

Abstract
Access to multifunctional hydrazones of relevance to dysiherbaine synthesis studies is described. Subsequent radical cyclizations of multifunctional hydrazones via a Si- and C-linked tethering strategy are shown to function effectively in 6-exo fashion. Conformational constraints are proposed to play a key role in suppressing unproductive premature reduction pathways. The stereochemical outcomes suggest that minimizing the dipole repulsion between neighboring C=N and C-O bonds favors a Cα-C(=N) dihedral angle placing the C=N bond axial within a chairlike transition state, in contrast to the usual Beckwith-Houk model.
Keywords: radical cyclization, stereoselectivity, aminosugars, dysiherbaine
1. Introduction
Amino alcohols and polyhydroxylated amines are important substructures of biologically active compounds, and include sphingolipids,1 azasugars2 and aminosugars.3,4 An interesting conjugate of amino acid and aminosugar functionality is found in the neuroexcitotoxin (-)-dysiherbaine (1, Scheme 1), which was isolated from the Micronesian marine sponge Dysidea herbacea in 1996.5 Neurotoxins are in high demand to study glutamate receptors and associated central nervous system function, and 1 is a non-NMDA glutamate agonist with 5-fold selectivity for KA vs. AMPA receptors.6 Synthetic studies toward 1 have led to total syntheses by Hatakeyama,7 Snider,8 Sasaki,9 and Chamberlin,10 as well as preparation of analogues.11 Through our prior studies of Si-tethered12 and acetal-tethered13 radical additions to imino compounds14 for synthesis of chiral hydroxyalkylamines, practical understanding of stereocontrol in these processes has been emerging. With predictions of relative configuration thus available, we became attracted to the prospect of testing the stereocontrol models on more complex substrates of relevance to synthesis of dysiherbaine and related compounds. The aminosugar 2 (Scheme 1) was judged to be an important subgoal to be pursued via allylic amine 3, which in turn could arise through stereocontrolled vinyl addition to imino compound 4.
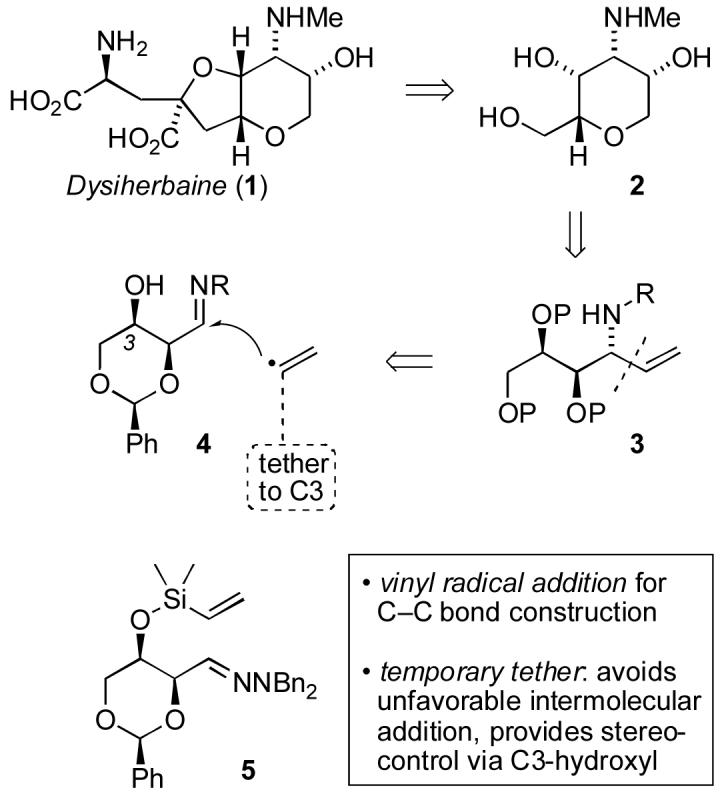
Scheme 1
The key C-C bond construction for synthesis of the aminosugar would entail stereocontrolled delivery of a tethered vinyl radical synthon via the 6-exo cyclization implied by structure 5. We have previously shown that this strategy offers high diastereoselectivity for Si-tethered vinyl group transfer to hydrazone acceptors via the 5-exo cyclization mode.12b-12e However, prior attempts in the 6-exo mode using vinylsilyl ethers linked to acyclic β-hydroxyhydrazones were previously found to suffer β-elimination during preparation of the substrates, premature reduction during thiyl-mediated vinyl addition, and poor diastereoselectivity.12e,12f It was not altogether clear whether these challenges could be met in this more highly functionalized system, but it was envisioned that the additional conformational constraints imposed by the benzylidene acetal of the cyclic substrate 5 could be beneficial. Tests of this hypothesis are reported herein. In the course of these studies, a modification of the currently accepted Beckwith-Houk stereocontrol model15 for 6-exo radical cyclization was required in order to accomodate unexpected results with α-alkoxyhydrazones.
2. Results and Discussion
2.1. Si-Tethered Cyclizations
Formation of the radical cyclization precursor 5 began with transformation of D-arabitol (Scheme 2) to the corresponding benzylidene derivative16 (mixture of regioisomers17) and oxidative cleavage with sodium periodate to afford threose derivative 6. Alternatively, reaction of D-galactose with benzaldehyde dimethyl acetal produced a single benzylidene derivative,18 then periodate cleavage furnished 6 in 67% yield for the two steps.19 Condensation of 6 with dibenzylhydrazine afforded hydroxyhydrazone 7, and installation of the silicon tethered radical precursor was achieved in excellent yields, furnishing the vinylsilyl ether 5.

Scheme 2
With silyl ether 5 in hand, the radical addition-cyclization using tin-free thiyl-mediated reaction conditions was next examined (Scheme 3).12b,12d Upon heating a mixture of 5, PhSH and AIBN in benzene, followed by exposure to fluoride ion, vinyl transfer from silicon to the imino carbon occurred to afford 8 in moderate yield. This result was a dramatic departure from our prior attempts with acyclic precursors in which the premature reduction predominated under these conditions.12f Even more surprisingly, only one diastereomer could be detected, whereas the only prior examples of 6-exo Si-tethered additions to C=N bonds were non-diastereoselective.12f

Scheme 3
Further optimization of the radical cyclization was attempted using different initiators (Scheme 3). Replacing AIBN with ABC (1,1-azodicyclohexanecarbonitrile) produced much lower yields (10%), as did photochemical activation of phenyl disulfide to directly generate thiyl radicals under non-reductive conditions. However, initiation with triethylborane/oxygen conditions afforded results similar to those obtained with AIBN.
The vinyl transfer process 5 → 8 is envisioned to involve the sequence of steps shown in Scheme 4, including (a) generation of PhS• upon heating PhSH in the presence of AIBN, (b) PhS• addition to the terminus of the vinyl group, (c) cyclization of the resulting carbon-centered radical 9, (d) H-atom abstraction from thiophenol by aminyl radical 10, (e) fluoride-induced cleavage of Si-O bond in 11, and (f) desilylative β-elimination with loss of PhS- from an intermediate of the type illustrated in hypervalent silicate 12 to reform the alkene in the observed product.

