

Abstract
Recent proteomic data indicate that a majority of the phosphorylated proteins in a eucaryotic cell contain multiple sites of phosphorylation. In many signaling events, a single kinase phosphorylates multiple sites on a target protein. Processive phosphorylation occurs when a protein kinase binds once to a substrate and phosphorylates all of the available sites before dissociating. In this review, we discuss examples of processive phosphorylation by serine/threonine kinases and tyrosine kinases. We describe current experimental approaches for distinguishing processive from non-processive phosphorylation. Finally, we contrast the biological situations that are suited to regulation by processive and non-processive phosphorylation.
1. Introduction
1.1 Multisite protein modification
The inventory of proteins in a eucaryotic cell is increased significantly by covalent modifications to amino acid sidechains or to the polypeptide backbone [1]. These posttranslational modifications represent the most common mechanism by which the functions of proteins can be altered. In many cases, multiple modifications (of the same chemical type or of different types) can occur on a single polypeptide, giving rise to a complex regulatory network that controls protein function. The multisite modification of such proteins has been referred to as a “molecular barcode.” According to this hypothesis, each combination of modifications on a protein conveys a unique meaning that can be interpreted by the cell [2, 3].
The barcode hypothesis was first advanced in the context of histone modification. The post-translational modification of histone proteins plays an important role in regulating heterochromatin-mediated silencing, chromosome segregation, and gene expression [3-8]. The N-terminal tails of the four core histone proteins (H2A, H2B, H3, and H4) have multiple sites that undergo lysine acetylation, lysine methylation, serine phosphorylation, and other types of covalent modification. Separate enzymes exist to catalyze the addition and removal of these modifications; thus, the modification pattern is dynamic and responsive to the status of the cell. The patterns of histone modifications are believed to guide the interactions between histones, chromatin, and non-histone regulatory proteins. Histone acetylation and methylation have recently been studied on a genomic scale [3, 9].
The p53 tumor suppressor also undergoes a complex set of modifications. In response to various genotoxic stresses such as DNA damage, nucleotide depletion, and hypoxia, p53 accumulates in the nucleus and undergoes activation, leading to growth arrest or apoptosis. The p53 protein is covalently modified on multiple sites at its N-terminus and C-terminus. The predominant modification at the N-terminus is phosphorylation, whereas the C-terminal modifications include phosphorylation, acetylation and sumoylation [10]. A variety of protein kinases have been shown to target p53, including DNA-activated protein kinase, members of the casein kinase family, mitogen-activated protein kinases (MAPKs), stress activated protein kinases, and protein kinase C [11, 12]. Cell signaling pathways that converge on p53 result in dynamic changes to the pattern of modification.
1.2 Multisite phosphorylation
Approximately one-third of all eucaryotic proteins are modified by phosphorylation on serine, threonine, or tyrosine during their lifetime in the cell. These phosphorylation events control a multitude of cellular functions [1, 13-15]. It is increasingly clear that the activities of many proteins are regulated by phosphorylation at more than one site. In a recent study using a mass spectrometric approach to identify and quantitate phosphorylation sites as a function of stimulus, time, and subcellular location, it was shown that a majority of proteins detected in response to stimulation with epidermal growth factor (EGF) were phosphorylated on multiple sites [16]. Of the proteins containing a phosphorylation site that responded to EGF, most (77%) contained an additional site that responded differently; thus, protein phosphorylation often plays distinct roles at different sites on a protein.
The stoichiometry of multisite phosphorylation can be remarkably high. The C-terminal repeat domain (CTD) of RNA polymerase II undergoes extensive phosphorylation and thereby serves as a binding scaffold for a variety of nuclear factors [17-19]. The CTD comprises approximately 25 to 52 tandem copies of the consensus repeat heptad YSPTSPS [20]. The CTD contains as many as 50 phosphorylated residues in vivo [18]. The predominant phosphorylation near the beginning of genes occurs on Ser5, whereas phosphorylation of Ser2 predominates near the end of genes [21]. This phosphorylation pattern is crucial in order to determine which factors associate with RNA Pol II.
In some cases of multisite phosphorylation, the sheer number of sites appears to be more important than the identities of the individual phosphorylated residues. Multisite phosphorylation of the yeast cyclin dependent kinase inhibitor Sic1 sets a threshold for the onset of DNA replication. Out of the nine possible phosphorylation sites on Sic1, six or more must be phosphorylated to promote binding to the ubiquitin ligase SCFCdc4, which then targets Sic1 for degradation [22, 23]. Strikingly, phosphorylation of any combination of six sites (but not five or fewer) appears to be sufficient for SCFCdc4 binding. In another example, multisite phosphorylation of Ste5, a MAPK scaffold protein, is essential for inhibition of the yeast mating pheromone pathway. These multiple phosphorylations (on eight Ser/Thr sites) inhibit membrane binding (and pheromone signaling) by repelling the negatively charged lipids of the inner leaflet of the plasma membrane. Thus, multisite phosphorylation of Ste5 provides a switch-like deactivation that is driven by bulk electrostatics [24].
1.3 Multisite phosphorylation by two or more kinases
There are numerous cases in which phosphorylation of a protein at one site increases its activity, while phosphorylation at a second site (by a second kinase) decreases its activity. For example, the activity of the cell cycle regulator Cdc25 is governed by antagonistic phosphorylation events. Cdc25 is a protein phosphatase that removes the inhibitory phosphates on Cdc2, thereby triggering entry into mitosis [25]. Phosphorylation of Ser216 on Cdc25 is catalyzed by the checkpoint control kinases CHK1 and CHK2 in response to DNA damage. Phosphorylation at Ser216 promotes binding to 14-3-3 proteins and cell cycle arrest at the G2/M checkpoint [26, 27]. In contrast, in the transition to mitosis, Cdc25 is phosphorylated at several alternate sites by the Cdc2/cyclin B complex (Thr48, Thr67, Ser122, Thr130, and Ser214) [28, 29]. These phosphorylations activate Cdc25, contributing to a feedback activation loop that facilitates the rapid onset of mitosis [25].
The activities of G-protein coupled receptors (GPCRs) are regulated by multiple Ser/Thr protein kinases. Agonist-induced desensitization of GPCRs involves phosphorylation of the intracellular portions of the receptors by G-protein coupled receptor kinases (GRKs), cAMP-dependent protein kinase (PKA), or protein kinase C (PKC) [30]. The GRK family consists of seven members, designated GRK1-7 [30]. GRK phosphorylation of GPCRs promotes binding of arrestin proteins, which block signaling to G-proteins and target GPCRs to clathrin-coated pits for internalization. Prevention of phosphorylation by GPCR truncation or mutation of the C-terminal serine/threonine rich segment, or by the use of kinase inhibitors (e.g., the PKA inhibitor H-89) can delay the onset of agonist-induced desensitization [30]. Recent studies have uncovered differential effects of GPCR phosphorylation at different sites. While GRK phosphorylation of β1-adrenergic receptor targets the protein to clathrin-coated pits, PKA phosphorylation triggers internalization through a pathway involving caveolae [31]. The β2-adrenergic receptor is phosphorylated by PKA at Ser262 in the third intracellular loop and by GRK at Ser355 and Ser256 in the C-terminus. The kinetics of PKA and GRK phosphorylation are different in this case, and the events are differentially affected by interfering with endocytosis [32]. PKA phosphorylation of β1- or β2-adrenergic receptors can also redirect the G protein coupling specificity, so that the receptors have reduced affinity for Gs and increased affinity for Gi. For β2-adrenergic receptor, this switch in coupling from Gs to Gi correlates with a shift from pro-apoptotic signaling to anti-apoptotic signaling [33-35].
