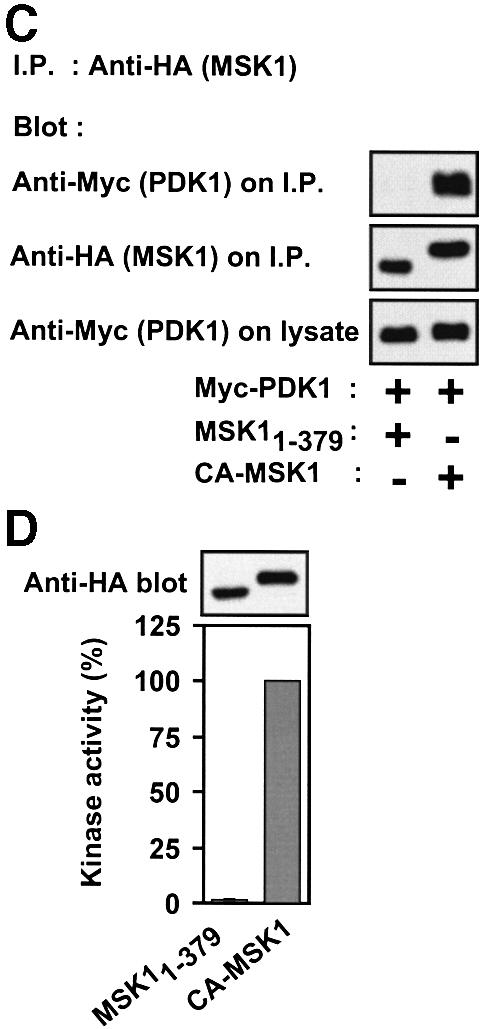
Fig. 11. Constitutively active mutants of RSK2 and MSK1. COS7 cells were transfected with plasmids expressing HA-tagged wild-type or mutant RSK2 or MSK1 and myc-tagged PDK1 as indicated in each panel. After 48 h and a final 3 h serum starvation period, cells were exposed, or not, to 20 nM EGF for 20 min and lysed. (A) RSK2 was precipitated from the cell lysates with Ab to the HA tag and subjected to kinase assay (lower panel). Data are expressed as a percentage of EGF-stimulated full-length RSK2 activity and are the mean ± SD of 3–4 independent experiments performed in duplicate The data of the following bars were different when compared by non-paired t-test: 3 versus 6 (p <0.05); 3 versus 7 (p <0.01). After the kinase assay, the precipitates were subjected to immunoblotting with Ab to the HA tag (upper panel). (B) MSK1 was precipitated from cell lysates with Ab to the HA tag, its kinase activity was determined and expressed as a percentage of EGF-stimulated wild-type MSK1 activity (lower panel). Data are the mean ± SD of four independent experiments performed in duplicate. After the kinase assay, MSK was subjected to immunoblotting with Ab to the HA tag (upper panel). (C) MSK1 mutants were precipitated from the cell lysates with Ab to the HA tag. Precipitates were subjected to immunoblotting with Ab to the myc tag in PDK1 or to the HA tag in MSK1. Equal expression of PDK1 in the cells was verified by subjecting pre-immunoprecipitation lysates to immunoblotting for the myc tag. The experiment was performed three times with similar results. (D) MSK1 mutants were precipitated from cell lysates with Ab to the HA tag and subjected to kinase assay (lower panel). Data are expressed as a percentage of CA-MSK1 activity and are the mean ± SD of three independent experiments performed in duplicate. After the kinase assay, the precipitates were subjected to immunoblotting with Ab to the HA tag (upper panel).
MSK1 contains a PDK1 consensus phosphorylation site in the NTK domain and a hydrophobic motif in the linker region. The deletion mutant MSK11–379, composed of the NTK domain and the linker truncated after the hydrophobic motif, was stimulated 14-fold by co-expression with PDK1, corresponding to twice the activity of full-length MSK1 in response to EGF (Figure 11B). However, compared with RSK21–389, MSK11–379 showed considerably less complex formation with PDK1 (data not shown). Replacement of the hydrophobic motif of MSK11–379 with that of PRK2 resulted in a mutant, CA-MSK1, with dramatically increased affinity for co-expressed PDK1 (Figure 11C) and a 50-fold increase in kinase activity compared with MSK11–379 (Figure 11D), most likely due to efficient recruitment of endogenous PDK1. The activity of CA-MSK1 was three times that of EGF-stimulated full-length MSK1.