Scheme 4
For subsequent elaboration toward aminosugars, direct dihydroxylation of hydrazine 8 was desirable, but allylic amines are troublesome substrates without prior acylation.20 Treatment of 8 with Boc2O furnished an O-acylated derivative, presumably 13 (Scheme 5), as benzoic anhydride also preferentially acylated the hydroxyl function in 8 to afford 14. Silica gel chromatography of the O-Boc derivative afforded 68% of cyclic carbamate 15.21 More importantly, when nOe data were obtained for the cyclic carbamate 15, the correlation was between Ha and Hc as depicted in Scheme 5, with no observed nOe between Ha and Hb. This offered strong evidence for the indicated stereostructure bearing the undesired configuration at the newly-formed amine center. Preliminary examination of dihydroxylation of 15 showed some promise,22 but was postponed pending further examination of the unexpected stereoselectivity in the radical addition.

Scheme 5
The foregoing results raised two fundamental questions of relevance to the understanding of radical cyclization to imino compounds. First, why are these thiyl-mediated 6-exo cyclizations with the Si-tethered vinyl group successful, where several other related 6-exo cyclizations have failed?12e,12f Second, why does the diastereoselectivity defy the usual predictions of the Beckwith-Houk model?
2.4. Factors Enabling the Si-Tethered 6-exo Cyclization
Previously, irreversible generation of more reactive vinyl radicals from a vinyl bromide was required for the Si-tethered 6-exo cyclization to hydrazones.12f In contrast, in this work compound 5 cyclizes smoothly in the 6-exo cyclization, even with the reversible thiyl addition method. Key structural differences are the presence of the alkoxy substituent adjacent to the imino acceptor carbon and conformational restrictions imparted by the benzylidene acetal ring (Figure 1). Are such conformational constraints sufficient to enable the 6-exo cyclization, or is some further effect of the α-oxygen required?
Figure 1.

Comparison of structural features in unsuccessful and successful 6-exo cyclizations.
In order to gain some further insight, a substrate was prepared which lacked the α-alkoxy substituent, yet maintained the conformational constraints of 5. Racemic β-hydroxyhydrazone 16 (Scheme 6) was obtained from the corresponding β-hydroxyester by reduction (DIBAL), condensation with Ph2NNH2 (β-hydroxy hydrazones prepared with Bn2NNH2 are more prone to β-elimination), and desilylation (TBAF). Silylation of 16 furnished cyclization precursor 17 in 88% yield. Upon exposure to the thiyl-mediated addition conditions there was no detectable cyclization. As in other unsuccessful 6-exo radical cyclizations,12f TLC indicated conversion of 17 to a new compound, presumably via addition of PhSH across the vinyl group (the new compound was converted back to hydroxyhydrazone 16 upon treatment with TBAF). This observation suggests that in this case the hydrogen atom abstraction from PhSH supercedes cyclization of the carbon-centered radical of the type illustrated in Figure 1. Thus the conformational constraint alone is insufficient to accelerate the 6-exo cyclization, and it can be proposed that the α-alkoxy substituent plays a more active role than previously expected.

Scheme 6
In prior work, the Beckwith-Houk model effectively predicted the configuration obtained in thiyl-mediated vinyl additions to hydrazones.12d,12e In the case of 5, the Beckwith-Houk model predicts transition state A (Figure 2) which minimizes strain by orienting substituents equatorially, including the C=N bond. However, the observed product configuration suggests a strong preference for transition state B with the C=N bond in an axial position. To explain these results, we suggest that the α-alkoxy substituent increases the energy of A through a vicinal dipole repulsion due to the gauche relationship of the C=N and C-O bonds. In transition state B, this dihedral angle is 180°, minimizing the dipole repulsion and thus favoring reaction through B to the observed configuration. To test this hypothesis, a diastereomeric hydrazone 19 (Scheme 7) was designed which would possess a gauche relationship with the neighboring C-O bond in both transition state models A’ and B’. All substituents could occupy an equatorial position in structure A’, so the Beckwith-Houk model would predict excellent stereocontrol. In contrast, our hypothesis incorporating the dipole repulsion predicts a lower diastereomeric ratio for this substrate.
Figure 2.

Transition state models for 6-exo cyclizations of diastereomeric hydrazones.

Scheme 7
The requisite hydrazone 20 was prepared from commercially available 4,6-O-benzylidene-D-glucose (Scheme 7) by periodate cleavage and condensation with Bn2NNH2 to afford crystalline 19, followed by silylation with chlorodimethylvinylsilane. Cyclization of 20 under the thiyl-mediated conditions resulted in a 53% yield of 21 with a diastereomeric ratio of 70:30.23 The complete stereocontrol from 5 changed to very weak diastereoselectivity from 20, consistent with the prediction in Figure 2. This provides experimental support for the hypothesized dipole repulsion modification to the Beckwith-Houk model.
2.2. Preliminary Study of C-Linked Cyclization
In light of the undesired configuration emanating from the Si-tethered cyclization above, a revised plan was formulated to explore the possibility of modified conditions to reverse the stereocontrol. Use of a Lewis acid for two-point binding between the C=N and C-O bonds could potentially restrict conformer populations to the gauche conformer in transition state A’ (Figure 3). Unfortunately, identifying a Lewis acid compatible with both the thiyl conditions and the Si tether functionality was non-trivial. Instead, a propargyl ether was selected as a C-linked radical precursor, with recognition of some advantages over the temporary Si-tether. First, the more robust ether linkage would potentially accomodate a variety of Lewis acids in the radical addition. Second, the alkyne could serve as a precursor of more reactive vinyl radicals such as C in the presence of Lewis acid-compatible tin reagents. Third, the pyran ring found in dysiherbaine would be formed directly in the cyclization, which could avoid subsequent steps to elaborate that ring. After a 6-exo radical cyclization via C and removal of tin, the exo-methylene would provide a handle for stereocontrolled hydroxyl introduction.
Figure 3.

Potential for use of Lewis acid chelation to override dipole repulsion.
Alkylation of 7 with propargyl bromide in the presence of sodium iodide gave high yields of propargyl ether 22 (Scheme 8), conveniently purified by recrystallization from a 3:1 mixture of hexane and ethyl acetate. Exposure to tributyltin hydride in the presence of AIBN led to smooth conversion to vinylstannane 23. Although the cyclization could be accomplished in benzene, DMF was preferred, offering a 90% yield over 12-24 h. Alternatively, microwave irradiation in DMF (0.5M) produced 23 in 10-15 minutes. Protodestannylation was achieved readily upon transmetallation with n-BuLi and treatment with propionic acid, used stoichiometrically to preserve the benzylidene acetal. Directly applying the protodestannylation to the crude cyclization product (after filtration through silica gel to remove excess tin hydride) furnished 24 in 80% yield for the two steps. It is noteworthy that this differentially functionalized compound is obtained with complete diastereoselectivity in 48% yield from D-galactose.

Scheme 8
Assignment of configuration of 24 by NMR was accomplished with COSY and NOESY experiments on the dibenzoyl derivative 26, prepared by successive hydrolysis and benzoylation (Scheme 9). The NOESY spectrum suggested the chair conformation indicated below, based on a crosspeak for H2-H6a. The equatorial orientation of H4 was then inferred from crosspeaks between H6e-H5Z and H5E-H4. No through-space interaction of H4 with H2 or H6a was detected.

