

Abstract
Segmented polyurethanes have been used extensively in implantable medical devices, but their tunable mechanical properties make them attractive for examining the effect of biomaterial modulus on engineered musculoskeletal tissue development. In this study a family of segmented degradable poly(esterurethane urea)s (PEUURs) were synthesized from 1,4-diisocyanatobutane, a poly(ε-caprolactone) (PCL) macrodiol soft segment and a tyramine-1,4-diisocyanatobutane-tyramine chain extender. By systematically increasing the PCL macrodiol molecular weight from 1100 to 2700 Da, the storage modulus, crystallinity and melting point of the PCL segment were systematically varied. In particular, the melting temperature, Tm, increased from 21 to 61°C and the storage modulus at 37°C increased from 52 to 278 MPa with increasing PCL macrodiol molecular weight, suggesting that the crystallinity of the PCL macrodiol contributed significantly to the mechanical properties of the polymers. Bone marrow stromal cells were cultured on rigid polymer films under osteogenic conditions for up to 14 days. Cell density, alkaline phosphatase activity, and osteopontin and osteocalcin expression were similar among PEUURs and comparable to poly(D,L-lactic-coglycolic acid). This study demonstrates the suitability of this family of PEUURs for tissue engineering applications, and establishes a foundation for determining the effect of biomaterial modulus on bone tissue development.
Keywords: Polycaprolatone, Tissue engineering, Polyurethane, Osteoblast, Modulus
1. Introduction
A primary limitation with allogeneic and synthetic materials as replacements for bone tissue function is that they eventually undergo mechanical failure. Tissue engineered bone is envisioned to circumvent this limitation by being degradable and – like autologous bone graft – capable of stimulating the natural tissue remodeling process [1,2]. Consequently, a variety of approaches have been undertaken that combine resorbable biomaterial scaffolds, bioactive factors and osteoprogenitor cells to produce engineered tissues that are capable of stimulating integration, vascular infiltration and tissue remodeling. Within this family of strategies, the biomaterial scaffold acts as a mechanically robust substratum to support osteoprogenitor cell adhesion, proliferation and differentiation. Importantly, evidence with skeletal muscle suggests that the modulus of the scaffold is critical for achievement of the target phenotype [3]. However, the effect of the mechanical properties of the biomaterial on bone tissue development has not been addressed.
Segmented polyurethanes are an ideal class of materials with which to characterize the effect of mechanical properties on tissue development. These polymers often are prepared via a two-step process that involves reacting a diisocyanate with a 1–5 kDa macrodiol to form an isocyanate-terminated prepolymer, and then chain-extending this prepolymer with a short-chain (<1 kDa) diamine or diol. By careful selection of the diisocyanate, chain extender and macrodiol components, a broad range of physical properties can be achieved. In general, polyurethanes are biocompatible and have been used in a variety of biomedical applications, including ligament and meniscus reconstruction [4,5], blood-contacting materials [6–8], infusion pumps [9], heart valves [10], insulators for pacemaker leads [11] and nerve guidance channels [12]. For tissue engineering applications a degradable polymer is desirable, and can be achieved by incorporating labile ester linkages into the polymer backbone [13,14]. Biodegradation to non-cytotoxic components may be promoted by the use of lysine ethyl ester diisocyanate (LDI) [15] or 1,4-diisocyanatobutane (BDI) [16] in place of methylene bisdiphenylisocyanate (MDI), which has been suggested to degrade into carcinogenic and mutagenic compounds [17].
In this study a family of degradable poly(esterurethane urea)s (PEUURs) were formed by reacting BDI with poly(ε-caprolactone) (PCL) macrodiols followed by chain-extension of the prepolymer with a tyramine (TyA)–BDI–TyA adduct [18]. In the solid state – or service window – the resultant polymer is expected to microphase separate into a soft, PCL-rich phase stabilized by van der Waals forces and a hard, BDI- and TyA-containing phase stabilized by hydrogen bonds. For this study the molecular weight of the PCL macrodiol was varied from 1100 to 2700 Da to produce a family of chemically similar polymers with hard segment contents of 20–40 wt.%. Increasing the PCL macrodiol molecular weight is also expected to increase the crystallinity of the soft segment. The resultant polymers were characterized by dynamic mechanical analysis (DMA), differential scanning calorimetry (DSC), wide-angle X-ray scattering (WAXS) and contact angle goniometry to determine how the physical properties of these PEUURs depend on the hard segment content.
To verify that these PEUURs are suitable for testing our hypothesis that bone tissue development is sensitive to biomaterial scaffold modulus, cell culture studies were performed on rigid polymer films. Bone marrow stromal cells (BMSCs) – a clinically attractive progenitor cell type [19] – were cultured under osteogenic conditions to verify that the polyurethanes support proliferation and osteoblastic differentiation, and to determine if cell behavior is sensitive to subtle differences in the chemistry of the PEUURs. Cell density, alkaline phosphatase (ALP) activity, and expression of osteopontin (OPN) and osteocalcin (OCN) were measured.
2 Materials and methods
2.1 Materials
All chemicals, including BDI, TyA, ε-caprolactone, poly(ε-caprolactone) diol (PCL diol, average molecular weight 2000 Da), 1,4-butanediol (BDO), diethyl ether and dibutyltin dilaurate (DBTDL), were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. Anhydrous (<50 ppm water) dimethyl formamide (DMF) was obtained from Acros Organics (Morris Plains, NJ). All chemical reagents were used as received except for PCL2000 and butanediol, which were dried for 24 h at 80°C under vacuum (10 mm Hg), and ε-caprolactone, which was dried over anhydrous MgSO4 prior to use. All cell culture materials were obtained from Fisher Scientific (Pittsburgh, PA) unless otherwise specified.
2.2 Synthesis of segmented poly(esterurethane urea) (PEUUR) elastomers
2.2.1 Chain extender synthesis
The diurea diol chain extender TyA.BDI.TyA (Fig. 1) was synthesized from TyA and BDI as described previously [18]. Briefly, tyramine was dissolved in DMF and placed in a round-bottom reaction flask, and the resultant solution was stirred with a magnetic stirring apparatus and heated to 50°C. The air space of the vessel was purged with argon, the diisocyanate added slowly and the vessel then purged again with argon. The reaction proceeded at 50°C with no catalyst for 24 h and the solids content in the reactor was controlled at 10 wt.%. Complete conversion of diisocyanate was verified by the disappearance of the NCO peak (2250–2270 cm−1) in the IR spectrum. The TyA.BDI.TyA chain extender was precipitated using diethyl ether and dried in a vacuum oven for 24 h at 80°C and 10 mm Hg absolute pressure to yield a fine powder. Because the reactivity of the amine is 1000–2000 times higher than that of phenol at 25°C [9], the 2:1 adduct is the sole reaction product in the absence of catalyst [18].