As an additional layer of complexity in GPCR phosphorylation, the various GRK isoforms appear to have distinct specificities, leading to different physiological outcomes. This has been demonstrated for the angiotensin II type 1A receptor, where arrestin proteins have a role in transducing signals independent of G proteins. RNA interference experiments showed that the signaling consequences of arrestin-receptor binding depended on which GRK isoform was responsible for phosphorylating the receptor [36]. Thus, GRK2 and GRK3 were primarily responsible for receptor endocytosis, while GRK5 and GRK6 were required for arrestin-mediated ERK activation. GPCRs contain many potential phosphorylation sites for these GRK isoforms, leading the authors to suggest that “the location of these phosphorylation sites may constitute a ‘bar code’ that instructs the bound β-arrestin as to its intended function.”
In some cases of multisite phosphorylation, the action of one protein kinase is required to “prime” phosphorylation at a nearby site by a second kinase. Glycogen synthase kinase (GSK3), an enzyme important in the insulin and Wnt signaling pathways, is a well-studied example of an enzyme that requires priming [14, 37]. GSK3 phosphorylates Ser/Thr residues in sequences that contain a phospho-Ser residue at the P+4 position (i.e., 4 residues to the C-terminal side). Thus, after Ser656 of glycogen synthase has been phosphorylated by casein kinase II, GSK3β sequentially phosphorylates Ser652, Ser648, Ser644, and Ser640; each phosphorylation depends on prior phosphorylation at the P+4 position [38, 39]. The crystal structure of GSK3 β provides an explanation for this unusual substrate specificity. Three basic residues in the substrate-binding cleft are positioned to interact with the negatively charged phosphoserine at the P+4 position [40, 41]. Casein kinase I (CK1) is another protein kinase that requires pre-phosphorylation of its substrate. CK1 phosphorylates serine residues that contain another phosphoserine at the P-3 position [42]. Several reviews have focused on phosphorylations by multiple kinases that are additive, hierarchical, or mutually exclusive [13-15].
1.4 Multisite phosphorylation by a single protein kinase
We will focus the remainder of this review on situations in which a protein is modified at multiple sites by a single protein kinase. Multisite phosphorylation by a kinase can be carried out by two basic mechanisms: (i) a processive mechanism, in which the kinase binds substrate and catalyzes all the phosphorylation events before dissociating from the fully phosphorylated product; or (ii) a non-processive (distributive) mechanism, in which each phosphorylation requires a separate binding event between the kinase and substrate (Figure 1). There are examples in the literature of both of these mechanisms of multisite phosphorylation. The dual phosphorylation of MAPKs by MAPK kinases occurs in a two step, distributive manner [43, 44]. Distributive phosphorylation of MAPKs is important for establishing, for example, the switch-like response observed in Xenopus oocytes in response to progesterone [45]. In the case of processive phosphorylation, Mayer et al. first showed that Src homology 2 (SH2) domains promote hyperphosphorylation of substrates by tyrosine kinases [46], and subsequent model experiments confirmed that Src family kinases can phosphorylate substrates containing an SH2 domain ligand in a processive manner [47]. In this review, we discuss specific examples of proteins that undergo a processive mechanism of multisite phosphorylation.
Figure 1. Mechanisms of multisite phosphorylation.
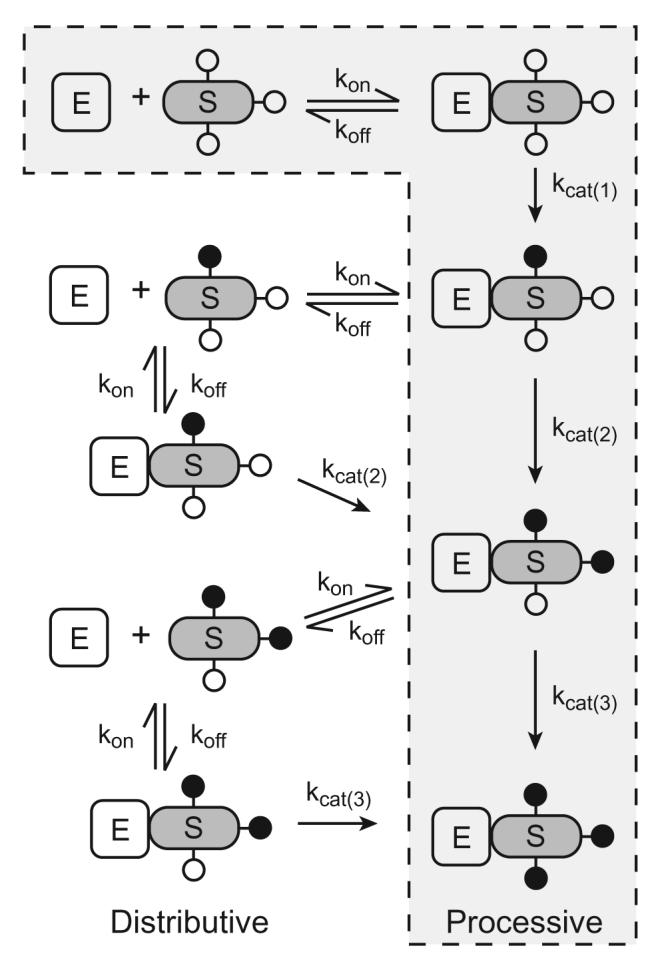
Multisite phosphorylation of a substrate (S) by a protein kinase (E) can be carried out by either a processive mechanism (outlined by a dotted line) or a distributive mechanism. For simplicity, multisite phosphorylation of a protein containing three phosphorylation sites is shown. Unphosphorylated sites are represented by open circles, and phosphorylated sites are represented by filled circles.
2. SR proteins
2.1 SR protein function and regulation
SR (serine/arginine rich) proteins belong to a family of nuclear splicing regulators that play crucial roles in both constitutive and alternative splicing [48-51]. These factors are assembled onto pre-mRNA and are essential for spliceosome assembly [52]. SR proteins are characterized by the presence of at least one RNA recognition motif (RRM) at the N-terminus, and a C-terminal RS domain rich in arginine and serine dipeptide repeats that is important for protein-protein interactions (Figure 2). Individual domains of SR proteins are important for their cellular localization and alternative splicing in vivo [53]. Although there is a degree of redundancy in SR protein function, different SR proteins have unique functions and distinct specificities [54, 55].