Scheme 9
The results suggest that the dipole repulsion stereocontrol model operative in the Si-tethered cyclizations also confers excellent selectivity in the propargyl ether cyclization. The greater stability of the propargyl ether radical precursor offers greater opportunities for potential modification of reaction conditions, for example with Lewis acidic additives.
4. Conclusions
A four-carbon trihydroxylated hydrazone prepared from arabitol or galactose undergoes highly stereoselective 6-exo radical cyclization onto the hydrazone unit; reversible thiyl radical addition to silicon-tethered vinyl group initiates the cyclization. Because the product configuration was opposite that predicted by the Beckwith-Houk model for application to the dysiherbaine aminosugar, interesting fundamental issues of stereocontrol emerged. In order to rationalize unexpected stereoselectivity, a dipole repulsion model is presented as a modification of the Beckwith-Houk model for α-alkoxyhydrazones. Cyclizations using stannyl addition to a propargylic ether were examined, resulting in a very efficient preparation of aminosugar 24 in 48% yield from D-galactose. Although these more robust substrates should be amenable to Lewis acid chelation and potential reversal of stereocontrol, preliminary experiments toward this end have not identified conditions for such a reversal. The dipole repulsion stereocontrol model, and studies designed to modulate it, may offer new possibilities for synthetic application of stereocontrolled radical additions to polyhydroxylated chiral amine targets.
5. Experimental Section
5.1. Materials and Methods
Reactions employed oven- or flame-dried glassware under nitrogen unless otherwise noted. THF, diethyl ether, benzene and toluene were distilled from sodium/benzophenone ketyl under argon. CH2Cl2 was distilled from CaH2 under argon or nitrogen. Alternatively, these solvents were purchased inhibitor-free and were sparged with argon and passed through columns of activated alumina prior to use (dropwise addition of blue benzophenone ketyl solution revealed the THF purified in this manner sustained the blue color more readily than the control sample purified by distillation). Nitrogen was passed successively through columns of anhydrous CaSO4 and R3-11 catalyst for removal of water and oxygen, respectively. All other materials were used as received from commercial sources unless otherwise noted. Thin layer chromatography (TLC) employed glass 0.25 mm silica gel plates with UV indicator. Flash chromatography columns were packed with 230-400 mesh silica gel as a slurry in the initial elution solvent. Gradient flash chromatography was conducted by adsorption of product mixtures on silica gel, packing over a short pad of clean silica gel as a slurry in hexane, and eluting with a continuous gradient from hexane to the indicated solvent. Radial chromatography refers to centrifugally accelerated thin-layer chromatography performed with a Chromatotron using commercially supplied rotors. Melting points are uncorrected. Nuclear magnetic resonance (NMR) data were obtained at operating frequencies of 500 or 300 MHz for 1H and 125 or 75 MHz for 13C and chemical shifts are reported in parts per million (ppm). Infrared spectra were recorded using a single beam FT-IR spectrophotometer by standard transmission methods or by use of an attenuated total reflectance (ATR) probe. Optical rotations were determined using a digital polarimeter operating at ambient temperature. Low resolution mass spectra were obtained using sample introduction by dip, liquid chromatography or gas chromatography. High resolution mass spectra and combustion analyses were obtained from external commercial and institutional services. Chromatographic diastereomer ratio analyses employed GCMS with 15 mL × 0.25 mm I.D X 0.25 μ F.T 5%-phenyl-95%-dimethylsiloxane column and helium as mobile phase or HPLC with Microsorb-MV Si 8um 100A or Chiralcel OD columns (2-propanol/hexane as mobile phase) or Chirex 3014 column (chloroform/hexane as mobile phase).
1,1-Dibenzyl-2-(((2S,4R,5R)-5-hydroxy-2-phenyl-1,3-dioxan-4-yl)methylene)hydrazine (7)
To a solution of aldehyde 624 (1.16 g, 5.58 mmol) in toluene (12 mL) was added dibenzylhydrazine (1.42 g, 6.70 mmol) and magnesium sulfate (30 mg). After 24 h, concentration and flash chromatography (5:1 to 1:1 hexanes/ethyl acetate) afforded 7 (2.20 g, 98%) as a colorless oil: [α]D24 = -41.3 (c 0.725, CHCl3); IR (film) 3452 (br), 3060, 2923, 2848, 1601, 1493, 1452, 1078, 1027 cm-1; 1H NMR (500 MHz, CDCl3) δ 7.51-7.49 (m, 2H), 7.36-7.21 (m, 13H), 6.63 (d, J = 4.8 Hz, 1H), 5.61 (s, 1H), 4.50 (d, J = 4.8 Hz, 1H), 4.40 (s, 4H), 4.28 (dd, J = 12.0, 1.5 Hz, 1H), 4.07 (d, J = 12.0 Hz, 1H), 3.77 (d, J = 7.3 Hz, 1H), 3.28 (d, J = 7.3 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 137.7, 136.9, 130.5, 128.9, 128.5, 128.1, 127.6, 127.2, 126.1, 101.4, 79.3, 72.1, 65.4, 57.4; MS (CI) m/z (relative intensity) 403 ([M+1]+, 100%), 385 (7%), 297 (24%), 279 (18%), 198 (15%); Anal. Calcd for C25H26N2O3: C, 74.60; H, 6.51; N, 6.96. Found: C, 74.36; H, 6.67; N, 6.62.
1,1-Dibenzyl-2-(((2S,4R,5R)-5-(dimethyl(vinyl)silyloxy)-2-phenyl-1,3-dioxan-4-yl) methylene)hydrazine (5)
To a solution of hydrazone 7 (77 mg, 0.19 mmol) triethylamine (0.08 mL, 0.57 mmol), and DMAP (3 mg) in THF (2 mL) was added dimethylvinylchlorosilane (0.08 mL, 0.57 mmol). After 4 h, concentration and flash chromatography (10:1 hexanes/ethyl acetate) afforded 5 (93 mg, 100%) as a colorless oil: [α]D24 = -22.5 (c = 0.855, CHCl3); IR (film) 3060, 2862, 1844, 1594, 1494, 1474, 1096, 1068, 840 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.55-7.52 (m, 2H), 7.38-7.22 (m, 13H), 6.69 (d, J = 6.2 Hz, 1H), 6.08-5.69 (m, 3H), 5.60 (s, 1H), 4.49 (dd, J = 6.2, 1.7 Hz, 1H), 4.36 (s, 4H), 4.10 (ABX, JAB = 12.2 Hz, JAX = 1.6 Hz, JBX = 1.5 Hz, Δν = 30.5 Hz, 2H), 3.71-3.68 (m, 1H), 0.09 (s, 3H), 0.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 138.2, 137.7, 137.4, 133.3, 131.5, 128.7, 128.5, 128.1, 127.7, 127.2, 126.3, 101.2, 81.2, 72.3, 67.0, 57.4, -1.4, -1.8; MS (EI) m/z 487 ([M+1]+, 90%), 403 (50%), 381 (75%), 351 (75%), 297 (80%), 279 (100%); Anal. Calcd for C29H34N2O3Si: C, 71.57; H, 7.04; N, 5.76. Found: C, 71.43; H, 7.18; N, 5.82.
