

Summary
Background:
Several checkpoint pathways employ Wee1-mediated inhibitory tyrosine phosphorylation of cyclin-dependent kinases (CDKs) to restrain cell-cycle progression. Whereas in vertebrates this strategy can delay both DNA replication and mitosis, in yeast cells only mitosis is delayed. This is particularly surprising because yeasts, unlike vertebrates, employ a single family of cyclins (B-type) and the same CDK to promote both S phase and mitosis. The G2-specific arrest could be explained in two fundamentally different ways: tyrosine phosphorylation of cyclin/CDK complexes could leave sufficient residual activity to promote S phase, or S phase-promoting cyclin/CDK complexes could somehow be protected from checkpoint-induced tyrosine phosphorylation.
Results:
We demonstrate that in Saccharomyces cerevisiae several cyclin/CDK complexes are protected from inhibitory tyrosine phosphorylation, allowing Clb5,6p to promote DNA replication and Clb3,4p to promote spindle assembly, even under checkpoint-inducing conditions that block nuclear division. In vivo, S phase-promoting Clb5p/Cdc28p complexes were phosphorylated more slowly and dephosphorylated more effectively than were mitosis-promoting Clb2p/Cdc28p complexes. Moreover, we show that the CDK inhibitor (CKI) Sic1p protects bound Clb5p/Cdc28p complexes from tyrosine phosphorylation, allowing the accumulation of unphosphorylated complexes that are unleashed when Sic1p is degraded to promote S phase. The vertebrate CKI p27Kip1 similarly protects Cyclin A/Cdk2 complexes from Wee1, suggesting that the antagonism between CKIs and Wee1 is evolutionarily conserved.
Conclusions:
In yeast cells, the combination of CKI binding and preferential phosphorylation/dephosphorylation of different B cyclin/CDK complexes renders S phase progression immune from checkpoints acting via CDK tyrosine phosphorylation.
Keywords: CDK, Wee1, Cdc25, cyclin, SWE1, MIH1, CLB5, CLB2, CDC28
Introduction
The execution of cell cycle events is driven by cyclin-dependent kinases (CDKs) in association with regulatory cyclin subunits [1]. Cyclin/CDK activity is regulated by stoichiometric binding of CDK inhibitors (CKIs), as well as inhibitory tyrosine phosphorylation by the Wee1 family of kinases and dephosphorylation by the Cdc25 family of phosphatases [1]. The balance between Wee1 and Cdc25 activities is controlled by cell cycle checkpoint pathways, thereby linking cell cycle progression to successful completion of key events [2-4].
In vertebrates, Wee1 can delay both DNA replication and mitosis when cells experience DNA damage. Phosphorylation of Cyclin E/Cdk2 complexes inhibits DNA replication, whereas phosphorylation of Cyclin B/Cdk1 complexes inhibits mitotic entry [5-9], suggesting that Wee1 has broad specificity for divergent complexes. In yeasts, both S phase and nuclear division are triggered by B cyclins associated with a single CDK [1], yet checkpoints acting through inhibitory CDK phosphorylation appear to arrest the cell cycle exclusively in G2 [10-12]. This surprising difference from vertebrates suggests that yeasts are more selective in how they deploy Wee1.
In the budding yeast S. cerevisiae, the CDK Cdc28p can be activated by six B cyclins, Clb1-6p. Although these cyclins display a significant degree of redundancy, they are expressed and act at different times in the cell cycle, with Clb5,6p primarily triggering S phase and Clb1,2p primarily triggering nuclear division [13]. A single Wee1-family protein, Swe1p, and a single Cdc25-family protein, Mih1p, control Cdc28p phosphorylation at tyrosine 19 (Y19) [11, 14]. They act in the morphogenesis checkpoint to block nuclear division (but not DNA replication) in response to perturbations of the cytoskeleton or bud formation [12, 15].
To address why Swe1p affects mitosis but not S phase, we have evaluated the degree to which different cyclin/Cdc28p complexes undergo tyrosine phosphorylation under various circumstances. Our results indicate that S and M phase promoting cyclin/Cdc28p complexes are differentially regulated by Swe1p and Mih1p, and that binding of the CKI Sic1p protects Clb5p/Cdc28p complexes from inhibitory phosphorylation.