Figure 2. Domain architecture of ASF/SF2.

Domain structure of the prototypical SR protein ASF/SF2 is shown. The two RNA recognition motifs are designated RRM1 and RRM2. A docking motif (D) for binding to SRPK1 is located between residues 184-197. The two serine-rich regions of the RS domain (RS1 and RS2) are indicated; individual phosphorylation sites are represented by black bars. SRPK1 phosphorylates serines in RS1, while Clk/Sty phosphorylates both RS1 and RS2.
Most SR proteins are localized completely in the nucleus, but some shuttle continuously between nucleus and cytoplasm [56]. The latter proteins are present in the cytoplasm only transiently and are mostly nuclear at steady state. Within the cell nucleus, splicing factors are typically present in 20-50 distinct domains called nuclear speckles [57]. Speckles are highly dynamic structures that respond to the activation of nearby genes [58]. Interphase SR proteins reside in nuclear speckles, and upon gene activation, serine phosphorylation of SR proteins causes them to relocalize to the sites of pre-mRNA synthesis where they participate in the splicing process [59, 60]. Both phosphorylation and dephosphorylation of SR proteins has been shown to be crucial for splicing. The hypophosphorylated forms of SR proteins such as 9G8 and ASF/SF2 (alternative splicing factor/splicing factor 2) exhibit higher affinity for the export receptor TAP, and consequently facilitate the export of mRNA out of the nucleus [61-65].
2.2 SR protein specific kinases and ASF/SF2
The C-terminal RS domain of SR proteins can be phosphorylated at multiple positions by SR protein specific kinases (SRPKs). SRPKs phosphorylate serine residues on SR proteins with a high degree of efficiency and specificity. A variety of SRPKs have been described, with SRPK1 and Clk/Sty being the most prominent [66-68]. Alternative splicing factor (ASF) is an SR protein that was initially purified as a specific factor in HEK293 cells that modulates alternate splicing [69]. Sequence analysis later revealed that ASF had also been isolated as a protein (designated SF2) that is required for 5' splice site cleavage and lariat formation, and that influences 5' splice site selection in HeLa cell extracts [70]. SRPK1 has a strong preference for phosphorylating Ser-Arg sites in ASF/SF2, while Clk/Sty can phosphorylate Ser-Arg, Ser-Lys, or Ser-Pro sites [67]. SRPK1 turns over ASF/SF2 much faster than Clk/Sty does. Tryptic phosphopeptide mapping showed that the sites on ASF/SF2 phosphorylated in vitro overlap with those phosphorylated in vivo, and overexpression of Clk/Sty causes a redistribution of SR proteins within the nucleus, resulting in the disassembly of nuclear speckles [66].
2.3 Processive phosphorylation of ASF/SF2
ASF/SF2 is a prototypical SR protein with two RNA recognition motifs at the N-terminus and an RS domain at the C-terminus, containing approximately 20 serine residues that are potential sites for phosphorylation (Figure 2). Recent reports have shown that ASF/SF2 is phosphorylated by SRPK1 and Clk/Sty kinases in a fully processive manner [71, 72]. SRPK1 binding to ASF/SF2 is dependent on the phosphorylation state of ASF/SF2 [73]. Serine phosphorylation within the Arg-Ser dipeptide produces a patch of alternating positively and negatively charged amino acids, which tether SRPK1 to ASF/SF2 [72]. The processive mechanism for ASF/SF2 was demonstrated using an elegant “start-trap” strategy [72] (Figure 3A). In this method, ATP and an SRPK1 inhibitor peptide were added simultaneously to the enzyme-substrate complex. If SRPK1 phosphorylated ASF/SF2 by a non-processive mechanism, the inhibitor would trap the free enzyme after dissociation from ASF/SF2, stopping the reaction after one round of phosphorylation. The results showed that the inhibitor peptide did not influence the rate or extent of ASF/SF2 phosphorylation, suggesting that SRPK1 remained locked on to ASF/SF2, allowing processive phosphorylation.
Figure 3. Experimental methods to determine the mechanism of multisite phosphorylation.
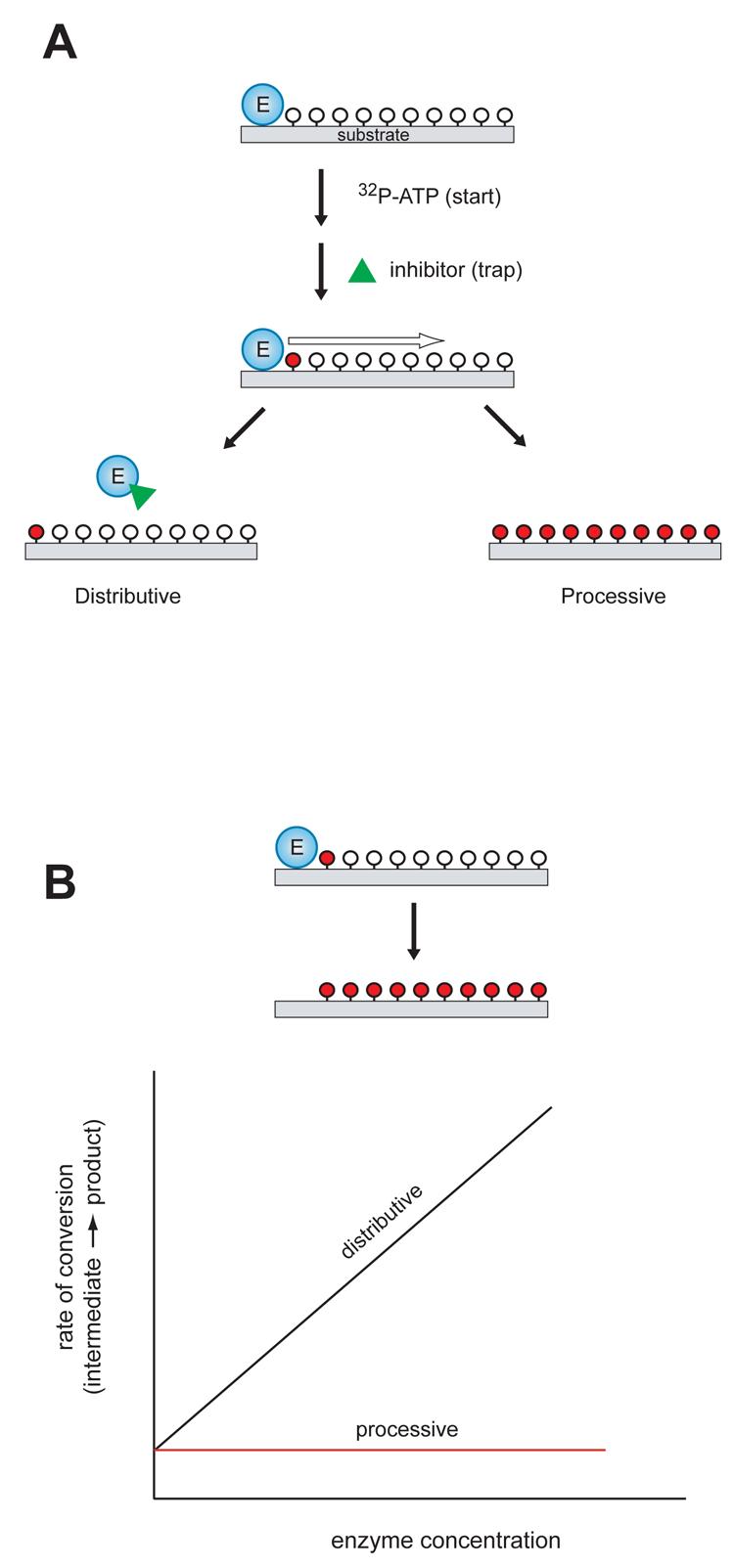
A, In the “start-trap” strategy, ATP is added to the enzyme-substrate complex together with an inhibitor that can trap the free enzyme (green triangle). If the enzyme phosphorylates the substrate by a non-processive mechanism, the inhibitor traps the free enzyme after dissociation from the substrate, stopping the reaction after one round of phosphorylation. In a processive mechanism, the inhibitor does not influence the rate or extent of substrate phosphorylation. Unphosphorylated sites are shown indicated by open circles; phosphorylated sites are indicated by red circles. B, Kinetic experiments with varying enzyme concentration. For processive phosphorylation, the enzyme and substrate remain bound during the multiple phosphorylation events, so the rate of progression from an intermediate phosphorylated form to fully phosphorylated product is independent of the enzyme concentration (red line). For a non-processive mechanism of phosphorylation, the rate of progression is dependent on enzyme concentration (black line). Unphosphorylated sites are shown indicated by open circles; phosphorylated sites are indicated by red circles.