We assessed the ability of the constitutively active mutants to phosphorylate histone H3, a recently proposed nuclear target of RSK2 and MSK1. In transfected COS7 cells, CA-RSK2 and CA-MSK1 were predominantly cytosolic, but the introduction of three nuclear localization sequences (NLS) at the N-terminus of these mutants resulted in complete nuclear localization, as analyzed by immunocytochemistry (data not shown). In transfected cells, NLS-CA-RSK2 and NLS-CA-MSK1 induced strong phosphorylation of histone H3 at Ser10, whereas kinase-deficient versions of these mutants induced no phosphorylation (Figure 12A). EGF-induced phosphorylation of histone H3 by wild-type RSK2 and MSK1 is shown for comparison. All constructs showed equal expression, except for the kinase-deficient mutants, which were somewhat less abundant (Figure 12B). Finally, we assayed in vivo phosphorylation of CREB at Ser133 by MSK1. As shown in Figure 12C, CREB phosphorylation was induced in cells transfected with NLS-CA-MSK1, but not with the kinase-deficient version. Vector-transfected cells show Ser133 phosphorylation by endogenous CREB-kinase activity in response to EGF.
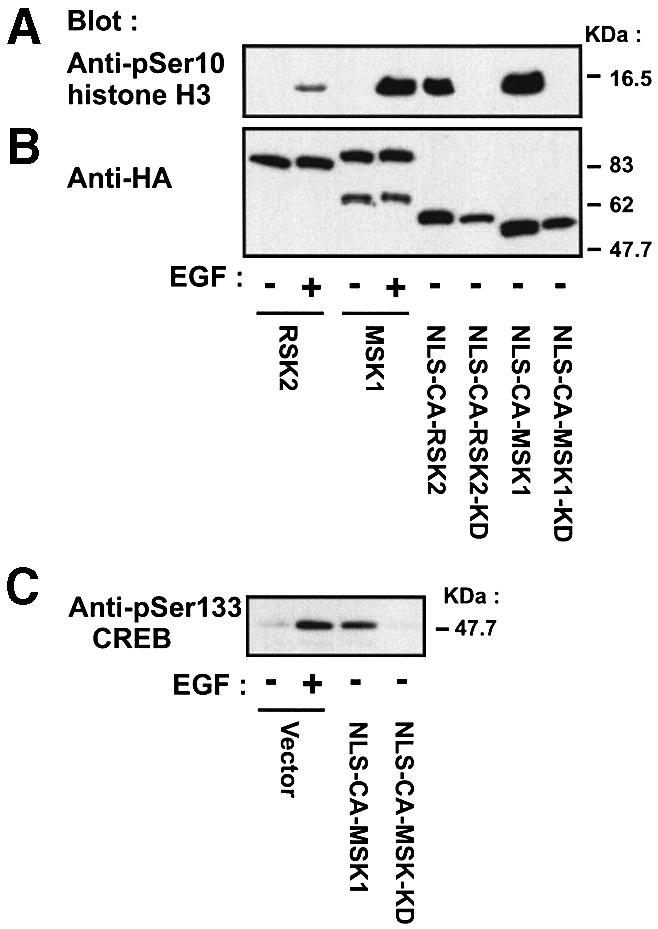
Fig. 12. In vivo phosphorylation of histone H3 and CREB by nuclear-targeted, constitutively active mutants of RSK2 and MSK1. COS7 cells were transfected with plasmids expressing HA-tagged wild-type or mutant RSK2 or MSK1. NLS, nuclear localization sequence. After 24 h and a final 3 h serum starvation period, cells were exposed, or not, to 20 nM EGF for 25 min. Cells were lysed in SDS–PAGE sample buffer and subjected to immunoblotting with Abs to histone H3 phosphorylated at Ser10 (A), to the HA tag (B) or to CREB phosphorylated at Ser133 (C).
Discussion
Recent studies have demonstrated a key role for PDK1 in the activation of several growth factor-activated protein kinases, including PKB, p70 S6 kinase, PKC, RSK and SGK, which regulate cellular survival, proliferation, protein synthesis and gene expression. These kinases are activated by distinct signaling pathways, but all require phosphorylation by PDK1 at a conserved serine/threonine in the activation loop. This has raised the issue of specificity in PDK1 action and why PDK1 is not stimulated by extracellular signals, in contrast to its substrates (discussed in Belham et al., 1999). Here, we report that PDK1 is recruited by a conserved motif in RSK2 in a phosphorylation-dependent manner, leading to stimulation of PDK1 kinase activity. This indicates that the catalytic activity of PDK1 is subject to regulation and suggests a substrate-directed targeting mechanism for specificity in PDK1 action.
Our results suggest a multi-step mechanism for the coordinated activation of PDK1 and RSK2 (Figure 13). The first step involves activation of ERK in response to an extracellular signal. ERK phosphorylates RSK2 at several sites, including Thr577 in the activation loop of the CTK, leading to autophosphorylation of RSK2 at Ser386 in the hydrophobic motif in the linker region.