Results
Actin depolymerization blocks nuclear division but not DNA replication or spindle formation
Stresses, mutations, and drugs that delay bud emergence also delay nuclear division in a Swe1p-dependent manner (reviewed in [4]). The duration of the delay depends on the specific stimulus, and the strongest documented response is a prolonged arrest following treatment with the actin depolymerizing drug Latrunculin (LAT) [15]. Although the arrest of nuclear division is well-documented, there has not been a detailed analysis of the timing of earlier cell cycle events in response to this stress. In cells synchronized by pheromone arrest and release, LAT treatment blocked both bud emergence and nuclear division (Fig. 1A), but had no effect on the timing of DNA replication (Fig. 1B) and caused only a small (though reproducible) delay in spindle assembly (Fig. 1C). The inability of LAT to block DNA replication was observed even in cells synchronized by centrifugal elutriation, in which a longer G1 leaves >30 min between LAT addition and S phase (Fig. S2). Thus, DNA replication is completely unaffected, and spindle assembly is only slightly affected, in response to the strongest available checkpoint stimulus.
Figure 1. Effect of actin depolymerization on DNA replication, spindle assembly, and nuclear division.
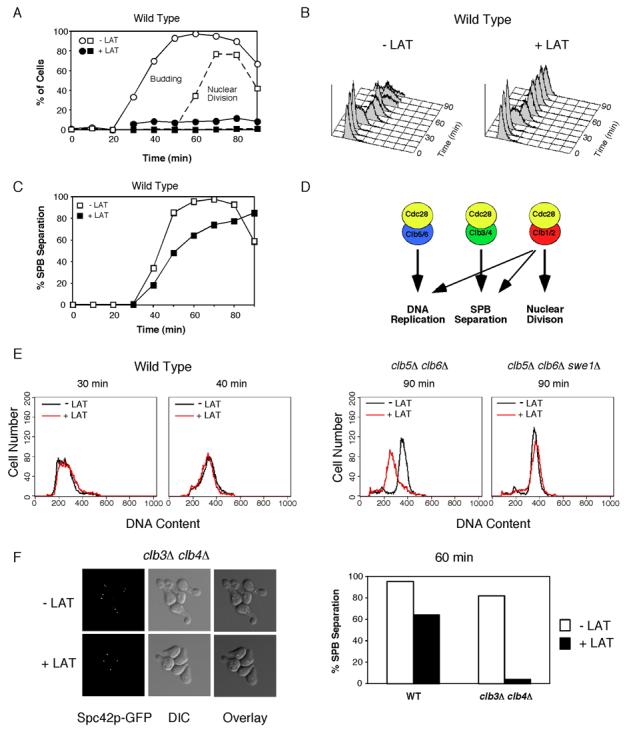
Cells were synchronized by pheromone arrest/release and part of the culture was treated with 150 μM LAT at 21 min after release from G1 arrest (just prior to budding). A) WT (DLY7974) cells were scored for budding (circles) and nuclear division (squares). The block of nuclear division was largely Swe1p dependent (Fig. S1A). B). LAT did not influence the timing of S phase, either in the presence or absence (Fig. S1B) of Swe1p as assessed by FACS analysis. C) Spindle formation was monitored by fluorescence microscopy to visualize the SPB marker Spc42-GFP. (n >200 cells). The small delay induced by LAT was largely Swe1p-dependent (Fig. S1C). D) Roles of different B-cyclin/Cdc28p complexes in the yeast cell cycle [18, 19, 21]. E) clb5Δ clb6Δ (DLY8300) and clb5Δ clb6Δ swe1Δ (DLY8963) cells were synchronized and DNA replication was monitored by flow cytometry (see Fig. S1D-E for synchrony data). Overlays of untreated (black) and LAT-treated (red) FACS profiles 90 min after release from G1 arrest. For comparison, WT FACS profiles during (30 min) and after (40 min) S phase are shown to illustrate lack of LAT effect in those cells (from data in B). F) WT (DLY7974) and clb3Δ clb4Δ (DLY8297) cells were synchronized and spindle formation was monitored as above (see Fig. S1G for synchrony data). Left, representative fluorescence and DIC images from clb3Δ clb4Δ cells. Right, >200 cells were scored for SPB separation at 60 min.