Mass spectrometric analysis showed that SRPK1 phosphorylates approximately 10 sites in the N-terminal half of the RS domain (RS1), while Clk/Sty phosphorylates the RS1 sites as well as the remaining serines in the C-terminal portion of the RS domain (RS2) [71]. Start-trap experiments showed that both SRPK1 and Clk/Sty kinases carry out phosphorylation of the RS domain in a processive manner. SRPK1 phosphorylates only the N-terminal region of the RS domain before dissociating, while Clk/Sty remains attached and phosphorylates the entire RS domain. For SRPK1, this substrate specificity is governed by a docking motif N-terminal to the RS domains, which restricts the number of serines in ASF/SF2 that can be phosphorylated [74] (Figure 2). In the cell, SRPK1 is found in the cytoplasm, while Clk/Sty is localized in the nucleus [66, 75-77]. The current model is that SRPK1 phosphorylates ASF/SF2 in the cytoplasm, triggering nuclear entry and recruitment to speckles. Subsequent phosphorylation by Clk/Sty promotes release of ASF/SF2 from speckles, at which point the protein can participate in splicing [74].
3. p130Cas
3.1 Cas structure and function
p130Cas (Crk-associated substrate, 130kDa) was first identified as a protein that is highly tyrosine phosphorylated in v-Src and v-Crk transformed cells and that forms stable complexes with these oncoproteins [78-80]. Cas is a multidomain protein containing an N-terminal SH3 domain and Pro-rich sequences, a large central substrate domain with 15 repeats of the motif YXXP, and a C-terminal Src binding region [81] (Figure 4). Mouse embryos lacking Cas die in utero due to defects in cardiovascular development, impaired actin stress fiber organization, disorganization of myofibrils and growth retardation [82]. Cas deficient fibroblasts contain disorganized actin filaments, and ectopic expression of Cas restores the actin stress fiber organization [82, 83]. Fibroblasts derived from Cas deficient mice are resistant to transformation by oncogenic Src, suggesting that Cas plays an important role downstream of Src in cell transformation.
Figure 4. Domain structure of p130Cas.

Cas contains an SH3 domain (SH3), proline-rich sequences (Pro), a substrate region containing 15 YXXP repeats, a serine-rich region (Ser), and a C-terminal Src binding site. The Src binding site contains a Pro-rich sequence that binds to the SH3 domain of Src, and a phosphorylated Tyr (Y668) that binds to the SH2 domain of Src. The positions of the four YQXP motifs and nine YDXP motifs are indicated.
The YXXP motifs in the substrate domain of Cas can be phosphorylated by several protein tyrosine kinases [46, 81, 84]. The phosphorylated YXXP motifs serve as ligands for the SH2 or phospho-Tyr binding (PTB) domains that are present in many signaling proteins. Cas interacts with the adaptor protein CrkII through SH2-phosphotyrosine interactions to promote cell migration via activation of Rac1 [85, 86]. The unique domain structure of Cas indicates a role in assembling signals from multiple SH2 containing molecules. Besides playing a role in the organization of the actin cytoskeleton and in promoting cell migration, Cas is involved in cell survival and prevention of apoptosis. A survival pathway in which Cas plays a role is the activation of c-Jun NH2 terminal kinase (JNK) via the small GTPases Ras and Rac1 [87].
Cas also plays an important role in microbial pathogenesis. Cas is a substrate of the protein tyrosine phosphatase YopH, an antiphagocytic factor of Y.pseudotuberculosis [88]. YopH is believed to de-phosphorylate adhesion proteins such as Cas, focal adhesion kinase (FAK), and paxillin, thereby preventing cytoskeletal rearrangements and inhibiting signaling pathways which lead to bacterial uptake.
A number of studies have examined the importance of the YXXP motifs in the various biological roles of Cas. In cells transformed by oncogenic Src, the YXXP motifs are crucial for invasiveness and the formation of podosomal structures [89, 90]. The YXXP motifs in the substrate domain of Cas can be divided into YDXP and YQXP motifs (Figure 4). The YDXP motifs are required for the ability of Cas to promote organization of the actin cytoskeleton. Cas mutants lacking the entire substrate domain or the YDXP motifs are defective in cell migration [85, 86]. Cas mutants containing single or double tyrosine mutations have migration activity that is comparable to wild-type Cas, and mutants containing as few as 6 tyrosines are able to promote cell migration to some extent [91]. A single YDXP tyrosine residue in the substrate domain of Cas, Tyr253, plays a crucial role in cell migration; however, it was dispensable for Src mediated anchorage independent growth [92]. Cas mutants containing mutations at multiple (7 or 12) tyrosine residues in the substrate domain are able to support Src-mediated anchorage independent growth, although these mutants are defective in cell migration [93]. Collectively, these studies suggest that phosphorylation of the YXXP motifs triggers different downstream pathways for cell migration, cytoskeletal rearrangement, and Src-mediated transformation.