1,1-Dibenzyl-2-((S)-1-((2S,4R,5R)-5-hydroxy-2-phenyl-1,3-dioxan-4-yl)allyl)hydrazine (8)
To a solution of hydrazone 5 (846 mg, 1.74 mmol) in refluxing deoxygenated cyclohexane (17.4 mL) was added a solution of AIBN (371 mg, 2.26 mmol) and thiophenol (0.55 mL, 5.2 mmol) in deoxygenated benzene (9.4 mL) over 12 h via syringe pump and heating was continued at reflux for 10 h. The solvent was replaced with THF (17.4 mL), and a solution of TBAF (1 M in THF, 3.83 mL, 3.83 mmol) was added. After 16 h, concentration and flash chromatography (20:1 to 1:1 hexane/ethyl acetate) afforded 8 (391 mg, 55%) as a colorless oil: [α]D23 = +28.0 (c 0.575, CHCl3); IR (film) 2973, 2361, 1655, 1531, 1453, 1339, 1148, 1114 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.48-7.22 (m, 15H), 5.66-5.49 (m, 1H), 5.47 (s, 1H), 5.35 (dd, J = 17.3, 2.0 Hz, 1H), 5.23 (dd, J = 10.0, 2.1 Hz, 1H), 4.02 (ABX, JAB = 11.9 Hz, JAX = 1.9 Hz, JBX = 1.2 Hz, Δν = 70.8 Hz, 2H), 3.71 (s, 4H), 3.67-3.65 (m, 2H), 3.50 (s, 1H), 1.60 (br s, 1H); 13C NMR (75 MHz, CDCl3) δ̤ppm̤137.8, 137.7, 136.8, 129.6, 129.0, 128.2, 128.1, 127.1, 126.1, 118.7, 101.5, 80.5, 72.8, 64.3, 62.5, 60.5; MS (EI) m/z (relative intensity) 431 ([M+1]+, 100%), 325 (60%), 301 (90%), 211 (60%).
1,1-Dibenzyl-2-((S)-1-((2S,4R,5R)-5-benzyloxy-2-phenyl-1,3-dioxan-4-yl)allyl)hydrazine (14)
To a solution of hydrazine 8 (219 mg, 0.509 mmol) in CH2Cl2 (25 mL) was added N,N-dimethylaminopyridine (93 mg, 0.76 mmol) followed by benzoic anhydride (173 mg, 0.764 mmol). After 3 h, additional benzoic anhydride was added (151 mg, 0.667 mmol). After another 4 h, concentration and flash chromatography (10:1 hexane/EtOAc) afforded 14 (326 mg, 100%) as a yellow oil. This material contained an unidentified benzoyl-derived compound in a ratio of ca. 1.5:1. IR (film) 3031, 1718, 1694, 1452, 1271, 1092 cm-1; 1H NMR (500 MHz, CDCl3) δ̣8.08-8.04 (m, 2H), 7.63-7.22 (m, 18H), 5.55 (s, 1H), 5.48-5.38 (m, 1H), 5.12 (d, J = 10.3 Hz, 1H), 5.03 (d, J = 17.1 Hz, 1H), 4.81 (s, 1H), 4.38 (d, J = 13.0 Hz, 1H), 4.04 (d, J = 12.7 Hz, 1H), 3.89 (d, J = 9.1 Hz, 1H), 3.76 (d, J = 8.6 Hz, 1H), 3.76 (d, J = 13.5 Hz, 2H), 3.69 (d, J = 13.4 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ̣171.1, 165.8, 137.9, 137.8, 135.9, 133.7, 133.2, 130.2, 129.9, 129.8, 129.6, 129.3, 129.2, 128.5, 128.4, 128.3, 128.0, 127.0, 126.4, 119.4, 101.5, 78.7, 69.6, 65.9, 62.0, 60.5 (includes 13C peaks for inseparable byproduct); MS (CI) m/z (relative intensity) 535 ([M+1+], 100%), 444 (16%), 429 (50%), 338 (35%), 211 (70%).
(2S,4aR,8S,8aR)-7-(dibenzylamino)-2-phenyl-8-vinyl-tetrahydro-[1,3]dioxino[4,5-e] [1,3]oxazin-6(7H)-one (15)
To a solution of hydrazine 8 (60 mg, 0.140 mmol) in CH2Cl2 (2 mL) was added pyridine (0.01 mL, 0.14 mmol) followed by Boc2O (31 mg, 0.14 mmol) at 0 °C. After 4 h, concentration and flash chromatography (5:1 hexanes/ethyl acetate) afforded 15 (43 mg, 68%) as a colorless solid; mp 166-167 °C; [α]D27 +104 (c 1.36, CHCl3); IR (film) 2864, 1701, 1385, 1260, 1102 cm-1; 1H NMR (500 MHz, CDCl3) δ 7.45-7.21 (m, 15H), 5.80 (ddd, J = 16.9 Hz, 9.7 Hz, 9.7 Hz, 1H), 5.40 (s, 1H), 5.25 (d, J = 10.0 Hz, 1H), 5.07 (d, J = 17.2 Hz, 1H), 4.57 (d, J = 13.4 Hz, 1H), 4.35 (d, J = 12.1 Hz, 1H), 4.31 (d, J = 12.1 Hz, 1H), 4.25 (d, J = 13.6 Hz, 1H), 3.88 (dd, J = 12.7, 1.4 Hz, 1H), 3.79 (dd, J = 3.7, 1.2 Hz, 1H), 3.74 (d, J = 12.0 Hz, 1H), 3.69 (d, J = 1.3 Hz, 1H), 3.09 (dd, J = 9.3, 3.7 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 151.3, 138.2, 137.5, 137.2, 134.0, 130.3, 129.1, 189.9, 128.2, 128.1, 128.0, 127.7, 127.2, 126.1, 119.0, 100.7, 73.2, 69.2, 68.2, 67.1, 60.8, 54.1; MS (CI) m/z (relative intensity) 457 ([M+1+], 100%), 397 (62%); Anal. Calcd for C28H28N2O4: C, 73.66; H, 6.18; N, 6.14; Found: C, 73.41; H, 6.26; N, 6.10.