Specific Cdc28p complexes trigger DNA replication and spindle assembly in LAT-treated cells
The six B cyclins in S. cerevisiae can be subdivided into three highly related pairs [13]. Clb5,6p are expressed in late G1 and trigger DNA replication [17, 18], Clb3,4p are expressed in S and G2/M [19, 20] and are thought to trigger spindle assembly, and Clb1,2p are expressed in G2/M and are required for nuclear division [20, 21] (Fig. 1D). Clb1,2p can also trigger DNA replication and spindle assembly when either of the other cyclin pairs are absent, albeit after a delay (Fig. 1D and Fig. S1E,G; note that this implies that Clb5,6p are normally rate-limiting for DNA replication and Clb3,4p are normally rate-limiting for spindle assembly). However, we found that clb5Δclb6Δ cells treated with LAT failed to replicate their DNA by 90 min (Fig. 1E), although some replication occurred by 120 min (Fig. S1F); this replication delay was Swe1p-dependent (Fig. 1E). In addition, clb3Δclb4Δ cells treated with LAT failed to separate their SPBs (Fig. 1F). Thus, Clb5,6p complexes remain able to promote DNA replication, and Clb3,4p complexes remain able to promote spindle assembly following LAT treatment. The Clb1,2p complexes, which can normally compensate for loss of these cyclins, can no longer do so (or do so only after a prolonged delay) under these checkpoint-inducing conditions.
Selective phosphorylation of Clb/Cdc28p complexes
Our findings suggest that the specific block of nuclear division in response to LAT reflects selective inhibition of Clb1,2p/Cdc28p while Clb3-6p/Cdc28p remain active. In the simplest scenario, selective inhibition could be due to selective phosphorylation by Swe1p. Previous work showed that Swe1p could phosphorylate Clb2p/Cdc28p but not Cln2p/Cdc28p complexes in vitro, providing a precedent for cyclin specificity [11]. However, Cln2p is a highly diverged G1 cyclin that is only distantly related to Clb1-6p [22]. To assess the phosphorylation state of Cdc28p associated with the more closely related Clb1-6p cyclins, we used TAP-tagged cyclins expressed from their endogenous promoters. Specific cyclin/Cdc28p complexes were isolated from lysates of asynchronous cultures, and the relative level of phosphorylation on the associated Cdc28p was evaluated by Western blot. Due to their low abundance, Cln3p, Clb1p, and Clb6p were omitted from the analysis. Individual blots were probed simultaneously with primary antibodies recognizing phospho-Y19-Cdc28p (pY19) or total Cdc28p (PSTAIRE), followed by fluorescently labeled secondary antibodies. No phosphorylation was detected in cells lacking Swe1p, confirming that the pY19 signal is specific for Swe1p-catalyzed Cdc28p phosphorylation (Fig. 2A). The ratio of pY19-to-PSTAIRE measured using a fluorescent scanner displayed a linear relation to the input phosphorylation ratio, validating the assay (Fig. 2B).
