

Summary
The Easson-Stedman hypothesis provided the rationale for the first studies of drug design for the α1-adrenergic receptor. Through chemical modifications of the catecholamine core structure, the need was established for a protonated amine, a β-hydroxyl on a chiral center, and an aromatic ring with substitutions capable of hydrogen bonding. After the receptors were cloned and three α1-adrenergic receptor subtypes were discovered, drug design became focused on the analysis of receptor structure and new interactions were uncovered. It became clear that α1 and β-adrenergic receptors did not share stringent homology in the ligand-binding pocket but this difference has allowed for more selective drug design. Novel discoveries on allosterism and agonist trafficking may be used in the future design of therapeutics with fewer side effects. This review will explore past and current knowledge of the structure-function of the α1-adrenergic receptor subtypes.
Art Hancock
This review is dedicated to the memory of Art Hancock because of his involvement in structure function studies of α1-adrenergic receptors (ARs) throughout his career. He performed early characterizations of the structure-function between α1- and α2-ARs by both pharmacological evaluation and modeling of the compounds (1). He led development of the first synthesized α1A-AR selective agonist, A-61603, highly used in academia (2). When the field was looking for an in vivo function for the new α1D-AR subtype, his work determined that this receptor mediated the contraction of the rat aorta (3). He studied the uroselectivity of α1-AR agonists and antagonists for the treatment of benign prostatic hyperplasia and stress urinary incontinence (4–9) and performed structure-function of bi-and tri-cyclic indoles as α1A-AR selective antagonists (10–11).
The Precloning Era
Easson-Stedman
Structure-activity relationships in the α1-ARs had their birthplace in the hypothesis of Easson and Stedman (12). This theory was an attempt to explain the functional differences between the enantiomers of biogenic amines (i.e. a pair of isomers that are mirror images of one another because of changes around a chiral center). Central to the hypothesis was a three-point attachment to a putative receptor binding pocket critical to the full function of a sympathomimetic amine composed of the (1) the basic nitrogen, (2) the hydroxyl group off the β-carbon, and (3) the phenyl group, where potent agonist activity is dependent on the presence of appropriate hydroxyl-substitutions (Fig 1). According to the Easson-Stedman hypothesis, potency is enhanced by the β-hydroxyl on the chiral carbon, and this has been confirmed to contribute about a 10–100 fold increased potency for the R (−) enantiomers. This hypothesis explained why the most potent α1-AR agonists are the R (−) enantiomers but only for phenethylamine-type agonists and not most imidazolines, which lack the β-hydroxyl and chiral center (Fig 2). Imidazolines that were synthesized to contain a β-hydroxyl at the chiral center actually had a decreased activity at α1-ARs instead of the predicted 10–100 fold increased activity (13).
Figure 1. The Easson-Stedman hypothesis.
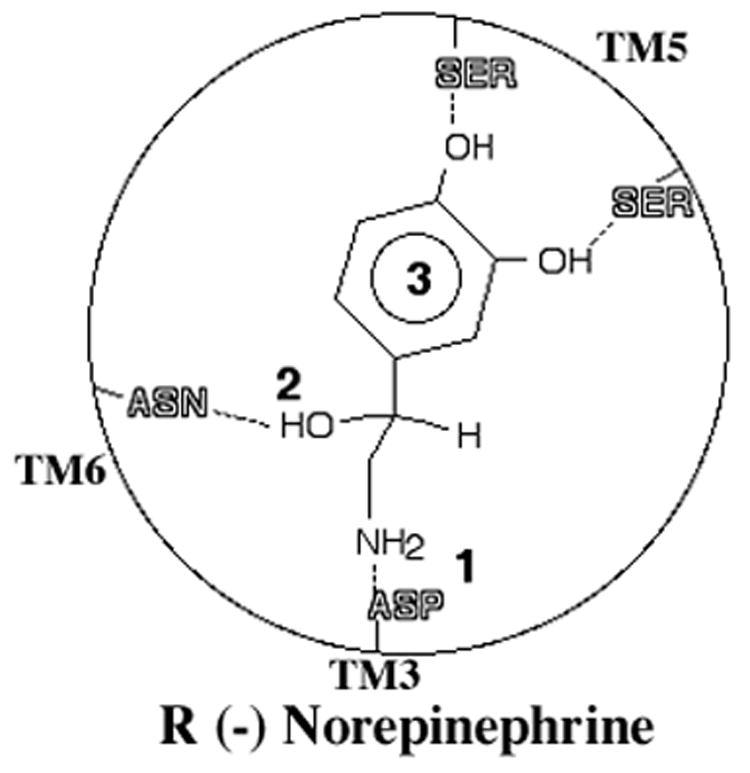
While the specific receptor residues were not known at the time of this hypothesis, they are included to show validity. The hypothesis states that catecholamines interact with the receptor in a three-point contact. 1. The protonated amine of norepinephrine interacts with an aspartic acid (ASP) residue in TM 3. 2. The β-hydroxyl on a chiral center interacts with an asparagine (ASN) in TM 6. 3. An aromatic ring with hydrogen bonding substitutions interacts with serines in TM 5.
Figure 2. Chemical structures for common α1-AR agonists.
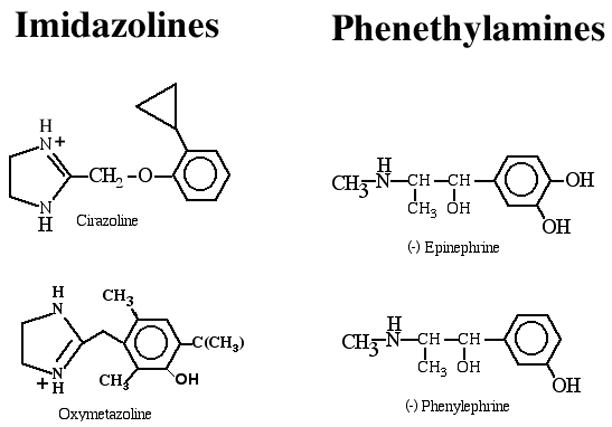
α1-AR agonists can be grouped into two classes: imidazolines (cirazoline and oxymetazoline) and phenethylamines ((−)epinephrine and (−) phenylephrine). Most imidazolines do not have chiral centers, and the positive charge on the nitrogen resonates between the two nitrogens in the imidazole ring.
Besides the obvious crucial role of the basic nitrogen atom in a biogenic amine, its position relative to the aromatic ring is important. Optimal agonist potency is obtained when the nitrogen is separated from a saturated six-member ring (14) by 3-carbon units (N+3 rule). Basic nitrogens can be contained within a cyclic structure, such as with imidazolines, but must retain a defined footprint relative to the aromatic ring. Quaternization of the nitrogen atom, or its replacement by a neutral atom such as oxygen, dramatically reduces its potency and intrinsic activity (15). In addition, substituents on the basic nitrogen are poorly tolerated in α1-ARs for either agonists or antagonists, usually being limited to one carbon length. However, they are well tolerated in β-ARs, with increasing steric bulk adjacent to the nitrogen being associated with increased antagonist potency or increased agonist selectivity (16).
The other critical component of agonist potency is the aromatic ring and its hydroxyl-substituents. Mutagenesis studies indicated the importance of an aromatic ring for agonist binding (17) as well as for agonist efficacy (18). Hydrogen-bonding substitutions on the aromatic ring can greatly influence both binding and functional agonism. Initially in the β2-AR, meta- and para-hydroxyl substitutions that mimic norepinephrine and epinephrine seemed essential for full agonism (19), but the α1-ARs were shown to be quite tolerant of the position and chemical group so long as hydrogen-bonding was capable at the meta-position of the ring (20). In addition, fluorine substitutions in the ortho-positions 2- and 6- in epinephrine can confer selectivity between α– and β-ARs (21).
Phenethylamines vs. Imidazolines
By the 1970s, Patil et al. (22) had shown the validity of the Easson-Stedman hypothesis for virtually all phenethylamines. Most imidazolines were found to have better selectivity for the α2-ARs (15). Of the notable exceptions, it was realized early that the way phenethylamines interacted with the α1-AR was different from the interactions with imidazolines (23–24). At the time, it was not known if this was due to different subtypes of α1-ARs or to the way they interacted with the same receptor. The work of Minneman and colleagues in the 1990s showed that most imidazolines had selectivity for the α1A-AR subtype (25). We also showed some years later that imidazolines (discussed in more detail later) interact with residues closer to the cell surface in the α1-AR binding pocket, much like α1-AR antagonists (26), which also explains why most imidazolines have poor intrinsic activity.