rac-2-((cis-2-Hydroxycyclohexyl)methylene)-1,1-diphenylhydrazine (16)
To a solution of ethyl cis-2-(tert-butyldimethylsilyloxy)cyclohexane carboxylate25 (1.52 g, 5.30 mmol) in CH2Cl2 (7 mL) was added diisobutylaluminum hydride (1.2 M in hexane, 5.3 mL, 6.4 mmol) over 30 min at -78 °C. After 2 h, the reaction was quenched with saturated aqueous sodium potassium tartrate (300 mL), extracted with CH2Cl2 (3 × 100 mL), dried over MgSO4 and concentrated. The residue (crude aldehyde) was dissolved in pyridine (5 mL). Diphenylhydrazine hydrochloride (1.29 g, 5.83 mmol) was added. After 16 h, the mixture was concentrated and purified by flash chromatography (10:1 petroleum ether/EtOAc) to afford the corresponding hydrazone (1.72 g, 80%) as a colorless oil: IR (film) 3060, 3027, 2931, 2856, 1591, 1494, 836 cm-1; 1H NMR (300 MHz, CHCl̤3δ 7.39-7.34 (m, 4H), 7.14-7.09 (m, 6H), 6.58 (d, J = 5.98 Hz, 1H), 3.99-3.94 (m, 1H), 2.48-2.40 (m, 1H), 1.88-1.26 (m, 8H), 0.75 (s, 9H), -0.03 (s, 3H), -0.18 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 144.3, 142.8, 129.5, 123.7, 122.4, 69.7, 46.0, 33.3, 25.7, 25.3, 24.7, 20.4, 17.9, -4.3, -5.1; MS (CI) m/z (relative intensity) 409 ([M+1]+, 84%), 277 (22%), 168 (100%); Anal. Calcd for C25H36N2OSi: C, 73.48; H, 8.88; N, 6.86. Found: C, 73.43; H, 8.99; N, 6.70. To a solution of the hydrazone obtained as described above (490 mg, 1.20 mmol) in THF (12 mL) was added tetrabutylammonium fluoride (1 M in THF, 1.4 mL, 1.4 mmol). After 30 min, the reaction was concentrated and filtered through silica gel, eluting with ethyl acetate. Concentration afforded 16 (280 mg, 79%) as a colorless oil: IR (film) 3432 (br), 2930, 2847, 1590, 1495, 1296, 1211 cm-1; 1H NMR (300 MHz, CHCl̤3δ 7.49-7.28 (m, 4H), 7.18-7.02 (m, 6H), 6.58 (d, J = 3.9 Hz, 1H), 4.26-4.21 (m, 1H), 2.45-2.28 (m, 1H), 1.94-1.22 (m, 9H); 13C NMR (75 MHz, CDCl3) δ 143.7, 142.2, 129.8, 124.2, 122.3, 68.0, 44.0, 32.1, 25.8, 25.0, 20.9; MS (CI) m/z (relative intensity) 317 ([M+Na]+, 15%), 295 ([M+1]+, 100%), 277 (12%), 168 (27%); HRMS (EI) calcd for C19H22N2O 294.1734, found 294.1732.
rac-2-((cis-2-(Dimethylvinylsilyloxy)cyclohexyl)methylene)-1,1-diphenylhydrazine (17)
To a solution of hydrazone 16 (90 mg, 0.306 mmol), triethylamine (0.05 mL, 0.34 mmol), N,N-dimethylaminopyridine (4 mg) in THF (3 mL) was added dimethylvinyl chlorosilane (0.05 mL, 0.34 mmol). After 4 h, the reaction mixture was concentrated and filtered through silica gel, eluting with ethyl acetate. Concentration afforded 17 (102 mg, 88%) as a colorless oil: IR (film) 2930, 2848, 1590, 1495, 1252, 1019 cm-1; 1H NMR (300 MHz, CHCl̤3δ 7.41-7.29 (m, 4H), 7.16-7.04 (m, 6H), 6.54 (d, J = 5.8 Hz, 1H), 6.07-5.80 (m, 2H), 5.63 (dd, J = 18.5, 5.8 Hz, 1H), 4.04-3.79 (m, 1H), 2.56-2.23 (m, 1H), 1.91-1.15 (m, 8H), 0.01 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 144.4, 143.0, 138.1, 132.5, 129.6, 123.7, 122.4, 70.2, 45.6, 33.2, 25.2, 24.6, 20.5, -1.4, -1.5; MS (EI) (relative intensity) m/z 378 (M+, 55%), 276 (8%), 195 (12%), 167 (100%); HRMS (EI) calcd for C23H30N2OSi 378.2129, found 378.2127.
1,1-Dibenzyl-2-(((2R,4S,5R)-5-hydroxy-2-phenyl-1,3-dioxan-4-yl)methylene)hydrazine (19)
To a mixture of 4,6-O-benzylidene-D-glucose (2.73 g, 13.1 mmol) and NaHCO3 (3.3 g, 39 mmol) was added a solution of NaIO4 (6.44 g, 30.1 mmol) in water (60 mL) over 30 min. After 2 h, the mixture was concentrated to a white solid which was triturated four times with hot EtOAc. The soluble portion was concentrated to afford the crude aldehyde, which was taken up in toluene (40 mL). To this solution was added dibenzylhydrazine (3.0 g, 14 mmol). After 36 h, concentration and flash chromatography (10:1 to 1:1 hexanes/ethyl acetate) afforded a solid which was recrystallized from diethyl ether/petroleum ether to afford 19 (3.83 g, 73%) as a colorless solid; mp 44.0-44.5 °C; [α]D24 -6.47 (c 0.665, CHCl3); IR (film) 3444 (br), 3030, 2852, 1955, 1601, 1392, 1075 cm-1; 1H NMR (500 MHz, CDCl3) δ̣7.48-7.25 (m, 15H), 6.64 (d, J = 2.35 Hz, 1H), 5.50 (s, 1H), 4.38 (s, 4H), 4.36 (dd, J = 10.8, 5.2 Hz, 1H), 4.19 (dd, J = 8.8, 2.7 Hz, 1H), 3.97 (ddd, J = 9.5, 9.0, 5.1 Hz, 1H), 3.70 (dd, 10.5 Hz, 10.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ̣37.4, 136.7, 129.0, 128.7, 128.3, 127.8, 127.5, 127.4, 126.1, 101.3, 80.4, 70.1, 65.2, 57.8; MS (CI) m/z (relative intensity) 403 ([M+1]+, 100%), 301 (100%); Anal. Calcd for C25H26N2O3: C, 74.60; H, 6.51; N, 6.96; Found: C, 74.87; H, 6.64; N, 6.89.
1,1-Dibenzyl-2-(((2R,4S,5R)-5-(dimethyl(vinyl)silyloxy)-2-phenyl-1,3-dioxan-4-yl) methylene)hydrazine (20)
To a solution of 19 (835 mg, 2.07 mmol), triethylamine (0.87 mL, 6.2 mmol) and N,N-dimethylaminopyridine (25 mg, 0.207 mmol) in THF (21 mL) was added dimethylvinylchlorosilane (0.86 mL, 6.2 mmol). After 4 h, the reaction mixture was filtered through silica gel, eluting with ethyl acetate. Concentration afforded 20 (96%) as a colorless oil: [α]D23 +3.77 (c 0.610, CHCl3); IR (film) 2922, 1596, 1493, 1452, 1098, 1012 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.47-7.44 (m, 2H), 7.36-7.19 (m, 13H), 6.43 (d, J = 6.44 Hz, 1H), 6.00-5.65 (m, 3H), 5.52 (s, 1H), 4.38 (ABq, J = 15.4 Hz, Δν = 16.1 Hz, 4H), 4.21-4.11 (m, 2H), 3.75 (ddd, J = 10.9, 10.0, 5.0 Hz, 1H), 3.61 (t, J = 10.3 Hz, 1H), 0.11 (s, 3H), 0.07 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 137.7, 137.5, 137.2, 133.7, 129.7, 128.8, 128.5, 128.2, 127.7, 127.2, 126.2, 100.8, 82.9, 71.6, 64.9, 57.6, -1.5, -1.7; MS (CI) m/z (relative intensity) 487 ([M+1]+, 100%), 403 (10%), 381 (14%); Anal. Calcd for C29H34N2O3Si: C, 71.57; H, 7.04; N, 5.76; Found: C, 71.52; H, 7.26; N, 5.75.