Fig. 13. Model for recruitment and coordinated activation of PDK1 and RSK2. (1) Quiescent cells contain a pre-formed complex of inactive RSK and ERK. (2) Stimulation of the Ras–ERK pathway results in ERK-catalyzed phosphorylation of the linker and the CTK, leading to autophosphorylation of RSK2 at Ser386 in the hydrophobic motif (blue box). (3) Phosphorylation of Ser386 in the hydrophobic motif creates a docking site that allows complex formation between PDK1 and RSK2. (4) Interaction of PDK1 with the Ser386-phosphorylated hydrophobic motif stimulates PDK1 activity and autophosphorylation. The combination of increased local concentration and kinase activity of PDK1 ensures efficient phosphorylation of Ser227, leading to full activation of RSK.
The second step is recruitment of PDK1 to RSK2. PDK1 was found to interact with the hydrophobic motif after its phosphorylation at Ser386, and dephosphorylation reversed the interaction. Thus, the hydrophobic motif of RSK functions as a phosphorylation-dependent docking site for PDK1 that is induced by the ERK signaling pathway. In unstimulated cells, RSK21–389 showed substantial phosphorylation at Ser386, whereas full-length RSK2 and RSK2375–740 did not, suggesting that the CTK domain masks the hydrophobic motif under basal conditions. Activation of the CTK domain and phosphorylation of Ser386 may result in exposure of the hydrophobic motif, enabling it to interact with PDK1. The identity of the kinase that phosphorylates Ser386 in RSK21–389 is currently enigmatic.
The third step involves activation and autophosphorylation of PDK1, which then phosphorylates Ser227 in the NTK domain. Phosphorylation at Ser227 and Ser369 (and possibly Thr365) may result in full activation of RSK2. However, it is possible that phosphorylation at Ser386 may also play a role in enhancing the activity of the NTK apart from creating a docking site for PDK1.
Interaction with the phosphorylated hydrophobic motif may induce a conformational change in PDK1 that stimulates its catalytic activity and autophosphorylation. Five phosphorylation sites have been identified in PDK1 transiently expressed in HEK 293 cells (Casamayor et al., 1999). Phosphorylation of PDK1 was not altered by treating the cells with insulin-like growth factor-I and four of the sites could be mutated without loss of activity. However, phosphorylation of Ser241, situated in the activation loop, was essential for activity and appeared to be catalyzed by PDK1 itself. The mechanism for PDK1 activation and autophosphorylation described in the present study is based on a stoichiometric interaction of PDK1 and RSK2. It would be interesting to map the phosphorylation sites in PDK1 co-expressed with RSK2 or incubated with pSer386 peptide in vitro and determine their function.
The proposed model represents a new mechanism for the regulation of PDK1, in which PDK1 is activated in coordination with its kinase substrate. In the case of RSK2, PDK1 may be activated, indirectly, by the Ras–ERK pathway. The mechanism also provides specificity to the action of PDK1, since PDK1 is selectively recruited to the kinase that is phosphorylated at the hydrophobic motif. Finally, the mechanism may be efficient, since it combines increased local concentration of PDK1 (mediated by the recruitment step) with increased kinase activity of PDK1 (induced by the interaction).
The model outlined in Figure 13 is simplified and does not fit with all data on RSK activation obtained here and in previous studies. For instance, in unstimulated cells, RSK is partially phosphorylated at Ser227 (Dalby et al., 1998; see also Figure 3). In addition, RSK isolated from unstimulated cells can be substantially activated by incubation with ERK in vitro (Zhao et al., 1996), indicating (at least some) pre-existing phosphorylation at Ser227. The discrepancy between these observations and the model in Figure 13 could be accounted for if phosphoSer227 is more resistant to dephosphorylation than the other phosphorylation sites. This would result in a fraction of RSK being phosphorylated at Ser227 in resting cells and thus capable of being activated by ERK. The mechanism outlined in Figure 13 may operate to replenish phosphorylation at Ser227 in the fraction of RSK that has been dephosphorylated at this site since the previous exposure to growth factor as well as in newly synthesized RSK.
The model proposed for RSK may be valid for other targets of PDK1. p70 S6 kinase forms a complex with PDK1 in co-transfected cells (Romanelli et al., 1999). The binding site has not been identified, but surface plasmon resonance analysis has shown that PDK1 exhibits higher affinity for a mutant of p70 S6 kinase with glutamic acid in place of Thr389 in the hydrophobic motif (equivalent to Ser386 of RSK2) than for wild-type p70 S6 kinase (Balendran et al., 1999b). Furthermore, a T389A mutant of p70 S6 kinase was not phosphorylated at the PDK1 site in the activation loop in vivo and was a poor in vitro substrate for PDK1 compared with a T389E mutant (Dennis et al., 1998). Similarly, the phosphorylation of SGK by PDK1 in vitro was greatly enhanced by mutation of Ser to Asp in the hydrophobic motif (Kobayashi and Cohen, 1999). These findings are consistent with the possibility that also in p70 S6 kinase and SGK, the hydrophobic motif functions to recruit and activate PDK1 in a phosphorylation-dependent manner. The hydrophobic motif of PRK2 has been isolated as a PDK1-interacting fragment (PIF) in a yeast two-hybrid screen (Balendran et al., 1999a). Recently, it was shown that interaction with PIF stimulated the kinase activity of PDK1 4-fold in vitro (Biondi et al., 2000). Furthermore, a hydrophobic pocket was identified in the catalytic domain of PDK1 that interacts with the aromatic residues of PIF. It is possible that this pocket may also constitute a binding site for RSK2 in PDK1. Several isotypes of PKC form a complex with and are phosphorylated by PDK1 in the activation loop (Le Good et al., 1998). PKCs of the atypical class have an acidic residue in the hydrophobic motif, whereas classical and novel PKCs have a phosphorylation site, which could function as a regulated docking site for PDK1. In contrast, PDK1 may not dock to the phosphorylated hydrophobic motif of PKB, since mutation of this site does not affect phosphorylation of the activation loop by PDK1 (Alessi et al., 1996).