Mutagenesis-Based Structure-Function
How many subtypes are there again?
Before the discussion of the structure-function of cloned receptors, there is a need to be clear about the classification, since this was once a matter of intense confusion. First characterized in isolated tissue, α1-ARs were initially divided into the α1A- and α1B-AR subtypes based upon the interpretation of two-site binding curves in rat brain to WB4101 and phentolamine, with the α1A-AR having a higher affinity for these ligands (27). The first α1-AR that was cloned was the α1B-AR, derived by the cloning of oligonucleotide probes based on peptide fragments of purified receptor (28). This receptor was correctly identified as the tissue-studied α1B-AR because of the cloned receptor’s low affinity for WB4101 and phentolamine. The confusion began when the next cloned receptor, isolated through homology screening, was designated as a novel subtype not previously identified in tissue, namely the α1C-AR (29). This expressed receptor had high affinity for typical α1A-AR ligands, and had this been the sole criterion for classification, the investigators would have labeled this clone the α1A-AR. However, contributing to its mis-classification was its isolation from a bovine cDNA library, and its expression in bovine tissues was not readily detected. Thus, not having α1A-AR-like tissue distribution, and in addition having sensitivity to chlorethylclonidine, an alkylating agent mistakenly thought at the time to be selective for the α1B-AR, led to the conclusion of a new subtype. Hence, this cloned receptor appeared to have unique properties not described before in tissue, and it appeared repeatedly in the literature as the α1C-AR. A few years later, two groups re-cloned the same receptor from a rat cDNA library, whose tissues were more thoroughly characterized for α1A-AR pharmacology, and they demonstrated that the mis-classified cloned α1C-AR really represented the pharmacologically-defined α1A-AR (30–31). During this mix-up with the α1C-AR, another receptor was cloned and was designated as the α1A-AR, because this subtype was not yet identified by cloning and the expressed receptor had high affinity for WB4101 and phenylephrine, both supposedly α1A-AR-selective ligands (32). In reality, this clone was the truly novel receptor subtype; when it was independently cloned, and more extensively analyzed to reveal novel pharmacology, it was named the α1D-AR (33). Therefore, all studies that cite the α1C-AR should be interpreted as really being about the α1A-AR. In summary, the designation α1C-AR is no longer used; the three α1-AR subtypes, characterized in both expressed systems and native tissues, are the α1A-, α1B-, and α1D-ARs (34).
α1-ARs: Not a copycat of β-AR agonism
From the first adrenergic receptor clone, the β2-AR, we learned that the receptor had a seven transmembrane (TM) topology like rhodopsin (35) and a ligand binding site within the hydrophobic core of the receptor (36). Substitution of the critical Asp in TM 3 indicated that this residue was involved in a salt-bridge contact with the protonated amine of the agonists, needed for both affinity and efficacy (37). Since they recognize the same endogenous catecholamines, most of the mutagenesis studies to determine the ligand-binding pocket in the β-ARs were expected to be equally translatable to the α1-ARs. This notion initially held true for the way the catechol hydroxyls interact with the receptor’s serine residues in TM 5 (19). The data suggested that catecholamine agonists interacted with the β-AR via two hydrogen bonds derived from phenyl-substitutions that were equal in contributing to affinity and efficacy, one between the hydroxyl side chain of Ser 204 in TM 5 for the meta-hydroxyl group of the agonist and a second being the hydroxyl side chain of Ser 207 in TM 5 for the para-hydroxyl group of the agonist. Our studies suggested that these interactions were not conserved in the α1-ARs, which could have been predicted since the equivalent position of Ser 204 in the β2-AR is not a hydroxyl in the α1A-AR (Table 1). We found that the meta-hydroxyl of the catecholamine preferentially interacts with Ser 188 in TM 5 for α1A-ARs, and it is this hydrogen bond interaction, and not that between the para-hydroxyl and Ser 192, that allows receptor activation. This residue alone accounts for essentially full agonism in the α1A-AR, whereas each serine accounts for roughly half of the activity in the β-AR. An interaction between the para-hydroxyl and Ser 192 (equivalent to Ser 207 in the β2-AR) has minimal contributions to receptor activation. Furthermore, since Ser 188 and Ser 192 are separated by three residues on TM 5 in the α1A-AR, whereas Ser 204 and Ser 207 of the β2-AR are separated by only two residues (Table 1), the orientation of the serines in the TM 5 helix are different, requiring the catechol ring in the α1-AR binding pocket to be rotated approximately 120 degrees relative to that in the β2-AR (Fig 3). This results in the meta-hydroxyl interaction in the β2-AR being closer to TM 6 in the binding pocket, while the meta-hydroxyl interaction in the α-AR is closer to TM 4 (38). Modeling also suggests that the catechol ring may be in a more planar orientation in the α1-ARs while it is skewed relative to the surface in the β2-ARs. Subsequent to our work, the β2-AR serine interaction with the catechol hydroxyls was revisited. Using the substituted cysteine accessibility method, Liapakis et al. (39) found that, in addition to Ser 204 and Ser 207, Ser 203 is also accessible in the binding-site crevice and that both Ser 203 and Ser 204 appear to interact with the meta-hydroxyl of catecholamines, perhaps through a bifurcated hydrogen bond. This revised binding scenario still maintains the skewed and rotated orientation of the catechol ring in the β2-AR relative to α1-ARs. Thus, our work has structurally explained previous drug-based studies that indicated that meta-substitutions on the aromatic ring were needed for essentially full agonism in the α1-ARs (20). In addition our studies also explain why 2- or 6-substituted fluorine compounds can discriminate between β- and α1-ARs, respectively, even though both are ortho-substitutions (21), because of the different orientations of the aromatic ring between the binding pockets of the two receptor families.
TABLE 1.
Sequence alignment between the α1A-AR and the β2-AR for the TM 5 serine residues (S) involved in hydrogen bonding with the catechol hydroxyls. The numbers refer to the number of amino acids in the primary sequence for each receptor.
| 188 | 192 | |||||||||||||||||||||||||||
| α1A-AR | Q | I | N | E | E | P | G | Y | V | L | F | S | A | L | G | S | F | Y | V | P | L | A | I | I | L | V | M | Y |
| 204 | 207 | |||||||||||||||||||||||||||
| β2-AR | D | F | F | T | N | Q | A | Y | A | I | A | S | S | I | V | S | F | Y | V | P | L | V | V | M | V | F | V | Y |
Figure 3. Serine-hydroxyl interactions between the β2- and α1A-AR.
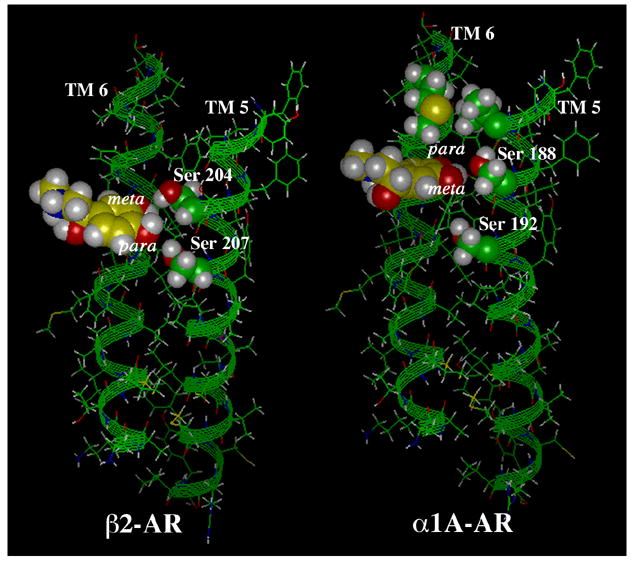
Ser 204 in the β2-AR interacts with the meta-hydroxyl while Ser 207 interacts with the para-hydroxyl of epinephrine, each contributing equally to agonism. This results in a skewed orientation of the catechol ring in the ligand binding pocket. However, in the α1A-AR, Ser 188 interacts with the meta-hydroxyl and was identified as the major interaction for agonism, resulting in a weak Ser 192 interaction with the para-hydroxyl near TM 6. This results in a more planar orientation of the catechol ring. Because the orientation of the serine residues on TM 5 is different (Ser 204 is closer to TM 6 while Ser 188 is closer to TM 4), this results in the catechol ring becoming rotated 120° in the α1A-AR relative to the β2-AR (note the position of the meta-hydroxyl). Reproduced with permission from (28).
