

Abstract
Although human polyomavirus JC (JCV) is known to cause progressive multifocal leukoencephalopathy (PML) in immunocompromised individuals, the mechanism by which, JCV crosses the blood-brain barrier (BBB) remains unclear. To test our hypothesis that cell-free JCV gains entry into the brain by infecting endothelial cells, we inoculated human brain microvascular endothelial (HBMVE) cells with 50 HAU (1.33±0.27×107 genome copies) of JCV(Mad1) and analyzed the expression of early and late viral genes and proteins by immunocytochemistry, quantitative real-time PCR (qPCR), quantitative real-time reverse transcriptase-PCR (qRT-PCR) and immunoprecipitation followed by Western blotting. JCV infected and replicated efficiently in HBMVE cells and produced infectious virions several hundred fold higher than the infecting inoculum. HBMVE cells in-vitro did not express serotonin receptor 2A (5HT2AR), and 5HT2AR blockers did not prevent JCV infection of HBMVE cells. Collectively, our data indicate that the productive in-vitro infection of HBMVE cells by JCV is independent of 5HT2AR.
Keywords: JCV, Human polyomavirus JC, Polyomaviridae, PML, HIV, AIDS, blood-brain barrier, HBMVE cells, human brain microvascular endothelial cells, serotonin receptor 2A, 5HT2AR, oligodendrocytes
INTRODUCTION
Progressive multifocal leukoencephalopathy (PML), a uniformly fatal, subacute demyelinating disease of the central nervous system (CNS), is caused by human polyomavirus JC (JCV) (Imperiale, 2001; Mazlo et al., 2001; Padgett et al., 1977), a member of the family Polyomaviridae, which has a naked icosahedral capsid and a circular double-stranded DNA genome of approximately 5.1 kb (Imperiale, 2001; Kim et al., 2001). Seroepidemiological studies indicate that JCV infects 60 to 80% of individuals (Knowles et al., 2003). Primary JCV infection, which is likely acquired via the oral or respiratory route during childhood, is typically subclinical (Monaco et al., 1996; Monaco et al., 1998). The primary sites of JCV infection and replication are unknown, although tonsillar tissue has been proposed (Monaco et al., 1996; Monaco et al., 1998). Virus-infected lymphocytes or cell-free virus presumably spread by the hematogenous route from the primary site to secondary sites, such as brain and kidney, to establish focal areas of virus persistence (Monaco et al., 1996; Monaco et al., 1998; Tornatore et al., 1992). JCV persists in the kidneys, brain, tonsils and lymphocytes of infected individuals, with or without PML (Dorries et al., 1994; Monaco et al., 1996; Monaco et al., 1998; Newman and Frisque, 1999; Tornatore et al., 1992; White et al., 1992), while pathology is confined to the CNS (Mazlo et al., 2001; Newman and Frisque, 1999). A neuropathological hallmark of PML is the destruction of oligodendrocytes, the myelin-forming cells in the brain, for which JCV has a strong tropism and which serve as the site of JCV replication in the brain.
The detection of JCV DNA in the brains of patients with non-neurological diseases and neurological disorders other than PML (Elsner and Dorries, 1992; Vago et al., 1996; White et al., 1992) suggests that immunosuppression is not a prerequisite for JCV to enter the CNS and that JCV migration across the blood-brain barrier (BBB) may occur early during the course of asymptomatic primary infection. Based on the demonstrated association between JCV and B cells, it has been proposed that JCV may traffic to the brain via infected B cells (Houff et al., 1988; Monaco et al., 1996; Monaco et al., 1998). JCV has occasionally been detected in the brain vascular endothelial cells of PML patients by in-situ hybridization (Dorries et al., 1979; von Einsiedel et al., 2004), and Fareed and colleagues (1978) have suggested that endothelial cells may support limited JCV replication. Thus, the most direct method to delineate the mechanism of JCV transmigration across the BBB is to study the replication potential of JCV in HBMVE cells, the most critical component of the BBB. Understanding the mechanisms underlying JCV trafficking across the BBB may assist in developing preventive or therapeutic strategies that limit JCV entry into the brain, resulting in the prevention of JCV persistence in the CNS.
JCV is proposed to initially engage the alpha 2-6–linked sialic acid receptor (Liu et al., 1998), followed by the interaction with the serotonin receptor 2A (5HT2AR) (Elphick et al., 2004) leading to virus internalization into glial cells by clathrin-dependent endocytosis (Baum et al., 2003). In a recent study, Elphick and colleagues demonstrated that several serotonin receptor antagonists blocked JCV entry and inhibited JCV infection of glial cells (Elphick et al., 2004). However, whether such blockers inhibit JCV trafficking across the BBB is unclear.
We hypothesized that during the viremic phase of asymptomatic infection, cell-free JCV infects HBMVE cells to cross the BBB and to establish latency in the brain. During immunosuppression, the latent JCV may be activated, resulting in lytic infection of the oligodendrocytes. In the first step toward dissecting the migration of JCV across the BBB, we employed primary HBMVE cells derived from adult brain to demonstrate replication of JCV(Mad 1). Our data indicate that JCV infects HBMVE cells in-vitro, HBMVE cells in-vitro do not express 5HT2AR, and JCV infection of HBMVE cells is independent of 5HT2AR.
RESULTS
Primary HBMVE cells retain their characteristics
Based on a total of 900 cells counted in three independent fields, more than 90% of HBMVE cells expressed vWF (Fig. 1A), whereas no vWF was detected in cells stained with only secondary antibody (Fig. 1B). It has been documented that HBMVE cells in culture gradually lose vWF expression in subsequent passages (Mukhtar et al., 2002). It is most likely that vWF negative cells may represent the subset of HBMVE cells that have lost their ability to express vWF.
Fig. 1. Primary HBMVE cells in-vitro express vWF.