Our results show that the NTK of MSK1 is activated by ectopic PDK1 and that an MSK1 mutant with increased affinity for PDK1 exhibits increased activity. This suggests that MSK1 may be regulated by PDK1 in a manner similar to RSK2. However, targeted disruption of the PDK1 gene in mouse embryonic stem cells has no effect on MSK1 activity, whereas activation of RSK is completely abolished (Williams et al., 2000). Thus, PDK1 is not rate limiting for phosphorylation of the NTK of MSK1, at least in embryonic stem cells.
In a recent study, a mutant of RSK2 was reported with disruption of a predicted autoinhibitory α-helix in the CTK, resulting in constitutive activity corresponding to 30% of EGF-stimulated wild-type RSK2 (Poteet-Smith et al., 1999). Furthermore, an active mutant of X.laevis RSK1 has been generated by deletion of the CTK and the first 44 residues of the N-terminal tail of NTK (Gross et al., 1999). A comparison of its activity with stimulated wild-type RSK1 was not reported, but expression of the mutant in Xenopus embryos was sufficient for induction of metaphase arrest. In the present study, we have generated mutants of RSK2 and MSK1 with constitutive activity 100–300% of that of wild-type enzymes in response to EGF. We have used these mutants to demonstrate that histone H3 is phosphorylated at Ser10 and CREB at Ser133 as a consequence of nuclear RSK2 or MSK1 activity independent of upstream signaling pathways.
The introduction of a constitutive docking site for the upstream activator PDK1 was an efficient means of activating RSK2 and MSK1 truncation mutants. By this strategy, we have also generated constitutively active full-length MSK1 and RSK1 NTK with activities higher than the EGF-stimulated wild-type enzymes (C.J.Jensen and M.Frödin, unpublished observation). The approach may, therefore, be generally applicable for the generation of constitutively active mutants of PDK1-regulated kinases.
Assembly of protein complexes via serine/threonine phosphorylation has emerged as a new regulatory mechanism in signal transduction. Proteins like 14-3-3 or protein modules like forkhead-associated (FHA) or tryptophan–tryptophan (WW) domains bind phosphoserines or phosphothreonines in a specific sequence context in kinases and transcription factors (reviewed in Yaffe and Cantley, 1999). The present study suggests that recognition of phosphoserine in the context of a hydrophobic motif leads to a reversible complex between PDK1 and RSK2, and possibly other PDK1 targets. The aromatic residues of the hydrophobic motif of RSK2 may interact with PDK1 via the PIF-binding pocket (Biondi et al., 2000). In this case, phosphoSer386 is predicted to lie outside the pocket. It will be important to identify the amino acids in PDK1 that interact with phosphoSer386 and determine how the phosphate group confers binding specificity.
Materials and methods
Phospho-specific Abs were from Upstate Biotechnology, except anti-pSer227 RSK2 Ab, which was generated as described (Merienne et al., submitted). S6 peptide (RRLSSLRA), CREBtide (EILSRRPSYRK) and Ser386 peptide (residues 373–396 of mouse RSK2: KKPPSANAHQLFRGFSFVAITSDDE, with the underlined serine phosphorylated in pSer386 peptide) were synthesized by K.J.Ross-Petersen, AS, Denmark. Anti-HA- and anti-myc-epitope Abs used for immunoprecipitation were from the 12CA5 and 9E10 mouse hybridoma cell lines, respectively. Anti-HA or anti-myc Abs for immunoblotting and Abs to the CTK domain of RSK2 were rabbit polyclonal Abs from Santa Cruz Biotechnologies. Other chemicals were from Sigma.
