

Abstract
A role for the transcription factor JunB in proliferation control was investigated in genetically modified mouse fibroblasts. Increased JunB expression induced high levels of the cyclin-dependent kinase inhibitor p16INK4a, leading to premature senescence in primary cells and reduced proliferation in 3T3 cells, whereas lack of JunB expression results in decreased p16 levels. Furthermore, JunB-mediated p16 induction in 3T3 cells completely abolished cyclin D-associated kinase activity, resulting in reduced pRb hyperphosphorylation and G1-phase extension. Moreover, three AP1-like binding sites were identified in the p16 promoter through which JunB directly activates p16 transcription. Elevated JunB expression in 3T3 cells also inhibited Ras- and Src-mediated transformation and tumour growth in vivo. The suppressive effect of JunB on cell proliferation was shown to be dependent on p16 since it did not occur in INK4a–/– fibroblasts that lack both p16 and p19ARF. These results demonstrate that p16 is a direct transcriptional target gene of JunB and identify JunB as a negative regulator of cell proliferation.
Keywords: AP-1/JunB/p16INK4a/proliferation/transformation
Introduction
The control of mammalian cell proliferation occurs mainly during the G1 phase of the cell cycle (Harper and Elledge, 1996). During this period, the G1-type cyclins bind and activate specific cyclin-dependent kinases (CDKs) 4–6 for D-type cyclins and CDK2 for cyclin E, thereby dictating when CDKs will phosphorylate appropriate substrates. The retinoblastoma protein pRb and related family members are critical targets for G1-type cyclin–CDK complexes (Weinberg, 1995). In mid-G1 phase, inactivating phosphorylation of pRb by cyclin D–CDK4–6 complexes and then later by cyclin E–CDK2 complexes enables transcriptional activation of genes required for S-phase entry and progression. Two families of cyclin-dependent kinase inhibitors (CKIs) block the activity of the G1-type cyclin–CDK complexes (Sherr and Roberts, 1995). CKIs of the INK4 family (p16INK4a, p15INK4b, p18INK4c and p19INK4d) specifically block the cyclin D–CDK4–6 complexes. Interestingly, the INK4a locus consists of two overlapping genes, each regulated by its own promoter, which through alternative reading frames encode for the two unrelated proteins p16INK4a and p19ARF (Serrano, 1997). In contrast to p16, p19ARF is not a CKI but is involved in cell cycle control by regulating the MDM2/p53 pathway (Haber, 1997). CKIs of the CIP/KIP family (p21CIP1, p27KIP1 and p57KIP2) inhibit cyclin E–CDK2 complexes as well as other cyclin–CDK complexes operating throughout the cell cycle. Recently, p21 and p27 were found to activate CDK4 by promoting cyclin D–CDK4 assembly, stability and nuclear localization (LaBaer et al., 1997; Cheng et al., 1999). In quiescent cells, CKIs are present in excess to cyclin–CDK complexes therefore maintaining the cells in a non-proliferating state. Mitogen stimulation leads to the sequential synthesis of G1-type cyclins and, as soon as the CKI inhibitory threshold is overcome, cyclin–CDK kinase activity appears and cells progress into S phase. CKIs are therefore critical mediators of antiproliferative signals that arrest the cell cycle and allow for processes such as DNA repair, terminal differentiation and senescence.
One of the immediate consequences of mitogenic signalling is the activation of the transcription factor AP-1 (activator protein-1), a collection of dimeric complexes composed of proteins from the Jun, Fos and ATF families, which convert extracellular signals into changes in gene expression (Angel and Karin, 1991). All Jun proteins (c-Jun, JunB and JunD) are similar in their primary structure and their DNA-binding specificity. However, JunB exhibits weaker DNA-binding and homodimerization affinity than c-Jun and was shown to act both as a transactivator and a transrepressor depending on the promoter context and on the heterodimerization partner (Deng and Karin, 1993; Hsu et al., 1993; Li et al., 1999). Interestingly, constitutive high JunB expression in transgenic mice does not lead to an obvious phenotype (Schorpp et al., 1996), while inactivation of junB results in an early embryonic lethality (Schorpp-Kistner et al., 1999).
The involvement of AP-1 in the transmission of proliferative signals is supported by the observation that microinjection of neutralizing antibodies directed against the different AP-1 components inhibits cell cycle progression (Kovary and Bravo, 1991). However, AP-1 activity is also associated with the induction of growth arrest, terminal differentiation and apoptosis (Karin et al., 1997). Analysis of fibroblasts either lacking or overexpressing one of the AP-1 components has recently shown that the different Jun family members may exert specific and distinct functions in controlling cell proliferation. A positive regulatory function of c-Jun on cell proliferation was established using c-jun–/– fibroblasts that displayed reduced proliferation (Johnson et al., 1993; Schreiber et al., 1999). Furthermore, the impaired G1–S phase progression in c-jun–/– cells was shown to be due to altered transcription of two cell cycle-regulating target genes, the tumour suppressor p53 and cyclin D1 (Schreiber et al., 1999; Wisdom et al., 1999). On the other hand, JunD was found to regulate fibroblast proliferation negatively by either establishing or maintaining the quiescent state of the cell cycle (Pfarr et al., 1994). In this study, we have specifically investigated the function of JunB in proliferation control using genetically modified mouse fibroblasts.
Results
JunB-expressing mouse embryonic fibroblasts prematurely enter senescence
To determine the effects of high JunB protein levels on cell proliferation, primary mouse embryonic fibroblasts (MEFs) were derived from E12.5 transgenic embryos expressing the murine junB cDNA under the control of the human ubiquitin C promoter (Schorpp et al., 1996). All junB transgenic (jB-Tg) MEFs expressed 5–10 times more JunB protein than the wild-type MEFs (Figure 1A). Until passage 4, no significant differences were found between the proliferation rates of wild-type and jB-Tg MEFs (Figure 1B). However, thereafter, the proliferation of jB-Tg MEFs was significantly reduced and by passage 6 all jB-Tg cells displayed characteristic features of senescence such as flattening, cytoplasmic enlargement, unresponsiveness to growth factors and expression of acidic β-galactosidase in ∼70–80% of the arrested cells (Figure 1B and C). These results indicate that jB-Tg MEFs undergo premature senescence in contrast to wild-type MEFs, which enter senescence by passage 8.

Fig. 1. JunB-expressing MEFs show premature entry into senescence. (A) Western blot analysis of JunB expression levels in wild-type (WT) and jB-Tg MEFs. The results from independent MEF lines of each genotype are shown. (B) Proliferation curves of WT and jB-Tg MEFs. The results from three independent MEF lines of each genotype are shown. (C) Senescence-associated β-galactosidase activity in passage 2, 4 and 6 WT and jB-Tg MEFs.