1,1-Dibenzyl-2-(1-((2R,4S,5R)-5-hydroxy-2-phenyl-1,3-dioxan-4-yl)allyl)hydrazine (21)
To a solution of hydrazone 20 (964 mg, 1.98 mmol) in refluxing deoxygenated benzene (20 mL) was added a solution of AIBN (325 mg, 1.98 mmol) and thiophenol (0.63 mL, 5.94 mmol) in deoxygenated benzene (8 mL) over 16 h. After an additional 10 h under reflux, tetrabutylammonium fluoride (1 M in THF, 4.4 mL, 4.4 mmol) was added and the mixture was allowed to cool to ambient temperature. After 24 h, concentration and flash chromatography (10:1 to 1:1 Hex/EtOAc) afforded 21 (452 mg, 53%, dr 70:30) as a colorless oil. Diastereomer 1: [α]D25 +7.0 (c 0.90, CHCl3); IR (film) 3580, 3384 (br), 3030, 2925, 2853, 1453, 1392, 1075 cm-1; 1H NMR (500 MHz, CDCl3) δ 7.48-7.30 (m, 15H), 5.90 (ddd, J = 17.8, 10.1, 7.7 Hz, 1H), 5.44 (s, 1H), 5.30 (d, J = 10.3 Hz, 1H), 5.29 (d, J = 17.1 Hz, 1H), 4.18 (dd, J = 10.5, 5.0 Hz, 1H), 3.81-3.65 (m, 7H), 3.47 (dd, J = 10.5 Hz, 10.5 Hz, 1H), 3.38 (br s, 1H), 2.94 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 137.8, 137.1, 135.4, 129.6, 128.9, 128.4, 128.2, 127.5, 126.2, 118.2, 101.4, 80.8, 70.8, 63.2, 63.0, 59.9; MS (EI) m/z (relative intensity) 430 (M+, 27%), 429 (100%), 339 (10%), 282 (45%), 251 (15%); Anal. Calcd for C27H30N2O3: C, 75.32; H, 7.02; N, 6.51; Found: C, 75.08; H, 7.28; N, 6.33. Diastereomer 2: [α]D25 = -4.6 (c 0.85, CHCl3); IR (film) 3399 (br), 3031, 2854, 1495, 1452, 1391, 1075 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.41-7.27 (m, 15H), 5.73 (ddd, J = 17.3, 10.2, 8.5 Hz, 1H), 5.29 (s, 1H), 5.24 (dd, J = 17.3, 1.0 Hz, 1H), 5.21 (dd, J = 10.2, 1.6 Hz, 1H), 4.23 (dd, J = 8.6, 3.1 Hz, 1H), 3.86 (d, J = 13.0 Hz, 2H), 3.70 (d, J = 13.1 Hz, 2H), 3.62-3.43 (m, 4H), 3.36 (dd, J = 8.4, 7.1 Hz, 1H), 2.94 (br, 1H); 13C NMR (125 MHz, CDCl3) δ 137.8, 136.7, 136.5, 129.7, 128.7, 128.6, 128.4, 128.1, 127.6, 125.9, 118.4, 100.4, 82.1, 65.7, 65.3, 60.9; MS (CI) m/z (relative intensity) 431 ([M+1]+, 100%), 301 (100%); Anal. Calcd for C27H30N2O3: C, 75.32; H, 7.02; N, 6.51; Found: C, 75.31; H, 7.20; N, 6.45.
1,1-Dibenzyl-2-(((2S,4R,5R)-2-phenyl-5-(prop-2-ynyloxy)-1,3-dioxan-4-yl)methylene) hydrazine (22)
To a mixture of potassium hydride (30% in mineral oil, 71 mg, 0.53 mmol) in THF (1 mL) was added a solution of 7 (192 mg, 0.48 mmol) in THF (1 mL) over 20 min. Sodium iodide (72 mg, 0.48 mmol) was added, followed by propargyl bromide (0.08 mL, 0.72 mmol). After 16 h, concentration and recrystallization from 3:1 hexanes/ethyl acetate afforded 22 (195 mg, 92%) as a light yellow solid: mp 49-50 °C; [α]D25 = -23.0 (c 0.95, CHCl3); IR (film) 3288, 3030, 2850, 2105, 1599, 1452, 1075 cm-1; 1H NMR (500 MHz, CDCl̤3δ 7.56-7.46 (m, 2H), 7.37-7.20 (m, 13H), 6.67 (d, J = 6.1 Hz, 1H), 5.63 (s, 1H), 4.58 (dd, J = 6.1, 1.8 Hz, 1H), 4.43 (dd, J = 12.5, 1.5 Hz, 1H), 4.42 (ABq, J = 15.4 Hz, Δν = 23.0 Hz, 4H), 4.04 (dd, J = 16.4, 2.4 Hz, 1H), 4.02 (dd, J = 12.5, 1.5 Hz, 1H), 3.89 (dd, J = 16.4, 2.3 Hz, 1H), 3.59 (d, J = 1.7 Hz, 1H), 2.30 (t, J = 2.4 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 137.9, 137.5, 131.1, 128.8, 128.5, 128.1, 127.7, 127.2, 126.2, 101.2, 80.4, 79.7, 74.5, 72.1, 68.8, 57.6, 57.0; MS (CI) m/z (relative intensity) 441 (M+, 100%), 335 (30%); Anal. Calcd for C28H28N2O3: C, 76.34; H, 6.41; N, 6.36; Found: C, 76.51; H, 6.37; N, 6.44.
1,1-Dibenzyl-2-((2S,4aR,8S,8aR)-2-phenyl-7-((tributylstannyl)methylene)-hexa-hydropyrano[3,2-d][1,3]dioxin-8-yl)hydrazine (23)
To a solution of 22 (54 mg, 0.123 mmol) in deoxygenated DMF (6.2 mL) at 80 °C was added a solution of AIBN (10 mg, 0.062 mmol), and tributyltin hydride (0.04 mL, 0.15 mmol) in deoxygenated DMF (0.5 mL) over 12 h via a syringe pump. After an additional 10 h at 80 °C, the mixture was partitioned between brine (20 mL) and diethyl ether (2 × 20 mL). The organic phase was washed with brine (2 × 40 mL), dried and concentrated. Flash chromatography (10:1 to 3:1 hexanes/ethyl acetate) afforded 23 (81 mg, 90%) as a light yellow oil: [α]D26 = -77.6 (c 0.650, CHCl3); IR (film) 2925, 2851, 1603, 1453, 1396, 1370, 1112, 1091, 1018 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.51-7.24 (m, 15H), 5.82 (s, 1H; satellite peaks indicated JSn-H = 60.4 Hz), 5.35 (s, 1H), 4.1 (d, J = 12.4 Hz, 1H), 3.95-3.82 (m, 4H), 3.75 (d, J = 1.3 Hz, 1H), 3.50-3.45 (m, 3H), 3.38 (dd, J = 12.4, 1.6 Hz, 1H), 2.47 (s, 1H), 2.15 (s, 1H), 1.60-1.38 (m, 6H), 1.36-1.18 (m, 6H), 0.87-0.80 (m, 15H); 13C NMR (75 MHz, CDCl3) δ 149.5, 138.5, 137.8, 130.1, 129.9, 128.8, 128.2, 128.1, 127.2, 126.5, 101.5, 75.4, 70.0, 69.8, 65.7, 63.9, 60.7, 29.1, 27.2, 13.7, 10.3; MS (CI) m/z (relative intensity) 733 ([M+1]+, 70%), 732 (90%), 442 (42%), 212 (100%); Anal. Calcd for C40H56N2O3Sn: C, 65.67; H, 7.72; N, 3.83. Found: C, 65.94; H, 7.98; N, 3.55.