Another area of interaction between the catecholamine and the receptor-binding pocket previously thought conserved between the β2-AR and α1-ARs is the aromatic contacts with the phenyl ring of agonists. Studies in the β2-AR established the role of two aromatic Phe residues in TM 6 (Phe 289 and 290 in the β2-AR, equivalent to Phe 310 and 311 in the α1B-AR) interacting with the phenyl ring of catecholamines but not antagonists (40). Interestingly, while these two Phe residues are strictly conserved (Table 2), their roles in agonist binding and activation are very different. In the α1B-AR, only Phe 310 is critically involved, both in forming an aromatic-aromatic interaction with the catecholamine phenyl ring and in receptor activation (Fig 4)(18). The altered catechol ring orientation demonstrated with the serine mutagenesis may also contribute to this aromatic agonist docking difference between β- and α1-ARs.
TABLE 2.
Sequence alignment between the α1B-AR and the β2-AR for aromatic residues in TM 6 involved in catecholamine binding and activation. The numbers refer to the number of amino acids in the primary sequence for each receptor.
| 310 | 311 | |||||||||||||||||||
| α1BAR | G | I | V | V | G | M | F | I | L | C | W | L | P | F | F | I | A | L | P | L |
| 289 | 290 | |||||||||||||||||||
| β2-AR | G | I | I | M | G | T | F | T | L | C | W | L | P | F | F | I | V | N | I | V |
Figure 4. Residues involved in agonist binding in the α1-ARs.
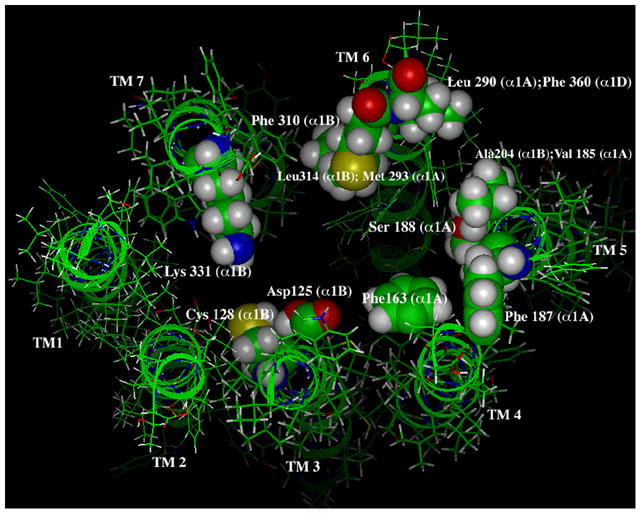
The view is looking down upon the extracellular face of the binding pocket. Mutagenesis studies have identified Asp 125 in TM 3 to interact with the protonated amine of catecholamines. Cys 128 in TM 3 is involved in agonist-selective signaling. Phe 163 in TM 4 is involved in aromatic interactions with the catechol ring as well as Phe 187 in TM 5. Ser 188 in TM 5 is involved in binding to the meta-hydroxyl of catecholamines. Ala 204 and Val 185 in TM 5 and Leu 314 and Met 293 in TM 6 are involved in conferring agonist selectivity between the α1-AR subtypes. Leu 290 and Phe 360 in TM 6 account for why the ability of the α1A-AR to accommodate bulky substituents in the para-position of catechol ring structures. Phe 310 in TM 6 interacts with the aromatic ring of catecholamines. Lys 331 in TM 7 is involved in α1-AR agonism by forming a salt-bridge to Asp 125 in TM 3. Numbering and residues are identified with their corresponding α1-AR subtypes in parentheses. Modeling was done independent of the rhodopsin structure and extracellular loops are removed for clarity.
The β-hydroxyl interaction?
The amino acid residue responsible for explaining the stereoselectivity of α1-AR agonists remains inconclusive. In early studies, it was proposed that Ser 165 in TM 4 of the β2-AR interacted with the hydroxyl on the β-carbon, and this was inferred to occur in the other adrenergic family members (40). However, it was modeling studies that suggested this interaction, as the data were inconclusive, as receptors mutated at this residue failed to be expressed. Another study in the α2-AR mutated the analogous Ser 165 to alanine, but this had no effects on dopamine binding (which lacks the β-hydroxyl), or on the enantiomers of norepinephrine or epinephrine (41), suggesting that Ser 165 is not the residue interacting with the β-hydroxyl. Another study mutated Ser 90 on TM 2 or Ser 419 on TM 7 to alanine, which produced a selective reduction in the affinity of the (−)-enantiomers of catecholamines for the α2A-AR, with no effect on the (+)-enantiomers or the corresponding β-desoxy analogs (42). However, modeling these interactions in the α2-AR is not consistent with other models and orients the agonist in unusual positions and at the opposite end of the binding pocket predicted for catecholamine agonists.
Yet another residue seems to be involved in the β-hydroxyl interaction in the β2-AR. While confirming that a functional mutation of Ser 165 did not affect stereoisomer binding, mutation of Asn 293 in TM 6 was shown to be involved in the interaction of the β-hydroxyl group of isoproterenol (43). In contrast, mutation of this corresponding area in the α2A-AR which contains three potential hydrogen bonding amino acids (Thr 393, Tyr 394, Thr 395) reduced the affinity of both the (−) and (+) enantiomers of catecholamines, indicating that these mutations are not selective for the (−)-enantiomers and are not likely to be involved in the β-hydroxyl group interaction (42). Yet another study in the α2A-AR has suggested that the β-hydroxyl in each of the R-isomers of phenethylamines forms a hydrogen bond to Asp 113 in TM 3, using docking simulations (44). One explanation for these discrepancies is that the β-hydroxyl interaction in each adrenergic receptor family is different or involves many residues, such that mutation of one or two is not sufficient for clear interpretation. The inconclusive nature of this residue(s) is likely to hold for any direct future studies in the α1-ARs.
Novel residues involved in agonism at α1-Ars
With the identification of residues in the α1-AR that contact the protonated amine (Asp in TM 3), the catechols (two Ser in TM 5) and phenyl ring (Phe in TM 6) all of the receptor contacts for epinephrine were thought to have been already identified (Fig 4). Since α1-AR ligands contain a high degree of aromatic character, we decided to explore novel aromatic residues in the α1-AR that might play a role in ligand recognition. Selecting aromatic residues in the putative α1-AR binding pocket that are not conserved in the β2-AR, we identified two phenylalanine residues (Phe-163, Phe-187) of the α1A-AR located near the surface of TM 4 and TM 5, respectively, that were obvious candidates for mutagenesis (Table 3)(Fig 4). These Phe163Gln and Phe187Ala mutations had greater impact on the binding properties of catecholamines and phenethylamines than on the imidazoline class of agonists, again suggesting that there are differences in the three-dimensional geometry of the agonist binding pocket recognized by these two drug classes (17). The potency but not the efficacy of epinephrine in stimulating inositol phosphates was reduced at the Phe163Gln/Phe187Ala mutation, suggesting that these residues were involved in affinity interactions and not signaling per se. Since these phenylalanine residues are not conserved in β2-ARs and α2-ARs, these results also again emphasize the differences in the agonist-binding pocket of adrenergic family members, despite the fact they all bind the same endogenous ligands. These two Phe residues are also not conserved in the other α1-AR subtypes, which suggests that these residues may be partly responsible for the increased binding affinity for agonists at the α1A-AR subtype compared to the α1B- or α1D-AR subtypes.
TABLE 3.
Sequence alignment between the α1A-AR and the β2-AR for aromatic residues in TMs 4 and 5 involved in catecholamine binding and activation. Phenylalanine (F) residues in the α1A-AR are near the surface of the receptor and not conserved in the β2-AR. Serine (S) residues in TM5 are bolded for comparison. The numbers refer to the number of amino acids in the primary sequence for each receptor.
| 183 | |||||||||||||||||||||
| TM4 α1AAR | W | G | F | L | P | G | I | S | I | V | L | S | L | V | W | V | C | L | L | A | |
| 190 | |||||||||||||||||||||
| β2-AR | H | M | Q | I | P | L | F | S | T | L | G | S | V | I | W | V | M | L | I | V | |
| 187 | |||||||||||||||||||||
| Tm5 α1AAR | G | Y | V | L | F | S | A | L | G | S | F | Y | V | P | L | A | I | I | L | V | M |
| 202 | |||||||||||||||||||||
| β2-AR | A | Y | A | I | A | S | S | I | V | S | F | Y | V | S | L | V | V | M | V | F | Y |
Interestingly, each of the binding contacts described above between receptor and agonist is more complex than a simple one-to-one independent interaction. In the α1-AR, mutations that contribute to constitutive activity and affects agonist binding are synergistic when mutations are combined (45). In the wild-type β2-AR, it has been shown that both binding and efficacy are synergistic with various types of phenylethylamine substitutions (46). These results are consistent with a mechanism in which each binding contact contributes to helical movements that act in a concerted fashion in agonist-induced activation; such synergism is predicted if multiple helix movements are involved in receptor activation.