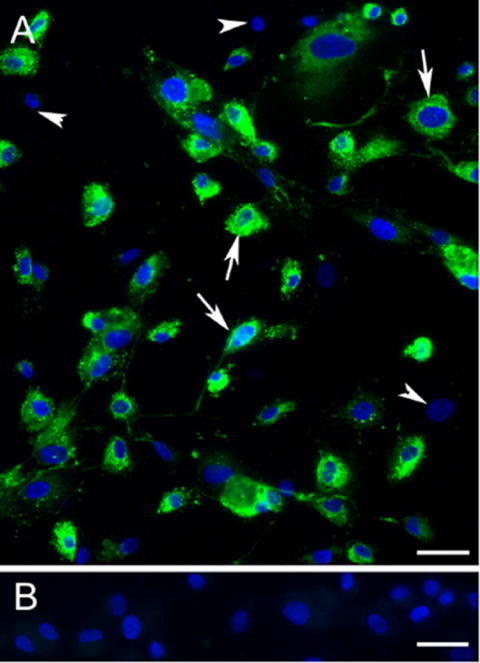
(A) Epifluorescence image demonstrate 90% of HBMVE cells expressing vWF (green) depicted by arrows, and cells lacking vWF expression depicted by arrowheads. The percentage of vWF positive cells, were quantitated by counting the number of fluorescent cells in a total of 900 cells selected from three random fields. (B) Immunolabel control cells stained with secondary antibody only and counterstained with bisbenzidine for nuclear stain (blue) did not show green fluorescence. Experiments were performed in duplicate. Scale bars 30 μm.
JCV efficiently replicates in HBMVE cells
JCV replication was observed in HBMVE cells. Based on epifluorescence and confocal microscopy, approximately 15% of HBMVE cells (vWF stained, green) expressed JCV T antigen (early protein) at day 10 post-inoculation. Of the total cells, 5% had strong immunoreactivity with anti-SV40 T antigen antibody which cross-reacts with JCV T antigen (pink color, Fig. 2A), whereas 10% had moderate immunoreactivity (purple color, Fig. 2A). Multiple nuclei were often observed in JCV-infected cells (arrowhead, Fig 2A). No staining was observed in JCV-infected HBMVE cells incubated with only secondary antibodies (Fig. 2B). Figures 2C and 2D show a cluster of four infected cell nuclei suggesting two subsequent nuclear divisions or fusion of the infected endothelial cells. Additionally, the infected HBMVE cells were frequently larger compared to the uninfected cells (Fig. 2A and 2D). Although T antigen staining was observed predominantly in the cell nucleus, we occasionally observed punctate staining in the cytoplasm of infected cells (Fig. 2E and 2F), most probably representing different stages of JCV replication (von Einsiedel et al., 2004). Moreover, both JCV T antigen (Fig. 2H) and VP-1 (Fig. 2G) proteins, were co-localized in the nucleus, suggesting productive JCV infection (Fig. 2I to 2J).
Fig. 2. Epifluorescence and confocal microscopy demonstrating presence of JCV T antigen and VP-1.
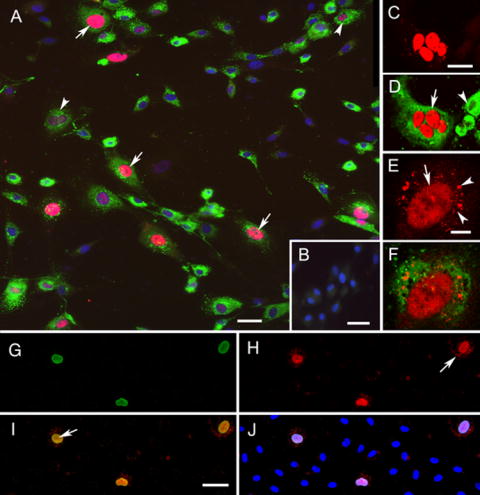
(A) Epifluorescence image demonstrating immunoreactivities for T antigen (red), vWF (green), and counterstaining for bisbenzidine to visualize cell nuclei (blue) in HBMVE cells infected with 50 HAU of JCV. Several HBMVE cells display strong immunoreactivity to T antigen in the cell nucleus, as indicated by the pink color (overlap of red and blue colors, arrows). HBMVE cells with moderate immunoreactivity for T antigen are indicated by the purple color (arrowheads). Note the bi-nucleation in the cell indicated by a arrowhead. (B) Immunolabel control without primary antibodies with bisbenzidine-counterstained cell nuclei. The secondary antibodies do not show significant background (green or red colors are not visible). (C) Confocal microscopy image showing four cell nuclei immunoreactive for T antigen (red). (D) The confocal image C, which additionally displays vWF immunoreactivity, strongly suggests that this infected HBMVE cell (arrow) recently underwent nuclear division or cell fusion. Compare the size of the infected HBMVE cell with the non-infected HBMVE cell (arrowhead). (E) The presence of T antigen immunoreactivity in both the nucleus (arrow) and cytoplasm (arrowheads) indicates different stages of viral replication in HBMVE cells. (F) The confocal image D, stained with anti vWF antibody (green). (G to J) JCV-infected HBMVE cells were simultaneously stained with antibodies against (G) VP-1 (green) and (H) SV-40 T antigen (red). (I) HBMVE cells expressed both VP-1 and T antigen (arrow) in their nuclei (merged image yellow color). Some JCV-infected cells only expressed T antigen in their cytoplasm (arrow in H). (J) shows triple merged image with VP-1, T antigen and nuclear staining. Scale bars for AC and I; 25 μm, and for E; 10 μm.
Based on the qPCR T antigen (TAg) genome data, approximately 5% of JCV was attached or internalized into HBMVE cells at 2 hr post-inoculation with 50 HAU of JCV. There was no difference in the JCV TAg or VP-1 genome copies recovered from HBMVE cells harvested on days 0 (2 hr) and 3 post-inoculation (data not shown). JCV TAg and VP-1 mRNA transcripts were detected as early as day 3 post-inoculation and increased exponentially thereafter and paralleled DNA replication (Fig. 3A-B). On day 20 post-inoculation, the total JCV TAg (2.72 × 109) and VP-1 (5.29 × 109) genome copies recovered from each 35-mm plate were approximately 204- and 395-fold higher than the mean infecting JCV dose, respectively. Moreover, T antigen protein was detected by immunoprecipitation and Western blot (WB) from JCV-infected HBMVE cells harvested on days 15 and 20 post-inoculation (Fig. 3C). Replication profiles of JCV in HBMVE cells and PHFG cells were very similar (Fig. 3D), suggesting that JCV replication in HBMVE cells is as efficient as in PHFG cells.
Fig. 3. JCV replicates in HBMVE cells.
