

Abstract
Celiac disease is an immune mediated enteropathy elicited by gluten ingestion. The disorder has a strong association with HLA-DQ2. This HLA molecule is involved in the disease pathogenesis by presenting gluten peptides to T cells. Blocking the peptide-binding site of DQ2 may be a way to treat celiac disease. In this study two types of peptide analogues, modeled after natural gluten antigens, were studied as DQ2 blockers. (a) Cyclic peptides. Cyclic peptides containing the DQ2-αI gliadin epitope LQPFPQPELPY were synthesized with flanking cysteine residues introduced and subsequently crosslinked via a disulfide bond. Alternatively, cyclic peptides were prepared with stable polyethylene glycol bridges across internal lysine residues of modified antigenic peptides such as KQPFPEKELPY and LQLQPFPQPEKPYPQPEKPY. The effect of cyclization as well as the length of the spacer in the cyclic peptides on DQ2 binding and T cell recognition was analyzed. Inhibition of peptide-DQ2 recognition by the T cell receptor was observed in T cell proliferation assays. (b) Dimeric peptides. Previously we developed a new type of peptide blocker with much enhanced affinity for DQ2 by dimerizing LQLQPFPQPEKPYPQPELPY through the lysine side chains. Herein, the effect of linker length on both DQ2 binding and T cell inhibition was investigated. One dimeric peptide analogue with an intermediate linker length was found to be especially effective at inhibiting DQ2 mediated antigen presentation. The implications of these findings for the treatment of celiac disease are discussed.
Keywords: Celiac disease, HLA-DQ2, gluten, gliadin, cyclic peptide, dimeric peptide, antigen presentation, inhibition
1. Introduction
Class II major histocompatibility complex (MHC) molecules are receptors expressed as αβ heterodimers on the surface of antigen presenting cells.1 They bind exogenous peptides (typically after sequential endocytosis, lysosomal assembly, and exocytosis) and present them to CD4+ T helper cells, a critical process in the mammalian adaptive immune response.1 Genetic susceptibility to autoimmune disease in human is often linked to variants of MHC class II molecules.2–4 In celiac disease more than 90% of the patients possess the HLA-DQ2 (DQA1*05/DQB1*02) molecule, while DQ8 (DQA1*03/DQB1*0302) is expressed by most of the remaining patients.5 DQ2 mediated presentation of gluten peptides (after regiospecific deamidation by extracellular transglutaminase 2) to CD4+ T cells is one of the key events in the pathogenesis of celiac disease.6 The current treatment of celiac disease is lifelong exclusion of gluten from the diet. To be on gluten free diet is a major challenge for the patient and there is therefore a need for therapeutic alternatives.
Interference with peptide antigen presentation has been explored as a treatment for autoimmune diseases such as type I diabetes, multiple sclerosis, experimental autoimmune encephalomyelitis, and rheumatoid arthritis. In principle, these studies fall into two categories based on the mode of action of the MHC binding ligands; a) altered peptide ligands7,8 or b) MHC blockers.9–11 An altered peptide ligand is specifically recognized by a T cell receptor (TCR), but it elicits a signal which is qualitatively different from that of the normal agonist peptide.7 An altered peptide ligand can induce anergy in T cells, and altered peptide ligands of this kind are termed MHC antagonists.12 The effect of an altered peptide ligand is specific to a given T cell receptor, and two different TCRs recognizing the same peptide-MHC complex may respond differently to the same altered peptide ligand.7 In contrast, peptide blockers act by out-competing the MHC binding of the agonist peptide. Ideally, these compounds should not serve as TCR ligands themselves. This can be achieved by steric principles, either via a bulky ligand which precludes docking of any TCR onto the ligand-MHC complex or via a small molecule inhibitor that eliminates the possibility of ligand-TCR interactions. The altered peptide ligand approach is less attractive for celiac disease therapy due to the multiplicity of gluten-derived epitopes as well as the polyclonal nature of T cells recognizing the same epitope (unpublished observations). We have previously reported that peptide blockers can be modeled after gluten epitopes by attachment of bulky groups (e.g. large functionalized alkyl groups) or by dimerization.13 Cyclization presents another way to prevent T cell recognition through steric hindrance.14–18 In this report, two types of hindered peptides – cyclic peptides and dimeric peptides – are systematically studied as DQ2 binding ligands and potential inhibitors of DQ2 mediated antigen presentation.
2. Results
2.1 Cyclic peptides containing disulfide linkage
Three Cys-containing variants of the peptide LQPFPQPELPY were synthesized, CGGLQPFPQPELPYGGCA (peptide 1), CGGGGLQPFPQPELPYGGCA (peptide 2), and CGGGGGGLQPFPQPELPYGGCA (peptide 3) (the DQ2-αI gliadin peptide is underlined, and the Gly linkers are shown in italics) (Figure 1). In each case the core peptide is flanked by Cys residues with variable Gly2, Gly4, or Gly6 spacers at the N-terminus making in total 4, 6, or 8 Gly residues between two Cys residues so as to systematically vary the torsional flexibility of the core peptide when subjected to intramolecular disulfide crosslinking. Following synthesis, the Cys residues of each peptide were allowed to crosslink in pH 7 phosphate buffer at 50°C with periodic vortexing in the absence of any reducing agent. Quantitative conversion of the linear peptides to cyclic peptides was verified by reverse phase HPLC, and the identity of the cyclic product was confirmed by MALDI-TOF MS, which revealed a loss of 2 units in m/z relative to the starting material. The DQ2 affinities of these peptides were assessed by measuring the IC50 values, with lower values corresponding to higher binding affinity to DQ2 (Figure 3). The three disulfide bridged cyclic peptides showed similar binding affinity to DQ2 as the reference acyclic peptide LQPFPQPELPY, demonstrating that the peptides still bind to DQ2 after cyclization.
Figure 1.
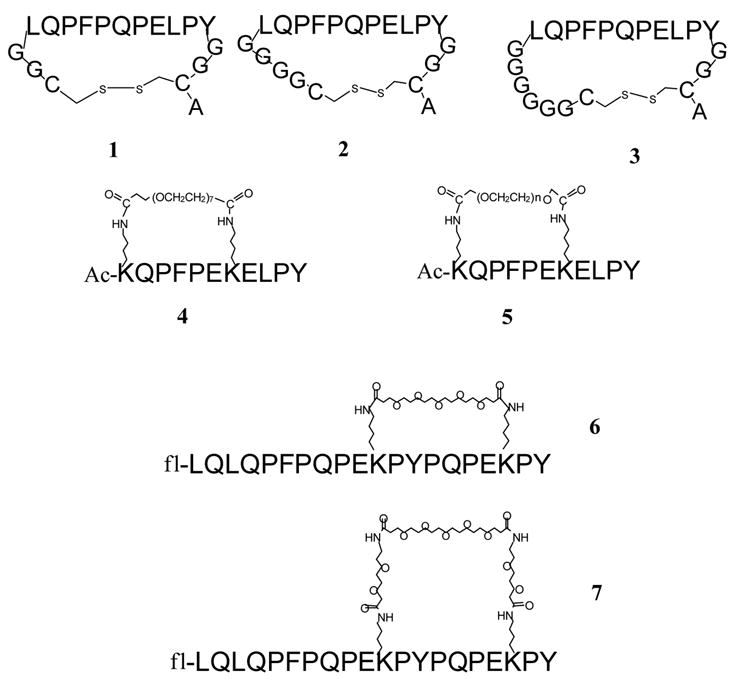
Structures of synthetic cyclic peptides. Peptide 5 represents a heterogenous mixture with n ~11. Ac denotes N terminal acetylation; fl denotes N terminal labeling with 5- (and 6-) carboxyfluorescein.