Agonist selectivity
In addition to general points of contact with catecholamine agonists, residues in the binding pocket can also contribute to the ability of the α1-AR subtypes to recognize synthetic agonists that are more valuable for drug design. The three cloned subtypes, although structurally similar, bind a series of synthetic ligands with different relative potencies. This is particularly true for the α1A-AR, which recognizes several agonists and antagonists with 10–100-fold higher affinity than the α1B- or α1D-AR subtypes. Using site-directed mutagenesis, selected putative ligand-binding residues in the α1B-AR were converted, either individually or in combination, to the corresponding residues in the α1A-AR. Mutation of only two such residues (out of approximately 172 amino acids in the TMs) converted the agonist binding profile of the α1B-AR to that of the α1A-AR. Over 80% of this conversion was due to an Ala204Val substitution in TM 5; the remainder was due to the additional Leu314Met substitution in TM 6, which was confirmed by the reversal mutation in the α1A-AR (Val185Ala and Met293Leu) (Fig 4)(47). These data again suggest that having large hydrophobic substitutions on the ortho 6-carbon position of the aromatic ring confers α1A-AR agonist selectivity. Indeed, in later studies in which Art Hancock contributed, imidazoles containing a bulky substituent at the ortho-position are α1A-AR selective agonists, as predicted (48).
In mutating residues between the α1A- and α1D-AR subtypes, one particular amino acid residue in TM 6, Leu 290 in the α1A-AR or Phe 360 in the α1D-AR, was found to be important in shaping this end of the agonist binding pocket, such that large hydrophobic molecules placed at the para-position of the phenyl ring of α1-AR ligands increased α1A-AR agonist selectivity (Fig 4)(49). Phe 360 in the α1D-AR effectively closed the binding pocket to para-substituted compounds, while Leu 290 in the α1A-AR can accommodate the increased bulk. From these data, in conjunction with macromolecular modeling of the ligand-binding pocket, a model has been developed (Fig 4). The data suggest that the importance of these residues for α1-AR agonist binding is due not only to interactions between their side chains and specific ligand moieties but also to critical interactions between these amino acids themselves.
Modulation of Agonist Activity: Allosterism
A potential mechanism to modulate agonist action at α1-ARs is though allosterism. It is becoming more evident that GPCRs possess extracellular and intracellular allosteric binding sites that can be recognized by a variety of small molecules. “Allosteric” modulators of GPCRs interact with binding sites on the receptor that are topographically distinct from the “orthosteric” site recognized by the receptor’s endogenous agonist. This has been most clearly documented for the muscarinic acetylcholine receptors (50–51). Allosteric modulators offer many advantages over classic ligands since they may provide greater receptor subtype selectivity and increased drug safety. Benzodiazepines, classic allosteric ligands at gamma-aminobutyric acid A (GABAA) receptors, may be potential allosteric modulators at α1-ARs. At transfected α1B-ARs and α1D-ARs, the maximal inositol phosphate response to phenylephrine was potentiated almost 2-fold by either midazolam or lorazepam at 100 uM. Diazepam, lorazepam, and midazolam all increased the maximal response of the partial agonist clonidine at α1A-ARs, whereas the response to the full agonist phenylephrine was unaltered or even inhibited (52). The potentiating actions of midazolam and its partial agonism at α1-ARs were blocked by the addition of prazosin but not by a GABAA receptor antagonist. There were also no GABAA receptor sites detected in the cell line. These studies show that benzodiazepines modulate the function of α1-ARs in a subtype-specific manner and may reveal an allosteric site on α1-ARs.
Other compounds reported to be allosteric modulators of α1-ARs include a peptide contained in the venom of the predatory marine snail Conus tulipa, rho-TIA. This peptide did not cross- react with α2-ARs. The unique allosteric antagonism of rho-TIA may allow the development of inhibitors that are highly subtype selective (53). A thiadiazole compound, SCH-202676 (N-(2,3-diphenyl-1,2, 4-thiadiazol-5-(2H)-ylidene)methanamine), has been identified as an inhibitor of both agonist and antagonist binding to many different GPCRs. SCH-202676 inhibited radioligand binding to a number of structurally distinct GPCRs, including the human μ-, δ-, and κ-opioid receptors, α- and β-ARs, M1 and M2 muscarinic, and dopaminergic D1 and D2 receptors (54), presumably via direct interaction with a structural motif common to a large number of GPCRs or by activation/inhibition of an unidentified accessory protein that regulates GPCR function. It had also been previously demonstrated that amiloride analogs interact with a well-defined allosteric site on the human α2A-AR, and the α1A-AR has now been shown to possess a similar site. Five analogs of amiloride interacted with the α1A-AR in a manner consistent with two allosteric sites, a much more complex interaction than was found for the α2A-AR (55).
Modulation of Agonist Activity: Agonist-Selective Signaling
A structure-function relationship that may have profound effects in future therapeutic applications is the phenomenon of agonist-selective signaling. According to the ternary complex model, receptors can exist in two affinity forms based upon their interaction with the G-protein, and the proportion of these forms correlates with the intrinsic activity of the agonist (56). The extended ternary complex model (57) states that the receptor exists in an equilibrium of two functionally distinct states: the inactive (R) and the active (R*) state (58–59); the R* state is specific for a G-protein and accounts for the effects of different classes of drugs on receptor signaling. An extension of this model suggests that each agonist is theoretically able to promote its own specific active-conformation of the receptor, leading to a limitless number of receptor conformations, Rn*. If we can understand how an agonist induces or stabilizes these conformations and determine which specific signals such conformations regulate, this may lead to the development of better targeted drugs with selective therapeutic effects.
α1-ARs can couple to multiple G-proteins, thus, potential for multiple receptor conformations selective for each G-protein (60). The first example of agonist-specific states in the α1-ARs was seen in a Cys128Phe mutation in TM 3 of the α1B-AR, one helix-turn below the critical Asp 125 involved in binding the protonated amine of the agonist (Fig 4). This mutation constitutively activated the receptor but the constitutive activation was selective for phospholipase C signaling but not signaling via phospholipase A2 (61). It was also found that phenethylamines, from full to partial agonists, were able to recognize this “selective active-state” as determined by enhanced binding and potency changes. However, imidazoline agonists did not change in either their binding or signaling characteristics, again suggesting that imidazolines were not binding the same way as epinephrine and were not able to recognize this specific activated conformation. Saturation mutagenesis of the Cys 128 site revealed various degrees of constitutive activity, with the greatest activity seen with large hydrophobic residues such as Phe, Trp, Tyr, and Met. These results suggest that these residues perturb a constraining factor, which we suggested to be a salt-bridge constraint between the critical Asp 125 in TM 3 and Lys 331 in TM 7 (Fig 4)(62). Modeling studies suggest that Cys 128 either projects into the ligand-binding pocket near this salt-bridge or perturbs the hydrophobic packing of the TM 2–TM 3 interface (Fig 4).
Interestingly, this Cys in TM 3 is strictly conserved in the β2-AR sequence, and the analogous Cys116Phe mutation was created in the β2-AR (63) and gave similar phenotypes. The β2-AR Cys116Phe mutant exhibited selective constitutive activity of the Na/H exchanger, NHE-1, without constitutively activating the Gαs/adenylate cyclase pathway. Analogous phenotypes have been seen when this region in TM 3 has been mutated in a variety of other GPCRs, such as the angiotensin receptor (Asn 111) (64), the CXCR4 chemokine receptor (Asn 119)(65), the platelet-activating factor (PAF) receptor (Asn 100)(66) and the bradykinin receptor (Asn 113)(67), suggesting that this region may be widely conserved target for in inducing agonist-specific conformations in GPCRs.