JunB-expressing 3T3 fibroblasts proliferate slowly due to an extended G1 phase
Both wild-type and jB-Tg MEF lines were established according to the 3T3 protocol and were readily immortalized after a crisis period of 20–30 days (data not shown). However, all immortalized jB-Tg 3T3 lines showed a pronounced proliferation defect with a doubling time of 43 ± 2.5 h, compared with 19.4 ± 1.3 h in wild-type controls (Figure 2A). Although jB-Tg 3T3 cells displayed a dose-dependent proliferative response to increased amounts of serum, their saturation densities were on average 50% of the wild-type densities at all serum concentrations tested (Figure 2B). As comparably low percentages of dead and apoptotic cells were found in exponentially growing wild-type and jB-Tg cells (Figure 2C), increased cell death is most probably not the reason for the difference observed in the proliferation rates. In contrast, significantly lower amounts of BrdU positive cells were found in asynchronously growing jB-Tg cultures compared with wild-type cultures (Figure 2D). These decreased numbers of S-phase cells correlated with the reduction in total cell numbers observed after 24, 48, 72 and 96 h of culture, which suggests an altered cell cycle progression in jB-Tg 3T3 fibroblasts. To determine which phase of the cell cycle is impaired in JunB-expressing fibroblasts, exponentially growing wild-type and jB-Tg cells were analysed by BrdU pulse–chase labelling over a 14 h period (Figure 2E). Although both wild-type and jB-Tg cells exited from S phase, passed through G2–M phase and entered the next G1 phase with similar kinetics, their subsequent cell cycle progression was different. Wild-type fibroblasts left G1 and entered the subsequent S phase after 8 h whereas jB-Tg fibroblasts remained in G1 phase and did not enter the subsequent S phase during the 14 h of this experiment. These results, therefore, suggest that jB-Tg 3T3 fibroblasts have an extended G1 phase.

Fig. 2. JunB-expressing 3T3 fibroblasts proliferate slowly with an extended G1 phase. (A) Proliferation curves of WT and jB-Tg 3T3 fibroblasts. The results from three independent 3T3 lines of each genotype are shown. (B) Saturation densities of WT and jB-Tg 3T3 fibroblasts under different fetal calf serum (FCS) concentrations. Values are the average ± SEM of two independent cell lines each measured in triplicate. (C) Numbers of dead cells (propidium iodide exclusion) and apoptotic cells (TUNEL assay) in exponentially growing WT and jB-Tg 3T3 fibroblasts. Values are expressed as a percentage of the total cell numbers and are the average ± SEM of two independent cell lines. (D) Comparative analysis of S-phase cell numbers and proliferation rates in WT and jB-Tg 3T3 fibroblasts. Cells were seeded at 0.4 × 106 cells per 10 cm dish and labelled for 1 h with 60 µM BrdU before harvesting after 24, 48, 72 and 96 h of culture. The total cell numbers were determined (top panel; y-axis, ×106) and the cells were analysed bytwo-parameter flow cytometry to determine the percentage of BrdU positive cells. (E) Analysis of cell cycle distribution in asynchronously growing WT and jB-Tg 3T3 fibroblasts. Cells were pulse-labelled for 1 h with 60 µM BrdU, chased for the periods of time indicated and analysed by two-parameter flow cytometry to determine the percentage of BrdU positive cells in S, G2/M and G1 phase. The numbers of BrdU positive cells obtained after the 1 h pulse were normalized to 100% in the two cell populations and correspond to 62% of the total cell numbers in WT cultures and 24% in jB-Tg cultures.
Specific induction of p16 INK4a in JunB-expressing fibroblasts
To identify the mechanism by which JunB could impair G1 to S phase progression, the expression levels of the different CKI family members were analysed (Figure 3A). Similar levels of the CIP/KIP proteins (p21, p27 and p57), the p21 transcriptional regulator p53, as well as the p15INK4b, p18INK4c and p19INK4d proteins were detected in all cell lines irrespective of their genotype. However, p16INK4a levels were dramatically elevated in all jB-Tg fibroblasts investigated whereas no changes were detected in the expression levels of p19ARF, the second member of the INK4a locus (Figure 3A). Furthermore, jB-Tg cells showed a 3- to 5-fold increase in p16 mRNA levels, suggesting that JunB induces p16 expression at the transcriptional level (Figure 3B). Since the 3T3 protocol was reported to affect either p53 function or the INK4a locus, all 3T3 cell lines used in this study were investigated for the functionality of these two pathways. Beside the junB–/– 3T3 clone that lost the entire INK4a locus during the immortalization process, none of the other wild-type and jB-Tg 3T3 cell lines (total of five) displayed detectable mutations, since all had wild-type functional p53 protein, expressed p16 and p19ARF and remained diploid (data not shown).

Fig. 3. Increased p16 expression levels in JunB-expressing fibroblasts. (A) Western blot analysis of CKI expression levels in WT and jB-Tg 3T3 fibroblasts. The results from two independent 3T3 lines of each genotype are shown. (B) Northern blot analysis of p16 (exon 1α probe) and GAPDH mRNA levels in WT and jB-Tg 3T3 fibroblasts. The results from two independent 3T3 lines of each genotype are shown. (C) Western blot analysis of JunB and p16 expression levels in an E12.5 wild-type embryo and wild-type MEFs at different passages. (D) Comparative analysis of p16 expression levels in WT and jB-Tg 3T3 cells and passage 3 MEFs. The results from two independent MEF lines of each genotype are shown. (E) Western blot analysis of JunB and p16 expression levels in junB–/– ES cells and junB–/– passage 3 MEFs. The monoclonal JunB antibody recognizes both fusion proteins obtained after single (JunB-neo) or double (JunB-hygro) targeting of the junB locus.
To establish a correlation between JunB and p16 levels, primary wild-type MEFs were investigated (Figure 3C). As previously reported, p16 levels progressively increased in culture with passage numbers to reach maximum levels when cells senesced at passage 8 (Palmero et al., 1997; Zindy et al., 1997). Interestingly, JunB displayed similar kinetics with barely detectable expression at early passages and with a progressive increase to reach maximal levels in senescing passage 8 MEFs. Since JunB-expressing primary cells displayed premature senescence, p16 expression levels were investigated in jB-Tg MEFs at passage 3 (Figure 3D). Although p16 protein already started to accumulate in passage 3 wild-type MEFs, jB-Tg MEFs displayed much higher p16 levels that appeared comparable to the ones observed in immortalized jB-Tg 3T3 fibroblasts. These results demonstrate that JunB expression parallels the increase in p16 levels in primary cells and suggest that the premature replicative senescence observed in jB-Tg MEFs could be due to elevated p16 expression.
Finally, two cell systems that lack JunB expression were analysed for p16 expression (Figure 3D). In ES cells, the loss of junB expression resulted in reduced p16 protein levels, an effect already detectable in junB+/– ES cells. Similarly, reduced p16 levels were found in four independently isolated junB–/– MEFs. Although decreased p16 levels did not noticeably increase the proliferation rates of junB–/– ES cells and MEFs, junB–/– MEFs entered senescence on average three passages later than wild-type MEFs (data not shown).