Fig. 1.

Synthesis scheme for forming TyA.BDI.TyA chain extender.
2.2.2 Polyester macrodiol synthesis
Three PCL macrodiols (1100, 1425 and 2700 Da) were synthesized from a BDO initiator and ε-caprolactone monomer using previously published techniques [20]. The molecular weight was controlled by varying the ratio of monomer to initiator. Briefly, the appropriate amounts of dried BDO, dried ε-caprolactone and stannous octoate catalyst (1000 ppm) were mixed in a 100 ml flask and heated under an argon atmosphere with mechanical stirring to 135°C. After a reaction time of 24 h, the mixture was removed from the oil bath. Nuclear magnetic resonance spectroscopy (NMR) was performed with a Bruker 300 MHz NMR (Bruker-Biospin, Billerica, MA) to verify the structure of the PCL macrodiol using deuterated dichloromethane as the solvent.
2.2.3 Prepolymer synthesis
Anhydrous DMF was charged to a round-bottom flask fitted with a condenser. BDI was added to the flask, which was then immersed in an oil bath at 70°C, purged with argon, and stirred with a Teflon blade stirrer turned by an electric motor. A solution of dried PCL macrodiol – synthesized with a molecular weight of 1100, 1425 or 2700 Da or purchased with a molecular weight of 2000 Da – was charged into the reactor by means of an addition funnel. The NCO:OH equivalent ratio of the prepolymer was equal to 2.0:1.0. (Note that the NCO:OH equivalent ratio is equal to the BDI:PCL macrodiol molar ratio because the BDI and PCL macrodiol each has a functionality of two.) The prepolymer content in the reactor was controlled at 6 wt.%. DBTDL was added to the flask at 1000 ppm and the reaction was allowed to proceed for 24 h. The reaction scheme is summarized in Fig. 2(a).
Fig. 2.
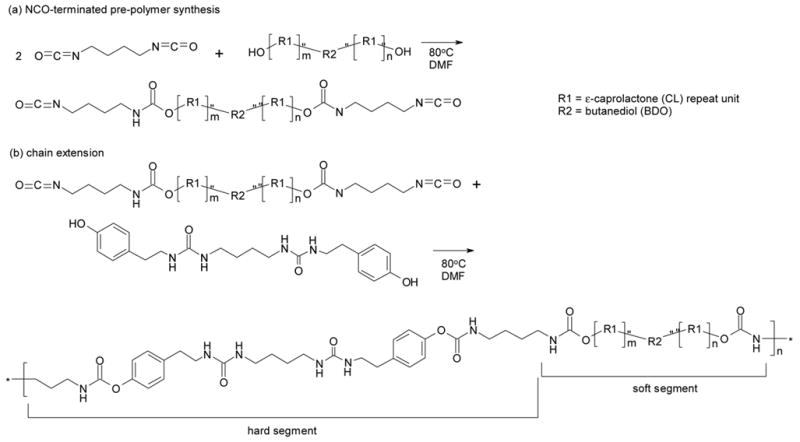
Synthesis scheme for forming segmented poly(esterurethane urea) elastomers. (a) Formation of PCL diisocyanate prepolymer. (b) Formation of PEUU by chain extending prepolymer.
2.2.4 Segmented PEUUR elastomer synthesis
A solution of chain extender in DMF was prepared at 50°C and added to the prepolymer in the reaction vessel. The NCO:OH equivalent ratio of the polyurethane was controlled at 1.03:1.0 and the polymer concentration was 3–6 wt.%. DBTDL was added to a concentration of 1000 ppm. The reaction was allowed to proceed at 70°C for 4 days. The polymer was then precipitated in diethyl ether and dried in a vacuum oven for 24 h at 80°C under 10 mm Hg vacuum. The reaction scheme is summarized in Fig. 2(b). The hard segment content was calculated from the reaction stoichiometry as the weight fraction of BDI and chain extender in the polymer.
2.3 Characterization of segmented PEUUR elastomers
2.3.1 Composition and molecular weight
NMR spectroscopy was performed using deuterated dimethylsulfoxide as the solvent to verify the structure of the chain extender and polymers. Number- and weight-average molecular weight of the polymers were measured by gel permeation chromatography (GPC) with a Waters Alliance GPC 2000 (Waters Corporation, Milford, MA) using DMF as the continuous phase, toluene as an internal standard and monodispersed polystyrene as the calibration standard.
2.3.2 Solvent-casting of PEUUR films
For the polymers synthesized from 1100, 1425 and 2000 weight-averaged molecular weight (Mw) PCL macrodiols, 3.0 wt.% solutions were prepared by dissolving the polymer in DMF at 60°C until the solution turned clear. A 2.1 wt.% solution was prepared for the PEUUR synthesized from 2700 Mw PCL. (This was the concentration at which the solution became clear.) The polymer solutions were then filtered and poured into teflon casting dishes. Films were dried at 30 kPa absolute pressure and 60°C for 4 days, then annealed for 24 h at 80°C (above the Tm for all of the PEUURs). The resultant films were analyzed by DSC, DMA and WAXS.
2.3.3 Differential scanning calorimetry
Experiments were conducted on a Seiko DSC 220C (Seiko Instruments, Japan) with an attached auto-cooler for precise temperature control. Solvent-cast samples (10–12.5 mg) were heated in a nitrogen atmosphere from −100 to 180°C at 20 °C min−1, held at 180°C for 5 min and then cooled to −100°C at 20 °C min−1. Samples were held at −100°C for 5 min and then heated again to 180°C at 20 °C min−1.
2.3.4 Thermal DMA
DMA was performed on a Seiko DMS 210 tensile module with an attached auto-cooler for precise temperature control. Rectangular samples measuring 10 mm in length, approximately 0.5 mm in thickness and 6.3–6.7 mm in width were cut from annealed films. In a nitrogen atmosphere films were cooled to −100°C and then uniaxially deformed in the linear viscoelastic region in tension mode at 1 Hz oscillating frequency. Temperature was increased from −100 to 180°C at a rate of 2 °C min−1.