3.2 Mechanism of Cas phosphorylation by Src
Mass spectrometry studies of Cas [92] and tryptic phosphopeptide mapping experiments [91] confirmed that the major sites for Src phosphorylation lie within the central substrate region. The majority of the YXXP motifs were phosphorylated in these studies. Synthetic peptides based on the Cas YXXP sequences were phosphorylated by Src to varying degrees; a peptide containing the sequence surrounding Tyr253 was the most efficiently phosphorylated in terms of kcat/Km. This same site was found to be crucial for Src-mediated migration in fibroblasts [92].
Kinetic studies with purified Cas have indicated that Src phosphorylates the YXXP motifs processively [93, 94]. Purified Src and Cas were incubated together in the presence of radioactively labeled ATP and analyzed by SDS-PAGE and autoradiography. Phosphorylated Cas migrated as a single sharp band with a much lower electrophoretic mobility in these experiments; the absence of intermediate forms suggested that Cas was fully phosphorylated [94]. Similar studies were performed with mutant forms of Cas. In one mutant, the Src SH2-binding motif at the C-terminus (Tyr668) was mutated to phenylalanine and in another mutant, the proline-rich Src SH3 domain ligand was mutated. The polyproline mutant of Cas was defective for Src phosphorylation in vitro, and when analyzed by SDS-PAGE, gave rise to diffuse, partially phosphorylated forms characteristic of non-processive phosphorylation. The Y668F mutant showed phosphorylation that was comparable to wild-type Cas. These experiments suggested that SH3 interactions were particularly important for processive phosphorylation [94].
Experiments with varying concentrations of Src and Cas were carried out to provide a more rigorous demonstration of processive phosphorylation [93, 94]. Pulse-chase experiments were used to measure rates of Cas phosphorylation at different concentrations of enzyme. The processive and non-processive mechanisms make different predictions about the outcome of these experiments (Figure 3B). For a processive mechanism of phosphorylation, since the enzyme and substrate remain bound during the multiple phosphorylation events, the rate of progression from an intermediate phosphorylated form to fully phosphorylated product is independent of the enzyme concentration. On the other hand, for a non-processive mechanism of phosphorylation, each phosphorylation depends on a separate binding event between enzyme and substrate, and consequently the rate of progression is dependent on enzyme concentration. Cas phosphorylation was independent of Src concentration, consistent with a processive mechanism [93]. A similar conclusion was reached from experiments with varying substrate concentrations [93, 94]. In the case of a processive mechanism of phosphorylation, formation of fully phosphorylated product follows classical Michaelis-Menten kinetics. In contrast, for a non-processive mechanism of phosphorylation (such as the dual phosphorylation of MAPKs), increases in substrate concentration result in decreased accumulation of fully phosphorylated product, due to the ability of partially phosphorylated species to act as competitive inhibitors [43, 44]. Cas phosphorylation by Src showed a Michaelian dependence on substrate concentration, consistent with a processive mechanism.
To test whether particular YXXP motifs were critical for multisite processive phosphorylation of Cas, single and multisite mutants were studied by a similar strategy. Four single-tyrosine mutant forms of Cas (Y253F, Y310F, Y366F, and Y414F) were phosphorylated normally by Src. Multisite mutants of Cas were created which lacked 7 or 12 tyrosines from the YXXP motifs in the substrate region. Experiments with varying concentrations of Src and Cas showed that these multisite mutants were phosphorylated processively by Src [93]. The results suggest that processive phosphorylation of Cas does not follow a defined sequence, and that the individual sites are not kinetically distinguishable.
4. Other examples of processive phosphorylation
4.1 Nonreceptor tyrosine kinase substrates
In addition to the example of Cas described above, other substrates for Src- and Abl-family kinases appear to be processively phosphorylated. CD3 is a complex of four distinct polypeptide chains that associate with the T-cell receptor (TCR) in T-lymphocytes. The lymphocyte-specific kinase Lck catalyzes phosphorylation of the TCR/CD3 associated ζ chain during T-cell activation. The three tandem immunoacceptor tyrosine based activation motifs (ITAMs) within the ζ chain are phosphorylated processively by Lck; mutant forms of Lck lacking a functional SH2 domain are unable to phosphorylate the ζ chain [95].
Abl tyrosine kinase catalyzes the processive phosphorylation of multiple tyrosine residues in the C-terminal repeat domain (CTD) of RNA polymerase II, while the CTD is not a substrate for Src [96]. Mutant forms of Abl in which the SH2 domain of Abl was deleted or replaced with the SH2 domain of Src were unable to phosphorylate the CTD. These results suggested that the Abl SH2 domain mediates the association of the partially phosphorylated CTD with the kinase, so that partially phosphorylated intermediates remain bound to the enzyme and only fully phosphorylated product is released. In this case as well as for the CD3 ζ chain, SH2 domain binding to phosphotyrosine residues is predicted to protect the substrates from dephosphorylation by phosphatases.
4.2 Semi-processive phosphorylation of Pho4 by Pho80/Pho85
In the yeast Saccharomyces cerevisiae, Pho4 is a transcription factor that responds to environmental levels of inorganic phosphate. Pho4 is phosphorylated by the cyclin-CDK complex Pho80/Pho85 as a consequence of the high affinity interaction between Pho4 and cyclin Pho80 [97]. At low phosphate levels, Pho4 is phosphorylated at low stoichiometry, resulting in the activation of transcription of phosphate responsive genes. At high phosphate levels, Pho4 is hyperphosphorylated by Pho80/Pho85, resulting in the inactivation of transcription and export of Pho4 out of the nucleus [98, 99].
Pho4 is phosphorylated by Pho80/Pho85 on five serine residues (referred as SP1, SP2, SP3, SP4, and SP6) that play distinct roles in the inactivation of Pho4 [100]. Kinetic studies and computer modeling showed that the multisite phosphorylation of Pho4 is semi-processive with a site preference [101]. Site SP6 has the highest probability of being phosphorylated first, followed by SP4. Sites SP2 and SP3 are probably phosphorylated last. By varying the molar ratios of Pho4 and Pho80/Pho85 and determining the concentrations of each phosphoform in the reaction, an average of 2.1 phosphorylation events per enzyme-substrate binding event was calculated. This suggested that the mechanism of phosphorylation is neither processive nor completely distributive; instead, it is partially processive due to a balance between the rates of Pho4 phosphorylation and enzyme-substrate dissociation.