JunB does not suppress cell proliferation in the absence of p16
To test directly whether increased p16 expression resulted from increased JunB expression, JunB was ectopically expressed in wild-type fibroblasts as well as in INK4a–/– fibroblasts that lack both p16 and p19ARF expression. INK4a–/– fibroblasts were chosen because no singular p16 knockout cells are currently available and because neither the increase nor the lack of JunB expression affected p19ARF levels (Figure 3A and data not shown). Wild-type cells infected with a JunB-expressing retrovirus (pB-junB; Matsuo et al., 2000) displayed a specific increase in p16 levels compared with cells infected with an empty virus (pB-puro) (Figure 4A). Furthermore, pB-junB-infected wild-type fibroblasts displayed reduced proliferation rates compared with control cells, which result from decreased numbers of S-phase cells as shown by the 40% reduction of BrdU incorporation in asynchronously growing pB-junB-infected wild-type cultures (Figure 4B and C). Consistently, flow cytometric analysis revealed an increased percentage of G1-phase cells in asynchronously growing pB-junB-infected wild-type populations (90 ± 0.2%) compared with control populations (77 ± 2.6%). In striking contrast, elevated JunB expression in INK4a–/– fibroblasts did not reduce the proliferation rates (Figure 4B), the BrdU uptake (Figure 4C) and the percentage of cells in G1 phase (65 ± 1.3% versus 67 ± 2.1%), strongly indicating that JunB cannot suppress proliferation in the absence of p16.
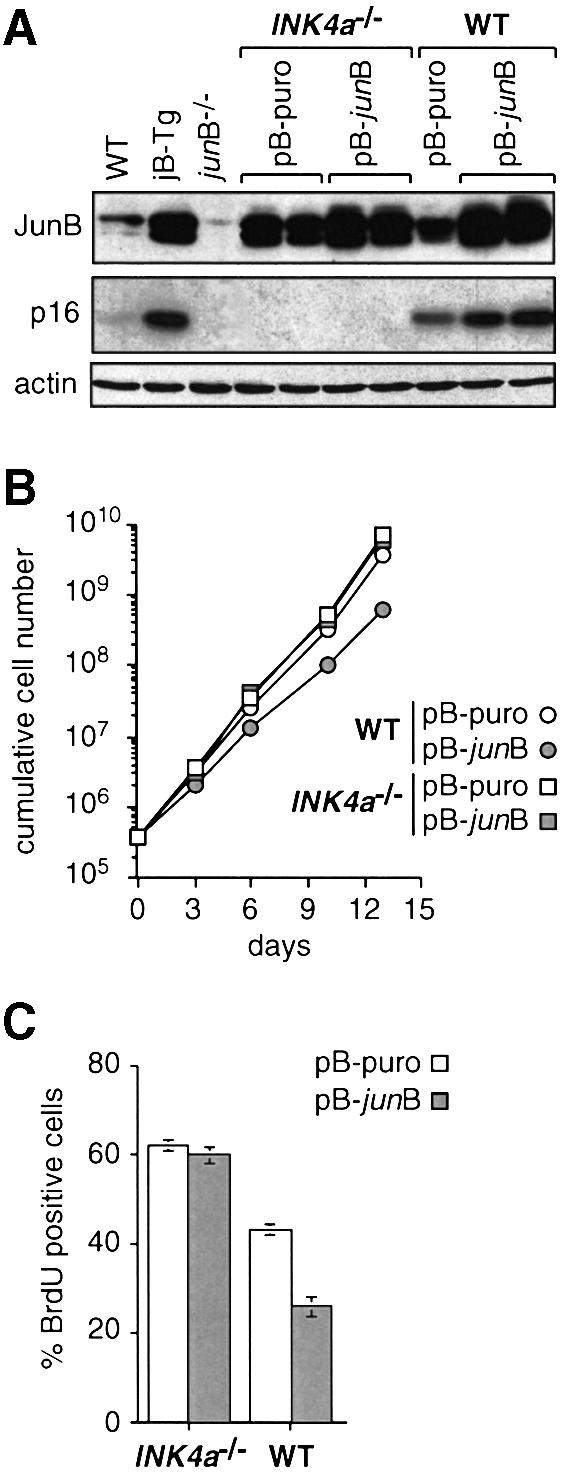
Fig. 4. The suppressive effect of JunB on cell proliferation is dependent on p16. (A) Western blot analysis of JunB and p16 expression levels in WT and INK4a–/– 3T3 fibroblasts infected with an empty retrovirus (pB-puro) or a JunB-expressing retrovirus (pB-junB). The results for two independent bulk cultures of each genotype are shown. JunB and p16 levels in non-infected WT, jB-Tg and junB–/– 3T3 fibroblasts are shown for comparison. (B) Proliferation curves of pB-puro- and pB-junB-expressing WT and INK4a–/– 3T3 fibroblasts. The results ± SD (bars included in the symbol when non-visible) from two independent bulk cultures of each genotype are shown. (C) Exponentially growing pB-puro- and pB-junB-expressing WT and INK4a–/– 3T3 fibroblasts were labelled for 1 h with 60 µM BrdU and analysed by two-parameter flow cytometry to determine the percentage of BrdU positive S-phase cells. The results ± SD from two independent bulk cultures of each genotype are shown.
JunB-mediated p16 induction abolishes cyclin D-associated kinase activity
To investigate the consequences of increased p16 levels in jB-Tg cells, the activity of the different G1-type cyclin–CDK complexes was analysed in serum-starved and serum-stimulated 3T3 fibroblasts. The basal level of cyclin D1 was found to be 2–3 times lower in serum-starved jB-Tg cells compared with wild-type cells (Figure 5A). However, upon serum stimulation, cyclin D1 induction appeared comparable in both cell types except that the maximal levels were delayed by 6 h in jB-Tg cells, peaking after 18 h compared with 12 h in wild-type cells. Wild-type and jB-Tg cells showed similar levels of expression and kinetics of induction of the other D-type cyclins and comparable amounts of CDK4 and CDK6 (Figure 5A and data not shown). However, no cyclin D1–CDK4–6 kinase activity was detected in jB-Tg fibroblasts throughout the entire kinetics, while wild-type cells displayed a time-dependent increase in cyclin D1–CDK4–6 kinase activity correlating with normal progression through the cell cycle (Figure 5B). Furthermore, jB-Tg cells did not show any increase in cyclin D2–CDK4–6 kinase activity throughout the entire kinetics (Figure 5C). Finally, neither wild-type nor jB-Tg cells expressed any detectable cyclin D3–CDK4–6 kinase activity (data not shown).

Fig. 5. Impaired cyclin D-associated kinase activity and reduced Rb phosphorylation in JunB-expressing 3T3 fibroblasts. Whole-cell extracts were prepared at the time points indicated post-serum stimulation from WT and jB-Tg 3T3 fibroblasts. (A) Western blot analysis of cyclin D1 and CDK4 expression levels. (B) Induction of cyclin D1-associated pRb kinase activity. The presence of CDK4 in the anti-cyclin D1 immunoprecipitate was confirmed by western blot analysis (ns, no substrate). (C) Induction of cyclin D2-associated pRb kinase activity. The presence of CDK4 in the anti-cyclin D2 immunoprecipitate was confirmed by western blot analysis. (D) Western blot analysis of cyclin E and CDK2 expression levels. (E) Induction of cyclin E–CDK2 kinase activity. The presence of CDK2 in the anti-cyclin E immunoprecipitate was confirmed by western blot analysis. (F) Western blot analysis of pRb expression and phosphorylation levels. A Molt-4 cell lysate was used as positive control (pc). (G) Western blot analysis of pRb expression levels in WT and jB-Tg 3T3 fibroblasts after serum starvation (s) or in an exponentially growing population (g). The results from two independent jB-Tg cell lines are shown.