2.3.5 Wide-angle X-ray scattering
Photographic flat wide-angle X-ray scattering studies were performed using a Philips PW 1720 X-ray diffractometer (Phillips Electronics, Eindhoven, The Netherlands) emitting Cu Kα radiation with a wavelength of 1.54 Å operating at 40 kV and 20 mA. The sample to film distance was set at 47.3 mm for all samples. Direct exposures were made using Kodak Biomax MS film in an evacuated sample chamber. X-ray exposures lasted 30 min.
2.4 Cell culture
2.4.1 Substrate preparation
PEUUR films – for cell culture studies and contact angle measurements – were prepared by spin-coating polymer solutions from DMF followed by annealing in order to achieve polymer morphologies similar to the cast films. Briefly, 18 mm diameter coverslips were sonicated in ethanol for 10 min and dried [21]. Next, 270 μl volumes of 3 wt.% PEUUR in DMF were deposited onto the coverslips and then spun at 2500 rpm for 30 s using a Model 1-EC101D-R485 spin-coater (Headway Research, Garland, TX) under ambient conditions. Control films were prepared by spin-coating 3 wt.% solutions of 75/25 poly(D,L-lactic-co-glycolic acid) (PLGA; Lactel Biodegradable Polymers, Birmingham, AL) in dichloromethane under the same conditions. PEUUR films were dried in a vacuum oven at 60°C for 72 h and then annealed at 80°C for 24 h. Advancing contact angles for spin-coated films were measured with a Ramé–Hart goniometer (Mountain Lakes, NJ). For each PEUUR, contact angles were measured using 3 μl drops of deionized water at four different locations on each of three films, as previously described [21]. For cell culture studies, substrates were placed in 12-well culture plates, sterilized under ultraviolet light for 12 h and incubated with 2 μg ml−1 human fibronectin (Sigma) in phosphate buffered saline (PBS) for 1 h prior to cell seeding.
2.4.2 BMSC culture
BMSCs were cultured from bone marrow explants taken from the tibias and femurs of 125–150 g male Sprague-Dawley rats (Harlan, Dublin, VA) in accordance with the Animal Care Committee at Virginia Tech [22,23]. Explants were dispersed in growth medium (α-MEM (Invitrogen, Gaithersburg, MD), 10% fetal bovine serum (Gemini, Calabasas, CA) and 1% antibiotic/antimycotic (Invitrogen)), and expanded for 14 days with medium changes every 3 or 4 days. After 14 days of primary expansion, cells were rinsed twice with PBS, lifted with trypsin/EDTA (Invitrogen) and seeded at 105 cells per well (corresponding to 6.7 × 104 cells per coverslip). On the following day, denoted as day 1, the medium was replaced with 2 ml differentiation medium (growth medium containing 0.13 mM ascorbate-2-phosphate, 2 mM β-glycerophosphate and 10 nM dexamethasone). Culture medium was changed every 3 or 4 days and cell layers were collected for analysis at days 7 and 14.
2.4.3 Cell number and ALP activity
Cell number was determined at days 7 and 14 by fluorometric measurement of total DNA using Hoechst 33258 dye [24,25]. ALP activity of cell layers was determined at day 14 using a commercially available kit (Biotron Diagnostics, Hemet, CA). Enzyme activity, defined as the rate of conversion of p-nitrophenol phosphate to p-nitrophenol, was determined colorimetrically and normalized by cell number [24].
2.4.4 OPN synthesis
Accumulation of OPN in cell layers was determined at day 14 by western blot analysis using rabbit anti-rat OPN antibody (Assay Designs, Ann Arbor, MI) followed by horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibody (Zymed, San Francisco, CA) and visualized by chemiluminescence [22]. Band densities were determined using Scion Image (Scion Corporation), and normalized by band densities for HRP-conjugated glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology, Santa Cruz, CA).
2.4.5 mRNA expression
RNA was isolated from cell layers using the RNeasy mini kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Next, 9 μl of RNA was reverse transcribed to cDNA using the Superscript kit (Invitrogen, Carlsbad, CA) with random hexamers as primers. Real time PCR amplification was performed using an ABI 7300 sequence detection system (Applied Biosciences, Foster, CA), SYBR green Master Mix (Applied Biosciences) and specific primers for β-actin (βA), OPN and OCN (Integrated Technologies, Coralville, IA) with previously published sequences [22]. Quantification of OPN and OCN gene expression was performed using the 2−ΔDelta;Ct method, with βA as the internal reference [26].
2.4.6 Statistics
Data were analyzed using a one-way analysis of variance and a 95% confidence criterion to test for differences between treatment groups. An asterisk denotes statistically significant differences between materials.
3. Results
3.1 Synthesis and characterization of segmented PEUUR elastomers
GPC analysis of the polymers indicated number-average and weight-average molecular weights of 25–36 and 36–64 kDa, respectively, and polydispersity indices of 1.3–1.8, which are typical for segmented polyurethanes (Table 1). DSC was performed to characterize the melting temperature and the relative crystallinity of the PCL microphase (Fig. 3). Comparison of the first heating curves (Fig. 3(a), solid lines) reveal a systematic increase in Tm from 21 to 61°C with increasing PCL macrodiol molecular weight, as expected. In addition, the size of the endothermic peak also systematically increased, suggesting an increase in crystallinity of the PCL phase with increasing soft segment content. This increase in crystallinity with PCL segment molecular weight is also supported by systematic increases in the size of the exothermic crystallization peak in the cooling curves (Fig. 3(b)) and the melting peak for the second heating curves (Fig. 3(a), dashed lines). Further, the sharper and more intense WAXS pattern for the 2700 Mw PCL relative to the 1100 Mw PCL also indicates an increase in PEUUR crystallinity with increasing macrodiol molecular weight (Fig. 4).
Table 1.
Physical characteristics of PEUURs
| PCL Mn (Da) | HS (wt.%) | Tg (°C) | Tm (°C) | Mn (kDa) | Mw (kDa) | θ (degrees) |
|---|---|---|---|---|---|---|
| 2700 | 20 | −55 | 61 | 35.4 | 47.0 | 74 ± 3 |
| 2000 | 26 | −52 | 42 | 36.4 | 64.4 | 83 ± 2 |
| 1425 | 35 | −52 | 34 | 25.1 | 36.1 | 79 ± 5 |
| 1100 | 39 | −52 | 21 | 34.8 | 53.3 | 78 ± 4 |
The theoretical hard segment content, HS, was predicted from the molecular weight of hard and soft segments. The glass transition temperature, Tg, and melting point, Tm, were determined from DSC curves. Number and weight-average molecular weights, Mn and Mw, respectively, were determined by GPC. Static contact angles, θ, were determined on spin-coated polymer films.