Figure 3.
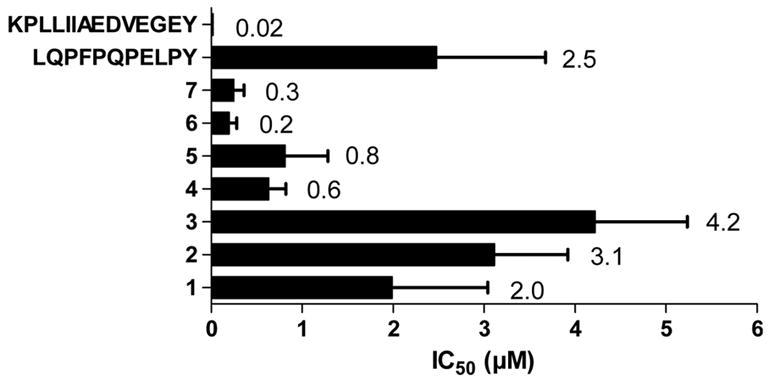
IC50 values for binding of cyclic peptides to DQ2. The results are shown as mean and SD of three experiments.
2.2 Internally bridged cyclic peptide analogues
Peptides 4 and 5 were designed based on a derivate of the DQ2-αI gliadin epitope, LQPFPQPELPY (nonamer core sequence underlined). The Gln residue in position 4 of the core sequence was substituted with Glu to increase the binding affinity (4-fold increase), and Lys residues were introduced in position -2 and 5, producing the sequence KQPFPEKELPY. Based on the X-ray crystal structure the introduction of Lys residues should minimally affect binding to DQ2.19 The side chains of these two Lys residues were crosslinked intramolecularly through bisfunctional PEG linkers (Figure 1). In addition, peptides 6 and 7 were also prepared; these peptides were based on a higher affinity 20-mer gluten peptide LQLQPFPQPELPYPQPELPY, which was deduced from the immunodominant 33-mer peptide.13 Two Leu residues were replaced by Lys residues in this peptide (Figure 1).
Peptides 4 and 5 were binding with similar affinities, which is notable as the linker length of peptide 5 is heterogenous. The mass spectrometry analysis of peptide 5 revealed presence of some dimeric species which may have biased the DQ2 binding analysis. Cyclic peptides 6 and 7 showed higher affinities to DQ2 than peptides 4 and 5 (Figure 3).
Peptides 1–7 were tested in a T cell proliferation assay for recognition by a DQ2-αI specific T cell clone TCC 430.1.142 (Figure 4). Surprisingly, peptide 6 is recognized at high concentrations. This fact is probably due to impurities in the peptide. The disulfide bridged peptides 1–3 were also recognized by the DQ2-αI specific T cell clone. The reason for this is probably that the disulfide bonds are unstable in the milieu of the cell culture. Peptides 4–7 were also tested for their ability to inhibit DQ2-mediated antigen presentation of the DQ2-γII gliadin epitope GIIQPEQPAQL to T cell clones derived from celiac disease patients (DQ2-γII specific) (Figure 5). As expected, peptides 6 and 7 showed the strongest inhibition among the cyclic peptides. Notably, these two peptides were even better inhibitors than the reference peptide KPLLIIAEDVEGEY (P198), which binds to DQ2 with lower IC50 values than either of the peptides. The two other cyclic peptides (4 and 5), based on shorter versions of gliadin epitopes, did not show appreciable inhibition (Figure 5). As a control, the inability of any of these peptides to inhibit DR3 mediated antigen presentation was verified (Figure 6).
Figure 4.
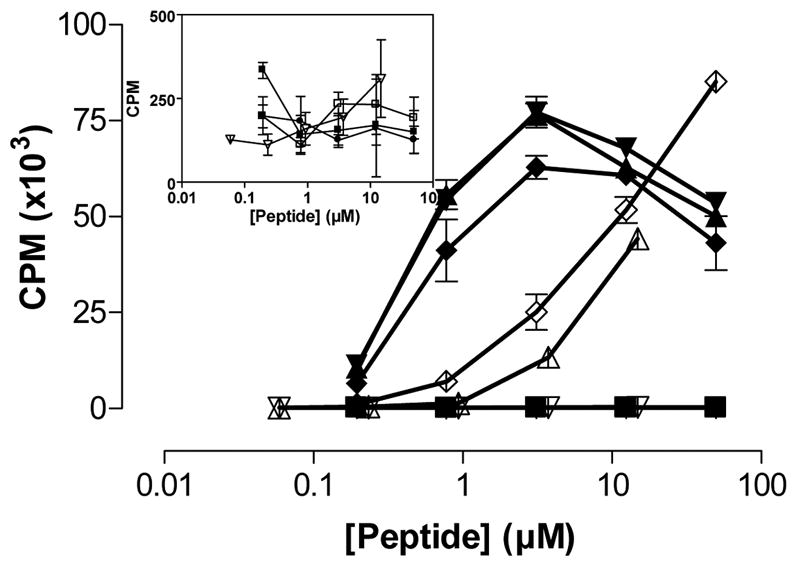
Recognition of cyclic peptides by T cells. The T cell clone 430.1.142 (specific for the DQ2-αI gliadin epitope) was stimulated by cyclic peptides harboring the DQ2-αI sequence. (■) P198, (▲) peptide 1, (▼) peptide 2, (◆) peptide 3, (●) peptide 4, (□) peptide 5, (△) peptide 6, (▽) peptide 7, (◇) LQPFPQPELPY. The inset shows CPM in lower ranges for non stimulatory cyclic peptides. P198: KPLLIIAEDVEGEY; Mycobacterium bovis 65 kDa heat shock protein 243-255Y. Mean and SD is shown.
Figure 5.