To understand how jB-Tg cells are still able to proliferate in the absence of any detectable D-type cyclin activities, the expression levels and the kinase activity of cyclin E, the second key G1 cyclin that is not a p16 target, were investigated. The basal levels of cyclin E and cyclin E–CDK2 kinase activity were found to be lower in serum-starved jB-Tg cells compared with wild-type cells, whereas no changes were detected in the levels of CDK2 (Figure 5D and E). However, upon serum stimulation, induction of both cyclin E expression and cyclin E–CDK2 kinase activity appeared comparable in both cell types except that the maximal levels of kinase activity were reached after 24 h of serum stimulation in jB-Tg cells, compared with 18 h in wild-type cells, a delay that is consistent with the slow proliferation rates of the cells. Finally, pRb phosphorylation upon serum stimulation was found to be strongly reduced in jB-Tg cells with only a minor increase in the hyperphosphorylated form of Rb detectable after 18 h (Figure 5F). In contrast, wild-type cells displayed a time-dependent increase in the pRB hyperphosphorylated form, starting after 6 h of serum stimulation, which correlates with normal progression through the cell cycle. Moreover, significantly lower amounts of pRb protein were found in exponentially growing jB-Tg cells compared with wild-type cells, while similar levels were detected in serum-starved cells from both genotypes (Figure 5F and G).
JunB transactivates the p16 promoter through three AP1-like binding sites
To study JunB-mediated p16 transcriptional regulation, a 1.2 kb 5′-fragment of the mouse p16 promoter was cloned into a luciferase reporter construct (p16-Luc) and transient transfection experiments were performed with wild-type, jB-Tg and junB–/– 3T3 fibroblasts (Figure 6A). Wild-type fibroblasts showed a moderate level of p16-Luc activity, consistent with the low levels of p16 mRNA and protein expressed in these cells. In contrast, jB-Tg cells exhibited three times higher p16 transcriptional activity whereas junB–/– cells displayed only half of the wild-type activity. Expression vectors for JunB (pB-junB) and c-Jun (pB-c-jun) were then co-transfected with the p16-Luc reporter construct (Figure 6A). Exogenous JunB expression in wild-type and junB–/– 3T3 cells increased p16 promoter activity to the levels obtained in jB-Tg cells with the reporter alone. Co-transfection of JunB in jB-Tg 3T3 cells, however, did not further increase the p16 promoter activity, suggesting that it had already reached saturation level. In contrast, co-transfection of c-Jun resulted in a slight decrease in p16 promoter activity in wild-type cells and in a strong inhibition of JunB-mediated p16 transactivation in jB-Tg 3T3 cells. Increased p16 promoter activity was also observed in c-jun–/– fibroblasts (data not shown), which further suggests that c-Jun represses p16 expression. Interestingly, while c-Jun levels in jB-Tg cells were similar to those in wild-type cells, junB–/– cells showed increased levels of c-Jun protein and AP-1 binding activity (data not shown).

Fig. 6. JunB mediates p16 transactivation through three AP-1-like binding sites. (A) Fibroblasts were co-transfected with 0.3 µg of the p16 promoter reporter construct together with 0.2 µg of empty plasmid (pB-puro) or plasmid expressing JunB (pB-junB) or c-Jun (pB-c-jun) and 0.2 µg of a LacZ-expressing vector. The mean ± SD of one representative experiment performed in triplicate is shown. (B) Gel-shift assays performed with jB-Tg nuclear extracts and 32P-labelled oligonucleotide encoding either a consensus AP-1 binding site or the three wild-type (A, B and C) or mutated (Amut., Bmut. and Cmut.) AP-1-like binding sites of the mouse p16 promoter. The arrowhead (SS) indicates the super-shift obtained with the JunB antibody. (C) Gel-shift assays performed with jB-Tg nuclear extracts, 32P-labelled oligonucleotide encoding a consensus AP-1 binding site and 100× cold competitors as indicated. (D) Fibroblasts were co-transfected with 0.3 µg of the depicted luciferase reporter constructs together with 0.2 µg of the LacZ-expressing vector. The results are normalized to the maximal stimulation obtained for the wild-type promoter in jB-Tg cells (100%) and are the mean ± SD of one representative experiment performed in triplicate. (E) Model for JunB-mediated p16 transcriptional regulation. In wild-type cells, JunB binds only site B, resulting in a basal level of promoter activity. In jB-Tg cells, JunB is bound to the three binding sites and the promoter activity is increased 3-fold. In junB–/– cells, only the JunB-interacting proteins are bound and the promoter activity is decreased 2-fold.
Sequence analysis of the mouse p16 promoter revealed five putative AP-1-like binding sites, and super-shift experiments performed with jB-Tg nuclear extracts demonstrated that three of them, termed A (TGACTGA, at –1189), B (TGACTTCA, at –783) and C (TGACACA, at –484), bind JunB (Figure 6B). Using wild-type nuclear extracts, JunB binding was detected only on site B and c-Jun binding on sites A and B (data not shown). Finally, these three sequences were able to compete out the AP-1 binding activity on a consensus AP-1 binding site (TGACTCA), thereby demonstrating that they are JunB/AP-1 binding sites (Figure 6C).
To determine whether the effect of JunB on p16 expression is mediated through these three AP-1-like sites, we mutated each of them by site-directed mutagenesis. These mutated sites lost JunB binding activity (Figure 6B) and failed to compete out AP-1 binding activity (Figure 6C). Luciferase reporter constructs containing all the combinations of wild-type and mutated sites were then generated and tested in transient transfections (Figure 6D). In jB-Tg fibroblasts, mutation of one or two of these binding sites decreased the expression of the p16-Luc reporter by ∼30% while mutation of all three abolished 70% of the promoter activity. Similar residual promoter activity was obtained with the triple mutant reporter construct in wild-type and junB–/– cells. In junB–/– cells, both the triple mutant and the wild-type reporter showed similar activity levels, therefore suggesting that these three binding sites mediate the entire JunB-induced p16 transactivation. Finally, in wild-type fibroblasts, a single mutation on site B gave similar residual promoter activity to the triple mutant, which, together with the data of the super-shift experiments, argues for a major role of site B in JunB-induced p16 transactivation. These results are summarized in Figure 6E, describing a model of how p16 promoter activity could be regulated by the amount of JunB protein expressed in the cell.