Fig. 3.
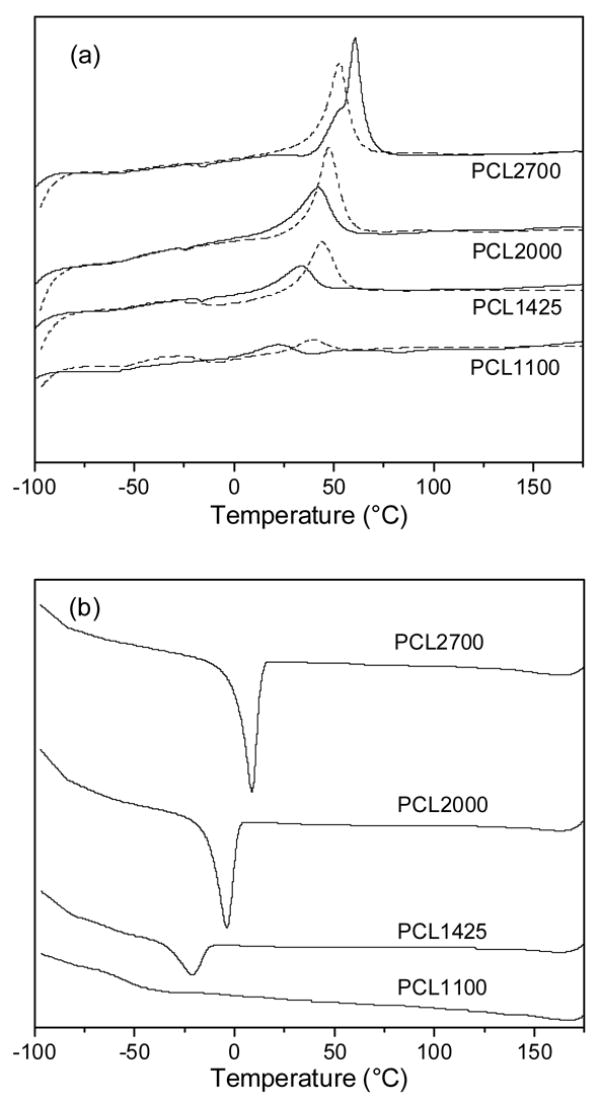
DSC analysis of polyurethanes: (a) heating curves; (b) cooling curve. Solid lines and dashed lines correspond to the first and second heating curves, respectively, of annealed polymer samples. A cooling curve was acquired between the first and second heating curves. Curves are offset vertically to permit visual comparison.
Fig. 4.

WAXS images of PEUURs synthesized from (a) 1100 Mw PCL and (b) 2700 Mw PCL.
DMA was performed on each material in order to monitor changes in the storage modulus (E′) and tan δ with temperature (Fig. 5). A decrease in E′ between −75 and −50°C was observed as the material was heated through the glass transition temperature (Tg). The value for Tg was reported as the location of the primary peak in Tan δ curve, which fell in the range of −55 to −50°C (Table 1) and is comparable to that of pure PCL (−63°C) [20]. A sharper decrease in E′ was observed in the temperature range of 0–60°C, and corresponds to melting of the PCL phase. The melting transitions in the DMA curves are consistent with the melting peaks observed by DSC (Fig. 3(a)), and the Tm decreases with decreasing PCL segment molecular weight. High molecular weight PCL has a melting temperature of 63°C, but melting point depression has been reported with decreasing PCL molecular weight [20]. The DMA curves also reveal that E′ increases systematically with PCL segment molecular weight below Tm, but decreases with increasing PCL segment molecular weight above Tm. This suggests that the high storage modulus observed at physiologic temperature (37°C) is primarily related to the crystallinity of the PCL segment. At this temperature storage moduli of 52, 49, 110 and 278 MPa were determined for PCL segments of 1100, 1425, 2000, and 2700, respectively (Table 1). Above the Tm for the PEUURs, tan δ remains relatively low, indicating that the polymer is not completely melted, and E′ increases systematically with hard segment content, suggesting that the storage modulus is dependent on microphase-separated hard domains.
Fig. 5.
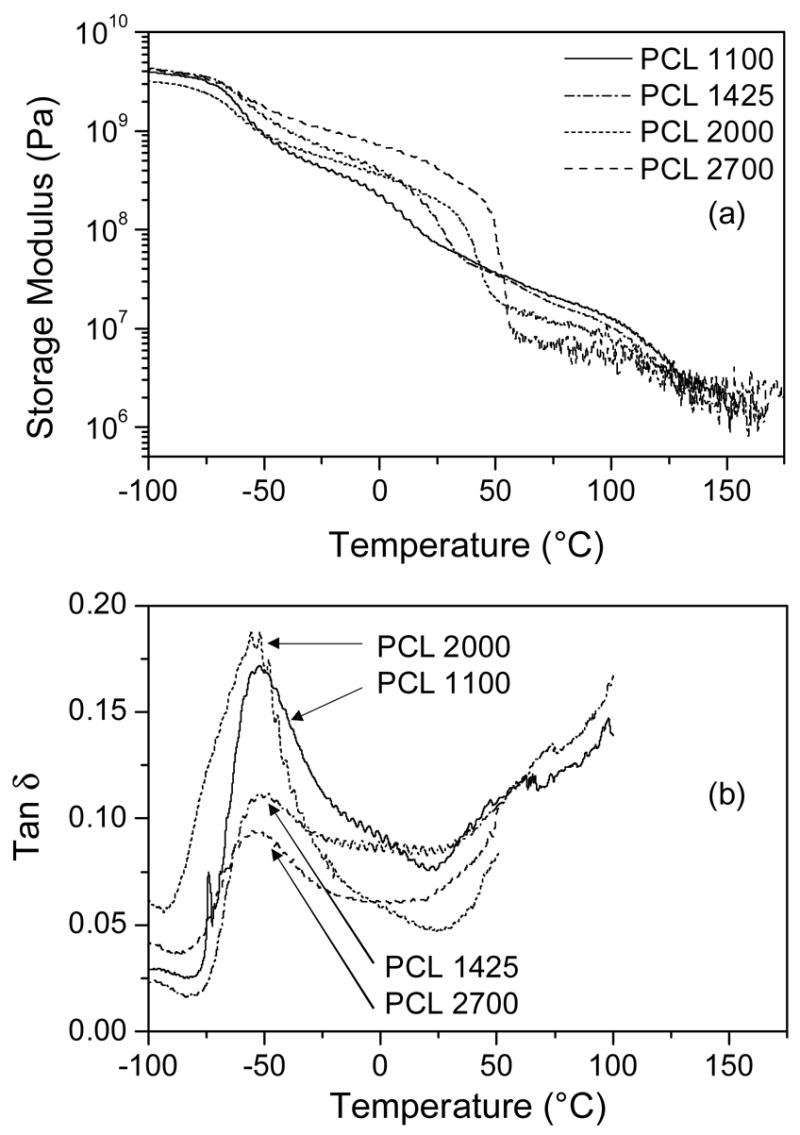
DMA analysis of polyurethanes: (a) storage modulus; (b) tan δ.