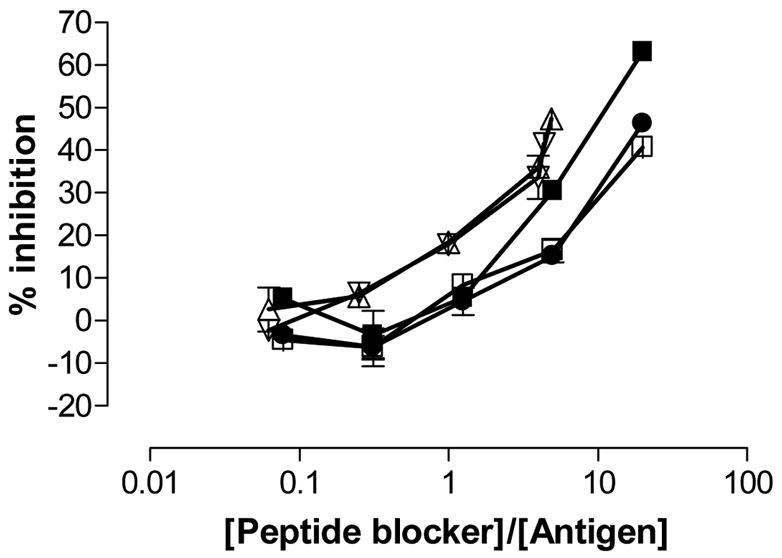
Inhibition of T cell proliferation by the blocking compounds 4, 5, 6, and 7. The T cell clone 437.1.3.17 (specific for the DQ2-γII gliadin epitope) was stimulated with 2.5 μM GIIQPEQPAQL (P1298, DQ2-γII gliadin epitope) in the presence of increasing concentration of peptide blockers (■) P198, (●) peptide 4, (□) peptide 5, (△) peptide 6, (▽) peptide 7. P198: KPLLIIAEDVEGEY; Mycobacterium bovis 65 kDa heat shock protein 243-255Y. Mean and range is shown.
Figure 6.
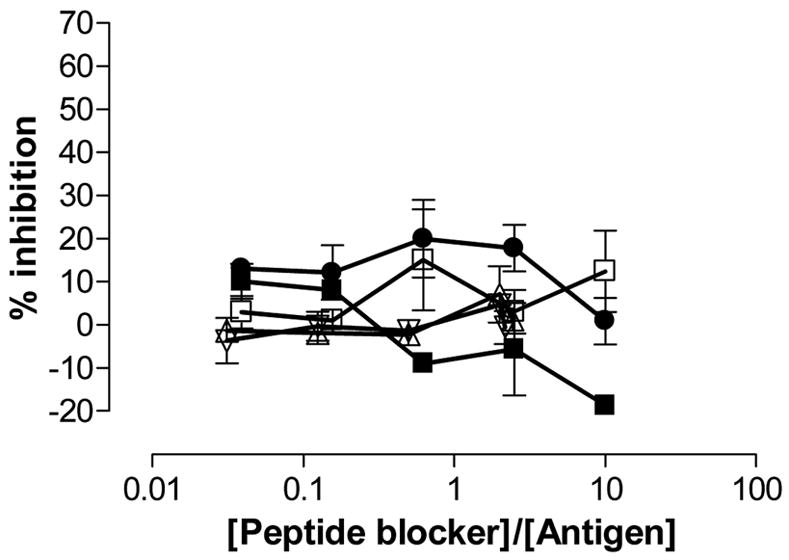
Inhibition of T cell proliferation by the blocking compounds 4, 5, 6, and 7. The T cell clone RN.46 (DR3-restricted) was stimulated with 5 μM KTIAYDEEARR (P261, DR3-restricted peptide; Mycobacterium tuberculosis 65 kDa heat shock protein 3–13) in the presence of increasing concentration of peptide blockers (■) P198, (●) peptide 4, (□) peptide 5, (△) peptide 6, (▽) peptide 7. P198: KPLLIIAEDVEGEY; Mycobacterium bovis 65 kDa heat shock protein 243-255Y. Mean and range is shown.
2.3 Dimeric peptides with various linker lengths
Peptide dimerization is another strategy that has been shown to inhibit proliferation of gluten sensitive T cell lines in the presence of antigen and DQ2 homozygous antigen presenting cells. For example, peptide 8 (Figure 2A) is one of the most potent inhibitors identified to date.13 We therefore systematically studied the effect of chain length between the two monomeric units of 8. Two derivatives of the high affinity gluten peptide LQLQPFPQPELPYPQPELPY, with L11K and L11C substitutions, were used as starting materials (Figure 2B). Previous studies had confirmed that modifications at this position do not affect its binding to DQ2.13 Seven dimeric analogues of 8 (peptides 9 to 15) were synthesized using the maleimidethiol reaction, succinimide-amine reaction or both (Figure 2C). The resulting dimeric peptides have PEG linkers (peptides 11, 12, 13, 14, and 15) or alkyl linkers (peptides 9 and 10) ranging from 13 to 89 atoms long between the α-carbon atoms of the bridging residues in the two monomers; this corresponds to extended linker lengths from 14Å to 98Å (Table 1). All reactions gave >70% yields, as verified by LC-MS (data not shown).
Figure 2.
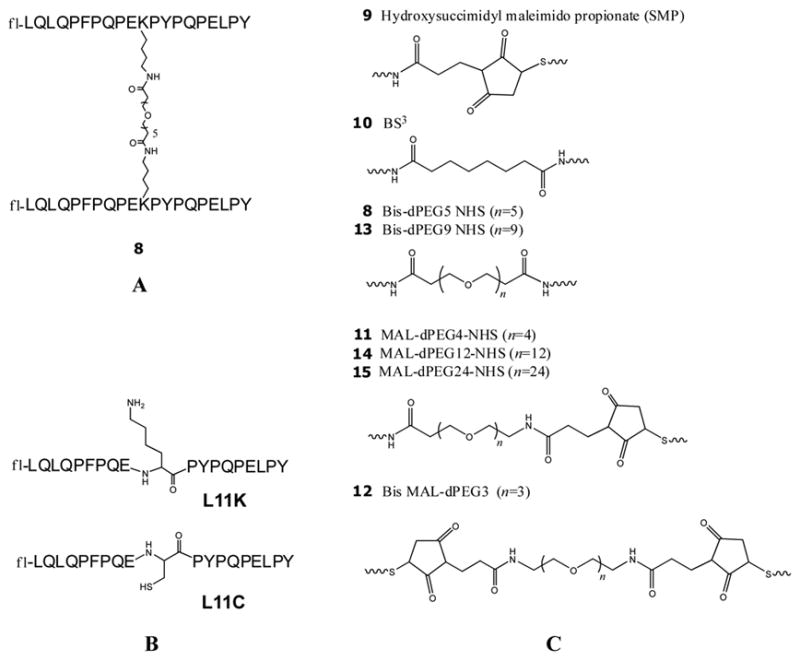
Dimeric peptides. (A) The structure of the reference dimeric peptide 8. (B) The structures of the modified monomers L11K and L11C. (C) The structures of the linkers in dimeric peptides 8–15.
Table 1.