Plasmid constructs
N-terminally HA-tagged mouse RSK2 and RSK2-K100A in pMT2 (Zhao et al., 1996) were provided by Christian Bjørbæk (Beth Israel Hospital, Boston, MA). N-terminally myc-tagged PDK151–556 in pCMV5 (Pullen et al., 1998) was kindly provided by Brian A.Hemmings (Friedrich Miescher Institute, Basel, Switzerland). Myc-PDK1 and myc-PDK1-KD (K111Q/D205N/D223N) in pCMV5 have been described (Jensen et al., 1999). pEXV3-MEK-S217E/S221E was kindly provided by Hugh Paterson (Institute of Cancer Research, London, UK). N-terminally HA-tagged human MSK1 was generated using PCR to amplify EST AA158571 from I.m.a.g.e. Consortium (HGMP Resource Centre, Hinxton, Cambridge, UK) with primers introducing a XhoI site and a SalI site in the 5′ and 3′ end, respectively, of the MSK1 coding region. RSK1 was removed from pMT2-HA-RSK1 (from Joseph Avruch, Massachusetts General Hospital, Boston, MA) by XhoI + SalI digestion and replaced with MSK1. Deletion mutants of RSK2 and MSK1 were generated by PCR amplification of the desired sequence using a 5′ end primer introducing a XhoI site and a 3′ end primer introducing a stop codon and a SalI site. RSK1 was removed from pMT2-HA-RSK1 by XhoI + SalI digestion and replaced with the PCR product. RSK21–389 S386D was generated likewise except that the 3′ end primer introduced an Asp at position 386. The same procedure was used to generate RSK21–389 with mutations T365E, S369E or T365E/S369E, except that the 3′ end primer encompassed the DraIII site at nucleotide 1123 of mouse RSK2 and the mutations. The PCR product was cloned into pMT2-HA-RSK21–389 digested with XhoI + DraIII. Point mutations in RSK21–389 S386A and RSK2-S386K, CA-MSK1-KD (D195A) were introduced using the QuickChange™ (Stratagene) mutagenesis procedure. CA-MSK1 was generated by two PCR products: A and B. A was generated by amplification of MSK1 with a 5′ end primer introducing a XhoI site in front of the MSK1 coding region and a 3′ end primer introducing a DraIII site at nucleotide 1094. B was generated by amplification of human PRK2 (kindly provided by Lawrence A.Quilliam, Indiana University School of Medicine, IN) with a 5′ end primer introducing a DraIII site in front of the PRK2 codon corresponding to residue 968 and a 5′ end primer with a KpnI site after the open reading frame. A and B were cut with XhoI + DraIII and DraIII + KpnI, respectively, and cloned into pMT2-HA vector after excision of RSK2 with XhoI + KpnI. The chimeric kinase consists of residues 1–365 of MSK1 followed by residues 968–985 of PRK2. pMT2-HA-CA-RSK2 and pMT2-HA-CA-RSK2-KD were generated similarly to CA-MSK1 and consist of residues 1–375 of RSK2 followed by residues 968–985 of PRK2. For the kinase-deficient version, RSK2-K100A was used as template in the PCR generating fragment A. pmt2-NLS-HA-CA-RSK2 was generated using three rounds of PCR with a common 3′ end primer encompassing RSK2 nucleotide 1131 and three overlapping 5′ end primers to introduce three repetitive nuclear localization sequences (DPKKKRKV) N-terminally to the RSK2 sequence. The last round of PCR introduced a 5′ end SalI site, and the PCR product was cut with SalI and DraIII and cloned into pMT2-HA-CA-RSK2 after excision of RSK2 with XhoI + DraIII. pMT2-HA-NLS-CA-RSK2-KD, pMT2-HA-NLS-CA-MSK1 and pMT2-HA-NLS-CA-MSK1-KD were generated by replacement of NLS-CA-RSK2 excised from pmt2-HA-NLS-CA-RSK2 using XhoI + DraIII with CA-RSK2-KD, CA-MSK1 or CA-MSK1-KD excised using XhoI + DraIII from pMT2-HA-CA-RSK2-KD, pMT2-HA-CA-MSK1 and pMT2-HA-CA-MSK1-KD, respectively. All mutations were confirmed by sequence analysis.
Transfection and immunoprecipitation
COS7 cells were cultured and transfected using lipofectamine reagent (Life Technologies Inc.) as described (Jensen et al., 1999). After washing with phosphate-buffered saline, cells were solubilized for 15 min in 500 µl of lysis buffer (0.5% Triton X-100, 150 mM NaCl, 50 mM Tris–HCl pH 7.4, 10% glycerol, 1 mM Na3VO4, 5 mM EDTA, 25 mM NaF, 10 nM calyculin A, 1 mM phenylmethylsulfonyl fluoride, 10 µM leupeptin, 10 µM pepstatin and 200 kallikrein inhibitor units/ml aprotinin) on ice. Subsequent manipulations were performed at 0–4°C. Cell extracts were clarified by centrifugation for 10 min at 14 000 g and the supernatant was incubated for 75 min with Ab with the addition of 20 µl of 50% protein A– or protein G–agarose beads (Amersham Pharmacia Biotech) during the final 25 min. Agarose beads–Ab complexes were precipitated by centrifugation, washed five times with lysis buffer, drained and dissolved in SDS–PAGE sample buffer (2% sodium dodecyl sulfate, 62 mM Tris pH 6.8, 10% glycerol, 5% 2-β-mercaptoethanol, 0.1% bromophenol blue). For kinase assays, the final two washes were with kinase assay buffer (30 mM Tris–HCl pH 7.4, 10 mM MgCl2, 1 mM dithiothreitol).