While mutational changes that induce specific conformations may not be indicative of native receptor conformations, one study in the α1-ARs has used a series of agonists to demonstrate the same concept of agonist-trafficking in a normal receptor. Agonist-selective signaling was demonstrated using norepinephrine and the meta- and para- isomers of octopamine, which are also referred to normetanephrine and para-hydroxyl analogs, respectively (68). The rank order of potencies of the three agonists was the same for all the three α1-AR subtypes when coupled to either the IP3 or arachidonic acid pathway: norepinephrine > meta-octopamine > para-octopamine. However, their efficacy to activate these pathways was different. Meta-octopamine was a partial agonist at the α1A-AR subtype, while para-octopamine was a full agonist when arachidonic acid release was measured. In contrast, at the α1B-AR subtype, the isomers switched efficacy, with meta-octopamine being the full agonist but para-octopamine only a partial agonist of arachidonic acid release. Since meta- or para-octopamine were not full agonists at accumulating inositol phosphates at any α1-AR subtype, these results suggest that these isomers induce or stabilize an α1-AR conformation that is better at coupling to arachidonic acid release than to inositol phosphates and that the conformations adopted by the α1-AR subtypes are different from each other. Therefore this study indicates that, at the level of the drug and independent of mutational changes in the receptor, agonist-selective signaling is possible.
Antagonist Binding
Although agonist binding in adrenergic receptors is now fairly well understood and involves residues located in TMs 3 through 6, there are few residues reported to be involved in antagonist binding. It had been speculated that antagonist binding is quite diverse depending upon the chemical structure of the antagonist, which can be quite different from agonist structures. In fact, a major docking site for antagonists has never been reported in any G-protein coupled receptor other than the α1-AR. Early studies involving chimeric receptors between the β1- and β2-AR failed to reveal discrete residues involved in antagonist binding (69–70), suggesting that global conformations and not single residues were responsible for antagonist affinity. However, we and others have defined discrete residues involved in α1-AR antagonist binding. Mutagenesis studies in our laboratory have revealed that the α1-AR subtype selectivity of two α1A-AR antagonists, phentolamine and WB4101, is conferred by interactions with three consecutive residues on the second extracellular loop (71). Three point mutations in the α1B-AR (Gly196Gln, Val197Ile, Thr198Asn) were additive in increasing affinity for phentolamine and WB4101 to those at the α1A-AR (Fig 5). Similar results on the role of the second extracellular loop on antagonist binding were also obtained for the 5-HT1D receptor (72), the opioid receptor (73) and the dopamine D2 receptor (74), suggesting that GPCRs in general may make use of extracellular loop 2 in antagonist binding. These observations indicate that, in contrast to agonist binding, which is localized closer to the interior core of the receptor, antagonists interact with residues closer to the extracellular surface of adrenergic receptors, above the plane of the agonist-binding pocket. The high-resolution structure of bovine rhodopsin indicates that the extracellular loop 2 also forms part of the binding site for retinal (75), suggesting that the involvement of extracellular loop 2 may be conserved across GPCRs and may be one component of an universal ligand-binding site.
Figure 5. Residues involved in antagonist binding in the α1-ARs.
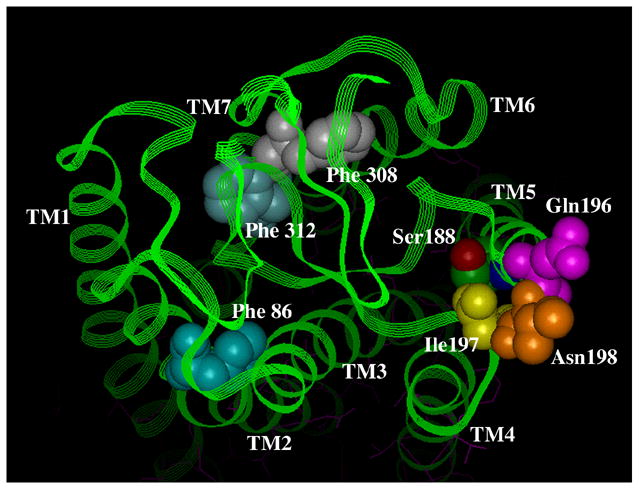
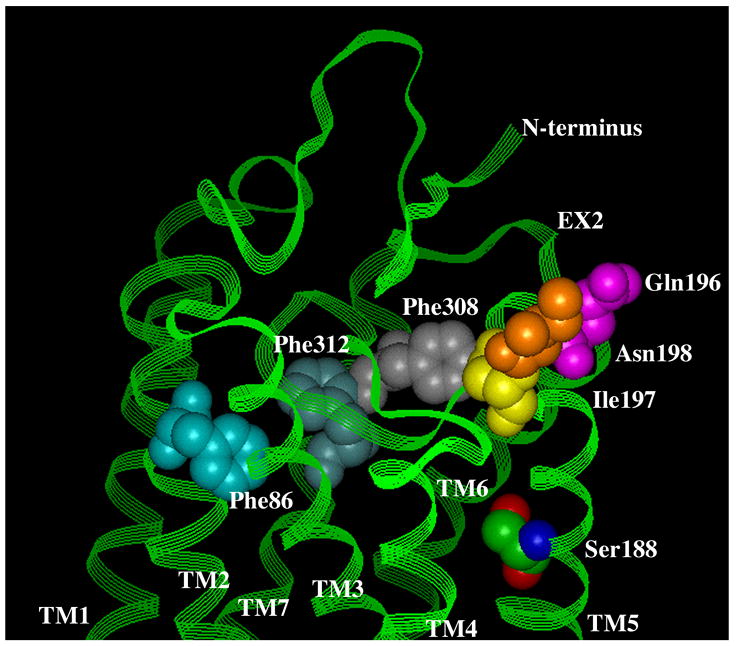
A. Surface view. B. Side view. Using the bovine rhodopsin α-carbon coordinates (64), residues involved in α1-AR antagonist binding were substituted in the corresponding positions of rhodopsin. Phe 86 in TM 2 discriminates the α1A-AR selective antagonist niguldipine as well as other dihydropyridine-type antagonists. Gln 196, Ile 197, and Asn 198, which are in the second extracellular loop, discriminate the α1A-AR selective antagonists phentolamine and WB4101. Phe 308 and Phe 312 in TM 7 are major aromatic contacts for most α1-AR antagonists as well as imidazoline-type agonists. Ser 188 in TM 5, which is involved in agonist binding, is shown for comparison of the depth of the antagonist-binding pocket. All residues are numbered according to the α1A-AR subtype.
Another major contact site for antagonists is found near the surface of TM 7, which interacts with both selective and non-selective antagonists. Early studies indicated that a single residue in TM 7 (Phe 412 in the α2-AR) conferred sensitivity to certain β1-AR antagonists (76). We identified two phenylalanine residues in TM 7 of the α1A-AR (Phe 312 and Phe 308) that are a major contributor to antagonist binding (Fig 5)(16). Mutation of either Phe 308 or Phe 312 resulted in significant losses of affinity (4–1200-fold) for the antagonists prazosin, WB4101, BMY7378, (+) niguldipine, and 5-methylurapidil, with no changes in affinity for phenethylamine-type agonists such as epinephrine, methoxamine, or phenylephrine. Interestingly, both residues were also involved in the binding of all imidazoline-type agonists such as oxymetazoline, cirazoline, and clonidine, confirming previous evidence that this class of ligands binds differently than phenethylamine-type agonists and that their binding may be more antagonist-like, which could also explain their partial agonist properties.
Other mutagenesis studies have indicated the importance of phenylalanine residues located close to the extracellular surface of TM 2 in antagonist binding at α1-ARs. Mutagenesis reveals that a phenylalanine residue (Phe 86) at the surface of TM 2 in the α1A-AR accounts for the α1A-AR versus α1D-AR selectivity of dihydropyridine antagonists such as niguldipine (Fig 5)(77). In fact, many α1-AR antagonists have bulky lipophilic substituents, such as that found on niguldipine, suggesting that additional parts of TMs 1 and 2 may be involved in antagonist binding and not just this one reported residue.
In modeling these interactions from various mutagenesis studies and using the current backbone structure of rhodopsin, we conclude that antagonist binding is docked higher in the pocket than agonist binding, closer to the extracellular surface, and may be skewed toward TM 7 (Fig 5). Modeling studies from other labs also suggest that the α1-AR antagonists, prazosin, tamsulosin and KMD-3213 all dock with amino acid residues near the extracellular surface, consistent with our hypothesis (78).