Summary of the DQ2 binding results of dimeric peptides 8–15.
| Peptides | Carbon atoms in linker | Putative length (Å) | Binding at pH 5.5 (%)a | Binding at pH 7.3 (%)a |
|---|---|---|---|---|
| 9 | 13 | 14.3 | 72.8 | 52.1 |
| 10 | 18 | 19.8 | 70.9 | 54.6 |
| 8 | 29 | 31.9 | 62.0 | 46.3 |
| 11 | 29 | 31.9 | 63.2 | 53.3 |
| 12 | 32 | 35.2 | 69.3 | 50.7 |
| 13 | 41 | 45.1 | 70.2 | 54.5 |
| 14 | 53 | 58.3 | 70.2 | 51.8 |
| 15 | 89 | 97.9 | 67.8 | 38.6 |
Measurements were made either at pH 5.5 or at pH 7.3. Recombinant DQ2 (4.7 μM) was mixed with fluorescein-conjugated peptide (0.185 μM) at 37°C for 5 h, and the amount of DQ2 bound peptide was calculated as the percentage (×100%) of total peptide. The results are presented as mean of the two experiments with errors <5% of the numerical values.
First, the binding of the dimeric peptides 8–15 was assessed in peptide exchange experiments using recombinant DQ2 and high performance size exclusion chromatography at pH 5.5 and pH 7.3 with a DQ2:peptide ratio of 25:1. The percentage of DQ2 bound fluorescent peptide is a measure of its DQ2 affinity. Dimeric peptides with varying linker lengths have comparably high affinity for DQ2, which achieved equilibrium after only 5 hours (Table 1). All dimeric peptides were able to form 2:1 complexes with two DQ2 molecules bound to one peptide at pH 5.5 (Table 2). These 2:1 complexes are observed only after 14 hours, suggesting that the binding of the second DQ2 molecule to a 1:1 DQ2-peptide complex is slow. At pH 7.3, no 2:1 complexes could be observed.
Table 2.
The 2:1 complex formation of two DQ2 molecules binding to one peptide
| Peptides | 2:1 complex formation at pH 5 (%)b |
|---|---|
| 9 | <10 |
| 10 | 18 |
| 8 | 22 |
| 11 | 21 |
| 12 | 22 |
| 13 | 26 |
| 14 | 25 |
| 15 | 26 |
Recombinant DQ2 (4.7 μM) was mixed with fluorescein-conjugated peptide (0.185 μM) at 37°C for 45 h at pH 5.5. The formation of 2:1 DQ2-peptide complex with two DQ2 molecules binding to one peptide was reported as the percentage among total DQ2-peptide complex, i.e. [2:1 complex] / ([2:1 complex] + [1:1 complex]) ×100%.
Second, in order to select effective blockers, the dimeric peptides 8–15 were screened to measure their potency to stimulate a gluten responsive T cell line P28 TCL2 which is derived from a celiac patient. Upon incubation of the dimeric peptides in Figure 2 with DQ2 expressing antigen presenting B cells overnight at 37°C at 10 μM or 50 μM concentrations, the extent of T cell stimulation was measured (Figure 7A). All the dimeric peptides containing cysteine-maleimide bonds stimulated T cell proliferation (peptides 9, 11, 12, 14, and 15), suggesting that the thiol-maleimide bond was not stable in this biological milieu. In contrast, peptides with amide crosslinkers (e.g. peptides 8, 10 and 13), showed lower T cell stimulation, suggesting their linkers were more metabolically stable than the cysteine-maleimide linkers, although the longest peptide 13 showed increased antigenicity at the highest concentration, 50 μM. Whether this was due to too much flexibility or metabolic instability or possible small amounts of monomeric peptide impurities (<5%) was not investigated.
Figure 7.
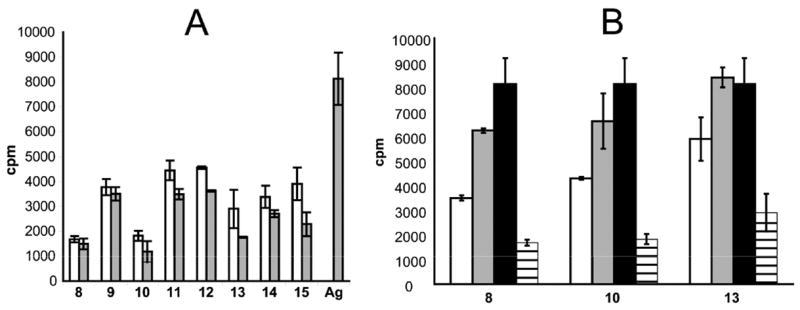
DQ2 dependent T cell stimulation of T cell line P28 TCL2 by dimeric peptides 8–15. (A) T cell stimulation caused by dimeric peptides 8–15 in the absence of antigen or by antigen peptide LQLQPFPQPELPYPQPELPY (Ag) at 50 μM or 10 μM. The background in the absence of blockers is 170 CPM. □ 50 μM dimeric peptide;
 10 μM dimeric peptide or antigen. (B) Blocking compounds 8, 10, and 13 block T cell proliferation caused by 10 μM antigen peptide LQLQPFPQPELPYPQPELPY in a dose dependent fashion. □ 50 μM dimeric peptide, 10 μM antigen;
10 μM dimeric peptide, 10 μM antigen; ■ 0 μM dimeric peptide, 10 μM antigen;▤50 μM dimeric peptide, 0 μM antigen.
10 μM dimeric peptide or antigen. (B) Blocking compounds 8, 10, and 13 block T cell proliferation caused by 10 μM antigen peptide LQLQPFPQPELPYPQPELPY in a dose dependent fashion. □ 50 μM dimeric peptide, 10 μM antigen;
10 μM dimeric peptide, 10 μM antigen; ■ 0 μM dimeric peptide, 10 μM antigen;▤50 μM dimeric peptide, 0 μM antigen.
Next, based on the above results, the T cell inhibitory effects of peptides 10 and 13 were tested and compared with peptide 8 which previously had demonstrated effect in this type of assay.13 All three peptides showed dose dependent inhibition of antigen presentation using fixed antigen presenting cells (Figure 7B).
Last, to test whether peptide 8 was able to inhibit antigen presentation in non-fixed antigen presenting cells, 25 μM peptide 8 was incubated with γ-irradiated DQ2 expressing B cells for 2 hours. The antigenic peptide was then added, and cells were incubated for another 10 hours. As shown in Figure 8, T cell proliferation was inhibited by the addition of peptide 8. Thus, DQ2 blockers of this type appear to be able to compete with antigenic peptides for surface HLA-DQ2 occupancy in both fixed and non-fixed antigen presenting cells.
Figure 8.