RSK and MSK assays
Agarose beads with immunoprecipitated kinase were drained with a syringe and resuspended in 20 µl of 1.5× kinase assay buffer. The kinase reaction was initiated by the addition of 10 µl (final concentrations) of ATP (300 µM, 0.2 µCi of [γ-32P]ATP) and 800 µM S6 peptide or CREBtide for RSK and MSK assays, respectively. After 10 min at 30°C (the reaction was linear with time), 20 µl of the supernatant were removed with a syringe (leaving behind precipitated kinase) and spotted onto phosphocellulose paper (Whatman p81), which was washed 5–6 times with 150 mM orthophosphoric acid. [32P]phosphate incorporated into protein substrate was quantified on a STORM™ PhosphorImager using ImageQuant™ software (Molecular Dynamics). A reaction blank (assay performed on non-transfected cells) was subtracted from all values.
PDK1 in vitro assay
Myc-tagged PDK1 was expressed in COS7 cells, immunoprecipitated with 9E10 Ab, whereafter agarose beads with kinase (10–30 ng per assay point) were drained with a syringe and resuspended in 20 µl of kinase assay buffer A. Thereafter, PDK1 was incubated at room temperature for 20 min with or without peptides (Ser386 peptide, S6 peptide) at 10 µM under vigorous shaking. Autophosphorylation was started by the addition of (final concentrations) 200 µM ATP (for mobility shift assay) or 20 µM ATP, including 15 µCi of [γ-32P]ATP (for autoradiographic assay) in 10 µl. In PDK1 kinase assays, the reaction was started by the addition of 2 µg of RSK21–373K100A or 0.5 µg of RSK21–360 or RSK21–360S227E and 20 µM ATP (15 µCi of [γ-32P]ATP). After 20 min of shaking at room temperature, reactions were stopped with SDS–PAGE sample buffer and subjected to SDS–PAGE followed by immunoblotting or autoradiography of the dried gel. RSK proteins were synthesized and purified from Escherichia coli as glutathione S-transferase (GST) fusions (Jensen et al., 1999). Prior to use, the proteins were digested with thrombin to remove GST.
Immunoblotting
Immunoblotting was performed as described (Jensen et al., 1999).
Acknowledgments
Acknowledgements
Birte Kofoed is thanked for expert technical assistance. This work was supported by grants from the Novo Nordisk Foundation, Denmark, the Danish Cancer Society, the Foundation for Medical Research in Copenhagen County, Greenland and Faeroe Islands, and the Danish Research Center for Growth and Regeneration.
References
- Alessi D.R., Andjelkovic,M., Caudwell,B., Cron,P., Morrice,N., Cohen,P. and Hemmings,B.A. (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J., 15, 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi D.R. et al. (1997a) 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr. Biol., 7, 776–789. [DOI] [PubMed] [Google Scholar]
- Alessi D.R., Kozlowski,M.T., Weng,Q.-P., Morrice,N. and Avruch,J. (1997b) 3-Phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylates and activates the p70 S6 kinase in vivo and in vitro.Curr. Biol., 8, 69–81. [DOI] [PubMed] [Google Scholar]
- Anderson K.E., Coadwell,J., Stephens,L.R. and Hawkins,P.T. (1998) Translocation of PDK-1 to the plasma membrane is important in allowing PDK-1 to activate protein kinase B. Curr. Biol., 8, 684–691. [DOI] [PubMed] [Google Scholar]
- Andjelkovic M. et al. (1997) Role of translocation in the activation and function of protein kinase B. J. Biol. Chem., 272, 31515–31524. [DOI] [PubMed] [Google Scholar]
- Balendran A., Casamayor,A., Deak,M., Paterson,A., Gaffney,P., Currie,R., Downes,C.P. and Alessi,D.R. (1999a) PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr. Biol., 9, 393–404. [DOI] [PubMed] [Google Scholar]
- Balendran A., Currie,R., Armstrong,C.G., Avruch,J. and Alessi,D.R. (1999b) Evidence that 3-phosphoinositide-dependent protein kinase-1 mediates phosphorylation of p70 S6 kinase in vivo at Thr-412 as well as Thr-252. J. Biol. Chem., 274, 37400–37406. [DOI] [PubMed] [Google Scholar]
- Belham C., Wu,S. and Avruch,J. (1999) PDK1—a kinase at the hub of things. Curr. Biol., 11, R93–R96. [DOI] [PubMed] [Google Scholar]
- Bhatt R.R. and Ferrell,J.E. (1999) The protein kinase p90 rsk as an essential mediator of cytostatic factor activity. Science, 286, 1362–1365. [DOI] [PubMed] [Google Scholar]