5. Biological importance of processive phosphorylation
Approximately 30% of all proteins in a eucaryotic cell are phosphorylated at any time, and most phosphoproteins are modified at more than one site [16, 102]. In cases where multisite phosphorylation is catalyzed by a single enzyme, the reaction can be processive or distributive; each mechanism is presumably fine-tuned to the biological system. The distributive phosphorylation of MAPKs by MAPKKs introduces a dependence on enzyme concentration [43, 44]. Thus, a small change in MAPKK concentration results in a large change in the proportion of phosphorylated MAPK, resulting in switch-like (or ultrasensitive) behavior. A similar switch-like response is observed in the multisite phosphorylation of the CDK inhibitor Sic1 [23]. These mechanisms are manifested in the decisive, all-or-none biological outcomes. Theoretical studies have augmented these conclusions. For a protein with multiple phosphorylation sites, the proportion of fully-phosphorylated substrate is maintained at low levels when the ratio of kinase to phosphatase activity lies below a threshold value, and this threshold increases as the number of phosphorylation sites increases [103]. True switch-like behavior requires additional factors, such as a disparity between the catalytic efficiencies of the kinase and phosphatase modifying a particular site. Another theoretical treatment of distributive phosphorylation showed that the order of phosphorylation is critical for the sensitivity and speed of response. Sequential phosphorylation (or dephosphorylation) gave rise to steeper response curves, whereas random phosphorylation gave rise to shallow response curves and faster kinetics [104].
In contrast, the lack of dependence on enzyme concentration in a processive mechanism suggests that processivity would be poorly suited to biological systems where switch-like behavior is required. The biological importance of processivity is presumably related to the need for a high rate of multisite phosphorylation. For Cas, this would permit faster assembly of Cas-Crk complexes and downstream signaling. Furthermore, whereas in a distributive mechanism partially phosphorylated intermediates are released which can act as competitive inhibitors, relatively few intermediates are released in a processive mechanism. The absence of potential competitive inhibitors means that a given signal can produce a significant population of highly phosphorylated substrate molecules. Thus, the processive mechanism may be found in situations where downstream signaling depends particularly on the fully phosphorylated forms of the target proteins.
Acknowledgments
This work was supported by National Institutes of Health Grant CA 58530 to W.T.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walsh CT, Garneau-Tsodikova S, Gatto GJ., Jr. Angew Chem Int Ed Engl. 2005;44(45):7342–7372. doi: 10.1002/anie.200501023. [DOI] [PubMed] [Google Scholar]
- 2.Yang XJ. Oncogene. 2005;24(10):1653–1662. doi: 10.1038/sj.onc.1208173. [DOI] [PubMed] [Google Scholar]
- 3.Martin C, Zhang Y. Nat Rev Mol Cell Biol. 2005;6(11):838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- 4.Polevoda B, Sherman F. J Biol Chem. 2000;275(47):36479–36482. doi: 10.1074/jbc.R000023200. [DOI] [PubMed] [Google Scholar]
- 5.Glozak MA, Sengupta N, Zhang X, Seto E. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Vogelauer M, Rubbi L, Lucas I, Brewer BJ, Grunstein M. Mol Cell. 2002;10(5):1223–1233. doi: 10.1016/s1097-2765(02)00702-5. [DOI] [PubMed] [Google Scholar]
- 7.Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Nature. 2000;406(6796):593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 8.Cheung WL, Briggs SD, Allis CD. Curr Opin Cell Biol. 2000;12(3):326–333. doi: 10.1016/s0955-0674(00)00096-x. [DOI] [PubMed] [Google Scholar]
- 9.Millar CB, Grunstein M. Nat Rev Mol Cell Biol. 2006;7(9):657–666. doi: 10.1038/nrm1986. [DOI] [PubMed] [Google Scholar]
- 10.Appella E, Anderson CW. Eur J Biochem. 2001;268(10):2764–2772. doi: 10.1046/j.1432-1327.2001.02225.x. [DOI] [PubMed] [Google Scholar]
- 11.Meek DW. Oncogene. 1999;18(53):7666–7675. doi: 10.1038/sj.onc.1202951. [DOI] [PubMed] [Google Scholar]
- 12.Meek DW. Cell Signal. 1998;10(3):159–166. doi: 10.1016/s0898-6568(97)00119-8. [DOI] [PubMed] [Google Scholar]
- 13.Holmberg CI, Tran SE, Eriksson JE, Sistonen L. Trends Biochem Sci. 2002;27(12):619–627. doi: 10.1016/s0968-0004(02)02207-7. [DOI] [PubMed] [Google Scholar]
- 14.Cohen P. Trends Biochem Sci. 2000;25(12):596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 15.Roach PJ. J Biol Chem. 1991;266(22):14139–14142. [PubMed] [Google Scholar]
- 16.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Cell. 2006;127(3):635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 17.Baskaran R, Chiang GG, Mysliwiec T, Kruh GD, Wang JY. J Biol Chem. 1997;272(30):18905–18909. doi: 10.1074/jbc.272.30.18905. [DOI] [PubMed] [Google Scholar]
- 18.Dahmus ME. J Biol Chem. 1996;271(32):19009–19012. doi: 10.1074/jbc.271.32.19009. [DOI] [PubMed] [Google Scholar]
- 19.Phatnani HP, Greenleaf AL. Genes Dev. 2006;20(21):2922–2936. doi: 10.1101/gad.1477006. [DOI] [PubMed] [Google Scholar]
- 20.Corden JL. Trends Biochem Sci. 1990;15(10):383–387. doi: 10.1016/0968-0004(90)90236-5. [DOI] [PubMed] [Google Scholar]
- 21.Komarnitsky P, Cho EJ, Buratowski S. Genes Dev. 2000;14(19):2452–2460. doi: 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deshaies RJ, Ferrell JE., Jr. Cell. 2001;107(7):819–822. doi: 10.1016/s0092-8674(01)00620-1. [DOI] [PubMed] [Google Scholar]
- 23.Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. Nature. 2001;414(6863):514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 24.Strickfaden SC, Winters MJ, Ben-Ari G, Lamson RE, Tyers M, Pryciak PM. Cell. 2007;128(3):519–531. doi: 10.1016/j.cell.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hutchins JR, Clarke PR. Cell Cycle. 2004;3(1):41–45. [PubMed] [Google Scholar]