1,1-Dibenzyl-2-((2S,4aR,8S,8aR)-7-methylene-2-phenyl-hexa-hydropyrano[3,2-d] [1,3]dioxin-8-yl)hydrazine (24)
A solution of 22 (344 mg, 0.781 mmol), AIBN (64 mg, 0.39 mmol), and tributyltin hydride (0.26 mL, 0.94 mmol) in deoxygenated DMF (35 mL) was heated at 80 °C for 16 h. The mixture was partitioned between brine (50 mL) and diethyl ether (2 × 50 mL). The organic phase was washed with brine (2 × 75 mL), dried and concentrated. The residue was dissolved in THF (16 mL) and n-butyllithium (1.6 M in THF, 1.22 mL, 1.95 mmol) was added at -78 °C. After 1 h propionic acid (0.12 mL, 1.64 mL) was added. Concentration and flash chromatography (10:1 to 1:1 hexanes/ethyl acetate) afforded 24 (265 mg, 80%) as a colorless solid: [α]D24 = -25.2 (c 1.10, CHCl3); IR (film) 3411, 3064, 3027, 2923, 2848, 1452, 1090, 1023 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.52-7.25 (m, 15H), 5.41 (s, 1H), 5.06 (s, 1H), 5.04 (s, 1H), 4.03 (s, 1H), 3.97 (d, J = 12.7 Hz, 2H), 3.90 (dd, J = 12.3, 1.3 Hz, 1H), 3.81 (dd, J = 2.2, 1.1 Hz, 1H), 3.54 (d, J = 2.4 Hz, 1H), 3.50 (d, J = 12.7 Hz, 2H), 3.44 (dd, J = 12.4, 1.8 Hz, 1H), 2.51 (d, J = 1.2 Hz, 1H), 2.41 (br s, 1H);1H NMR (600 MHz, C6D̤6δ 7.73-7.66 (m, 2H), 7.32-6.92 (m, 13H), 5.53 (s, 1H), 4.77 (s, 1H), 4.76 (s, 1H), 4.14 (dd, J = 12.2, 1.3 Hz, 1H), 4.00 (s, 2H), 3.90-3.88 (m, 1H), 3.67 (d, J = 12.6 Hz, 2H), 3.53 (d, J = 2.3 Hz, 1H), 3.43 (dd, J = 12.2, 1.8 Hz, 1H), 3.18 (d, J = 12.6 Hz, 2H), 2.62 (d, J = 1.3 Hz, 1H), 1.88 (s, 1H); 13C NMR (75 MHz, CDCl3) δ ppm 140.8, 138.2, 137.7, 130.0, 128.7, 128.2, 128.0, 127.3, 126.3, 115.2, 101.2, 75.2, 70.2, 68.1, 66.0, 60.9, 60.0; MS (EI) m/z (relative intensity) 442 (M+, 3%), 351 (5%), 211 (50%), 91 (50%); Anal. Calcd for C28H30N2O3: C, 75.99; H, 6.83; N, 6.33. Found: C, 75.63; H, 6.81; N, 6.13.
(2R,3R)-4-(2,2-Dibenzylhydrazinyl)-2-(hydroxymethyl)-5-methylene-tetrahydro-2H-pyran-3-ol (25)
To a solution of 24 (65 mg, 0.15 mmol) in methanol (0.7 mL) was added p-toluenesulfonic acid (7 mg). After 1 h, concentration and flash chromatography (1:1 to 1:3 hexanes/ethyl acetate) afforded 25 (40 mg, 75%) as a yellow oil: [α]D23 = -49.5 (c 0.810, CHCl3); IR (film) 3583, 3415 (br), 2924, 1493, 1452, 1067, 920 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.39-7.25 (m, 10H), 5.11 (s, 1H), 5.05 (s, 1H), 4.02-3.86 (m, 4H), 3.68 (dd, J = 9.3, 2.7 Hz, 1H), 3.48-3.25 (m, 5H), 2.95-2.90 (m, 1H), 2.33 (s, br, 1H), 2.29 (d, J = 9.2 Hz, 1H), 1.64 (t, J = 6.3 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ̣141.2, 137.9, 129.9, 128.2, 127.2, 116.9, 73.8, 69.7, 68.9, 64.0, 62.4, 61.0; MS (CI) m/z (relative intensity) 377 ([M+Na]+, 60%), 355 ([M+1]+, 100%), 355 (10%), 301 (15%); Anal. Calcd for C21H26N2O3: C, 71.16; H, 7.39; N, 7.90; Found: C, 71.66; H, 7.69; N, 7.85.
1,1-Dibenzyl-2-((2R,3R)-3-benzoyloxy-2-(benzoyloxymethyl)-5-methylene tetrahydro-2H-pyran-4-yl)hydrazine (26)
To a solution of 25 (72 mg, 0.203 mmol), triethylamine (0.07 mL, 0.81 mmol), N,N-dimethylaminopyridine (10 mg) and 4Å MS in CH2Cl2 (2 mL) was added benzoyl chloride (0.09 mL, 0.81 mmol). After 12 h, the reaction was poured into brine (10 mL), extracted with CH2Cl2 (10 mL × 2), dried, concentrated and flash chromatography (1:1 petroleum ether: EtOAc) to afford 26 (111 mg, 97%) as a light yellow solid; mp 144-147 °C; IR (film) 3583, 3064, 3031, 2847, 1722, 1712, 1451, 1268, 1111 cm-1; 1H NMR (300 MHz, CDCl̤3δ 8.42-6.88 (m, 20H), 5.39 (d, J = 2.2 Hz, 1H), 5.08 (s, 1H), 4.96 (s, 1H), 4.19 (dd, J = 11.6, 8.2 Hz, 1H), 4.15 (d, J = 11.7 Hz, 1H), 4.05 (d, J = 11.9 Hz, 1H), 4.02 (d, J = 11.9 Hz, 2H), 3.87 (dd, J = 11.6, 3.5 Hz, 1H), 3.69 (d, J = 2.5 Hz, 1H), 3.49 (d, J = 12.5 Hz, 2H), 3.32 (dd, J = 8.0, 3.3 Hz, 1H), 2.45 (br s, 1H); 13C NMR (125 MHz, CDCl3) δ 166.2, 165.7, 140.8, 137.9, 133.0, 132.8, 130.1, 130.0, 129.9, 129.8, 129.7, 128.4, 128.3, 128.2, 127.3, 115.8, 70.7, 70.6, 68.3, 64.9, 60.9, 59.2; MS (EI) m/z (relative intensity) 563 (M+, 1%), 429 (25%), 325 (100%), 254 (25%), 221 (20%); HRMS (EI) calcd for C35H34N2O5 562.2469, found 562.2473.