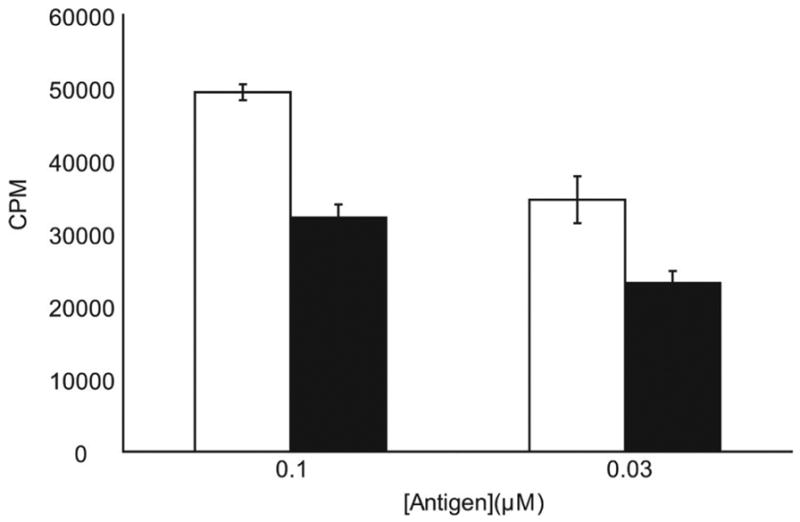
Dimeric peptide 8 shows blocking efficacy in non-fixed, γ-irradiated antigen presenting cells. HLA-DQ2 expressing B cells were γ-irradiated and incubated with 25 μM dimeric peptide 8 for 2 hours before the addition of antigen peptide LQLQPFPQPELPYPQPELPY. After an additional 10 hours, the peptides were removed and P28 TCL2 T cells were added to assess the quantity of antigen peptide-DQ2 complexes on the B cell surface. □ Antigen only; ■ Antigen + peptide 8
3. Discussion
Our studies have explored the potential of using cyclic and dimeric peptides as peptide blockers to prevent DQ2 mediated antigen recognition by T cell receptors that are uniquely found in small intestinal mucosa of celiac disease patients. The lengths of spacers in cyclic peptides and linkers in dimeric peptides were varied to investigate their effects on DQ2 binding affinity and T cell recognition.
Interference of MHC class II restricted antigen presentation by altered peptide ligands, or peptide or peptidomimetic analogues have been explored as possible therapeutic options in autoimmune diseases.8–11,13–16,18,20–25 This principle has not been successful mainly due to poor bioavailability and phamacokinetics of peptidic compounds.20 In celiac disease this should be of a less problem as the therapeutic compound can be orally administered to the intestinal surface, either before or in conjunction with gluten ingestion. Peptide based therapeutics are also susceptible to proteolysis; this challenge is particularly acute in celiac disease where the compounds must be administered in the upper digestive tract with intense proteolytic activity. Encouragingly, the T cell epitopes of gluten in celiac disease are intrinsically stable towards proteolysis in the gastrointestinal tract.26,27 Therefore, using the T cell epitope sequences as a starting point for design of peptide blockers can ameliorate this problem.
Therapeutic interference with MHC based peptide presentation can be done via altered peptide ligands (antagonistic peptides) or peptide blockers. Peptide antagonists are inappropriate for effective treatment of celiac disease due to the vast heterogeneity of gluten epitopes and gluten reactive TCRs. However, one can take advantage of gluten epitopes by transforming them into MHC blocking compounds. In addition to proteolytic stability, there are several additional advantages to designing DQ2 blockers by mimicking naturally occurring gluten antigens. The structural features in prototypical antigenic gluten peptides that contribute to DQ2 binding as well as T cell receptor recognition have been extensively investigated, and are known to be orthogonal.13 This orthogonality enables the design of blockers that hinder TCR recognition while maintaining or even strengthening DQ2 interactions. Blockers that are close structural analogues of disease-related antigenic peptides are likely to retain the pharmacokinetic properties of the antigens themselves. Because gluten antigens are consumed as part of the human diet, drugs modeled after natural gluten peptides may also be able to reach the target tissues of the small intestine via oral administration. Gluten peptide mimetics could preserve the natural DQ2 specificity of the antigens themselves,13 thus minimizing potential cross-reactivity with other MHC alleles. Cyclization represents an attractive strategy for the design of MHC class II inhibitors. Cyclic peptides are proteolytically more stable compared to their linear counterparts.16 When it comes to receptor binding, peptide cyclization brings two conflicting structural features into play. On one hand, the conformational constraints imposed by cyclization might hinder binding of the epitope to the receptor/the MHC class II molecule. On the other hand, the entropic constraint due to cyclization could lock the peptide into its bound conformation, thereby enhancing affinity for its receptor. A favorable conformation is typically a polyproline II helix for epitopes bound to MHC class II molecules.19,28 To test whether cyclization affects peptide-DQ2 interaction, cyclic peptides containing the DQ2-αI peptide LQPFPQPELPY were synthesized through disulfide linkage with various lengths of linkers. Disulfide bonding is a convenient way for constructing cyclic peptides to test the effect of cyclization on DQ2 binding, but as the disulfide cyclized peptides appeared to be unstable in the cell culture conditions employed, these peptides could not be used for testing the effect on prevention of T cell activation. For this purpose, we generated more stable cyclic peptides by connecting two internal lysine residues via bis-functional polyethylene glycol (PEG) linkers. These compounds were active as MHC blockers, and two of them based on a 20-mer gliadin peptide are among the best DQ2 blockers we have observed. Still we believe the efficacies of these are insufficient to give in vivo effect much because their binding affinities are too low. Gliadin T cell peptide epitopes are in general mediocre binders to DQ2,29 and we hope that by specifically introducing anchor residues optimal for DQ2, compounds of therapeutic potential may be developed.
Inspired by the success of synthetic multivalent ligands in generation of high affinity binding interaction in other biological systems,30–34 we initiated the strategy of dimerizing antigenic ligand through the residue in the middle of the sequence.13 The first dimer peptide demonstrated surprisingly elevated binding affinity to DQ2 and surpassingly effective inhibition of antigen presentation.13 This enhancement of binding affinity could be either caused by multivalency effect (i.e. one peptide binding to two DQ2 molecules) or by proximity effect (i.e. the clustering of multiple binding epitopes increases the local concentration of binding registers). In the case of proximity effect a 1:1 stoichiometry of ligand-receptor interaction will be observed as opposed to the multivalency effect where a 2:1 stoichiometry will be seen. The current work explored dimeric peptides by varying the linker length between the two monomeric units. We observed two binding phases in the binding interaction between these dimeric peptides and DQ2 at pH 5.5 using high-performance size exclusion chromatography. In the first phase (5 hr), 1:1 binding stoichiometry to DQ2 was observed with negligible 2:1 complex formation, even when the linker between the two monomeric units was as long as 100 Å. The binding equilibrium was reached after the first phase. In the second phase which lasts for more than 40 hr, 1:1 DQ2-peptide complex slowly converts to 2:1 DQ2-peptide complex with ~20% conversion achieved after 48 hr (Table 2). The dimer peptides with longer linker length tend to show higher percentage of 2:1 DQ2-peptide complex formation in the second phase, i.e. about 10% in the case of peptide 9 and about 26% in the case of peptide 15. Interestingly, this two phase binding kinetics was not observed at pH 7.3; no 2:1 DQ2-peptide complexes was observed after 48 hr. The observation that this enhanced affinity is primary caused by the fast phase at acidic pH and that no 2:1 complexes were found at neutral pH disfavors the multivalency mechanism. Rather, proximity effect caused by the clustering of multiple epitopes might explain this property.34 When two binding monomeric units are brought in vicinity an enhanced binding affinity can be achieved. Owing to the promising enhanced binding affinity, synthesis of dimeric, multimeric or even dendritic peptides carrying multiple binding epitopes seem to be an effective venue in the search of DQ2 blockers.