- 26.Kumagai A, Yakowec PS, Dunphy WG. Mol Biol Cell. 1998;9(2):345–354. doi: 10.1091/mbc.9.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H. Science. 1997;277(5331):1501–1505. doi: 10.1126/science.277.5331.1501. [DOI] [PubMed] [Google Scholar]
- 28.Strausfeld U, Fernandez A, Capony JP, Girard F, Lautredou N, Derancourt J, Labbe JC, Lamb NJ. J Biol Chem. 1994;269(8):5989–6000. [PubMed] [Google Scholar]
- 29.Izumi T, Maller JL. Mol Biol Cell. 1993;4(12):1337–1350. doi: 10.1091/mbc.4.12.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lefkowitz RJ. Trends Pharmacol Sci. 2004;25(8):413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Rapacciuolo A, Suvarna S, Barki-Harrington L, Luttrell LM, Cong M, Lefkowitz RJ, Rockman HA. J Biol Chem. 2003;278(37):35403–35411. doi: 10.1074/jbc.M305675200. [DOI] [PubMed] [Google Scholar]
- 32.Iyer V, Tran TM, Foster E, Dai W, Clark RB, Knoll BJ. Br J Pharmacol. 2006;147(3):249–259. doi: 10.1038/sj.bjp.0706551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lefkowitz RJ, Pierce KL, Luttrell LM. Mol Pharmacol. 2002;62(5):971–974. doi: 10.1124/mol.62.5.971. [DOI] [PubMed] [Google Scholar]
- 34.Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, Houslay MD. Proc Natl Acad Sci U S A. 2003;100(3):940–945. doi: 10.1073/pnas.262787199. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Martin NP, Whalen EJ, Zamah MA, Pierce KL, Lefkowitz RJ. Cell Signal. 2004;16(12):1397–1403. doi: 10.1016/j.cellsig.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 36.Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, Lefkowitz RJ. Proc Natl Acad Sci U S A. 2005;102(5):1442–1447. doi: 10.1073/pnas.0409532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woodgett JR. Semin Cancer Biol. 1994;5(4):269–275. [PubMed] [Google Scholar]
- 38.Wang Y, Roach PJ. J Biol Chem. 1993;268(32):23876–23880. [PubMed] [Google Scholar]
- 39.Fiol CJ, Wang A, Roeske RW, Roach PJ. J Biol Chem. 1990;265(11):6061–6065. [PubMed] [Google Scholar]
- 40.Dajani R, Fraser E, Roe SM, Young N, Good V, Dale TC, Pearl LH. Cell. 2001;105(6):721–732. doi: 10.1016/s0092-8674(01)00374-9. [DOI] [PubMed] [Google Scholar]
- 41.ter Haar E, Coll JT, Austen DA, Hsiao HM, Swenson L, Jain J. Nat Struct Biol. 2001;8(7):593–596. doi: 10.1038/89624. [DOI] [PubMed] [Google Scholar]
- 42.Flotow H, Graves PR, Wang AQ, Fiol CJ, Roeske RW, Roach PJ. J Biol Chem. 1990;265(24):14264–14269. [PubMed] [Google Scholar]
- 43.Burack WR, Sturgill TW. Biochemistry. 1997;36(20):5929–5933. doi: 10.1021/bi970535d. [DOI] [PubMed] [Google Scholar]
- 44.Ferrell JE, Jr., Bhatt RR. J Biol Chem. 1997;272(30):19008–19016. doi: 10.1074/jbc.272.30.19008. [DOI] [PubMed] [Google Scholar]
- 45.Ferrell JE, Jr., Machleder EM. Science. 1998;280(5365):895–898. doi: 10.1126/science.280.5365.895. [DOI] [PubMed] [Google Scholar]
- 46.Mayer BJ, Hirai H, Sakai R. Curr Biol. 1995;5(3):296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- 47.Scott MP, Miller WT. Biochemistry. 2000;39(47):14531–14537. doi: 10.1021/bi001850u. [DOI] [PubMed] [Google Scholar]
- 48.Zahler AM, Lane WS, Stolk JA, Roth MB. Genes Dev. 1992;6(5):837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- 49.Fu XD. Rna. 1995;1(7):663–680. [PMC free article] [PubMed] [Google Scholar]
- 50.Valcarcel J, Green MR. Trends Biochem Sci. 1996;21(8):296–301. [PubMed] [Google Scholar]
- 51.Bourgeois CF, Lejeune F, Stevenin J. Prog Nucleic Acid Res Mol Biol. 2004;78:37–88. doi: 10.1016/S0079-6603(04)78002-2. [DOI] [PubMed] [Google Scholar]
- 52.Shen H, Green MR. Mol Cell. 2004;16(3):363–373. doi: 10.1016/j.molcel.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 53.Caceres JF, Misteli T, Screaton GR, Spector DL, Krainer AR. J Cell Biol. 1997;138(2):225–238. doi: 10.1083/jcb.138.2.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J, Takagaki Y, Manley JL. Genes Dev. 1996;10(20):2588–2599. doi: 10.1101/gad.10.20.2588. [DOI] [PubMed] [Google Scholar]
- 55.Zahler AM, Neugebauer KM, Lane WS, Roth MB. Science. 1993;260(5105):219–222. doi: 10.1126/science.8385799. [DOI] [PubMed] [Google Scholar]
- 56.Caceres JF, Screaton GR, Krainer AR. Genes Dev. 1998;12(1):55–66. doi: 10.1101/gad.12.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spector DL. Annu Rev Cell Biol. 1993;9:265–315. doi: 10.1146/annurev.cb.09.110193.001405. [DOI] [PubMed] [Google Scholar]
- 58.Misteli T, Caceres JF, Spector DL. Nature. 1997;387(6632):523–527. doi: 10.1038/387523a0. [DOI] [PubMed] [Google Scholar]
- 59.Misteli T, Caceres JF, Clement JQ, Krainer AR, Wilkinson MF, Spector DL. J Cell Biol. 1998;143(2):297–307. doi: 10.1083/jcb.143.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Misteli T. Curr Biol. 1999;9(6):R198–200. doi: 10.1016/s0960-9822(99)80128-6. [DOI] [PubMed] [Google Scholar]
- 61.Tenenbaum SA, Aguirre-Ghiso J. Mol Cell. 2005;20(4):499–501. doi: 10.1016/j.molcel.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lin S, Xiao R, Sun P, Xu X, Fu XD. Mol Cell. 2005;20(3):413–425. doi: 10.1016/j.molcel.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 63.Huang Y, Yario TA, Steitz JA. Proc Natl Acad Sci U S A. 2004;101(26):9666–9670. doi: 10.1073/pnas.0403533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang Y, Gattoni R, Stevenin J, Steitz JA. Mol Cell. 2003;11(3):837–843. doi: 10.1016/s1097-2765(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 65.Xiao SH, Manley JL. Embo J. 1998;17(21):6359–6367. doi: 10.1093/emboj/17.21.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. Embo J. 1996;15(2):265–275. [PMC free article] [PubMed] [Google Scholar]
- 67.Colwill K, Feng LL, Yeakley JM, Gish GD, Caceres JF, Pawson T, Fu XD. J Biol Chem. 1996;271(40):24569–24575. doi: 10.1074/jbc.271.40.24569. [DOI] [PubMed] [Google Scholar]