JunB suppresses v-ras- and v-src-induced cellular transformation and tumour growth
Finally, we investigated whether oncogenic transformation is affected by high JunB expression since this cellular process critically depends on cell proliferation and is often associated with increased AP-1 activity. Wild-type and jB-Tg 3T3 fibroblasts were infected with an empty retrovirus or retroviruses expressing activated Ras and Src oncogenes. As shown in Figure 7A for v-ras-infected fibroblasts, both wild-type and jB-Tg cells exhibited the typical morphology of transformed cells such as increased light refraction and an elongated shape. Loss of contact inhibition is a characteristic of oncogenic transformation that can be monitored in vitro by a focus formation assay (Figure 7B). Wild-type and jB-Tg fibroblasts infected with an empty virus remained contact-inhibited and did not show any signs of oncogenic transformation. Expression of activated Ras and Src in wild-type cultures efficiently induced loss of contact inhibition as shown by the high number of foci. In contrast, jB-Tg cultures exhibited greatly reduced numbers of foci after v-ras and v-src infection. Out of five jB-Tg cell lines tested, two exhibited no signs of focus formation whereas the three others showed a 60–70% decrease. To test whether JunB can still suppress cell transformation in the absence of p16, pB-puro- and pB-junB-infected INK4a–/– fibroblasts were used in the same assay (Figure 7B). The lack of both p16 and p19ARF expression has already been reported to cause spontaneous transformation and to increase susceptibility to oncogenic transformation by Ras (Serrano et al., 1996). Consistently, control INK4a–/– cells infected with an empty virus readily formed numerous foci, which increased in numbers following v-ras and v-src infection. Most importantly, both control INK4a–/– and JunB-expressing INK4a–/– fibroblasts displayed similar numbers of foci after infection with v-ras, v-src and empty virus, indicating that JunB cannot suppress cell transformation in the absence of p16.

Fig. 7. JunB suppresses v-ras- and v-rc-induced tumour growth. (A) Phase-contrast photographs of WT and jB-Tg 3T3 fibroblasts infected with empty-neo or v-ras-expressing retroviruses. (B) Focus formation assay 2 weeks after infection with empty-neo or v-ras and v-src-expressing retroviruses. (C) v-ras-infected cells were injected subcutaneously into nude mice and the individual tumours were dissected and weighed after 2 weeks. One representative tumour of each genotype is shown in the upper part and the weight of each tumour is depicted in the lower part.
The ability of the infected cells to form tumours when injected into immunosuppressed mice was analysed next (Figure 7C). Neither wild-type nor jB-Tg cells infected with an empty virus formed tumours, whereas small tumours were obtained with INK4a–/– cells at 5 weeks post-injection. However, within 2 weeks, mice injected with v-ras-infected wild-type fibroblasts developed large tumours (120 ± 40 mg) while v-ras-infected jB-Tg fibroblasts from two independent cultures gave rise to only very small tumours (15 ± 7 mg). In addition, these small tumours did not increase in size when evaluated at 5 weeks post-injection. The lack of p16 and p19ARF expression resulted in increased tumour mass since tumours derived from v-ras-infected INK4a–/– fibroblasts were found to be significantly larger (214 ± 106 mg) than those derived from v-ras-infected wild-type fibroblasts. Similar results were obtained for tumours derived from v-src-infected fibroblasts, i.e. dramatically reduced jB-Tg-derived tumours and enlarged INK4a–/–-derived tumours (data not shown). These results demonstrate that elevated JunB expression suppresses both neoplastic transformation and tumour growth induced by v-ras and v-src. Finally, no changes in tumour size were observed between tumours derived from v-ras-infected control INK4a–/–- and JunB-expressing INK4a–/– fibroblasts, indicating that JunB cannot suppress tumour growth in the absence of p16 (Figure 7C).
Discussion
In this study, we have shown for the first time that JunB suppresses cell proliferation by direct transcriptional activation of the CKI p16INK4a, thereby providing a novel molecular link between AP-1 complexes and the cell cycle machinery. Although the levels of p16 protein do not fluctuate in response to mitogenic stimulation, their control is critical in determining the length of the G1 phase of the cell cycle and consequently the proliferation rates of the cells (Harper and Elledge, 1996).
A correlation between JunB and p16 levels is first observed in fibroblastic cell lines derived from transgenic mice that constitutively express high junB proteins. In both jB-Tg primary MEFs and immortalized 3T3 cells, a specific increase in p16 expression is found, which correlates with impaired proliferation. Primary jB-Tg MEFs display decreased proliferation rates after 4–5 passages and undergo premature senescence compared with wild-type MEFs. The CKI p16 has been directly implicated in inducing cellular senescence based on the observations that p16 levels are rapidly increasing when primary cells approach senescence (Palmero et al., 1997; Zindy et al., 1997) and that INK4a–/– MEFs do not senesce (Serrano et al., 1996). Similarly, JunB levels rapidly increase in wild-type MEFs when cells approach senescence. These results suggest that JunB contributes to establish p16 levels during senescence and further support the conclusion that elevated p16 levels are causal to the premature onset of replicative senescence in jB-Tg MEFs. In immortalized jB-Tg 3T3 fibroblasts, the proliferation defect results from a complete inhibition of cyclin D-associated kinase activity, which, in turn, leads to reduced pRb hyperphosphorylation and to an extension of the G1 phase of the cell cycle. Since p16 specifically inhibits the cyclin D–CDK4–6 complexes, its presence at high levels is likely to be the cause of the alteration in the cyclin D–pRb cascade that affects S-phase transition in jB-Tg 3T3 fibroblasts. Furthermore, ectopic JunB expression in wild-type fibroblasts results in increased p16 levels and reduces proliferation. Most importantly, proliferation rates are unchanged following ectopic JunB expression in p16-deficient INK4a–/– fibroblasts, thereby directly demonstrating that the suppressive effect of JunB on cell proliferation cannot occur in the absence of p16. INK4a–/– fibroblasts also lack p19ARF but since the expression of this cell cycle regulator is not affected by JunB, the lack of p16 appears likely to be the cause for the absence of a JunB-mediated effect in INK4–/– cells. However, it would be important to confirm these data in a pure p16 knockout background when such a cell system becomes available. The correlation between JunB and p16 levels is also confirmed in cells lacking JunB since decreased p16 protein levels are found in both junB–/– ES cells and junB–/– MEFs. However, none of these cell types exhibits increased proliferation rates. Proliferation of junB–/– ES cells might not be affected because undifferentiated ES cells do not express D-type cyclins and are therefore insensitive to p16 levels (Savatier et al., 1996). The unaffected proliferation rates of junB–/– MEFs may simply reflect the fact that p16 levels are reduced and not abolished as in INK4a–/– MEFs. However, in agreement with decreased p16 levels, junB–/– MEFs do undergo a delayed senescence.