Measurements of advancing contact angles on spin-coated PEUUR films show a small but systematic increase in angle with PCL macrodiol Mw from 1100 to 2000 (Table 1), consistent with an increase in the content of the hydrophobic soft segment. However, the increase in advancing contact angle with PCL macrodiol Mw could also be a consequence of increasing surface roughness [27]. In contrast, the PEUUR with the highest PCL macrodiol Mw demonstrated the lowest contact angle, which suggests that microphase separation and crystallization of PCL segments may partition the hydrophilic TyA.BDI.TyA segments to the surface. By comparison, spin-coated PLGA (control) films exhibited an advancing contact angle of 74 ± 2, which is very similar to the PEUUR synthesized from 2700 Mw PCL.
3.2 BMSC proliferation and differentiation on PEEUR films
BMSCs were seeded onto fibronectin-coated PEUUR films and cultured under osteogenic conditions. They were collected at days 7 and 14 to assay for cell number and ALP activity, and at days 14 and 21 to assay for OPN and OCN expression. Cell number increased from 7 to 14 days on all surfaces, indicating that the PEUURs support cell adhesion and proliferation (Fig. 6), but cell numbers were not statistically different between PEUURs and the PLGA control. Similarly, ALP activity at day 14 was not statistical different between the PEUURs and PLGA (Fig. 7). Analysis of OPN accumulation into cell layers – as determined by Western blot analysis at day 14 – indicated that OPN was statistically lower on the PEUUR containing PCL 2700 (Fig. 8). However, OPN mRNA expression by PCR at days 14 and 21 indicated no statistically significant differences between the polymers (Fig. 9(a)). Likewise, analysis of OCN, another osteoblastic marker, indicated no differences between the polymers (Fig. 9(b)). Together, these data suggest that the PEUURs support osteoblastic differentiation to the same extent as PLGA. However, analysis of OCN synthesis and mineral deposition – definitive markers of osteoblastic maturation – are necessary to demonstrate differentiation.
Fig. 6.

Cell number on PEUUR films at 7 and 14 days. PLGA films were used as a reference. Data are mean ± standard error for n = 4 samples.
Fig. 7.

ALP activity of BMSCs on PEURR films at 14 days. PLGA films were used as a reference. Data are mean ± standard error for n = 4 samples. Activity per cell was determined by normalizing activity per cell layer by cell number.
Fig. 8.

OPN protein content of BMSC cell layers on PEURR films at 14 days. (a) OPN protein bands visualized by chemiluminescence. (b) Density of OPN bands normalized by GAPDH band density. PLGA films were used as a reference. Data are mean ± standard error for n = 8 samples. An asterisk denotes statistically different level of protein content with respect to PLGA (p<0.05).
Fig. 9.

(a) OPN and (b) OCN mRNA expression of BMSCs on PEURR films at 14 and 21 days. Expression was normalized to PLGA films at day 14. Data are mean ± standard error for n = 4 samples.
4. Discussion
In this study a family of segmented PEUURs was synthesized using PCL macrodiols and a novel TyA.BDI.TyA chain extender. By systematically varying the molecular weight of the PCL segment, the storage modulus, the soft segment Tm and the degree of crystallinity were systematically varied. In particular, the modulus at 37°C increased from 49 to 278 MPa with increasing PCL macrodiol Mw. Cell culture studies using BMSCs showed that all PEUURs supported cell viability, proliferation and osteoblastic differentiation.
One of the underlying motivations for this study was the development of segmented polyurethanes that degrade to non-toxic decomposition products. Polyurethanes and polyureas synthesized from aromatic diisocyanates, such as MDI, exhibit microphase-separated morphologies, ordered hard domains and useful mechanical properties [7]. In recent studies, polyureas synthesized from MDI, a diamine chain extender, and a PCL530 soft segment have been reported to degrade to non-cytotoxic decomposition products in vivo [28–30]. However, other studies have suggested that polyurethanes based on such aromatic polyisocyanates degrade in vivo to carcinogenic and mutagenic compounds [31–33]. Therefore, due to the potentially cytotoxic degradation products associated with MDI, we and others have sought to synthesize biodegradable segmented PEUUR elastomers from aliphatic polyisocyanates such as hexamethylene diisocyanate (HDI), BDI [14,16,18,34–38] and lysine diisocyanate [37,39,40]. To date, segmented PEUUR elastomers incorporating a PCL soft segment and aliphatic diisocyanates have been synthesized from a variety of chain extenders, including an adduct of butanediol and BDI [16,34,35], putrescine [14,36,41], lysine ethyl ester [14], and an adduct of phenylalanine and cyclohexanedimethanol [37,38,40]. In a previous study, we described the synthesis and characterization of a segmented PEUUR elastomer incorporating a hard segment comprising an adduct of tyramine and BDI [18]. This chain extender was designed with specific structural features to promote ordering of microphase-separated hard domains, including phenyl groups to induce π bond stacking and urea groups to establish bidentate hydrogen bonding [7].
However, thermal phase transitions of the hard segment – definitive evidence of microphase separation – were not detected by DSC (Fig. 3) in this study. We conjecture that the phase transitions for the hard segment lie above the decomposition temperature, which is consistent with observations for polyureas [7,42]. However, we note two indirect signs of phase separation of the hard segment. First, at temperatures greater than the Tm of the PCL segment the materials have moduli ranging from 10 to 40 MPa (Fig. 5), the moduli increase with increasing hard segment content and tan δ is relatively low. In the absence of hard segment ordering, the materials would be anticipated to have mechanical properties resembling a polymer melt (e.g. a storage modulus several orders of magnitude lower).