- Biondi R.M., Cheung,P.C.F., Casamayor,A., Deak,M., Currie,R.A. and Alessi,D.R. (2000) Identification of a pocket in the PDK1 kinase domain that interacts with PIF and the C-terminal residues of PKA. EMBO J., 19, 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonni A., Brunet,A., West,A.E., Datta,S.R., Takasu,M.A. and Greenberg,M.E. (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science, 286, 1358–1362. [DOI] [PubMed] [Google Scholar]
- Casamayor A., Morrice,N.A. and Alessi,D.R. (1999) Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo.Biochem. J., 342, 287–292. [PMC free article] [PubMed] [Google Scholar]
- Currie R.A., Walker,K.S., Gray,A., Deak,M., Casamayor,A., Downes,C.P., Cohen,P., Alessi,D.R. and Lucocq,J. (1999) Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem. J., 337, 575–583. [PMC free article] [PubMed] [Google Scholar]
- Dalby K.N., Morrice,N., Caudwell,F.B., Avruch,J. and Cohen,P. (1998) Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J. Biol. Chem., 273, 1496–1505. [DOI] [PubMed] [Google Scholar]
- Deak M., Clifton,A.D., Lucocq,L.M. and Alessi,D.R. (1998) Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38 and may mediate activation of CREB. EMBO J., 17, 4426–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis P.B., Pullen,N., Pearson,R.B., Kozma,S.C. and Thomas,G. (1998) Phosphorylation sites in the autoinhibitory domain participate in p70 (s6k) activation loop phosphorylation. J. Biol. Chem., 273, 14845–14852. [DOI] [PubMed] [Google Scholar]
- Dutil E.M., Toker,A. and Newton,A.C. (1998) Regulation of conventional protein kinase C isozymes by phosphoinositide-dependent kinase 1 (PDK-1). Curr. Biol., 8, 1366–1375. [DOI] [PubMed] [Google Scholar]
- Fisher T.L. and Blenis,J. (1996) Evidence for two catalytically active kinase domains in pp90rsk. Mol. Cell. Biol., 16, 1212–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frödin M. and Gammeltoft,S. (1999) Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell. Endocrinol., 151, 65–77. [DOI] [PubMed] [Google Scholar]
- Gavin A.C. and Nebreda,A.R. (1999) A MAP kinase docking site is required for phosphorylation and activation of p90 (rsk)/MAPKAP kinase-1. Curr. Biol., 9, 281–284. [DOI] [PubMed] [Google Scholar]
- Gross S.D., Schwab,M.S., Lewellyn,A.L. and Maller,J.L. (1999) Induction of metaphase arrest in cleaving Xenopus embryos by the protein kinase p90Rsk. Science, 286, 1365–1367. [DOI] [PubMed] [Google Scholar]
- Jensen C.J., Buch,M.-B., Krag,T.O., Hemmings,B.A., Gammeltoft,S. and Frödin,M. (1999) 90 kDa ribosomal S6 kinase (RSK) is phosphorylated and activated by 3-phosphoinositide-dependent protein kinase-1 (PDK1). J. Biol. Chem., 274, 27168–27176. [DOI] [PubMed] [Google Scholar]
- Joel P.B., Smith,J., Sturgill,T.W., Fisher,T.L., Blenis,J. and Lannigan,D.A. (1998) pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol. Cell. Biol., 18, 1978–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T. and Cohen,P. (1999) Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem. J., 339, 319–328. [PMC free article] [PubMed] [Google Scholar]
- Le Good J.A., Ziegler,W.H., Parekh,D.B., Alessi,D.R., Cohen,P. and Parker,P.J. (1998) Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science, 281, 2042–2045. [DOI] [PubMed] [Google Scholar]
- Nakajima T., Fukamizu,A., Takahashi,J., Gage,F.H., Fisher,T., Blenis,J. and Montminy,M.R. (1996) The signal-dependent coactivator CBP is a nuclear target for pp90RSK. Cell, 86, 465–474. [DOI] [PubMed] [Google Scholar]
- Nebreda A.R. and Gavin,A.C. (1999) Cell survival demands some Rsk. Science, 286, 1309–1310. [DOI] [PubMed] [Google Scholar]