Arguments have been made that structure-function studies on GPCRs are inconsequential since pharmaceutical companies have relied upon screening large chemical libraries to obtain novel compounds. However, after years of screening, no compounds with 1,000 fold or more selectivity between the adrenergic receptor subtypes have been discovered. This degree of selectivity is needed to prevent cross talk between receptor subtypes. In reality, novel pharmacophores may be obtained through screening but structure function knowledge is still needed to enhance selectivity. Structure function work will have a future impact on drug design as the more selective we design therapeutics, the less side effects that will occur.
Footnotes
In dedication:I feel very honored to write this review in tribute to a man who noticed and supported young scientists. Art would come to my posters as a postdoc, and he has followed my career ever since. He always had a kind word and optimistic spirit, and he will be sorrowfully missed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hancock AA, Kyncl JJ, Martin YC, DeBernardis JF. Differentiation of α-adrenergic receptors using pharmacological evaluation and molecular modeling of selective adrenergic agents. J Recept Res. 1988;8(1–4):23–46. doi: 10.3109/10799898809048976. [DOI] [PubMed] [Google Scholar]
- 2.Knepper SM, Buckner SA, Brune ME, DeBernardis JF, Meyer MD, Hancock AA. A-61603, a potent α1-adrenergic receptor agonist, selective for the α1A receptor subtype. J Pharmacol Exp Ther. 1995 Jul;274(1):97–103. [PubMed] [Google Scholar]
- 3.Buckner SA, Oheim KW, Morse PA, Knepper SM, Hancock AA. α1-adrenoceptor-induced contractility in rat aorta is mediated by the α1D subtype. Eur J Pharmacol. 1996;297:241–8. doi: 10.1016/0014-2999(95)00755-5. [DOI] [PubMed] [Google Scholar]
- 4.Brune ME, Katwala SP, Milicic I, Buckner SA, Ireland LM, Kerwin JF, Jr, et al. Effects of selective and nonselective α1-adrenoceptor antagonists on intraurethral and arterial pressures in intact conscious dogs. Pharmacology. 1996;53:356–68. doi: 10.1159/000139451. [DOI] [PubMed] [Google Scholar]
- 5.Hancock AA, Buckner SA, Brune ME, Esbenshade TA, Ireland LM, Katwala S, et al. Preclinical pharmacology of fiduxosin, a novel α1-adrenoceptor antagonist with uroselective properties. J Pharmacol Exp Ther. 2002;300:478–86. doi: 10.1124/jpet.300.2.478. [DOI] [PubMed] [Google Scholar]
- 6.Brune ME, O’Neill AB, Gauvin DM, Katwala SP, Altenbach RJ, Brioni JD, et al. Comparison of α1-adrenoceptor agonists in canine urethral pressure profilometry and abdominal leak point pressure models. J Urol. 2001;166:1555–9. [PubMed] [Google Scholar]
- 7.Hancock AA, Brune ME, Witte DG, Marsh KC, Katwala S, Milicic I, et al. Actions of A-131701, a novel, selective antagonist for α1A compared with α1B adrenoceptors on intraurethral and blood pressure responses in conscious dogs and a pharmacodynamic assessment of in vivo prostatic selectivity. J Pharmacol Exp Ther. 1998;285:628–42. [PubMed] [Google Scholar]
- 8.Brune ME, Katwala SP, Milicic I, Witte DG, Kerwin JF, Jr, Meyer MD, et al. Effect of fiduxosin, an antagonist selective for α1A- and α1D-adrenoceptors, on intraurethral and arterial pressure responses in conscious dogs. J Pharmacol Exp Ther. 2002;300:487–94. doi: 10.1124/jpet.300.2.487. [DOI] [PubMed] [Google Scholar]
- 9.Witte DG, Brune ME, Katwala SP, Milicic I, Stolarik D, Hui YH, et al. Modeling of relationships between pharmacokinetics and blockade of agonist-induced elevation of intraurethral pressure and mean arterial pressure in conscious dogs treated with α1-adrenoceptor antagonists. J Pharmacol Exp Ther. 2002;300:495–504. doi: 10.1124/jpet.300.2.495. [DOI] [PubMed] [Google Scholar]
- 10.Meyer MD, Altenbach RJ, Basha FZ, Carroll WA, Condon S, Elmore SW, et al. Structure-activity studies for a novel series of tricyclic substituted hexahydrobenz[e]isoindole α1A adrenoceptor antagonists as potential agents for the symptomatic treatment of benign prostatic hyperplasia (BPH) J Med Chem. 2000;43:1586–603. doi: 10.1021/jm990567u. [DOI] [PubMed] [Google Scholar]
- 11.Meyer MD, Altenbach RJ, Bai H, Basha FZ, Carroll WA, Kerwin JF, Jr, et al. Structure-activity studies for a novel series of bicyclic substituted hexahydrobenz[e]isoindole α1A adrenoceptor antagonists as potential agents for the symptomatic treatment of benign prostatic hyperplasia. J Med Chem. 2001;44:1971–85. doi: 10.1021/jm000541z. [DOI] [PubMed] [Google Scholar]
- 12.Easson LH, Stedman E. Studies on the relationship between chemical constitution and physiological action: Molecular dissymmetry and physiological activity. Biochem J. 1933;27:1257–66. doi: 10.1042/bj0271257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruffolo RR, Jr, Waddell JE. Aromatic and benzylic hydroxyl substitution of imidazolines and phenethylamines: differences in activity at α1 and α2 adrenergic receptors. J Pharmacol Exp Ther. 1983;224:559–66. [PubMed] [Google Scholar]
- 14.DeMarinis RM, Bryan WM, Shah DH, Hieble JP, Pendleton RG. α-adrenergic agents. 1. Direct-acting α1 agonists related to methoxamine. J Med Chem. 1981;24:1432–7. doi: 10.1021/jm00144a012. [DOI] [PubMed] [Google Scholar]
- 15.Ruffolo RR, Jr, Rice PJ, Patil PN, Hamada A, Miller DD. Differences in the applicability of the Easson-Stedman hypothesis to the α1- and α2-adrenergic effects of phenethylamines and imidazolines. Eur J Pharmacol. 1983;86:471–5. doi: 10.1016/0014-2999(83)90199-1. [DOI] [PubMed] [Google Scholar]
- 16.Labrid C, Rocher I, Guery O. Structure-activity relationships as a response to the pharmacological differences in β-receptor ligands. Am J Hypertens. 1989;2(11 Pt 2):245S–251S. doi: 10.1093/ajh/2.11.245s. [DOI] [PubMed] [Google Scholar]
- 17.Waugh DJ, Zhao MM, Zuscik MJ, Perez DM. Novel aromatic residues in transmembrane domains IV and V involved in agonist binding at α1A-adrenergic receptors. J Biol Chem. 2000;275:11698–705. doi: 10.1074/jbc.275.16.11698. [DOI] [PubMed] [Google Scholar]
- 18.Chen S, Xu M, Lin F, Lee D, Riek P, Graham RM. Phe310 in transmembrane VI of the α1B-adrenergic receptor is a key switch residue involved in activation and catecholamine ring aromatic bonding. J Biol Chem. 1999;274:16320–30. doi: 10.1074/jbc.274.23.16320. [DOI] [PubMed] [Google Scholar]
- 19.Strader CD, Candelore MR, Hill WS, Sigal IS, Dixon RA. Identification of two serine residues involved in agonist activation of the β-adrenergic receptor. J Biol Chem. 1989;264:13572–8. [PubMed] [Google Scholar]
- 20.Kaiser C, Schwartz MS, Colella DF, Wardell JR., Jr Adrenergic agents. 3. Synthesis and adrenergic activity of some catecholamine analogs bearing a substituted sulfonyl or sulfonylalkyl group in the meta position. J Med Chem. 1975;18:674–83. doi: 10.1021/jm00241a006. [DOI] [PubMed] [Google Scholar]
- 21.Adejare A, Gusovsky F, Padgett W, Creveling CR, Daly JW, Kirk KL. Syntheses and adrenergic activities of ring-fluorinated epinephrines. J Med Chem. 1988;31:1972–7. doi: 10.1021/jm00118a019. [DOI] [PubMed] [Google Scholar]