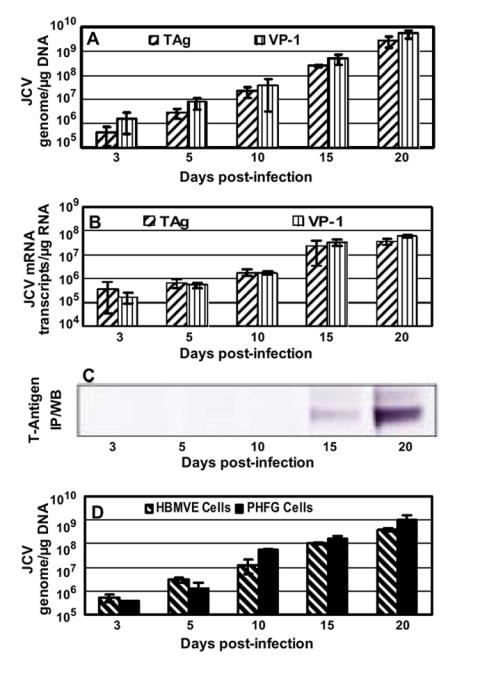
HBMVE cells grown in 35-mm plates in duplicate were infected with JCV(Mad1) for 2 hr and viral genome copies and mRNA transcripts production were monitored over a period of 20 days by qPCR and qRT-PCR, respectively. (A) Viral genome copies increased rapidly from day 3 to day 20 post-inoculation. (B) JCV TAg and VP-1 mRNA transcripts increased in parallel with viral genome copies, and (C) JCV T protein was detected in JCV-infected HBMVE cells by immunoprecipitation and Western blotting with anti-SV40 T antigen mouse mAb. (D) For comparison, HBMVE and PHFG cells in 35-mm plates were infected with 50 HAU of JCV, and JCV TAg genome was quantitated by qPCR. There was no significant difference in JCV replication in HBMVE and PHFG cells. JCV genome or mRNA transcripts were expressed as copies per μg DNA or RNA, respectively, and data are representative of two independent experiments with duplicate samples in each experiment. Error bars represent SD.
To further verify that JCV infection of HBMVE cells resulted in production of infectious virions, we inoculated naïve PHFG cells with HBMVE cell lysates collected at day 20 post-inoculation and analyzed these for viral DNA and RNA expression. Our data clearly demonstrated that the virions produced in HBMVE cell were infectious and JCV produced in HBMVE cells replicated efficiently in naïve PHFG cells (Fig. 4A and 4B).
Fig. 4. JCV replication in HBMVE cells produces infectious virions.
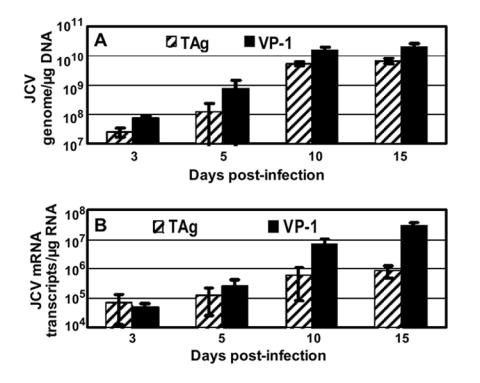
PHFG cells grown in 35-mm plates in duplicate were inoculated with day 20 post-inoculation lysates from JCV-infected HBMVE cells, and were analyzed for the expression of (A) JCV genome, and (B) JCV mRNA transcripts. JCV TAg and VP-1 mRNA transcripts increased in parallel to genome copy numbers.
JCV infection of HBMVE cells occurs independent of 5HT2AR
Recently, 5HT2AR was reported to be associated with JCV infection of glial cells (Elphick et al., 2004). Since JCV replicated efficiently in HBMVE cells, we next examined whether JCV employs 5HT2AR to infect HBMVE cells. 5HT2AR gene and protein expression was analyzed in uninfected PHFG, HBMVE and HBCA cells by RT-PCR and WB, respectively. While PHFG cells expressed 5HT2AR Mrna transcripts (Fig. 5A) and protein (Fig. 5B), HBMVE and HBCA cells lacked the expression of 5HT2AR (Fig. 5A and 5B). Moreover, there was no significant difference (Fig. 5B) in the JCV replication in HBMVE cells untreated or exposed continuously to chlorpromazine (which inhibits both serotonin and dopamine receptors), 5-hydroxytryptamine sulfate (which down regulates serotonin receptors), kentaserine (which blocks serotonin receptors 2A and 2C), or ritanserine (which blocks serotonin receptors 2A, 2B and 2C) (Elphick et al., 2004), suggesting that JCV infection of HBMVE cells is independent of 5HT2AR.
Fig. 5. HBMVE cells in culture do not express serotonin receptor 2A and serotonin receptor blockers did not inhibit JCV replication in HBMVE cells.
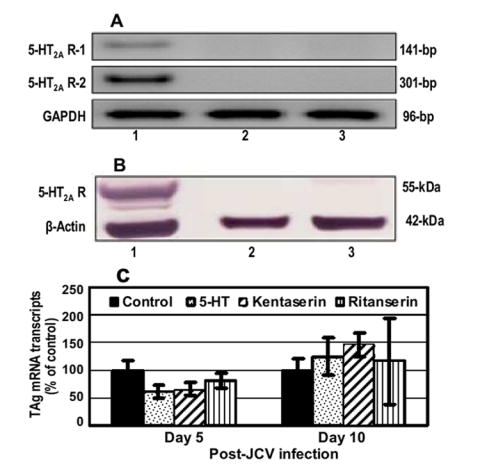
RNA and total protein were extracted from PHFG, HBMVE and HBCA cells, lanes 1, 2 and 3, respectively. (A) cDNA was synthesized and expression of 5HT2AR was analyzed by two independent primers (table 1). Housekeeping gene GAPDH was used as a positive control. (B) Total cellular protein extract from PHFG, HBMVE and HBCA cells were analyzed for the expression of 5-HT2AR or housekeeping protein, β-actin by Western blotting using polyclonal 5-HT2AR and monoclonal β-actin antibodies, respectively. (C) JCV replication in HBMVE cells was quantitated by measuring JCV TAg mRNA transcript by qRT-PCR. JCV replication was expressed as percentage of control.
DISCUSSION
The mechanism of JCV trafficking across the BBB remains poorly understood. Of the potential pathways JCV might employ to cross the BBB, virus-infected B cells have been proposed (Houff et al., 1988; Monaco et al., 1996; Monaco et al., 1998), similar to human immunodeficiency virus type 1 (HIV) and simian immunodeficiency virus (SIV) which gain entry into the brain via infected monocytes (Trojan horse) (Georgsson, 1994). However, other potential routes of transmigration of JCV have not been explored. It is not yet clear whether JCV infects HBMVE cells productively or latently, like SIV (Mankowski et al., 1994) and HIV (Liu et al., 2002; Moses et al., 1993), respectively, to cross the BBB via transcytosis or initially compromises the BBB and crosses it via the paracellular pathway. To address this issue, we employed primary HBMVE cells to study the replication of JCV. We found that JCV productively infects HBMVE cells in-vitro and JCV infection of HBMVE cells in-vitro is independent of 5HT2AR.