- Palmer P., Gavin,A.C. and Nebreda,A.R. (1998) A link between MAP kinase and p34cdc2/cyclin B during oocyte maturation: p90rsk phosphorylates and inactivates the p34cdc2 inhibitory kinase Myt1. EMBO J., 17, 5037–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J., Leong,M.L., Buse,P., Maiyar,A.C., Firestone,G.L. and Hemmings,B.A. (1999) Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J., 18, 3024–3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R.B., Dennis,P.B., Han,J.W., Williamson,N.A., Kozma,S.C., Wettenhall,R.E. and Thomas,G. (1995) The principal target of rapamycin-induced p70s6k inactivation is a novel phosphorylation site within a conserved hydrophobic domain. EMBO J., 14, 5279–5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierrat B., da Silva Correia,J., Mary,J.-L., Tomás-Zuber,M. and Lesslauer,W. (1998) RSK-B, a novel ribosomal S6 kinase family member, is a CREB kinase under dominant control of p38α mitogen-activated protein kinase (p38αMAPK). J. Biol. Chem., 273, 29661–29671. [DOI] [PubMed] [Google Scholar]
- Poteet-Smith C.E., Smith,J.A., Lannigan,D.A., Freed,T.A. and Sturgill,T.W. (1999) Generation of constitutively active p90 ribosomal S6 kinase in vivo. Implications for the mitogen-activated protein kinase-activated protein kinase family. J. Biol. Chem., 274, 22135–22138. [DOI] [PubMed] [Google Scholar]
- Pullen N., Dennis,P.B., Andjelkovic,M., Dufner,A., Kozma,S.C., Hemmings,B.A. and Thomas,G. (1998) Phosphorylation and activation of p70s6k by PDK1. Science, 279, 707–710. [DOI] [PubMed] [Google Scholar]
- Richards S.A., Fu,J., Romanelli,A., Shimamura,A. and Blenis,J. (1999) Ribosomal S6 kinase 1 (RSK1) activation requires signals dependent on and independent of the MAP kinase ERK. Curr. Biol., 9, 810–820. [DOI] [PubMed] [Google Scholar]
- Romanelli A., Martin,K.A., Toker,A. and Blenis,J. (1999) p70 S6 kinase is regulated by protein kinase Cζ and participates in a phosphoinositide 3-kinase-regulated signalling complex. Mol. Cell. Biol., 19, 2921–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassone-Corsi P., Mizzen,C.A., Cheung,P., Crosio,C., Monaco,L., Jacquot,S., Hanauer,A. and Allis,C.D. (1999) Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science, 285, 886–891. [DOI] [PubMed] [Google Scholar]
- Smith J.A., Poteet-Smith,C.E., Malarkey,K. and Sturgill,T.W. (1999) Identification of an extracellular signal-regulated kinase (ERK) docking site in ribosomal S6 kinase, a sequence critical for activation by ERK in vivo.J. Biol. Chem., 274, 2893–2898. [DOI] [PubMed] [Google Scholar]
- Stephens L. et al. (1998) Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-triphosphate-dependent activation of protein kinase B. Science, 279, 710–714. [DOI] [PubMed] [Google Scholar]
- Stokoe D., Stephens,L.R., Copeland,T., Gaffney,P.R., Reese,C.B., Painter,G.F., Holmes,A.B., McCormick,F. and Hawkins,P.T. (1997) Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science, 277, 567–570. [DOI] [PubMed] [Google Scholar]
- Thomson S., Clayton,A.L., Hazzalin,C.A., Rose,S., Barratt,M.J. and Mahadevan,L.C. (1999) The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J., 18, 4779–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivier E., De Cesare,D., Jacquot,S., Pannetier,S., Zackai,E., Young,I., Mandel,J.L., Sassone-Corsi,P. and Hanauer,A. (1996) Mutations in the kinase Rsk-2 associated with Coffin–Lowry syndrome. Nature, 384, 567–570. [DOI] [PubMed] [Google Scholar]
- Vik T.A. and Ryder,J.W. (1997) Identification of serine 380 as the major site of autophosphorylation of Xenopus pp90rsk. Biochem. Biophys. Res. Commun., 235, 398–402. [DOI] [PubMed] [Google Scholar]
- Williams M.R., Arthur,J.S.C., Balendran,A., Van der Kaay,J., Poli,V., Cohen,P. and Alessi,D.R. (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr. Biol., 10, 439–448. [DOI] [PubMed] [Google Scholar]
- Xing J., Ginty,D.D. and Greenberg,M.E. (1996) Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science, 273, 959–963. [DOI] [PubMed] [Google Scholar]
- Yaffe M.B. and Cantley,L.C. (1999) Grabbing phosphoproteins. Nature, 402, 30–31. [DOI] [PubMed] [Google Scholar]
- Yntema H.G. et al. (1999) A novel ribosomal S6-kinase (RSK4; RPS6KA6) is commonly deleted in patients with complex X-linked mental retardation. Genomics, 62, 332–343. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Bjorbaek C. and Moller,D.E. (1996) Regulation and interaction of pp90rsk isoforms with mitogen-activated protein kinases. J. Biol. Chem., 271, 29773–29779. [DOI] [PubMed] [Google Scholar]