- 22.Patil PN, Miller DD, Trendelenburg U. Molecular geometry and adrenergic drug activity. Pharmacol Rev. 1974;26:323–92. [PubMed] [Google Scholar]
- 23.Ruffolo RR, Jr, Yaden EL, Waddell JE, Dillard RD. Receptor interactrions of imidazolines. VI. Significance of carbon bridge separating phenyl and imidazoline rings of tolazoline-like α-adrenergic imidazolines. J Pharmacol Exp Ther. 1980;214:535–40. [PubMed] [Google Scholar]
- 24.Hieble JP, DeMarinis RM, Matthews WD. Evidence for and against heterogeneity of α1-adrenoceptors. Life Sci. 1986;38:1339–50. doi: 10.1016/0024-3205(86)90466-2. [DOI] [PubMed] [Google Scholar]
- 25.Minneman KP, Theroux TL, Hollinger S, Han C, Esbenshade TA. Selectivity of agonists for cloned α1-adrenergic receptor subtypes. Mol Pharmacol. 1994;46:929–36. [PubMed] [Google Scholar]
- 26.Waugh DJJ, Gaivin RJ, Zuscik MJ, Gonzalez-Cabrera P, Ross SA, Yun J, et al. Phe308 and Phe312 in TM VII are major sites of α1-Adrenergic Receptor Antagonist Binding: Imidazoline Agonists Bind Like Antagonists. J Biol Chem. 2001;276:25366–25371. doi: 10.1074/jbc.M103152200. [DOI] [PubMed] [Google Scholar]
- 27.Morrow AL, Creese I. Characterization of α1-adrenergic receptor subtypes in rat brain: a reevaluation of [3H]WB4104 and [3H]prazosin binding. Mol Pharmacol. 1986;29:321–30. [PubMed] [Google Scholar]
- 28.Cotecchia S, Schwinn DA, Randall RR, Lefkowitz RJ, Caron MG, Kobilka BK. Molecular cloning and expression of the cDNA for the hamster α1-adrenergic receptor. Proc Natl Acad Sci U S A. 1988;85:7159–63. doi: 10.1073/pnas.85.19.7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwinn DA, Lomasney JW, Lorenz W, Szklut PJ, Fremeau RT, Jr, Yang-Feng TL, et al. Molecular cloning and expression of the cDNA for a novel α1-adrenergic receptor subtype. J Biol Chem. 1990;265:8183–9. [PubMed] [Google Scholar]
- 30.Perez DM, Piascik MT, Malik N, Gaivin RJ, Graham RM. Cloning, expression and tissue distribution of the rat homolog of the bovine α1C-adrenergic receptor provide evidence for its classification as the α1A-subtype. Mol Pharmacol. 1994;46:823–831. [PubMed] [Google Scholar]
- 31.Laz TM, Forray C, Smith KE, Bard JA, Vaysse PJ, Branchek TA, et al. The rat homologue of the bovine α1C-adrenergic receptor shows the pharmacological properties of the classical α1A subtype. Mol Pharmacol. 1994;46:414–22. [PubMed] [Google Scholar]
- 32.Lomasney JW, Cotecchia S, Lorenz W, Leung WY, Schwinn DA, Yang-Feng TL, et al. Molecular cloning and expression of the cDNA for the α1A-adrenergic receptor. The gene for which is located on human chromosome 5. J Biol Chem. 1991;266:6365–9. [PubMed] [Google Scholar]
- 33.Perez DM, Piascik MT, Graham RM. Solution-phase library screening for the identification of rare clones: Isolation of an α1D-adrenergic receptor cDNA. Mol Pharmacol. 1991;40:876–883. [PubMed] [Google Scholar]
- 34.Hieble JP, Bylund DB, Clarke DE, Eikenburg DC, Langer SZ, Lefkowitz RJ, et al. International Union of Pharmacology. X. Recommendation for nomenclature of α1-adrenoceptors: consensus update. Pharmacol Rev. 1995;47:267–70. [PubMed] [Google Scholar]
- 35.Dixon RA, Kobilka BK, Strader DJ, Benovic JL, Dohlman HG, Frielle T, et al. Cloning of the gene and cDNA for mammalian β-adrenergic receptor and homology with rhodopsin. Nature. 1986;321:75–9. doi: 10.1038/321075a0. [DOI] [PubMed] [Google Scholar]
- 36.Dixon RA, Sigal IS, Rands E, Register RB, Candelore MR, Blake AD, et al. Ligand binding to the β-adrenergic receptor involves its rhodopsin-like core. Nature. 1987;326:73–7. doi: 10.1038/326073a0. [DOI] [PubMed] [Google Scholar]
- 37.Strader CD, Sigal IS, Register RB, Candelore MR, Rands E, Dixon RA. Identification of residues required for ligand binding to the β-adrenergic receptor. Proc Natl Acad Sci U S A. 1987;84:4384–8. doi: 10.1073/pnas.84.13.4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hwa J, Perez DM. The unique nature of the serine residues involved in α1-adrenergic receptor binding and activation. J Biol Chem. 1996;271:6322–6327. doi: 10.1074/jbc.271.11.6322. [DOI] [PubMed] [Google Scholar]
- 39.Liapakis G, Ballesteros JA, Papachristou S, Chan WC, Chen X, Javitch JA. The forgotten serine. A critical role for Ser-203 5.42 in ligand binding to and activation of the β2-adrenergic receptor. J Biol Chem. 2000;275:37779–88. doi: 10.1074/jbc.M002092200. [DOI] [PubMed] [Google Scholar]
- 40.Dixon RA, Sigal IS, Strader CD. Structure-function analysis of the β-adrenergic receptor. Cold Spring Harb Symp Quant Biol. 1988;53(Pt 1):487–97. doi: 10.1101/sqb.1988.053.01.056. [DOI] [PubMed] [Google Scholar]
- 41.Hehr A, Hieble JP, Li YO, Bergsma DJ, Swift AM, Ganguly S, et al. Ser165 of transmembrane helix IV is not involved in the interaction of catecholamines with the α2A-adrenoceptor. Pharmacology. 1997;55:18–24. doi: 10.1159/000139508. [DOI] [PubMed] [Google Scholar]
- 42.Hieble JP, Hehr A, Li YO, Ruffolo RR., Jr Molecular basis for the stereoselective interactions of catecholamines with α-adrenoceptors. Proc West Pharmacol Soc. 1998;41:225–8. [PubMed] [Google Scholar]
- 43.Wieland K, Zuurmond HM, Krasel C, Ijzerman AP, Lohse MJ. Involvement of Asn-293 in stereospecific agonist recognition and in activation of the β2-adrenergic receptor. Proc Natl Acad Sci U S A. 1996;93:9276–81. doi: 10.1073/pnas.93.17.9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nyronen T, Pihlavisto M, Peltonen JM, Hoffren AM, Varis M, Salminen T, et al. Molecular mechanism for agonist-promoted α2A-adrenoceptor activation by norepinephrine and epinephrine. Mol Pharmacol. 2001;59:1343–54. doi: 10.1124/mol.59.5.1343. [DOI] [PubMed] [Google Scholar]
- 45.Hwa J, Gaivin R, Porter JE, Perez DM. Synergism of constitutive activity in α1-adrenergic receptor activation. Biochemistry. 1997;36:633–9. doi: 10.1021/bi962141c. [DOI] [PubMed] [Google Scholar]
- 46.Liapakis G, Chan WC, Papadokostaki M, Javitch JA. Synergistic contributions of the functional groups of epinephrine to its affinity and efficacy at the β2 -adrenergic receptor. Mol Pharmacol. 2004;65:1181–90. doi: 10.1124/mol.65.5.1181. [DOI] [PubMed] [Google Scholar]
- 47.Hwa J, Graham RM, Perez DM. Identification of critical determinants of α1-adrenergic receptor subtype selective agonist binding. J Biol Chem. 1995;270:23189–95. doi: 10.1074/jbc.270.39.23189. [DOI] [PubMed] [Google Scholar]
- 48.Altenbach RJ, Khilevich A, Kolasa T, Rohde JJ, Bhatia PA, Patel MV, et al. Synthesis and structure-activity studies on N-[5-(1H-imidazol-4-yl)-5,6,7,8-tetrahydro-1 naphthalenyl] methanesulfonamide, an imidazole-containing α1A-adrenoceptor agonist. J Med Chem. 2004;47:3220–35. doi: 10.1021/jm030551a. [DOI] [PubMed] [Google Scholar]
- 49.McCune D, Gaivin R, Rorabaugh B, Perez D. Bulk is a determinant of oxymetazoline affinity for the α1A-adrenergic receptor. Receptors and Channels. 2004;10:109–16. doi: 10.1080/10606820490514923. [DOI] [PubMed] [Google Scholar]