In summary, herein we presented alternative approaches to systematically study hindered peptide analogues for the purpose of DQ2 blockage. The current strategies provide understanding of cyclic and dimeric peptide analogues in both DQ2 binding interaction and inhibition of antigen presentation, which might assists further pharmacological studies in more disease related physiological conditions, and inspire the development of more feasible drug leads in celiac disease as well as other autoimmune diseases.
4. Experimental
4.1 Peptide synthesis, labeling and purification
Peptides used in this study were synthesized primarily using Boc/HBTU chemistry starting from N-α-t-Boc-L-aminoacyl-phenylacetamidomethyl (PAM) resin. While still attached to the resin, peptides were labeled at their N-termini with 5- (and 6-) carboxyfluorescein, 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride (EDC·HCl), and 1-hydroxy-7-azabenzotriazole (HOAt) in 1:1:1 ratio in dimethyl formamide (DMF) as the solvent.13,35 Following cleavage of the peptidyl resin in trifluoroacetic acid/trifluoromethanesulfonic acid/thioanisole (TFA/TFMSA/thioanisole 10:1:1, v/v/v) for 4 h, the crude peptides were precipitated in cold ether and dissolved in 1:1 v/v acetonitrile/water. The peptides were purified by reverse-phase HPLC on a semi-preparative C18 column using a water-acetonitrile gradient in 0.1% (v/v) TFA. The identity and purity of the peptides were confirmed by liquid chromatography coupled electrospray mass spectrometry (LC-MS). The peptides were lyophilized and stored at −20°C. Prior to use, peptide stock solutions were prepared in 10 mM PBS with 0.02% sodium azide.
Peptides 4 and 5 (Figure 1) were synthesized using an automated synthesizer (Multipep, Intavis AG) with Fmoc/PyBOP chemistry starting from TentaGEL SRAM resin. Before cleavage of peptides from the resin, the N-termini were acetylated and the peptides were cyclized. The protecting group on Lys residues, 1-(4,4-dimethyl-2,6-dioxycyclohex-1-ylidine)ethyl (Dde) was selectively removed with 1% hydrazine in DMF. Peptide 4 was cyclized with Bis-dPEG7-acid (HO2CCH2CH2(CH2CH2O)7CO2H) (Quanta BioDesign) while peptide 5 was cyclized with Poly(ethylene glycol)bis(carboxymethyl)ether (HO2CCH2(OCH2CH2)nOCH2CO2H) average Mn ~600 (Aldrich). Cyclization was achieved by mixing peptidyl resin in DMF with either of the dicarboxylic acids (2 eq), PyBOP (4 eq) and diisopropylethylamine (DIPEA) (8 eq). This mix was incubated overnight, after which a negative ninhydrin test was obtained. Peptides were deprotected and cleaved from the polymer support by treatment with 95% TFA, 2.5% triisopropylsilan and 2.5% water for 4 hours. Peptides were precipitated by the addition of cold tert-butyl methyl ether, dissolved in water and lyophilized. The peptides were analyzed by reverse phase HPLC and MALDI-TOF mass spectrometry. The peptides were aliquoted and lyophilized before storage at −20°C. It should be noted that peptide 5 represents a mixture of peptides with different lengths of the linker, due to the polydispersity of the PEG linker used for synthesis.
Cyclic peptides with disulfide bridges (1, 2 and 3 in Figure 1) were prepared in pH 7 phosphate buffer at 50°C with periodic vortexing. Internally bridged cyclic peptides 4 and 5 in Figure 1 were cyclized as described above. Cyclic peptides 6 and 7 were prepared by incubating purified carboxyfluorescein labeled LQLQPFPQPEKPYPQPEKPY peptide with bis-dPEG N-hydroxylsuccinimide ester at a 1:1 ratio in anhydrous DMF containing 5% v/v DIPEA. The cyclized product was confirmed by LC-MS. The concentrations of carboxyfluorescein labeled peptides were determined by UV-Vis spectrophotometry at 495 nm using the absorption coefficient factor 80,200 cm−1M−1.
Dimeric peptides were synthesized from pure monomeric carboxyfluorescein labeled peptides L11C or L11K (Figure 2B). Monomeric subunits (L11C-L11C, L11C-L11K, or L11K-L11K depending on the functional groups of the linkers) were mixed with bis-functional linkers purchased from Quanta Biodesign or Pierce Biotechnology, (e.g. hydroxysuccimidyl maleimido propionate (SMP), bis(sulfosuccinimidyl)suberate (BS3), bis-dPEG5-NHS, bis-dPEG9-NHS, MAL-dPEG4-NHS, MAL-dPEG12-NHS, MAL-dPEG24-NHS, and bis-MAL-dPEG3, where NHS denotes N-hydroxylsuccinimide ester, and MAL denotes maleimide), at 1:1:0.9 ratio (monomer1:monomer2:crosslinker) in anhydrous DMF with 5% v/v DIPEA. For monitoring the formation of dimeric peptides as well as purification of the desired product, C18 reverse phase HPLC was used. The dimeric product peaks eluted approximately 2 min after the monomeric starting material, as confirmed by LC-MS. The concentration of each fluorescent dimeric peptide was quantified by using the absorption coefficient factor 160,400 cm−1M−1 at 495 nm.
4.2 Peptide exchange assay
Peptide exchange assays were conducted as previously described.13 In brief, soluble recombinant DQ2 molecules with a gliadin epitope fused to the N-terminus of the β-chain were expressed and purified. Prior to use in exchange experiments, recombinant DQ2 molecules were treated with ~2% w/w thrombin in PBS pH 7.3 at 0°C for 2 h to release the covalently linked epitope for peptide exchange measurements. Thrombin treated DQ2 was incubated with fluorescein-conjugated peptides in a 25:1 ratio (i.e. 4.7 μM DQ2 with 0.185μM fluorescent peptide) at 37°C in a 1:1 mixture of PBS buffer (10 mM sodium phosphate, 150 mM NaCl, pH 7.3, supplemented with 0.02% NaN3) and McIlvaine’s citrate-phosphate buffer (pH 5 or pH 7) such that the final pH was either 5.5 or 7.3, respectively. Peptide binding was measured by high performance size exclusion chromatography (HPSEC) coupled with fluorescence detection with excitation at 495 nm and emission at 520 nm. The DQ2-peptide 1:1 complex eluted at ~8.5 min, with free peptides emerging ~2 min later. When present, the 2:1 DQ2-peptide complex eluted ~0.5 min before the 1:1 complex. Peak areas corresponding to the DQ2-peptide complex and the free peptide were used to calculate the fractional yield of the DQ2-fluoresceinated peptide complex. At least two independent measurements were conducted, with an error <5%.