Ectopic expression of p16 has been shown to induce G1 arrest in cell lines containing functional pRb (Serrano, 1997). In contrast, the elevated p16 levels induced by exogenous junB expression do not fully arrest jB-Tg 3T3 cells and, more surprisingly, do not affect cyclin E–CDK2 kinase activity. This result is striking since active forms of cyclin D–CDK4–6 complexes contain the CKIs p21 and p27, which are released upon p16 inhibition of the cyclin D–CDK4–6 complexes. As a consequence, the free p21 and p27 block the activity of cyclin E–CDK2 complexes, thereby arresting the cells in G1 (Sherr and Roberts, 1995; LaBaer et al., 1997; Cheng et al., 1999). In jB-Tg cells, release of p21/p27 only delays the induction of cyclin E–CDK2 kinase activity, thereby contributing to the G1-phase extension and reduced proliferation. Another intriguing observation is the reduced pRb protein levels found in exponentially growing jB-Tg 3T3 fibroblasts. However, since this effect is not observed in serum-arrested jB-Tg cells, it is most probably a secondary consequence of the slow proliferation rates of the cells rather than a direct effect of JunB on pRb expression. Moreover, mutations in the p16/Rb pathway, mainly deletions of the entire INK4a locus, and mutations of the tumour suppressor gene p53 are the two major known events leading to cellular immortalization and cancer development (Harper and Elledge, 1996). We therefore expected that in jB-Tg cells p16 and/or p53 would have been inactivated during immortalization. However, we found that jB-Tg MEFs became immortalized with similar kinetics to wild-type MEFs and that spontaneously immortalized jB-Tg 3T3 cells maintained a functional p53 and INK4a locus.
Accumulation of p16 mRNA and protein has been reported in response to various stimuli or conditions such as cellular senescence and overexpression of oncogenic Ras in rodent cells or inactivation of pRb in human cells (Serrano, 1997). More recently, Jacobs et al. (1999) have shown that the polycomb-group gene bmi-1 is a repressor of the entire INK4a locus that regulates the expression of both p16 and p19ARF proteins. In this study, we have identified JunB as one of the first transcription factors that directly regulates the p16 promoter. We also propose a mechanism explaining how p16 transcription levels might be determined by the amount of JunB protein expressed in the cell (Figure 6E). In wild-type cells, where JunB is expressed at a low level, only one site (B) is occupied. Interaction of JunB with other(s), as yet unknown, transcription factor(s)/co-factor(s) results in a basal level of p16 transactivation. Since p16 mRNA and protein are remarkably stable, with a half-life of >24 h for the mRNA and ranging from 8 to 18 h for the protein, this low level of transactivation is sufficient to ensure a constant low level of p16 protein throughout the cell cycle. When JunB levels rise, the two other AP-1-like sites (A and C) are occupied, resulting in a 3-fold increase in p16 promoter activity. Owing to the high stability of the mRNA and protein, this minor increase in transcription produces a robust and constant accumulation of p16 protein. Conversely, in the absence of JunB protein none of these sites are occupied and the low residual transactivation is ensured by the associated partner(s). Co-transfection experiments have shown that c-Jun contributes to p16 regulation since c-Jun represses the basal p16 transcriptional activity and inhibits JunB-mediated p16 transactivation. Interestingly, elevated c-Jun protein levels are found in junB–/– cells that display reduced p16 transactivation. However, the multiple DNA–protein complexes obtained on each of these AP-1-like sites suggest that the other(s) JunB heterodimerization partner(s) may differ from the usual Fos and ATF family members. Finally, in light of the recently published paper by Li et al. (1999), it will be interesting to investigate the importance of JunB phosphorylation in p16 regulation.
JunB has been postulated to be a growth arrest mediator since increased junB expression was found in different cell types such as myeloid cells or T helper cells undergoing terminal differentiation (Lord et al., 1990; Li et al., 1999). Moreover, treating fibroblasts as well as human mammary carcinoma cells and neuronal cells with junB-specific antisense oligonucleotides led to increased cell proliferation (Schlingensiepen et al., 1993). However, in none of these cell types has a clear molecular mechanism been described, and the inhibitory effect of JunB on cell proliferation has rather been linked to its ability to decrease AP-1 transactivation. Besides this indirect effect, which may contribute to inhibition of cell proliferation by reducing the transactivation potential of positive regulators such as c-Jun, we show that JunB can also directly control the expression level of at least one very important negative regulator of the cell cycle, p16. In a recent paper, Bakiri et al. (2000) have identified a second target gene for JunB in the same regulatory pathway, since they could demonstrate that JunB negatively regulates the cyclin D1 promoter. In agreement with their observations, we detected reduced steady-state levels and delayed serum induction of cyclin D1 in jB-Tg cells, which may also contribute to their proliferation defect. Since Bakiri et al. (2000) were using two different immortalized cell lines that happen to lack the entire INK4a locus, the effect of JunB was only observed on cyclin D1 regulation with consequently a less dramatic reduction in the proliferation rates. Nevertheless, these two convergent findings support the notion that JunB, in contrast to c-Jun, is a negative regulator of cell proliferation and show that JunB can directly transactivate the expression of some critical cell cycle regulators.
JunB is widely expressed throughout embryonic development and its expression is maintained at a low level in adult tissues (Wilkinson et al., 1989; Schorpp et al., 1996; Schorpp-Kistner et al., 1999). In contrast, p16 is not expressed during embryonic development and only low levels of mRNA have been detected by RT–PCR in the lung, spleen and testis of old mice (Zindy et al., 1997). Furthermore, INK4a–/– mice develop normally and do not show obvious abnormalities in adulthood besides a higher incidence of tumour development (Serrano et al., 1996). Interestingly, although junB–/– embryos are severely retarded in growth and development, leading to an early embryonic lethality at ∼E9.5, cellular proliferation is apparently not impaired (Schorpp-Kistner et al., 1999). It is therefore likely that other JunB target genes, different from p16, are involved in mediating JunB functions during embryogenesis. In adult mice, JunB and p16 are co-expressed in all organs where p16 is present, which suggests that in the organs where junB is expressed but p16 is not, other regulatory mechanisms, as yet unknown, are able to down-regulate p16 expression. To corroborate the findings in fibroblasts with other cell types in vivo, p16 expression levels were analysed by RT–PCR in different organs of junB transgenic mice. Interestingly, the spleen of junB transgenic mice showed significantly increased p16 mRNA levels, and preliminary analysis of junB transgenic B and T cells in vitro indicates reduced proliferation upon mitogenic stimulation (data not shown). These observations therefore suggest that the JunB/p16 pathway may well act in vivo to regulate lymphocyte proliferation and we are presently analysing whether junB transgenic mice are immune-deficient.