Although controversial, available data suggest that JCV can be detected in the brains of immunocompetent individuals, as well as in the brains of patients with neurological disorders other than PML (Elsner and Dorries, 1992; Ferrante et al., 1995; Vago et al., 1996; White et al., 1992), suggesting that immunodeficiency is not a absolute prerequisite for migration of JCV across the BBB. Instead, JCV may cross the BBB during asymptomatic primary infection. However, there are no data on the trafficking of JCV across the BBB. To delineate the mechanism of JCV transmigration across the BBB, it is important to study the replication potential of JCV in HBMVE cells. JCV is well known to have a limited host range, and productive infection of HBMVE cells has not been demonstrated. Our data, based on expression of JCV early and late transcripts by qRT-PCR, T antigen and VP-1 proteins by immunocytochemistry and T antigen protein by WB, conclusively demonstrate that JCV productively infects HBMVE cells. Moreover, we demonstrated that the lysates from day 20 JCV-infected HBMVE cells were infectious in PHFG cells, which strongly support that infectious JCV virions were produced in HBMVE cells. Since JCV is known to have a very limited host range, it was surprising, that side-by-side infection of HBMVE and PHFG cells with the same inoculum size of JCV resulted in a similar pattern of JCV DNA replication, suggesting that HBMVE cells may be a suitable alternative to PHFG cells in future studies on the molecular pathogenesis of PML.
JCV is reported to cause the death of infected oligodendrocytes and astrocytes by apoptosis and necrosis, respectively (Richardson-Burns et al., 2002; Seth et al., 2004). However, we did not observe any gross or microscopic cytopathic effect in JCV-infected HBMVE cells in culture, although, by immunocytochemistry the infected cells appeared to be slightly larger than uninfected cells and more often had clusters of nuclei at day 10 post-inoculation, suggesting more frequent nuclear division or fusion of JCV-infected HBMVE cells. Polyomavirus T antigen is a known inducer of S phase (Imperiale, 2001), and JCV infection may induce S phase progression of HBMVE cells leading to more frequent nuclear division.
Several viruses, including measles (Andres et al., 2003), human cytomegalovirus (HCMV) (Fish et al., 1998) and SIV (Mankowski et al., 1994), are known to productively infect microvascular endothelial cells in-vitro and in-vivo. Although JCV was occasionally detected in the vascular endothelial cells of PML patients by in-situ hybridization (Dorries et al., 1979; von Einsiedel et al., 2004), to our knowledge, this is the first study demonstrating that JCV can infect and efficiently replicate in HBMVE cells, the most critical component of the BBB. It is possible that JCV infection of HBMVE cells in-vivo does not result in productive infection, and the level of antigen expression is often lower than the detectable threshold. Non-productive infection or infection in the absence of cytopathology provides the most favourable environment for establishment of persistent JCV infection, and HBMVE cells in-vivo may be an important site of JCV latency in immunocompetent individuals.
A recent study demonstrated that the chimera polyomavirus JC (Mad-1/SVEΔ) consisting of JCV-SV40 promoter–enhancer sequences in the backbone of JCV coding region sequences employs the 5HT2AR to infect SVG-A cells, a subclone of transformed human fetal glial cells by an origin-defective SV40 mutant (Elphick et al., 2004). Antibodies and blockers against 5HT2AR specifically inhibited JCV infection of SVG-A cells, and insertion of the 5HT2AR gene in receptor-negative and JCV-resistant HeLa cells restored susceptibility (Elphick et al., 2004). To oursurprise, 5HT2AR blockers did not inhibit JCV replication in HBMVE cells. Moreover, mRNA and protein expression data indicated that HBMVE cells in-vitro do not express 5HT2AR, which is consistent with the published literature (Cohen et al., 1999; Ullmer et al., 1995). This inconsistency may have resulted from the use of different cell types. While Elphick et al (2004) have used SVG-A cells and JCV chimera (Mad-1/SVEΔ), we employed primary HBMVE cells and prototype JCV(Mad1), which is predominantly found among brains of PML patients (Dubois et al., 2001). It is possible that JCV may employ receptors other than 5HT2AR to infect HBMVE cells. Interestingly, several viruses, such as HIV and measles, infect HBMVE cells even though HBMVE cells do not express known cellular receptors for these viruses (Andres et al., 2003; Argyris et al., 2003).
At this point, we conjecture that cell-free JCV may cross the BBB in-vivo by infecting HBMVE cells. Our data indicate that HBMVE cells do not express 5HT2AR, and 5HT2AR blockers may not prevent the entry of JCV in the brain. Studies are now underway to delineate the molecular mechanisms of cell-free JCV transmigration across the in-vitro BBB.
MATERIALS AND METHODS
Virus and Primary cell cultures
JCV(Mad1) was propagated in primary human fetal glial (PHFG) cells, purified and quantitated by the HA assay and real time PCR (Chapagain et al., 2006), and the transcriptional control region sequences were reconfirmed by cloning and sequencing, as described previously (Fernandez-Cobo et al., 2001).
Low-passage primary HBMVE cells (Cat #ACBRI 376 and 401), isolated from adult human brain cortex and expressing ≥95% von Willebrand Factor (vWF), were purchased from Cell Systems Corporation (Kirkland, WA), and propagated on attachment factor (Cat #4Z0-210) coated T75 flasks with CSC-Complete medium (Cat #Z0-500) at 37°C, 5% CO2 and 100% humidity, according to the manufacturer’s protocol. All experiments were performed with cells between passages 6 to 8. After obtaining approval from the institutional review board of the Kapi‘olani Medical Center for Women and Children, Honolulu, Hawai‘i, PHFG cells were isolated from therapeutically aborted 10- to 14-week-old fetal brains, as described previously (Chapagain et al., 2006; Padgett et al., 1977).