4.3 Competitive DQ2-Peptide Binding Assay
Detergent-solubilized DQ2 molecules were purified from HLA homozygous (DQA1*0501/DQB1*0201) Epstein-Barr virus-transformed B lymphoblastoid cell lines as previously described.36 The indicator peptide (KPLLIIAEDVEGEY; Mycobacterium bovis 65-kDa heat shock protein 243–255Y) was 125I-labeled by the chloramine-T method.37 The labeled indicator peptide (30,000 cpm; 1–5 nM) and various concentrations of unlabeled peptides were incubated overnight with 70–200 nM DQ2 at 37°C in the presence of a cocktail of protease inhibitors at pH 5.2. After incubation, complexes of peptide and DQ2 molecules were separated from unbound peptides on Sephadex G-50 (Amersham Biosciences AB) spin columns as described previously.38 Radioactivity was measured, and the concentrations of competing peptides required to give 50% inhibition of binding of the indicator peptide (IC50) were calculated. The IC50 values were determined by three 3-fold titration experiments.
4.4 T cell reagents
Gluten reactive T cell lines (TCL) and T cell clones (TCC) established from small intestinal biopsies of celiac disease patients were used.29,39 This includes the T cell line P28 TCL2 (which is reactive to DQ2-αI DQ2-αII and DQ2-αIII epitopes of the deamidated 33-mer peptide) and the T cell clones TCC 437.1.3.17 (specific for the DQ2-γII epitope) and TCC 430.1.142 (specific for the DQ2-αI epitope). In addition a DR3-restricted T cell clone, TCC RN.46 (specific for the 3–13 epitope of Mycobacterium tuberculosis heat shock protein 65, KTIAYDEEARR), was used.
4.5 T cell proliferation assays
T cell proliferation assays were performed mainly as described previously.13 HLA-DR3/DQ2 homozygous Epstein-Barr virus transformed B-lymphoblastoid cell lines (VAVY or CD114) were used as antigen presenting cells. The cells were fixed with 1% paraformaldehyde for 10 min or with 0.05% glutaraldehyde for 90 s using 0.2 M glycine to quench the reaction. The antigen presenting cells were incubated with the appropriate peptides overnight in 60 or 65 μl media containing 10% fetal bovine serum/2% human serum or 15% human serum, penicillin and streptomycin at a cell density of 2×106 cells/ml in 96-well flat bottom plates. The next day, the volume was doubled to yield a cell density of 1×106 cells/ml, and the cells were seeded in duplicates of 50 μl each in a U-bottom 96-well plate. An equal volume of T cells (50 μl of 1×106 cells/ml) was added to each well, and cells were incubated at 37°C and 5% CO2 for 48 h, at which time 0.5 μCi/well of [methyl-3H] thymidine (Amersham, TRK120) or 1 μCi/well of [methyl-3H]thymidine (Hartmann Analytic) was added. Cells were incubated for an additional 12–16 h and then frozen or harvested directly. After thawing, incorporated thymidine was collected on a filter mat (Wallac) using a Tomtec cell harvester, and counted using a Wallac 1205 Betaplate or Wallac 1450 MicroBeta TriLux liquid scintillation counter. For testing of whether the cyclic peptides which harbored the DQ2-αI sequence were recognized by a DQ2-αI-specific T cell clone, the same assay was performed except that triplicate wells containing 7.5×104 antigen presenting cells were prepulsed overnight with peptide in a volume of 100 μl before 5×104 T cells per well in 50 μl were added the following day.
For T cell assays using γ-irradiated B-lymphoblastoid cells, VAVY cells were γ-irradiated (12,000 rads) with a cesium irradiator, resuspended to 2×106 cells/ml, and incubated with or without 25 μM peptide 8 for 2 hours in a flat bottom 96-well plate. Antigen peptide LQLQPFPQPELPYPQPELPY was then added at 0.1 or 0.03 μM and incubated for an additional 10 hours. The volume was doubled and each well was transferred into an Eppendorf tube. The cells were centrifuged at 800g for 3 minutes at 4°C. Next, the supernatant was aspirated, the cells were resuspended to 1×106 cells/ml, and the T cell proliferation protocol described above was used to measure 3H-thymidine incorporation into P28 TCL2 T cells.
Acknowledgments
This work was supported by a grant from the NIH (R21 DK65965 to C.K.) and by grants from the Research Council of Norway, the Rikshospitalet-Radiumhospitalet Medical Center and the Fulbright Fellowship Program to L.M.S. M. Siegel is a recipient of a predoctoral fellowship from the Stanford-NIH Biotechnology Training Grant, and E. Bergseng is recipient of a fellowship from the Norwegian Foundation for Health and Rehabilitation (EXTRA fund).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tiwari J, Terasaki P. HLA and disease association. Springer-Verlag; New York: 1985. [Google Scholar]
- 2.Nepom BS. Clin Immunol Immunopathol. 1993;67:S50–S55. doi: 10.1006/clin.1993.1084. [DOI] [PubMed] [Google Scholar]
- 3.Todd JA, Acha-Orbea H, Bell JI, Chao N, Fronek Z, Jacob CO, McDermott M, Sinha AA, Timmerman L, Steinman L, McDevitt HO. Science. 1988;240:1003–1009. doi: 10.1126/science.3368786. [DOI] [PubMed] [Google Scholar]
- 4.Jones EY, Fugger L, Strominger JL, Siebold C. Nat Rev Immunol. 2006;6:271–282. doi: 10.1038/nri1805. [DOI] [PubMed] [Google Scholar]
- 5.Sollid LM, Thorsby E. Gastroenterology. 1993;105:910–922. doi: 10.1016/0016-5085(93)90912-v. [DOI] [PubMed] [Google Scholar]
- 6.Sollid LM. Nat Rev Immunol. 2002;2:647–655. doi: 10.1038/nri885. [DOI] [PubMed] [Google Scholar]
- 7.Sloan-Lancaster J, Allen PM. Annu Rev Immunol. 1996;14:1–27. doi: 10.1146/annurev.immunol.14.1.1. [DOI] [PubMed] [Google Scholar]