The oncogenic potential of the Jun family members has been extensively studied and efficient neoplastic transformation of rodent cells has been found to require cooperation with activated oncogenes such as Ras and Src (Angel and Karin, 1991). In cooperation with oncogenic Ras, the different Jun proteins display distinct transforming capacities: c-Jun, a positive regulator of cell proliferation, transforms very efficiently (Schütte et al., 1989a), while JunD, a negative regulator of cell proliferation, antagonizes ras-induced transformation (Pfarr et al., 1994). JunB, similarly to JunD, displays a poor transforming activity and was found to inhibit c-Jun/ras-induced transformation (Schütte et al., 1989b). Here, we show using jB-Tg fibroblasts that elevated JunB expression greatly reduces both ras- and src-mediated cell transformation. Since p16 has been shown to play a major inhibitory function in ras-mediated oncogenic transformation (Serrano, 1997), increased p16 levels are likely to account for the impaired ras-mediated transformation in jB-Tg cells. Although p16 involvement in src-mediated oncogenic transformation is not as well documented, increased p16 levels are also likely to account for the impaired src-induced transformation. Furthermore, the suppressive effect of JunB on both ras- and src-mediated transformation does not occur in INK4a–/– fibroblasts. However, since the respective contribution of p16 and p19ARF to oncogenic transformation is not yet understood, this experiment does not allow us to clearly attribute to p16 the absence of a JunB-mediated effect in INK4a–/– fibroblasts. Finally, we show that elevated JunB expression also suppresses ras- and src-induced tumour growth in vivo, thereby identifying JunB as an anti-oncogene. Additional in vivo evidence for such a tumour suppressive function of JunB comes from the analysis of mice specifically lacking junB expression in the myeloid lineage, since absence of JunB leads to increased myeloid cell proliferation and to the development of a myeloid leukaemia (E.Passegué, W.Jochum and E.F.Wagner, manuscript in preparation). Future experiments will address whether the JunB/p16 pathway is important in suppressing tumour growth in other murine cancer models and whether the suspected tumour suppressive function of JunB can be extended to human cells and disease.
Materials and methods
Cell cultures and proliferation assays
All wild-type and jB-Tg MEFs were isolated from individual E12.5 fetuses obtained from intercrosses between junB transgenic mice (Schorpp et al., 1996). The junB–/– 3T3 clone was derived from a single E8.5 fetus obtained from intercrosses between junB+/– mice and all junB–/– MEFs from individual E12.5 rescued fetuses obtained by micro-injection of junB–/– ES cells into tetraploid wild-type blastocysts (Schorpp-Kistner et al., 1999). All cell lines were immortalized from individual primary cultures according to the 3T3 protocol (Todaro and Green, 1963). INK4a–/– fibroblasts were obtained from C.Sherr (Howard Hughes Medical Institute, Memphis, TN). Fibroblasts were grown at 37°C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum, 2 mM glutamine, 1% penicillin/streptomycin and 0.1 mM β-mercaptoethanol. For fibroblast proliferation, 3.75 × 105 cells were plated per 25 cm2 flask and passaged every 3–4 days at the same density to determine the cumulative cell number. Senescence-associated β-galactosidase staining was performed as previously described (Dimri et al., 1995).
Retroviral infections
Viral supernatants were generated by standard methods. Fibroblasts were plated at 3 × 105 per 10 cm dish and infected the next day with viral supernatant containing 8 µg/ml polybrene. The selection was started 48 h post-infection using 2.5 µg/ml puromycin or 1 mg/ml G418. For transformation assays, the following retroviral supernatants were used: Ψ2-she-ras [LTR-Ha-ras], SR1 [LTR-neo TK-v-src] and N2/Ψ2 [LTR-neo] (Boulter and Wagner, 1988). For focus formation assays, cells were cultured in the same dishes without selection for 2 weeks, washed with PBS, fixed with methanol and stained with 0.2% methylene blue. For tumorigenicity assays, 1 × 106 exponentially growing G418-resistant cells were subcutaneously injected into the scapular region of 5-week-old anaesthetized nude mice (two injection sites per mouse).
Flow cytometry analysis
Propidium iodide staining and propidium iodide exclusion were performed according to standard procedures. TUNEL staining (In Situ Cell Death Detection Kit, Boehringer Mannheim) and anti-BrdU-FITC staining (Becton Dickinson) were carried out according to the manufacturer’s instructions. Data were collected and analysed with a Becton Dickinson FACScan and CellQuest software.
Protein and RNA analysis
Immunoblot analyses were performed according to standard procedures using 20 µg of nuclear proteins or 30 µg of whole cell extracts. For kinase assays, 500 µg of whole cell extracts were immunoprecipitated with cyclin D1– or cyclin E–agarose conjugate antibodies, and immune complexes were incubated with glutathione S-transferase (GST)–Rb (C-pocket 769–921; Santa Cruz) or histone H1 (Boehringer Mannheim) substrates as described (Matsushime et al., 1994). The JunB monoclonal antibody and the p19ARF polyclonal antibody were generous gifts from D.Lallemand (Pasteur Institute, Paris) and C.Sherr (Howard Hughes Medical Institute, Memphis, TN), respectively. The p53, pRb and actin antibodies were obtained from Medac, PharMingen and Sigma, respectively. All the other antibodies were obtained from Santa Cruz. Total RNA was isolated using Trizol reagent (Gibco-BRL) and northern blot analyses were performed using 20 µg of total RNA according to standard procedures.
Reporter gene assays
A 1.2 kb 5′-fragment of the mouse p16 promoter was cloned by PCR and inserted into the pGL3 plasmid to generate the reporter p16-Luc. Site-directed mutagenesis (QuickChange™ Site-directed Mutagenesis kit; Stratagene) was performed to mutate the individual AP-1-like binding sites A (5′-TGACTGA-3′ to 5′-aaACTGA-3′), B (5′-TGA CTTCA-3′ to 5′-aaACTTCA-3′) and C (5′-TGACACA-3′ to 5′-aaACACA-3′). Transfections were performed in 24-well plates using LipofectaminePlus (Gibco BRL). Cells were harvested 24 h post-transfection and assayed for luciferase and β-galactosidase activities according to standard procedures.
Gel-shift experiments
Binding reactions were performed for 1 h on ice in the presence of 10 µg of nuclear extracts, 1 µg of poly(dI–dC), 15 000 c.p.m. of 32P-end-labelled oligonucleotide and competitor where specified. For super-shift experiments, nuclear extracts were pre-incubated with the antibody for 1 h on ice prior to addition of the 32P-labelled probe. Analyses of oligonucleotide bound complexes were performed according to standard procedures.
Acknowledgments
Acknowledgements
We are grateful to G.Peters for providing reagents and sequence information on the p16 promoter prior to publication, to D.Lallemand and M.Yaniv for the JunB monoclonal antibody, to M.Roussel and C.Sherr for the INK4a–/– cells and p19ARF antibody, to K.Matsuo for the pBabe-junB plasmid and to A.Fleishmann and P.Steinlein for help with cell transformation experiments and flow cytometry, respectively. We thank P.Angel, M.Schorpp-Kistner, K.Nasmyth and H.Beug for critical reading of the manuscript and W.Jochum, K.Matsuo and L.Stingl for fruitful discussions and assistance. This work was partly supported by the Austrian Research Foundation and by a TMR-network grant from the European Economic Community.