Characterization of HBMVE cells
Cultured HBMVE cells grown on coverslips were washed twice with 1X PBS and fixed in 4% paraformaldehyde. After permeabilization with 0.5% Triton X100 and pre-incubation in PBS-0.2% gelatin, HBMVE cells were incubated at room temperature with primary rabbit polyclonal antibody against vWF (1:500 dilution) (Cat #ab6994, Abcam, Cambridge, MA) for 2 hr, washed and incubated with fluorescein isothiocyanate (FITC)-conjugated donkey anti-rabbit (1:100) (Cat #N1034V, Amersham, Pittsburgh, PA) secondary antibody for 40 min, followed by three washes with PBS. Cells were counterstained with 2μg/mL bisbenzidine (Cat #H33258, Sigma Aldrich, St. Louis, MO) for 5 min to visualize cell nuclei, and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Fluorescent cells were examined using a Zeiss Confocal Pascal equipped with a Zeiss Axiovert 200 microscope, or a Axiocam MRm camera mounted on a Zeiss Axiovert 200 microscope, both equipped with appropriate fluorescence filters and objectives.
JCV infection of HBMVE cell
On each attachment factor-coated 35-mm culture plate, 105 HBMVE cells were seeded and grown to 70-80% confluency, and inoculated with 50 HAU containing approximately 1.33±0.27 × 107 genome copies of JCV(Mad1). After adsorption for 2 hr, the medium was aspirated; plates were washed three times with 1X PBS, and then incubated with 2 mL of fresh medium. The medium was changed every 2 days, and cells were harvested for DNA, RNA and protein extraction on days 3, 5, 10, 15, and 20 post-inoculation. For comparison, PHFG cells seeded on 35-mm plates were inoculated with the same dose (50 HAU) of prototype JCV(Mad1), and cells were harvested, and DNA was extracted at the same time points post-inoculation, as mentioned above. Additionally, for enumerating the number of T antigen expressing cells, HBMVE cells were grown on attachment factor-coated coverslips, infected with 50 HAU of JCV and on day 10 post-inoculation, fixed and permeabilized as described above. The coverslips were stained with anti-vWF antibody (see above), and mouse monoclonal anti-SV40 T antigen (1:400) (Cat #DP02, EMD Biosciences, San Diego, CA), which cross reacts with JCV T antigen protein, for 2 hr at room temperature, washed and incubated with biotinylated anti-mouse IgG from sheep (Cat# RPN 1001, Amersham, 1:250) and FITC-conjugated donkey anti-rabbit (Cat #RPN1034, Amersham, 1:250) for 40 min, washed and further incubated for 40 min with streptavidin-Texas red (1:250) (Cat #RPN1233, Amersham, Buckinghamshire, England). Moreover, JCV-infected HBMVE cells were simultaneously incubated with mouse monoclonal antibody against SV-40 and rabbit polyclonal anti-VP-1 antibody (1:1500) (Chang et al., 1996) for 2 hr, washed and incubated with secondary antibodies as above. Finally the cells were counterstained with bisbenzidine for visualizing cell nuclei, and fluorescent cells were visualized with epifluorescence or confocal microscopy as described above.
DNA and RNA extraction
Uninfected and JCV-infected HBMVE cells in 35-mm plates were washed twice with 1X PBS and were either frozen for DNA extraction or lysed with 350 μL of RLT plus buffer (Cat #74134, Qiagen) for RNA extraction and stored at -80°C. 100 μL of DNA was extracted with the Qiagen QIAprep® Spin Miniprep Kit (Cat #27104) from each 35-mm plate, according to the manufacturer’s protocol. Cells lysed with RLT plus buffer were thawed, homogenized by passing through a QIAshredder spin column (Cat #79654) and 50 μL RNA was extracted with RNeasy® Plus kit (Cat #74134, Qiagen). Genomic DNA was eliminated by employing gDNA eliminator column, and by treating the RNA in the spin column with DNase (Cat #79254, Qiagen) for 30 min.
Quantitation of viral DNA and mRNA
JCV DNA or cDNA synthesized from 0.5 μg total RNA with Bio-Rad’s iScript® cDNA synthesis kit (Bio-Rad Inc., Hercules, CA) were amplified and quantitated in the Bio-Rad iCycler iQ™ Multicolor Real-Time PCR Detection System using 2 μL of template DNA/cDNA, Bio-Rad 2X iQ™ SYBER® Green supermix and 12.5 pmol each of forward and reverse primers specific for JCV TAg (Chapagain et al., 2006; Ryschkewitsch et al., 2004) and VP-1 genes (table 1), in a final reaction volume of 20 μL. Thermal cycling conditions are outlined in table 1 (Chapagain et al., 2006; Ryschkewitsch et al., 2004). Real-time PCR amplification data was analyzed using the Bio-Rad iCycler iQ™ Multicolor Real-Time PCR Optical System Software Version 3.1. A standard curve for the quantitation of JCV was constructed using serial dilutions of the linearized JCV(Mad1) plasmid. The dynamic range of detection was determined by preparing 10-fold serial dilutions of JCV plasmid DNA (pDNA) in the range of 1.0×106 to 1.0×101 copies of JCV pDNA in each PCR template. All experiments were repeated at least twice and each sample was processed in duplicate. Copies of JCV genome or cDNA (mRNA transcript) in experimental samples were calculated from the standard curve and were expressed as copies of viral genome or mRNA transcript in one μg of DNA or RNA respectively. The reliability of qPCR and qRT-PCR was defined by calculating coefficients of variation (Ct) values of replicates of standard curve dilutions (Chapagain et al., 2006).
Table 1.