Second, for microphase-separated materials, each phase exhibits its own Tg. For phase-mixed systems, the value of Tg is related to the Tg of the individual components by the Fox equation [43,44]:
where subscripts 1 and 2 represent the individual segments, and Mi represents the mass fraction of segment i. Thus, a systematic shift in Tg with soft segment content would be consistent with the existence of a phase mixed system. However, the Tg for the PCL segment is similar to that for pure PCL (−63°C [20]) and does not vary with PCL content (Table 2), suggesting that the system is microphase-separated.
Table 2.
Glass transition temperatures, melting temperatures and Young’s modulus reported for PCL-based segmented PEUUR elastomers
| Polymer | Tg (°C) | Tm (°C) | Modulus (MPa) |
|---|---|---|---|
| BDI/BDA/PCL1250 [36] | −37 | A | 54 |
| BDI/BDA/PCL2000 [36] | −53 | 40 | 78 |
| BDI/Lys/PCL1250 [36] | −40 | A | 14 |
| BDI/Lys/PCL2000 [36] | −54 | 45 | 38 |
| LDI/Phe/PCL530 [38] | −6 | A | 6.6 |
| LDI/Phe/PCL1250 [38] | −34 | 43 | 54 |
| LDI/Phe/PCL2000 [38] | −52 | 45 | 82 |
| BDI/BDA/PCL2000 [34] | −57 | 20 | 52 |
| HDI/BDA/PCL2000 [34] | −51 | 22 | 38 |
| LDI/BDA/PCL2000 [34] | −52 | 41 | 40 |
| BDI/BDO.BDI.BDO/PCL2000 [16] | −54 | 18 | 70 |
An ‘a’ denotes amorphous polymers that did not exhibit a PCL melting temperature.
The thermal and mechanical properties of a broad variety of segmented PEUUR elastomers, which incorporate a PCL soft segment, are summarized in Table 2. We note that materials previously formed from a 2000 Mw PCL macrodiol exhibit glass transition temperatures ranging from −51 to −54°C, which is quite close to the value of −52°C measured in this study. However, materials incorporating a 1250 Mw PCL soft segment are reported to exhibit Tg values in the range from −34 to −40°C, suggesting a greater degree of phase mixing in these materials relative to the 2000 Mw PCL materials. It is interesting to note that the PEUURs in this study formed from 1100 and 1425 Mw PCL exhibited Tg values of −52°C. These data imply that the extent of microphase separation is not as sensitive to PCL molecular weight when tyramine-based hard segments are employed. In particular, we conjecture that the relatively large and hydrophilic TyA.BDI.TyA chain extender used in this study may drive microphase separation even when the PCL Mw is relatively low.
All four of the tyramine-based materials exhibit melting transitions ranging from 21 to 61°C (see Table 1), associated with the melting of the PCL diol soft segment. As predicted by the theory of melting point depression [43], the melting temperature decreases with decreasing PCL molecular weight. The 2000 Mw PCL PEUUR examined in this study had a Tm of 42°C, which falls within the narrow range of values (40–45°C) reported for other 2000 Mw PCL PEUUR elastomers (Table 2). In contrast, PEUURs synthesized from lower Mw PCL exhibit a broad range of properties: LDI/Phe/PCL1250 material melts at 43°C [38], while the BDI/BDA/PCL1250 and BDI/Lys/PCL1250 materials are amorphous [36]. Although we did not examine a material with 1250 Mw PCL, we note that our material formed from 1425 and 1100 Mw PCL fall within this range of properties, with melting temperatures of 34 and 21°C, respectively.
The storage moduli (at 37°C) of the materials prepared from 1100, 1425 and 2000 Mw PCL were in the range from 49 to 110 MPa, which is comparable to the values of Young’s modulus ranging from 14 to 82 MPa reported for other PCL-based segmented PEUUR elastomers (Table 2). The significantly higher modulus of the 2700 Mw PCL-based PEUUR (278 MPa) is attributed to the higher crystallinity of this PCL soft segment. At 37°C, the soft segment of this material is substantially below its melting temperature (61°C) and therefore semi-crystalline. This observation is also supported by the WAXS data. The significant change in the modulus observed over the temperature range of 20–60°C implies that over this temperature range the mechanical properties are dominated by the PCL soft segment.
Concurrent with the physical analysis of the PEUURs, biochemical analysis of BMSC was undertaken to verify biocompatibility of the polymers and to determine how systematic changes in soft segment content altered proliferation and development of the osteoblastic phenotype. It has been demonstrated that proliferation and phenotypic behavior of osteoblasts is sensitive to both interfacial chemistry and surface roughness. Gross changes in the chemistry at the biomaterial interface have been shown to alter surface hydrophilicity, protein adsorption curves and adsorbed protein conformation [45,46], and these factors likely act in concert to alter cell morphology, adhesion, proliferation and mineralization [47,48]. Concurrently, surface roughness. which can be introduced by annealing of polymer blends. has been shown to affect cell morphology, cell density and alkaline phosphatase activity [27,49]. Contact angle measurements (Table 1), which are sensitive to surface hydrophobicity, roughness and chemical heterogeneity, indicated only modest differences in the interfacial properties of the annealed polymer films. Although such differences may have influenced OPN deposition (Fig. 8), we note that cell proliferation as well as other biochemical markers of osteoblastic differentiation were not affected.
This research project establishes that a series of segmented PEUURs can be synthesized from PCL (soft) and TyA.BDI.TyA (hard) segments, exhibit a broad range of storage moduli at 37°C, and support proliferation and osteoblastic differentiation of bone marrow stromal cells. Our next step will be to use these PEUURs to form porous foam scaffolds with similar architectures but differing compressive moduli. Because we have shown here that BMSC density, ALP activity and mRNA expression of OPN and OCN are insensitive to variations in PEUUR hard segment content, we will use these foam scaffolds to determine how BMSC properties vary with scaffold modulus.
5. Conclusions
A series of poly(esterurethane-urea)s were synthesized from TyA.BDI.TyA and PCL macrodiols of molecular weights ranging from 1100 to 2700 with hard segment contents of 20–40 wt.%. DMA showed the materials had storage moduli ranging from 49 to 278 MPa at 37°C, and DSC and WAXS showed that storage modulus correlated with crystallinity of the soft (PCL) phase. BMSCs cultured on the different films exhibited similar cell densities, ALP activities, and mRNA expression of OCN and OPN to conventional PLGA, indicating the suitability of these materials for bone tissue engineering applications.