References
- Angel P. and Karin,M. (1991) The role of Jun, Fos and the AP-1 complex in cell proliferation and transformation. Biochim. Biophys. Acta, 1072, 129–157. [DOI] [PubMed] [Google Scholar]
- Bakiri L., Lallemand,D., Bossy-Wetzel,E. and Yaniv,M. (2000) Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J., 19, 2056–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter C.A. and Wagner,E.F. (1988) The effect of v-src expression on the differentiation of embryonal carcinoma cells. Oncogene, 2, 207–214. [PubMed] [Google Scholar]
- Cheng M., Olivier,P., Diehl,J.A., Fero,M., Roussel,M.F., Roberts,J.M. and Sherr,C.J. (1999) The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J., 18, 1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng T. and Karin,M. (1993) Jun B differs from c-Jun in its DNA-binding and dimerization domains, and represses c-Jun by formation of inactive heterodimers. Genes Dev., 7, 479–490. [DOI] [PubMed] [Google Scholar]
- Dimri G.P. et al. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl Acad. Sci. USA, 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber D.A. (1997) Splicing into senescence: the curious case of p16 and p19ARF. Cell, 91, 555–558. [DOI] [PubMed] [Google Scholar]
- Harper J.W. and Elledge,S.J. (1996) Cdk inhibitors in development and cancer. Curr. Opin. Genet. Dev., 6, 56–64. [DOI] [PubMed] [Google Scholar]
- Hsu J.C., Cressman,D.E. and Taub,R. (1993) Promoter-specific transactivation and inhibition mediated by JunB. Cancer Res., 53, 3789–3794. [PubMed] [Google Scholar]
- Jacobs J.L., Kieboom,K., Marino,S., DePinho,R.A. and Van Lohuizen,M. (1999) The oncogene and polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature, 397, 164–168. [DOI] [PubMed] [Google Scholar]
- Johnson R.S., Van Lingen,B., Papaioannou,V.E. and Spiegelman,B.M. (1993) A null mutation at the c-jun locus causes embryonic lethality and retarded cell growth in culture. Genes Dev., 7, 1309–1317. [DOI] [PubMed] [Google Scholar]
- Karin M., Liu,Z.G. and Zandi,E. (1997) Ap-1 function and regulation. Curr. Opin. Cell Biol., 9, 240–246. [DOI] [PubMed] [Google Scholar]
- Kovary K. and Bravo,R. (1991) The jun and fos protein families are both required for cell cycle progression in fibroblasts. Mol. Cell. Biol., 11, 4466–4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBaer J., Garrett,M.D., Stevenson,L.F., Slingerland,J.M., Sandhu,C., Chou,H.S., Fattaey,A. and Harlow,E. (1997) New functional activities for the p21 family of CDK inhibitors. Genes Dev., 11, 847–862. [DOI] [PubMed] [Google Scholar]
- Li B., Tournier,C., Davis,R.J. and Flavell,R.A. (1999) Regulation of IL-4 expression by the transcription factor JunB during T helper cell differentiation. EMBO J., 18, 420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord K.A., Hoffmann-Libermann,B. and Libermann,D.A. (1990) Complexity of the immediate early response of myeloid cells to the terminal differentiation and growth arrest including ICAM-1, JunB and histone variants. Oncogene, 5, 387–396. [PubMed] [Google Scholar]
- Matsuo K., Owens,J.M., Tonko,M., Elliot,C., Chambers,T.J. and Wagner,E.F. (2000) Fosl1 is a transcriptional target of c-Fos during osteoclast differentiation. Nature Genet., 24, 184–187. [DOI] [PubMed] [Google Scholar]
- Matsushime H., Quelle,D.E., Shurtleff,S.A., Shibuya,M., Sherr,C.J. and Kato,J.Y. (1994) D-type cyclin-dependent kinase activity in mammalian cells. Mol. Cell. Biol., 14, 2066–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmero I., McConnell,B., Parry,D., Brookes,S., Hara,E., Bates,S., Jat,P. and Peters,G. (1997) Accumulation of p16/INK4a in mouse fibroblasts as a function of replicative senescence and not of retinoblastoma gene status. Oncogene, 15, 495–503. [DOI] [PubMed] [Google Scholar]
- Pfarr C.M., Mechta,F., Spyrou,G., Lallemand,D., Carillo,S. and Yaniv,M. (1994) Mouse JunD negatively regulates fibroblast growth and antagonizes transformation by ras. Cell, 76, 747–760. [DOI] [PubMed] [Google Scholar]
- Savatier P., Lapillonne,H., Van Grunsven,L.A., Rudkin,B.B. and Samarut,J. (1996) Withdrawal of differentiation inhibitory activity/leukemia inhibitory factor up-regulates D-type cyclins and cyclin-dependent kinase inhibitors in mouse embryonic stem cells. Oncogene, 12, 309–322. [PubMed] [Google Scholar]
- Schlingensiepen K.H., Schlingensiepen,R., Kunst,M., Klinger,I., Gerdes,W., Seifert,W. and Brysch,W. (1993) Opposite functions of junB and c-jun in growth regulation and neuronal differentiation. Dev. Genet., 14, 305–312. [DOI] [PubMed] [Google Scholar]
- Schorpp M., Jäger,R., Schellander,K., Schenkel,J., Wagner,E.F., Weiher,H. and Angel,P. (1996) The human ubiquitin C promoter directs high ubiquitous expression of transgenes in mice. Nucleic Acids Res., 24, 1787–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorpp-Kistner M., Wang,Z.-Q., Angel,P. and Wagner,E.F. (1999) JunB is essential for mammalian placentation. EMBO J., 18, 934–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber M., Kolbus,A., Piu,F., Szabowsky,A., Möhle-Steinlein,U., Tian,J., Karin,M., Angel,P. and Wagner,E.F. (1999) Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev., 13, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütte J., Minna,J.D. and Birrer,M.J. (1989a) Deregulated expression of human c-jun transforms primary rat embryo cells in cooperation with an activated c-Ha-ras gene and transforms Rat-1a cells as a single gene. Proc. Natl Acad. Sci. USA, 86, 2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütte J., Viallet,J., Nau,M., Segal,S., Federiko,J. and Minna,J. (1989b) JunB inhibits and c-Fos stimulates the transforming and transactivating activities of c-Jun. Cell, 59, 987–997. [DOI] [PubMed] [Google Scholar]
- Serrano M. (1997) The tumour suppressor protein p16INK4a. Exp. Cell Res., 237, 7–13. [DOI] [PubMed] [Google Scholar]
- Serrano M., Lee,L.-W., Chin,L., Cordon-Cardo,C., Beach,D. and DePinho,R.A. (1996) Role of the INK4a locus in tumor suppression and cell mortality. Cell, 85, 27–37. [DOI] [PubMed] [Google Scholar]
- Sherr C.J. and Roberts,J.M. (1995) Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev., 9, 1149–1163. [DOI] [PubMed] [Google Scholar]
- Todaro G.J. and Green,H. (1963) Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol., 17, 299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg R.A. (1995) The retinoblastoma protein and cell cycle control. Cell, 81, 323–330. [DOI] [PubMed] [Google Scholar]
- Wilkinson D.G., Sangita,B., Ryseck,R.P. and Bravo,R. (1989) Tissue-specific expression of c-jun and junB during organogenesis in the mouse. Development, 106, 465–471. [DOI] [PubMed] [Google Scholar]
- Wisdom R., Johnson,R.S. and Moore,C. (1999) c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J., 18, 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F., Quelle,D.E., Roussel,M. and Sherr,C.J. (1997) Expression of the p16/INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene, 15, 203–211. [DOI] [PubMed] [Google Scholar]