Primer Sequences and Cycling Conditions
| Target Gene
(GenBank Acc. No) |
Nucleotide
Position |
Primer Sequence (5′-3′) | Amplicon
Size |
PCR Cycling Conditions |
|---|---|---|---|---|
| JCV (J02226) | ||||
|
|
|
|
|
|
| JCV TAg (F)a | 4298-4320 | AGA GTG TTG GGA TCC TGT GTT TT | 95°C 4 min; 95°C 10s, 60°C 30s, 40 cycles; | |
| JCV TAg (R) a | 4375-4352 | GAG AAG TGG GAT GAA GAC CTG TTT | 78-bp | C55°C to 95°C 10 s, 80 cycles |
|
|
|
|
|
|
| JCV VP-1 (F) a | 1853-1875 | CAT GAC AAT GGT GCA GGG AAG CC | 95°C 4 min; 95°C 10s, 58°C 30s, 40 cycles; | |
| JCV VP-1 (R) a | 2032-2012 | CGC CTT GTG CTC TGT GTT CAT | 180-bp | C55°C to 95°C 10 s, 80 cycles |
|
| ||||
| 5HT2AR (NM000621) | ||||
|
|
|
|
|
|
| 5HT2AR-1 (F)b | 951-970 | GGC ACA CGG GCC AAA TTA GC | 95°C 4 min; 90°C 20s, 58°C 20s, 72°C 30s; | |
| 5HT2AR-1 (R)b | 1091-1070 | TTG CTC ATT GCT GAT GGA CTG C | 141-bp | 35 cycles; 72°C 10 min |
|
|
|
|
|
|
| 5HT2AR-2 (F)b | 793-817 | AGG TCT TTA AGG GGA GTT GCT T | 95°C 90s; 95°C 30s, 68°C 60s, 45 cycles; | |
| 5HT2AR-2 (R)b | 1093-1071 | TTT TGC TCA TTG CTG ATG GAC TG | 301-bp | 68°C 10 min |
|
| ||||
| GAPDH (BC025925) | ||||
|
|
|
|
|
|
| GAPDH (F) a,b | 5-26 | AGT TAG CCG CAT CTT CTT TTG C | 95°C 4 min; 90°C 20s, 58°C 20s, 72°C 30s, | |
| GAPDH (R) a,b | 100-78 | CAA TAC GAC CAA ATC CGT TGA CT | 96-bp | 35 cycles; 72°C 10 min |
Real Time PCR primers for quantitating JCV and GAPDH
RT-PCR primers for 5HT2AR and GAPDH
Melt curve
Immunoprecipitation and Western immunoblot for detection of JCV T antigen
HBMVE cells infected with 200 HAU of JCV(Mad1) in each 100-mm plate were washed twice with cold PBS and lysed with 500 μL NP40 lysis buffer, consisting of 50 mM Tris pH 7.5, 120 mM NaCl, 0.5% NP-40, and 1% protease inhibitor cocktail (Cat #P8340, Sigma-Aldrich) (Tyagarajan and Frisque, 2006). Total cellular protein was separated by centrifuging the lysate for 30 min at 10,000×g at 4°C. Protein concentration was determined by the Bradford technique (Bio-Rad), and T antigen from 250 μg of total protein extracts was immunoprecipitated using 60 μL of Protein G Plus/Protein A Agarose suspension (Cat #IP05) and 10 μL (2 μg) of anti-SV40 T antigen mouse mAb (Cat #DP02, EMD Biosciences, San Diego, CA), which recognizes JCV T antigen protein, and incubated overnight at 4°C. The suspension was then spun down, washed three times in resuspension buffer, dissolved in sample buffer (NuPAGE 10% reducing buffer and 4X loading dye, Cat #NP0007, Invitrogen) and was heated at 95°C for 10 min. In order to separate out the beads, the suspension was centrifuged at 10,000×g for 5 min and 20 μL of supernatant was fractionated on 4-12% NuPAGE Bis-Tris gradient gels (Cat #NP0322, Invitrogen). Proteins were transferred by electro-blotting onto nitrocellulose filters (Cat #162-0112, Bio-Rad) and blocked for 2 hr in 10% skim milk in 1X PBS with 0.1% Tween (PBST). Filters were incubated overnight at 4°C with anti-SV40 T antigen mouse primary antibody (1:1,000 dilution), washed three times with PBST for 30 min and incubated with goat alkaline phosphatase (AP)-conjugated anti-mouse IgG secondary antibody (1:3,000 dilution) for 2 hr at room temperature. After three vigorous washes with PBST, the membranes were developed using a color development kit (Cat #170-6532, Bio-Rad).
Effect of serotonin blockers on JCV infection of HBMVE cells
HBMVE cells in 35-mm plates in duplicate, were first incubated for 1 hr either with medium alone or medium containing chlorpromazine (1.0 μM), 5-hydroxytryptamine sulfate (1.0 μM), kentaserine (0.1 μM) or ritanserine (0.1 μM) (Sigma-Aldrich) (Elphick et al., 2004), and inoculated with JCV in the continuous presence of each drug. JCV TAg mRNA transcript copies detected in drug-treated JCV-infected HBMVE cells, were expressed as percentage of JCV-infected drug naïve HBMVE cells in 35-mm plates.
Detection of 5HT2AR expression by RT-PCR
RNA was extracted from HBMVE, HBCA and PHFG cells and one μg of RNA was employed for cDNA synthesis. 5HT2AR gene was amplified using two independent primer pairs 5HT2AR-1 and 5HT2AR-2 (table 1) in a GeneAmp Thermal Cycler 9700 (Perkin-Elmer, Wellesley, MA) with 4 μL of cDNA in a 20-μL reaction mixture containing 0.5 U AmpliTaq Gold (Applied Biosystems, Foster City, CA), PCR buffer with 1.5 mM MgCl2, 0.2 mM dNTPs and 0.4 μM of each primer. Thermal cycling conditions are outlined in table 1 (Verheggen et al., 2004). Simultaneously, PCR was performed using primers specific for housekeeping gene GAPDH (table 1), which was used as an internal control. The amplicons were electrophoresed on a 2% agarose gel, and the ethidium bromide fluorescence was visualized after scanning with a Bio-Rad Molecular Phosphorimager. Additionally, amplicons were gel purified using Qiagen Gel Purification Kit (Qiagen) according to the manufacturer’s instructions and directly sequenced in both directions using PCR primers on an automated sequencer (Model 373A, Applied Biosystems, Foster City, CA). Based on the NCBI Genomic BLAST program, the 254-bp amplicon sequence was identical to the 5HT2AR sequence deposited in the GenBank (accession number NM000621) (data not shown).
Detection of 5HT2AR expression by Western immunoblot
Total cellular protein extract (40 μg), fractioned on a 4-12% gradient SDS polyacrylamide gel, was transferred onto 0.2-μm nitrocellulose filters (Bio-Rad) and non-specific binding sites were blocked with 5% skim milk in 1X PBS-Tween (0.05%). Membranes were further incubated with anti-5HT2AR mouse polyclonal antibody (1 µg/mL) (Cat #556236, BD Pharmingen™, San Diego, CA) or 1:5,000 diluted mouse anti-β-actin monoclonal antibody (Cat #A5316, Sigma-Aldrich) overnight at 4°C, followed by incubation with AP-conjugated goat anti-mouse IgG antibody (Cat #170-6520, Bio-Rad) for 2 hr at room temperature and developed with AP-conjugate substrate kit (Bio-Rad).
Acknowledgments
This work was supported by U.S. Public Health Service grants from the Collaborative Neurological Sciences Program (S11 NS041833) and the Specialized Neuroscience Research Program (U54 NS039406, U54 NS041833), National Institute of Neurological Disorders and Stroke, as well as from the Research Centers in Minority Institutions Program (G12 RR003061) and Centers of Biomedical Research Excellence (P20 RR018727), National Center for Research Resources, National Institutes of Health.