Supplementary Material
Acknowledgment
We thank NIH (NIGMS, GM-67183) for generous support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kolter T, Sandhoff K. Angew. Chem. Int Ed. 1999;38:1532–1568. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1532::AID-ANIE1532>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 2.Reviews:Heightman TD, Vasella AT. Angew. Chem. Int. Ed. 1999;38:750–770. doi: 10.1002/(SICI)1521-3773(19990315)38:6<750::AID-ANIE750>3.0.CO;2-6.; (b) Bols M. Acc. Chem. Res. 1998;31:1–8. [Google Scholar]; (c) Chapleur Y, editor. Carbohydrate Mimics. Weinheim; Wiley-VCH: 1998. [Google Scholar]; (d) Nash RJ, Watson AA, Asano N. In: Alkaloids: Chemical and Biological Perspectives. Pelletier SW, editor. Pergamon; Tarrytown, New York: 1996. pp. 345–376. [Google Scholar]
- 3.Arcamone F, Penco S. In: Anthracycline and Anthracenedione-Based Anticancer Agents. Lown JW, editor. Elsevier; New York: 1988. [Google Scholar]
- 4.ReviewHauser FM, Ellenberger SR. Chem. Rev. 1986;86:35–67.
- 5.Sakai R, Kamiya H, Murata M, Shimamoto K. J. Am. Chem. Soc. 1997;119:4112–4116. [Google Scholar]
- 6.For studies of activity of (-)-dysiherbaine see:Sanders J, Ito K, Settimo L, Pentikainen O, Shoji M, Sasaki M, Johnson M, Sakai R, Swanson GJ. Pharmacol. Exp. Ther. 2005;314:1068–1078. doi: 10.1124/jpet.105.086389.; Sanders J, Pentikäinen O, Settimo L, Pentikäinen U, Shoji M, Sasaki M, Sakai R, Johnson M, Swanson G. Mol. Pharmacol. 2006;69:1849–1860. doi: 10.1124/mol.106.022772. [DOI] [PubMed] [Google Scholar]
- 7.Masaki H, Maeyama J, Kamada K, Esumi T, Iwabuchi Y, Hatakeyama S. J. Am. Chem. Soc. 2000;122:5216–5217. [Google Scholar]
- 8.Snider BB, Hawryluk NA. Org. Lett. 2000;2:635–638. doi: 10.1021/ol991393d. [DOI] [PubMed] [Google Scholar]
- 9.Sasaki M, Koike T, Sakai R, Tachibana K. Tetrahedron Lett. 2000;41:3923–3926. [Google Scholar]
- 10.Phillips D, Chamberlin AR. J. Org. Chem. 2002;67:3194–3201. doi: 10.1021/jo0107610. [DOI] [PubMed] [Google Scholar]
- 11.(a) Cohen J, Limon A, Miledi R, Chamberlin AR. Bioorg. Med. Chem. 2006;16:2189–2194. doi: 10.1016/j.bmcl.2006.01.047. [DOI] [PubMed] [Google Scholar]; (b) Shoji M, Akiyama N, Tsubone K, Lash L, Sanders J, Swanson G, Sakai R, Shimamoto K, Oikawa M, Sasaki M. J. Org. Chem. 2006;71:5208–5220. doi: 10.1021/jo0605593. [DOI] [PubMed] [Google Scholar]; (c) Shoji M, Shiohara K, Oikawa M, Sakai R, Sasaki M. Tetrahedron Lett. 2005;46:5559–5562. [Google Scholar]; (d) Tamura O, Shiro T, Ogasawara M, Toyao A, Ishibashi H. J. Org. Chem. 2005;70:4569–4577. doi: 10.1021/jo040296h. [DOI] [PubMed] [Google Scholar]; (e) Kang S, Lee Y. Synlet. 2003;993:994. [Google Scholar]; (e) Miyata O, Iba R, Hashimoto J, Naito T. Org. Biomol. Chem. 2003;1:772–774. doi: 10.1039/b212556k. [DOI] [PubMed] [Google Scholar]; (f) Huang J, Xu K, Loh T. Synthesi. 2003;755:764. [Google Scholar]; (g) Sakai R, Koike T, Sasaki M, Shimamoto K, Oiwa C, Yano A, Suzuki K, Tachibana K, Kamiya H. Org. Lett. 2001;3:1479–1482. doi: 10.1021/ol015798l. [DOI] [PubMed] [Google Scholar]; (h) Naito T, Nair J, Nishiki A, Yamashita K, Kiguchi T. Heterocycle. 2000;53:2611–2615. [Google Scholar]; (e) Sasaki M, Maruyama T, Sakai R, Tachibana K. Tetrahedron Lett. 1999;40:3195–3198. [Google Scholar]
- 12.(a) Friestad GK. Org. Lett. 1999;1:1499–1501. [Google Scholar]; (b) Friestad GK, Massari SE. Org. Lett. 2000;2:4237–4240. doi: 10.1021/ol0067991. [DOI] [PubMed] [Google Scholar]; (c) Friestad GK, Jiang T, Fioroni GM. Tetrahedron: Asymmetry. 2003;14:2853–2856. [Google Scholar]; (d) Friestad GK, Massari SE. J. Org. Chem. 2004;69:863–875. doi: 10.1021/jo035405r. [DOI] [PubMed] [Google Scholar]; (e) Friestad GK, Jiang T, Mathies AK. Org. Lett. 2007;9:777–780. doi: 10.1021/ol063010z. [DOI] [PubMed] [Google Scholar]; (f) Friestad GK, Jiang T, Mathies AK. Tetrahedron. 2007;63:3964–3972. doi: 10.1016/j.tet.2007.06.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friestad GK, Fioroni GM. Org. Lett. 2005;7:2393–2396. doi: 10.1021/ol050663r. [DOI] [PubMed] [Google Scholar]
- 14.Reviews of radical addition to C=N bonds:Friestad GK. Tetrahedron. 2001;57:5461–5496.; (b) Bertrand M, Feray L, Gastaldi S. Comptes Rend. Acad. Sci. Paris, Chimie. 2002;5:623–638. [Google Scholar]; (c) Miyabe H, Ueda M, Naito T. Synlett. 2004:1140–1157. [Google Scholar]
- 15.Beckwith ALJ, Schiesser CH. Tetrahedron. 1985;41:3925–3941. [Google Scholar]; Spellmeyer DC, Houk KN. J. Org. Chem. 1987;52:959–974. [Google Scholar]
- 16.Haskins W, Hann R, Hudson C. J. Am. Chem. Soc. 1943;65:1663–1667. [Google Scholar]
- 17.A mixture of 5- and 6-membered acetals was present, from which the desired regioisomer was obtained in 27% yield after recrystallization.
- 18.Patroni J, Stick R, Skelton B, White A. Aust. J. Chem. 1988;41:91–102. [Google Scholar]
- 19.On larger scale, the arabitol route was preferred due to the cumbersome purification of the 4,6-O-benzylidene-D-galactopyranose
- 20.Xu Z, Johannes C, La D, Hofilena G, Hoveyda A. Tetrahedron. 1997;53:16377–16390. [Google Scholar]
- 21.TLC showed complete conversion, and the 1H NMR spectrum of the crude reaction mixture was consistent with O-acylation. However, 30% recovery of 8 was observed after chromatography
- 22.Treatment of 15 with OsO4/NMO under standard conditions afforded a substance with 1H NMR spectrum consistent with 4:1 mixture of diastereomeric diols
- 23.Attempts to form the cyclic carbamate from 21 for nOe analysis were unsuccessful
- 24.Schmidt R, Maier T. Carbohydr. Res. 1988;174:169–179. [Google Scholar]
- 25.Panunzio M, Camerini R, Pachera R, Donati D, Marchioro C, Perboni A. Tetrahedron: Asymmetry. 1996;7:2929–2938. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.