Acknowledgments
The authors would like to thank Dr Garth L. Wilkes, from the Department of Chemical Engineering at Virginia Tech, for assistance with the DSC, DMA and WAXS measurements and helpful conversations on materials characterization. In addition, the authors thank Thomas C. Ward, from the Department of Chemistry at Virginia Tech, for the use of goniometer and spin-coater. This project was funded by the National Institutes of Health (R21 AR015945).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Crane GM, Ishaug SL, Mikos AG. Bone tissue engineering. Nature Med. 1995;1:1322–1324. doi: 10.1038/nm1295-1322. [DOI] [PubMed] [Google Scholar]
- 2.Langer R, Vacanti JP. Tissue engineering. Science. 1993;260:920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 3.Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 4.Gisselfaelt K, Edberg B, Flodin P. Synthesis and properties of degradable poly(urethane urea)s to be used for ligament reconstructions. Biomacromolecules. 2002;3:951–958. doi: 10.1021/bm025535u. [DOI] [PubMed] [Google Scholar]
- 5.Spaans CJ, Belgraver VW, Rienstra O, De Groot JH, Veth RPH, Pennings AJ. Solvent-free fabrication of micro-porous polyurethane–amide and polyurethane–urea scaffolds for repair and replacement of the knee-joint meniscus. Biomaterials. 2000;21(23):2453–2460. doi: 10.1016/s0142-9612(00)00113-7. [DOI] [PubMed] [Google Scholar]
- 6.Lelah MD, Cooper JL. Polyurethanes in Medicine. Boca Raton, FL: CRC Press; 1987. [Google Scholar]
- 7.Oertel G. Polyurethane Handbook. Berlin: Hanser Gardner Publications; 1994. [Google Scholar]
- 8.Thomas V, Kumari TV, Jayabalan M. In vitro studies on the effect of physical cross-linking on the biological performance of aliphatic poly(urethane urea) for blood contact applications. Biomacromolecules. 2001;2:588–596. doi: 10.1021/bm010044f. [DOI] [PubMed] [Google Scholar]
- 9.Szycher M. Szycher’s Handbook of Polyurethanes. Boca Raton: CRC Press; 1999. [Google Scholar]
- 10.Hoffman D, Gong G, Pinchuk L, Sisto D. Safety and intracardiac function of a silicone–polyurethane elastomer designed for vascular use. Clin Mater. 1993;13:95–110. doi: 10.1016/0267-6605(93)90095-o. [DOI] [PubMed] [Google Scholar]
- 11.Capone CD. Biostability of a non-ether polyurethane. J Biomat Appl. 1992;7:108–129. doi: 10.1177/088532829200700202. [DOI] [PubMed] [Google Scholar]
- 12.Borkenhagen M, Stoll RC, Neuenschwander P, Suter UW, Aebischer P. In vivo performance of a new biodegradable polyester urethane system used a nerve guidance channel. Biomaterials. 1998;19:2155–2165. doi: 10.1016/s0142-9612(98)00122-7. [DOI] [PubMed] [Google Scholar]
- 13.de Groot JH, Zijlstra FM, Kuipers HW, Pennings AJ, Klompmaker J, Veth RP, Jansen HW. Meniscal tissue regeneration in porous 50/50 copoly(L-lactide/epsilon-caprolactone) implants. Biomaterials. 1997;18:613–622. doi: 10.1016/s0142-9612(96)00169-x. [DOI] [PubMed] [Google Scholar]
- 14.Guan J, Sacks MS, Beckman EJ, Wagner WR. Synthesis, characterization, and cytocompatibility of elastomeric, biodegradable poly(ester–urethane)ureas based on poly(caprolactone) and putrescine. J Biomed Mater Res. 2002;61:493–503. doi: 10.1002/jbm.10204. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J-Y, Beckman EJ, Hu J, Yang GG, Agarwal S, Hollinger JO. Synthesis, biodegradability, and biocompatibility of lysine diisocyanate–glucose polymers. Tissue Eng. 2002;8:771–785. doi: 10.1089/10763270260424132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spaans CJ, de Groot JH, Dekens FG, Pennings AJ. High molecular weight polyurethanes and a polyurethane urea based on 1,4-butate diisocyanate. Polymer Bulletin. 1998;41:131–138. [Google Scholar]
- 17.Szycher M. Biostability of polyurethane elastomers: a critical review. Journal of Biomaterials Applications. 1988;3:297. doi: 10.1177/088532828800300207. [DOI] [PubMed] [Google Scholar]
- 18.Guelcher SA, et al. Sythesis of biocompatible segemented polyurethanes from aliphatic diisocyanates and diurea diol chain extenders. Acta Biomateriala. 2005;1:471–484. doi: 10.1016/j.actbio.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 19.Bianco P, Riminucci M, Gronthos S, Robey PG. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19(3):180–192. doi: 10.1634/stemcells.19-3-180. [DOI] [PubMed] [Google Scholar]
- 20.Sawhney AS, Hubbell JA. Rapidly degraded terpolymers of D,L-lactide, glycolide, and e-caprolactone with increased hydrophilicity by copolymerization with polyethers. J Biomed Mater Res. 1990;24:1397–1411. doi: 10.1002/jbm.820241011. [DOI] [PubMed] [Google Scholar]
- 21.Badami AS, Kreke MR, Thompson MS, Riffle JS, Goldstein AS. Effect of fiber diameter on spreading, proliferation, and differentiation of osteoblastic cells on electrospun poly(lactic acid) substrates. Biomaterials. 2006;27:596–606. doi: 10.1016/j.biomaterials.2005.05.084. [DOI] [PubMed] [Google Scholar]
- 22.Kreke MR, Huckle WR, Goldstein AS. Fluid flow stimulates expression of osteopontin and bone sialoprotein by bone marrow stromal cells in a temporally dependent manner. Bone. 2005;36:1047–1055. doi: 10.1016/j.bone.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Porter RM, Huckle WR, Goldstein AS. Effect of dexamethasone withdrawal on osteoblastic differentiation of bone marrow stromal cells. J Cell Biochem. 2003;90(1):13–22. doi: 10.1002/jcb.10592. [DOI] [PubMed] [Google Scholar]