We thank Dr. Deching Chang and Dr. Richard Frisque for the generous gift of VP-1 antibody and JCV(Mad1) respectively. We also thank Dr. Frisque for critical comments on a previous version of this manuscript, Dr. Satoru Arai and the Greenwood Molecular Biology Facility, UHM for sequencing. Ms. Juliene Co, Ms. Ushii Gurjav, Ms. Laarni Sumibicay, Ms. Yeung Lo, and the staff and students of the Retrovirology Research Laboratory provided technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andres O, Obojes K, Kim KS, ter Meulen V, Schneider-Schaulies J. CD46- and CD150-independent endothelial cell infection with wild-type measles viruses. J Gen Virol. 2003;84(Pt 5):1189–97. doi: 10.1099/vir.0.18877-0. [DOI] [PubMed] [Google Scholar]
- Argyris EG, Acheampong E, Nunnari G, Mukhtar M, Williams KJ, Pomerantz RJ. Human immunodeficiency virus type 1 enters primary human brain microvascular endothelial cells by a mechanism involving cell surface proteoglycans independent of lipid rafts. J Virol. 2003;77(22):12140–51. doi: 10.1128/JVI.77.22.12140-12151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum S, Ashok A, Gee G, Dimitrova S, Querbes W, Jordan J, Atwood WJ. Early events in the life cycle of JC virus as potential therapeutic targets for the treatment of progressive multifocal leukoencephalopathy. J Neurovirol. 2003;9(Suppl 1):32–7. doi: 10.1080/13550280390195342. [DOI] [PubMed] [Google Scholar]
- Chang D, Liou ZM, Ou WC, Wang KZ, Wang M, Fung CY, Tsai RT. Production of the antigen and the antibody of the JC virus major capsid protein VP1. J Virol Methods. 1996;59(12):177–87. doi: 10.1016/0166-0934(96)02039-3. [DOI] [PubMed] [Google Scholar]
- Chapagain ML, Nguyen T, Bui T, Verma S, Nerurkar VR. Comparison of real-time PCR and hemagglutination assay for quantitation of human polyomavirus JC. Virol J. 2006;3:3. doi: 10.1186/1743-422X-3-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Z, Bouchelet I, Olivier A, Villemure JG, Ball R, Stanimirovic DB, Hamel E. Multiple microvascular and astroglial 5-hydroxytryptamine receptor subtypes in human brain: molecular and pharmacologic characterization. J Cereb Blood Flow Metab. 1999;19(8):908–17. doi: 10.1097/00004647-199908000-00010. [DOI] [PubMed] [Google Scholar]
- Dorries K, Johnson RT, ter Meulen V. Detection of polyoma virus DNA in PML-brain tissue by (in situ) hybridization. J Gen Virol. 1979;42(1):49–57. doi: 10.1099/0022-1317-42-1-49. [DOI] [PubMed] [Google Scholar]
- Dorries K, Vogel E, Gunther S, Czub S. Infection of human polyomaviruses JC and BK in peripheral blood leukocytes from immunocompetent individuals. Virology. 1994;198(1):59–70. doi: 10.1006/viro.1994.1008. [DOI] [PubMed] [Google Scholar]
- Dubois V, Moret H, Lafon ME, Brodard V, Icart J, Ruffault A, Guist’hau O, Buffet-Janvresse C, Abbed K, Dussaix E, Ingrand D. JC virus genotypes in France: molecular epidemiology and potential significance for progressive multifocal leukoencephalopathy. J Infect Dis. 2001;183(2):213–217. doi: 10.1086/317927. [DOI] [PubMed] [Google Scholar]
- Elphick GF, Querbes W, Jordan JA, Gee GV, Eash S, Manley K, Dugan A, Stanifer M, Bhatnagar A, Kroeze WK, Roth BL, Atwood WJ. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306(5700):1380–3. doi: 10.1126/science.1103492. [DOI] [PubMed] [Google Scholar]
- Elsner C, Dorries K. Evidence of human polyomavirus BK and JC infection in normal brain tissue. Virology. 1992;191(1):72–80. doi: 10.1016/0042-6822(92)90167-n. [DOI] [PubMed] [Google Scholar]
- Fareed GC, Takemoto KK, Gimbrone MA., Jr . Interaction of simian virus 40 and human papovaviruses, BK and JC, with human vascular endothelial cells. In: Schlessinger D, editor. Microbiology-1978. American Society of Microbiology; Washington, DC: 1978. pp. 427–431. [Google Scholar]
- Fernandez-Cobo M, Jobes DV, Yanagihara R, Nerurkar VR, Yamamura Y, Ryschkewitsch CF, Stoner GL. Reconstructing population history using JC virus: Amerinds, Spanish, and Africans in the ancestry of modern Puerto Ricans. Hum Biol. 2001;73(3):385–402. doi: 10.1353/hub.2001.0032. [DOI] [PubMed] [Google Scholar]
- Ferrante P, Caldarelli-Stefano R, Omodeo-Zorini E, Vago L, Boldorini R, Costanzi G. PCR detection of JC virus DNA in brain tissue from patients with and without progressive multifocal leukoencephalopathy. J Med Virol. 1995;47(3):219–25. doi: 10.1002/jmv.1890470306. [DOI] [PubMed] [Google Scholar]
- Fish KN, Soderberg-Naucler C, Mills LK, Stenglein S, Nelson JA. Human cytomegalovirus persistently infects aortic endothelial cells. J Virol. 1998;72(7):5661–8. doi: 10.1128/jvi.72.7.5661-5668.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgsson G. Neuropathologic aspects of lentiviral infections. Ann N Y Acad Sci. 1994;724:50–67. doi: 10.1111/j.1749-6632.1994.tb38895.x. [DOI] [PubMed] [Google Scholar]
- Houff SA, Major EO, Katz DA, Kufta CV, Sever JL, Pittaluga S, Roberts JR, Gitt J, Saini N, Lux W. Involvement of JC virus-infected mononuclear cells from the bone marrow and spleen in the pathogenesis of progressive multifocal leukoencephalopathy. N Engl J Med. 1988;318(5):301–5. doi: 10.1056/NEJM198802043180507. [DOI] [PubMed] [Google Scholar]
- Imperiale MJ. The human polyomaviruses: An overview. In: Khalili K, Stoner GL, editors. Human Polyomaviruses Molecular and Clinical Perspectives. Wiley-Liss, Inc; New York: 2001. pp. 53–71. [Google Scholar]
- Kim H-S, Henson JW, Frisque RJ. Transcription and replication in the human polyomaviruses. In: Khalili K, Stoner GL, editors. Human Polyomaviruses. Wiley-Liss, Inc; New York: 2001. pp. 73–126. [Google Scholar]