- 50.May LT, Christopoulos A. Allosteric modulators of G-protein-coupled receptors. Curr Opin Pharmacol. 2003;3:551–6. doi: 10.1016/s1471-4892(03)00107-3. [DOI] [PubMed] [Google Scholar]
- 51.May LT, Avlani VA, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Curr Pharm Des. 2004;10:2003–13. doi: 10.2174/1381612043384303. [DOI] [PubMed] [Google Scholar]
- 52.Waugh DJ, Gaivin RJ, Damron DS, Murray PA, Perez DM. Binding, partial agonism, and potentiation of α1-adrenergic receptor function by benzodiazepines: A potential site of allosteric modulation. J Pharmacol Exp Ther. 1999;291:1164–71. [PubMed] [Google Scholar]
- 53.Sharpe IA, Thomas L, Loughnan M, Motin L, Palant E, Croker DE, et al. Allosteric α1-adrenoreceptor antagonism by the conopeptide rho-TIA. J Biol Chem. 2003;278:34451–7. doi: 10.1074/jbc.M305410200. [DOI] [PubMed] [Google Scholar]
- 54.Fawzi AB, Macdonald D, Benbow LL, Smith-Torhan A, Zhang H, Weig BC, et al. SCH-202676: An allosteric modulator of both agonist and antagonist binding to G protein-coupled receptors. Mol Pharmacol. 2001;59:30–7. doi: 10.1124/mol.59.1.30. [DOI] [PubMed] [Google Scholar]
- 55.Leppik RA, Mynett A, Lazareno S, Birdsall NJ. Allosteric interactions between the antagonist prazosin and amiloride analogs at the human α1A-adrenergic receptor. Mol Pharmacol. 2000;57:436–45. doi: 10.1124/mol.57.3.436. [DOI] [PubMed] [Google Scholar]
- 56.De Lean A, Stadel JM, Lefkowitz RJ. A ternary complex model explains the agonist-specific binding properties of the adenylate cyclase-coupled β-adrenergic receptor. J Biol Chem. 1980;255:7108–17. [PubMed] [Google Scholar]
- 57.Samama P, Cotecchia S, Costa T, Lefkowitz RJ. A mutation-induced activated state of the β2-adrenergic receptor. Extending the ternary complex model. J Biol Chem. 1993;268:4625–36. [PubMed] [Google Scholar]
- 58.Kenakin T. Trends Pharmacol Sci. Vol. 16. 1995. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals; pp. 232–23. [DOI] [PubMed] [Google Scholar]
- 59.Kenakin T. Agonist-specific receptor conformations. Trends Pharmacol Sci. 1997;18:416–417. doi: 10.1016/s0165-6147(97)01127-9. [DOI] [PubMed] [Google Scholar]
- 60.Perez DM, DeYoung MB, Graham RM. Coupling of expressed α1B- and α1D-adrenergic receptors to multiple signaling pathways is both G-protein and cell-type specific. Mol Pharmacol. 1993;44:784–795. [PubMed] [Google Scholar]
- 61.Perez DM, Hwa J, Gaivin R, Mathur M, Brown F, Graham RM. Constitutive Activation of a Single Effector Pathway: Evidence for Multiple Activation States of a G-Protein-Coupled Receptor. Mol Pharmacol. 1996;49:112–122. [PubMed] [Google Scholar]
- 62.Porter J, Hwa J, Perez DM. Activation of the α1B-adrenergic receptor is initiated by disruption of an inter-helical salt-bridge constraint. J Biol Chem. 1996;271:28318–28323. doi: 10.1074/jbc.271.45.28318. [DOI] [PubMed] [Google Scholar]
- 63.Zuscik M, Porter J, Gaivin R, Perez DM. Identification of a conserved switch residue responsible for selective constitutive activation of the β2-adrenergic receptor. J Biol Chem. 1998;273:3401–3407. doi: 10.1074/jbc.273.6.3401. [DOI] [PubMed] [Google Scholar]
- 64.Noda K, Feng Y-H, Liu X-P, Saad Y, Husain A, Karnik SS. The active state of the AT1 angiotensin receptor is generated by angiotensin II induction. Biochemistry. 1996;35:16435–16442. doi: 10.1021/bi961593m. [DOI] [PubMed] [Google Scholar]
- 65.Zhang WB, Navenot JM, Haribabu B, Tamamura H, Hiramatu K, Omagari A, et al. A point mutation that confers constitutive activity to CXCR4 reveals that T140 is an inverse agonist and that AMD3100 and ALX40-4C are weak partial agonists. J Biol Chem. 2002;277:24515–21. doi: 10.1074/jbc.M200889200. [DOI] [PubMed] [Google Scholar]
- 66.Ishii I, Izumi T, Tsukamoto H, Umeyama H, Ui M, Shimizu T. Alanine exchanges of polar amino acids in the transmembrane domains of a platelet-activating factor receptor generate both constitutively active and inactive mutants. J Biol Chem. 1997;272:7846–54. doi: 10.1074/jbc.272.12.7846. [DOI] [PubMed] [Google Scholar]
- 67.Marie J, Koch C, Pruneau D, Paquet JL, Groblewski T, Larguier R, et al. Constitutive activation of the human bradykinin B2 receptor induced by mutations in transmembrane helices III and VI. Mol Pharmacol. 1999;55:92–101. doi: 10.1124/mol.55.1.92. [DOI] [PubMed] [Google Scholar]
- 68.Richardson J, Chatwin H, Hirasawa A, Tsujimoto G, Evans PD. Agonist-specific coupling of a cloned human α1A-adrenoceptor to different second messenger pathways. Naunyn Schmiedebergs Arch Pharmacol. 2003;367:333–41. doi: 10.1007/s00210-003-0703-x. [DOI] [PubMed] [Google Scholar]
- 69.Dixon RA, Hill WS, Candelore MR, Rands E, Diehl RE, Marshall MS, et al. Genetic analysis of the molecular basis for β-adrenergic receptor subtype specificity. Proteins. 1989;6:267–74. doi: 10.1002/prot.340060309. [DOI] [PubMed] [Google Scholar]
- 70.Marullo S, Emorine LJ, Strosberg AD, Delavier-Klutchko C. Selective binding of ligands to β1, β2 or chimeric β1/β2-adrenergic receptors involves multiple subsites. EMBO J. 1990;9:1471–6. doi: 10.1002/j.1460-2075.1990.tb08264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao MM, Hwa J, Perez DM. Identification of critical extracellular loop residues involved in α1-adrenergic receptor subtype-selective antagonist binding. Mol Pharmacol. 1996;50:1118–26. [PubMed] [Google Scholar]
- 72.Wurch T, Colpaert FC, Pauwels PJ. Chimeric Receptor Analysis of the Ketanserin Binding Site in the Human 5-Hydroxytryptamine1D Receptor: Importance of the Second Extracellular Loop and Fifth Transmembrane Domain in Antagonist Binding. Mol Pharmacol. 1998;54:1088–1096. doi: 10.1124/mol.54.6.1088. [DOI] [PubMed] [Google Scholar]
- 73.Li X, Varga EV, Stropova D, Zalewska T, Malatynska E, Knapp RJ, et al. δ-Opioid receptor: the third extracellular loop determines naltrindole selectivity. Eur J Pharmacol. 1996;300:R1–R2. doi: 10.1016/0014-2999(96)00098-2. [DOI] [PubMed] [Google Scholar]
- 74.Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc Natl Acad Sci U S A. 2004;101:440–5. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 76.Suryanarayana S, Daunt DA, Von Zastrow M, Kobilka BK. A point mutation in the seventh hydrophobic domain of the α2 adrenergic receptor increases its affinity for a family of β receptor antagonists. J Biol Chem. 1991;266:15488–15492. [PubMed] [Google Scholar]
- 77.Hamaguchi N, True TA, Saussy DL, Jr, Jeffs PW. Phenylalanine in the second membrane-spanning domain of α1A-adrenergic receptor determines subtype selectivity of dihydropyridine antagonists. Biochemistry. 1996;35:14312–14317. doi: 10.1021/bi961024e. [DOI] [PubMed] [Google Scholar]
- 78.Ishiguro M, Futabayashi Y, Ohnuki T, Ahmed M, Muramatsu I, Nagatomo T. Identification of binding sites of prazosin, tamsulosin and KMD-3213 with α1-adrenergic receptor subtypes by molecular modeling. Life Sci. 2002;71:2531–41. doi: 10.1016/s0024-3205(02)02077-5. [DOI] [PubMed] [Google Scholar]