- 24.Goldstein AS. Effect of seeding osteoprogenitor cells as dense clusters on cell growth and differentiation. Tissue Eng. 2001;7:817–827. doi: 10.1089/107632701753337753. [DOI] [PubMed] [Google Scholar]
- 25.Ishaug SL, Crane GM, Miller MJ, Yasko AW, Yaszemski MJ, Mikos AG. Bone formation by three-dimensional stromal osteoblast culture in biodegradable polymer scaffolds. J Biomed Mater Res. 1997;36(1):17–28. doi: 10.1002/(sici)1097-4636(199707)36:1<17::aid-jbm3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 26.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 27.Lim JY, Hansen JC, Siedlecki CA, Runt J, Donahue HJ. Human foetal osteoblastic cell response to polymer-demixed nanotopographic interfaces. J R Soc Interface. 2005;2:97–108. doi: 10.1098/rsif.2004.0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liljensten E, et al. Studies of polyurethane urea bands for ACL reconstruction. J Material Sci: Mater Med. 2002;13:351–359. doi: 10.1023/a:1014380332762. [DOI] [PubMed] [Google Scholar]
- 29.Gisselfaelt K. Structure dependent chemical and biological interactions of poly(urethane urea)s. Göteborg: Chalmers University of Technology; 2002. [Google Scholar]
- 30.Gisselfaelt K, Edberg B, Flodin P. Synthesis and properties of degradable poly(urethane urea)s to be used for ligament reconstructions. Biomacromolecules. 2002;3:951–958. doi: 10.1021/bm025535u. [DOI] [PubMed] [Google Scholar]
- 31.Blais P. Letter to the editor. J Appl Biomater. 1990;1:197. [Google Scholar]
- 32.Coury A. In: Biomaterials Science: An Introduction to Materials in Medicine. Ratner B, Hoffman A, Schoen F, Lemons J, editors. Boston, MA: Elsevier Academic Press; 2004. pp. 411–430. [Google Scholar]
- 33.Szycher M, Siciliano A. An assessment of 2,4-TDA formation from Surgitek polyurethane foam under stimulated physiological conditions. J Biomater Appl. 1991;5:323–336. doi: 10.1177/088532829100500404. [DOI] [PubMed] [Google Scholar]
- 34.de Groot JH, de Vrijer R, Wildeboer BS, Spaans CJ, Pennings AJ. New biomedical polyurethane ureas with high tear strengths. Polymer Bulletin. 1997;38:211–218. [Google Scholar]
- 35.Spaans CJ, de Groot JH, Belgraver VW, Pennings AJ. A new biomedical polyurethane with a high modulus based on 1,4-butanediisocyanate and e-caprolactone. J Mater Sci Mater Med. 1998;9:675–678. doi: 10.1023/a:1008922128455. [DOI] [PubMed] [Google Scholar]
- 36.Guan J, Sacks MS, Beckman EJ, Wagner WR. Biodegradable poly(ether ester urethane)urea elastomers based on poly(ether ester) triblock copolymers and putrescine: synthesis, characterization and cytocompatibility. Biomaterials. 2004;25:85–96. doi: 10.1016/s0142-9612(03)00476-9. [DOI] [PubMed] [Google Scholar]
- 37.Skarja GA, Woodhouse KA. Synthesis and characterization of degradable polyurethane elastomers containing an amino-acid based chain extender. J Biomat Sci Polym Ed. 1998;9:271–295. doi: 10.1163/156856298x00659. [DOI] [PubMed] [Google Scholar]
- 38.Skarja GA, Woodhouse KA. Structure–property relationships of degradable polyurethane elastomers containing an amino acid-based chain extender. J Appl Polym Sci. 2000;75:1522–1534. [Google Scholar]
- 39.Bruin P, Veenstra GJ, Nijenhuis AJ, Pennings AJ. Design and synthesis of biodegradable poly(ester-urethane) elastomer networks composed of non-toxic building blocks. Makromol Chem, Rapid Commun. 1988;9:589–594. [Google Scholar]
- 40.Fromstein JD, Woodhouse KA. Elastomeric biodegradable polyurethane blends for soft tissue applications. Journal of Biomaterials Science Polymer Edition. 2002;13:391–406. doi: 10.1163/156856202320253929. [DOI] [PubMed] [Google Scholar]
- 41.Stankus JJ, Guan J, Wagner WR. Fabrication of biodegradable elastomeric scaffolds with sub-micron morphologies. J Biomed Mater Res. 2004;70A:603–614. doi: 10.1002/jbm.a.30122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herrington R, Hock K, editors. Flexible Polyurethane Foams. Freeport, TX: The Dow Chemical Company; 1997. [Google Scholar]
- 43.Sperling LH. Introduction to Physical Polymer Science. Hoboken, NJ: Wiley-Interscience; 2006. [Google Scholar]
- 44.Fox TG. Bull Am Phys Soc. 1956;1:123. [Google Scholar]
- 45.Keselowsky BG, Collard DM, García AJ. Surface chemistry modulates fibronectin conformation and directs integrin binding and specificity to control cell adhesion. J Biomed Mater Res. 2003;66A:247–259. doi: 10.1002/jbm.a.10537. [DOI] [PubMed] [Google Scholar]
- 46.Michael KE, Vernekar VN, Keselowsky BG, Meredith JC, Latour RA, García AJ. Adsorption-induced conformational changes in fibronectin due to interactions with well-defined surface chemistries. Langmuir. 2003;19:8033–8040. [Google Scholar]
- 47.Scotchford CA, Cooper E, Leggett GJ, Downes S. Growth of human osteoblast-like cells on alkanethiol on gold self-assembled monolayers: The effect of surface chemistry. J Biomed Mater Res. 1998;41:431–442. doi: 10.1002/(sici)1097-4636(19980905)41:3<431::aid-jbm13>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 48.Keselowsky BG, Collard DM, García AJ. Surface chemistry modulates focal adhesion composition and signaling through chages in integrin binding. Biomaterials. 2004;25:5947–5954. doi: 10.1016/j.biomaterials.2004.01.062. [DOI] [PubMed] [Google Scholar]
- 49.Meredith JC, Sormana JL, Keselowsky BG, García AJ, Tona A, Karin A, Amis EJ. Combinatorial characterization of cell interaction with polymer surfaces. J Biomed Mater Res. 2003;66A:483–490. doi: 10.1002/jbm.a.10004. [DOI] [PubMed] [Google Scholar]