- Knowles WA, Pipkin P, Andrews N, Vyse A, Minor P, Brown DW, Miller E. Population-based study of antibody to the human polyomaviruses BKV and JCV and the simian polyomavirus SV40. J Med Virol. 2003;71(1):115–23. doi: 10.1002/jmv.10450. [DOI] [PubMed] [Google Scholar]
- Liu CK, Wei G, Atwood WJ. Infection of glial cells by the human polyomavirus JC is mediated by an N-linked glycoprotein containing terminal alpha(2-6)-linked sialic acids. J Virol. 1998;72(6):4643–9. doi: 10.1128/jvi.72.6.4643-4649.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B, Roberts J, Pushkarsky T, Bukrinsky M, Witte M, Weinand M, Fiala M. Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway. J Virol. 2002;76(13):6689–700. doi: 10.1128/JVI.76.13.6689-6700.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankowski JL, Spelman JP, Ressetar HG, Strandberg JD, Laterra J, Carter DL, Clements JE, Zink MC. Neurovirulent simian immunodeficiency virus replicates productively in endothelial cells of the central nervous system in vivo and in vitro. J Virol. 1994;68(12):8202–8. doi: 10.1128/jvi.68.12.8202-8208.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazlo M, Ressetar HG, Stoner GL. The neuropathology and pathogenesis of progressive multifocal leukoencephalopathy. In: Khalili K, Stoner GL, editors. Human Polyomaviruses, Molecular and Clinical Perspectives. Wiley-Liss, Inc; New York: 2001. pp. 257–335. [Google Scholar]
- Monaco MC, Atwood WJ, Gravell M, Tornatore CS, Major EO. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: implications for viral latency. J Virol. 1996;70(10):7004–12. doi: 10.1128/jvi.70.10.7004-7012.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco MC, Jensen PN, Hou J, Durham LC, Major EO. Detection of JC virus DNA in human tonsil tissue: evidence for site of initial viral infection. J Virol. 1998;72(12):9918–23. doi: 10.1128/jvi.72.12.9918-9923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses AV, Bloom FE, Pauza CD, Nelson JA. Human immunodeficiency virus infection of human brain capillary endothelial cells occurs via a CD4/galactosylceramide-independent mechanism. Proc Natl Acad Sci U S A. 1993;90(22):10474–8. doi: 10.1073/pnas.90.22.10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtar M, Harley S, Chen P, BouHamdan M, Patel C, Acheampong E, Pomerantz RJ. Primary isolated human brain microvascular endothelial cells express diverse HIV/SIV-associated chemokine coreceptors and DC-SIGN and L-SIGN. Virology. 2002;297(1):78–88. doi: 10.1006/viro.2002.1376. [DOI] [PubMed] [Google Scholar]
- Newman JT, Frisque RJ. Identification of JC virus variants in multiple tissues of pediatric and adult PML patients. J Med Virol. 1999;58(1):79–86. [PubMed] [Google Scholar]
- Padgett BL, Rogers CM, Walker DL. JC virus, a human polyomavirus associated with progressive multifocal leukoencephalopathy: additional biological characteristics and antigenic relationships. Infect Immun. 1977;15(2):656–62. doi: 10.1128/iai.15.2.656-662.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson-Burns SM, Kleinschmidt-DeMasters BK, DeBiasi RL, Tyler KL. Progressive multifocal leukoencephalopathy and apoptosis of infected oligodendrocytes in the central nervous system of patients with and without AIDS. Arch Neurol. 2002;59(12):1930–6. doi: 10.1001/archneur.59.12.1930. [DOI] [PubMed] [Google Scholar]
- Ryschkewitsch C, Jensen P, Hou J, Fahle G, Fischer S, Major EO. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. J Virol Methods. 2004;121(2):217–21. doi: 10.1016/j.jviromet.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Seth P, Diaz F, Tao-Cheng JH, Major EO. JC virus induces nonapoptotic cell death of human central nervous system progenitor cell-derived astrocytes. J Virol. 2004;78(9):4884–91. doi: 10.1128/JVI.78.9.4884-4891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornatore C, Berger JR, Houff SA, Curfman B, Meyers K, Winfield D, Major EO. Detection of JC virus DNA in peripheral lymphocytes from patients with and without progressive multifocal leukoencephalopathy. Ann Neurol. 1992;31(4):454–62. doi: 10.1002/ana.410310426. [DOI] [PubMed] [Google Scholar]
- Tyagarajan SK, Frisque RJ. Stability and function of JC virus large T antigen and T’ proteins are altered by mutation of their phosphorylated threonine 125 residues. J Virol. 2006;80(5):2083–91. doi: 10.1128/JVI.80.5.2083-2091.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullmer C, Schmuck K, Kalkman HO, Lubbert H. Expression of serotonin receptor mRNAs in blood vessels. FEBS Lett. 1995;370(3):215–21. doi: 10.1016/0014-5793(95)00828-w. [DOI] [PubMed] [Google Scholar]
- Vago L, Cinque P, Sala E, Nebuloni M, Caldarelli R, Racca S, Ferrante P, Trabottoni G, Costanzi G. JCV-DNA and BKV-DNA in the CNS tissue and CSF of AIDS patients and normal subjects. Study of 41 cases and review of the literature. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;12(2):139–46. doi: 10.1097/00042560-199606010-00006. [DOI] [PubMed] [Google Scholar]
- Verheggen R, Meier A, Werner I, Wienekamp A, Kruschat T, Brattelid T, Levy FO, Kaumann A. Functional 5-HT receptors in human occipital artery. Naunyn Schmiedebergs Arch Pharmacol. 2004;369(4):391–401. doi: 10.1007/s00210-004-0878-9. [DOI] [PubMed] [Google Scholar]
- von Einsiedel RW, Samorei IW, Pawlita M, Zwissler B, Deubel M, Vinters HV. New JC virus infection patterns by in situ polymerase chain reaction in brains of acquired immunodeficiency syndrome patients with progressive multifocal leukoencephalopathy. J Neurovirol. 2004;10(1):1–11. doi: 10.1080/13550280490269691. [DOI] [PubMed] [Google Scholar]
- White FA, III, Ishaq M, Stoner GL, Frisque RJ. JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol. 1992;66(10):5726–34. doi: 10.1128/jvi.66.10.5726-5734.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]