- 8.de Haan EC, Moret EE, Wagenaar-Hilbers JPA, Liskamp RMJ, Wauben MHM. Mol Immunol. 2005;42:365–373. doi: 10.1016/j.molimm.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 9.Adorini L, Muller S, Cardinaux F, Lehmann PV, Falcioni F, Nagy ZA. Nature. 1988;334:623–625. doi: 10.1038/334623a0. [DOI] [PubMed] [Google Scholar]
- 10.Sakai K, Zamvil SS, Mitchell DJ, Hodgkinson S, Rothbard JB, Steinman L. Proc Natl Acad Sci U S A. 1989;86:9470–9474. doi: 10.1073/pnas.86.23.9470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hurtenbach U, Lier E, Adorini L, Nagy ZA. J Exp Med. 1993;177:1499–1504. doi: 10.1084/jem.177.5.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Magistris MT, Alexander J, Coggeshall M, Altman A, Gaeta FCA, Grey HM, Sette A. Cell. 1992;68:625–634. doi: 10.1016/0092-8674(92)90139-4. [DOI] [PubMed] [Google Scholar]
- 13.Xia J, Siegel M, Bergseng E, Sollid LM, Khosla C. J Am Chem Soc. 2006;128:1859–1867. doi: 10.1021/ja056423o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tselios T, Daliani I, Deraos S, Thymianou S, Matsoukas E, Troganis A, Gerothanassis I, Mouzaki A, Mavromoustakos T, Probert L, Matsoukas J. Bioorg Med Chem Lett. 2000;10:2713–2717. doi: 10.1016/s0960-894x(00)00556-4. [DOI] [PubMed] [Google Scholar]
- 15.Tselios T, Apostolopoulos V, Daliani I, Deraos S, Grdadolnik S, Mavromoustakos T, Melachrinou M, Thymianou S, Probert L, Mouzaki A, Matsoukas J. J Med Chem. 2002;45:275–283. doi: 10.1021/jm0102147. [DOI] [PubMed] [Google Scholar]
- 16.Matsoukas J, Apostolopoulos V, Kalbacher H, Papini AM, Tselios T, Chatzantoni K, Biagioli T, Lolli F, Deraos S, Papathanassopoulos P, Troganis A, Mantzourani E, Mavromoustakos T, Mouzaki A. J Med Chem. 2005;48:1470–1480. doi: 10.1021/jm040849g. [DOI] [PubMed] [Google Scholar]
- 17.Bouvier M, Wiley DC. Proc Natl Acad Sci U S A. 1996;93:4583–4588. doi: 10.1073/pnas.93.10.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berezhkovskiy L, Pham S, Reich EP, Deshpande S. J Pept Res. 1999;54:112–119. doi: 10.1034/j.1399-3011.1999.00084.x. [DOI] [PubMed] [Google Scholar]
- 19.Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM. Proc Natl Acad Sci U S A. 2004;101:4175–4179. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishioka GY, Adorini L, Guery JC, Gaeta FCA, Lafond R, Alexander J, Powell MF, Sette A, Grey HM. J Immunol. 1994;152:4310–4319. [PubMed] [Google Scholar]
- 21.Lamont AG, Powell MF, Colon SM, Miles C, Grey HM, Sette A. J Immunol. 1990;144:2493–2498. [PubMed] [Google Scholar]
- 22.Jones AB, Acton JJ, Rivetna MN, Cummings RT, Cubbon RM, Nichols EA, Schwartz CD, Wicker LS, Hermes JD. Bioorg Med Chem Lett. 1999;9:2109–2114. doi: 10.1016/s0960-894x(99)00333-9. [DOI] [PubMed] [Google Scholar]
- 23.Jones AB, Acton JJ, Adams AD, Yuen W, Nichols EA, Schwartz CD, Wicker LS, Hermes JD. Bioorg Med Chem Lett. 1999;9:2115–2118. [PubMed] [Google Scholar]
- 24.Falcioni F, Ito K, Vidovic D, Belunis C, Campbell R, Berthel SJ, Bolin DR, Gillespie PB, Huby N, Olson GL, Sarabu R, Guenot J, Madison V, Hammer J, Sinigaglia F, Steinmetz M, Nagy ZA. Nat Biotechnol. 1999;17:562–567. doi: 10.1038/9865. [DOI] [PubMed] [Google Scholar]
- 25.Bolin DR, Swain AL, Sarabu R, Berthel SJ, Gillespie P, Huby NJS, Makofske R, Orzechowski L, Perrotta A, Toth K, Cooper JP, Jiang N, Falcioni F, Campbell R, Cox D, Gaizband D, Belunis CJ, Vidovic D, Ito K, Crowther R, Kammlott U, Zhang XL, Palermo R, Weber D, Guenot J, Nagy Z, Olson GL. J Med Chem. 2000;43:2135–2148. doi: 10.1021/jm000034h. [DOI] [PubMed] [Google Scholar]
- 26.Shan L, Molberg Ø, Parrot I, Hausch F, Filiz F, Gray GM, Sollid LM, Khosla C. Science. 2002;297:2275–2279. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- 27.Shan L, Qiao SW, Arentz-Hansen H, Molberg Ø, Gray GM, Sollid LM, Khosla C. J Proteome Res. 2005;4:1732–1741. doi: 10.1021/pr050173t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Strominger JL, Wiley DC. Proc Natl Acad Sci U S A. 1996;93:734–738. doi: 10.1073/pnas.93.2.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiao SW, Bergseng E, Molberg Ø, Jung G, Fleckenstein B, Sollid LM. J Immunol. 2005;175:254–261. doi: 10.4049/jimmunol.175.1.254. [DOI] [PubMed] [Google Scholar]
- 30.Kiessling LL, Gestwicki JE, Strong LE. Curr Opin Chem Biol. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 31.Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR. Nature. 2000;403:669–672. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- 32.Mammen M, Choi SK, Whitesides GM. Angew Chem Int Edit. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 33.Sucheck SJ, Wong AL, Koeller KM, Boehr DD, Draker K, Sears P, Wright GD, Wong CH. J Am Chem Soc. 2000;122:5230–5231. [Google Scholar]
- 34.Mangold SL, Cloninger MJ. Org Biomol Chem. 2006;4:2458–2465. doi: 10.1039/b600066e. [DOI] [PubMed] [Google Scholar]
- 35.Xia J, Sollid LM, Khosla C. Biochemistry. 2005;44:4442–4449. doi: 10.1021/bi047747c. [DOI] [PubMed] [Google Scholar]
- 36.Johansen BH, Buus S, Vartdal F, Viken H, Eriksen JA, Thorsby E, Sollid LM. Int Immunol. 1994;6:453–461. doi: 10.1093/intimm/6.3.453. [DOI] [PubMed] [Google Scholar]
- 37.Greenwood FC, Hunter WM. Biochem J. 1963;89:114. doi: 10.1042/bj0890114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buus S, Stryhn A, Winther K, Kirkby N, Pedersen LO. Biochim Biophys Acta. 1995;1243:453–460. doi: 10.1016/0304-4165(94)00172-t. [DOI] [PubMed] [Google Scholar]
- 39.Siegel M, Bethune MT, Gass J, Ehren J, Xia J, Johannsen A, Stuge TB, Gray GM, Lee PP, Khosla C. Chem Biol. 2006;13:649–658. doi: 10.1016/j.chembiol.2006.04.009. [DOI] [PubMed] [Google Scholar]