- 68.Gui JF, Tronchere H, Chandler SD, Fu XD. Proc Natl Acad Sci U S A. 1994;91(23):10824–10828. doi: 10.1073/pnas.91.23.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ge H, Manley JL. Cell. 1990;62(1):25–34. doi: 10.1016/0092-8674(90)90236-8. [DOI] [PubMed] [Google Scholar]
- 70.Krainer AR, Conway GC, Kozak D. Cell. 1990;62(1):35–42. doi: 10.1016/0092-8674(90)90237-9. [DOI] [PubMed] [Google Scholar]
- 71.Velazquez-Dones A, Hagopian JC, Ma CT, Zhong XY, Zhou H, Ghosh G, Fu XD, Adams JA. J Biol Chem. 2005;280(50):41761–41768. doi: 10.1074/jbc.M504156200. [DOI] [PubMed] [Google Scholar]
- 72.Aubol BE, Chakrabarti S, Ngo J, Shaffer J, Nolen B, Fu XD, Ghosh G, Adams JA. Proc Natl Acad Sci U S A. 2003;100(22):12601–12606. doi: 10.1073/pnas.1635129100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koizumi J, Okamoto Y, Onogi H, Mayeda A, Krainer AR, Hagiwara M. J Biol Chem. 1999;274(16):11125–11131. doi: 10.1074/jbc.274.16.11125. [DOI] [PubMed] [Google Scholar]
- 74.Ngo JC, Chakrabarti S, Ding JH, Velazquez-Dones A, Nolen B, Aubol BE, Adams JA, Fu XD, Ghosh G. Mol Cell. 2005;20(1):77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 75.Prasad J, Manley JL. Mol Cell Biol. 2003;23(12):4139–4149. doi: 10.1128/MCB.23.12.4139-4149.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sacco-Bubulya P, Spector DL. J Cell Biol. 2002;156(3):425–436. doi: 10.1083/jcb.200107017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang J, Xiao SH, Manley JL. Genes Dev. 1998;12(14):2222–2233. doi: 10.1101/gad.12.14.2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matsuda M, Mayer BJ, Fukui Y, Hanafusa H. Science. 1990;248(4962):1537–1539. doi: 10.1126/science.1694307. [DOI] [PubMed] [Google Scholar]
- 79.Sakai R, Iwamatsu A, Hirano N, Ogawa S, Tanaka T, Mano H, Yazaki Y, Hirai H. Embo J. 1994;13(16):3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reynolds AB, Kanner SB, Wang HC, Parsons JT. Mol Cell Biol. 1989;9(9):3951–3958. doi: 10.1128/mcb.9.9.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bouton AH, Riggins RB, Bruce-Staskal PJ. Oncogene. 2001;20(44):6448–6458. doi: 10.1038/sj.onc.1204785. [DOI] [PubMed] [Google Scholar]
- 82.Honda H, Oda H, Nakamoto T, Honda Z, Sakai R, Suzuki T, Saito T, Nakamura K, Nakao K, Ishikawa T, Katsuki M, Yazaki Y, Hirai H. Nat Genet. 1998;19(4):361–365. doi: 10.1038/1246. [DOI] [PubMed] [Google Scholar]
- 83.Honda H, Nakamoto T, Sakai R, Hirai H. Biochem Biophys Res Commun. 1999;262(1):25–30. doi: 10.1006/bbrc.1999.1162. [DOI] [PubMed] [Google Scholar]
- 84.Ruest PJ, Shin NY, Polte TR, Zhang X, Hanks SK. Mol Cell Biol. 2001;21(22):7641–7652. doi: 10.1128/MCB.21.22.7641-7652.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Huang J, Hamasaki H, Nakamoto T, Honda H, Hirai H, Saito M, Takato T, Sakai R. J Biol Chem. 2002;277(30):27265–27272. doi: 10.1074/jbc.M203063200. [DOI] [PubMed] [Google Scholar]
- 86.Klemke RL, Leng J, Molander R, Brooks PC, Vuori K, Cheresh DA. J Cell Biol. 1998;140(4):961–972. doi: 10.1083/jcb.140.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Almeida EA, Ilic D, Han Q, Hauck CR, Jin F, Kawakatsu H, Schlaepfer DD, Damsky CH. J Cell Biol. 2000;149(3):741–754. doi: 10.1083/jcb.149.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Black DS, Bliska JB. Embo J. 1997;16(10):2730–2744. doi: 10.1093/emboj/16.10.2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brabek J, Constancio SS, Siesser PF, Shin NY, Pozzi A, Hanks SK. Mol Cancer Res. 2005;3(6):307–315. doi: 10.1158/1541-7786.MCR-05-0015. [DOI] [PubMed] [Google Scholar]
- 90.Brabek J, Constancio SS, Shin NY, Pozzi A, Weaver AM, Hanks SK. Oncogene. 2004;23(44):7406–7415. doi: 10.1038/sj.onc.1207965. [DOI] [PubMed] [Google Scholar]
- 91.Shin NY, Dise RS, Schneider-Mergener J, Ritchie MD, Kilkenny DM, Hanks SK. J Biol Chem. 2004;279(37):38331–38337. doi: 10.1074/jbc.M404675200. [DOI] [PubMed] [Google Scholar]
- 92.Goldberg GS, Alexander DB, Pellicena P, Zhang ZY, Tsuda H, Miller WT. J Biol Chem. 2003;278(47):46533–46540. doi: 10.1074/jbc.M307526200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Patwardhan P, Shen Y, Goldberg GS, Miller WT. J Biol Chem. 2006;281(30):20689–20697. doi: 10.1074/jbc.M602311200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pellicena P, Miller WT. J Biol Chem. 2001;276(30):28190–28196. doi: 10.1074/jbc.M100055200. [DOI] [PubMed] [Google Scholar]
- 95.Lewis LA, Chung CD, Chen J, Parnes JR, Moran M, Patel VP, Miceli MC. J Immunol. 1997;159(5):2292–2300. [PubMed] [Google Scholar]
- 96.Duyster J, Baskaran R, Wang JY. Proc Natl Acad Sci U S A. 1995;92(5):1555–1559. doi: 10.1073/pnas.92.5.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kaffman A, Herskowitz I, Tjian R, O'Shea EK. Science. 1994;263(5150):1153–1156. doi: 10.1126/science.8108735. [DOI] [PubMed] [Google Scholar]
- 98.Ogawa N, DeRisi J, Brown PO. Mol Biol Cell. 2000;11(12):4309–4321. doi: 10.1091/mbc.11.12.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schneider KR, Smith RL, O'Shea EK. Science. 1994;266(5182):122–126. doi: 10.1126/science.7939631. [DOI] [PubMed] [Google Scholar]
- 100.Komeili A, O'Shea EK. Science. 1999;284(5416):977–980. doi: 10.1126/science.284.5416.977. [DOI] [PubMed] [Google Scholar]
- 101.Jeffery DA, Springer M, King DS, O'Shea EK. J Mol Biol. 2001;306(5):997–1010. doi: 10.1006/jmbi.2000.4417. [DOI] [PubMed] [Google Scholar]
- 102.Mann M, Ong SE, Gronborg M, Steen H, Jensen ON, Pandey A. Trends Biotechnol. 2002;20(6):261–268. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- 103.Gunawardena J. Proc Natl Acad Sci U S A. 2005;102(41):14617–14622. doi: 10.1073/pnas.0507322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Salazar C, Hofer T. Febs J. 2007;274(4):1046–1061. doi: 10.1111/j.1742-4658.2007.05653.x. [DOI] [PubMed] [Google Scholar]