Figure 2. Selective phosphorylation of B cyclin/Cdc28p complexes.
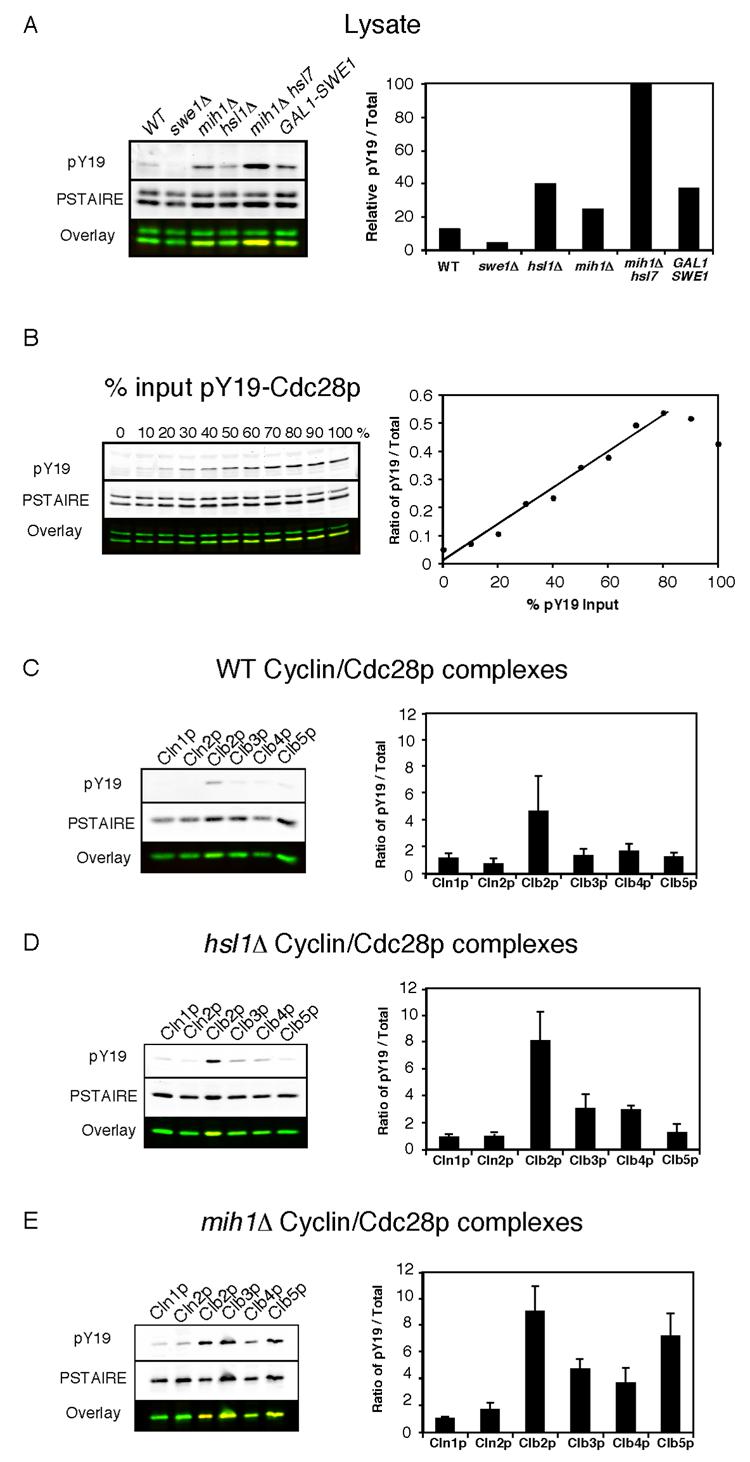
A) Y19 phosphorylation of the complete cellular pool of Cdc28p from WT (DLY7598), swe1Δ (DLY7712), hsl1Δ (DLY7944), mih1Δ (DLY7854), mih1Δ hsl7-OFF (DLY8197), and GAL1-SWE1 (DLY7946) strains. The first four strains were harvested during exponential growth, and the other strains were arrested by addition of dextrose (lane 5) or galactose (lane 6) as described in the Materials and Methods. The graph shows the ratio of phosphorylated (pY19)-to-total Cdc28p (PSTAIRE) for each strain, normalized to the level in mih1Δ hsl7-OFF arrested cells (this is the maximal experimentally obtained phosphorylation, but does not represent 100% phosphorylated Cdc28p because only B cyclin-bound Cdc28p is phosphorylated). Note that the anti-PSTAIRE antibody recognizes Pho85p (upper band) as well as Cdc28p (lower band) in whole cell lysates. B) Linearity of the Cdc28p phosphorylation measurement. Lysate from swe1Δ cells (DLY7712), lacking Cdc28p Y19 phosphorylation, was mixed with an increasing proportion of lysate from arrested mih1Δ hsl7-OFF cells (DLY8197), in which Cdc28p is maximally phosphorylated (the total amount of protein was equal in each lane). The ratio of pY19-to-total Cdc28p signal was plotted against the percentage of input lysate from the mih1Δ hsl7-OFF strain. C-E) The indicated cyclin/Cdc28p complexes were isolated from the following strains harvested during exponential growth: C) WT (DLY7646, DLY7647, DLY7598, DLY7648, DLY7599, DLY7662); D) hsl1Δ (DLY7735, DLY7718, DLY7873 DLY7770, DLY7749); E) mih1Δ (DLY7857, DLY7717, DLY7854, DLY7858, DLY7860, DLY7786). The ratio of pY19-to-total Cdc28p was quantitated for both the purified complexes and the complete cellular pool of Cdc28p in lysates for each experiment. To account for gel-to-gel variability, the results from individual experiments were normalized by dividing the ratio for each purified complex by the average ratio obtained from lysates used in that experiment. The average normalized ratios for each cyclin/Cdc28p complex are displayed in arbitrary units as mean +/− SEM (n = 3). (pY19, red; PSTAIRE, green)
In wild-type cells, there was a low basal level of Cdc28p Y19 phosphorylation (Fig. 2A), which was only detectable on Clb2p complexes (Fig. 2C). Thus, phosphorylation is indeed selective for Clb2p/Cdc28p under unperturbed conditions. Because of the large number of cells required for this assay, assessing cyclin-associated Cdc28p phosphorylation following LAT treatment was not economically feasible. However, we could mimic partial checkpoint activation by deleting HSL1, a negative regulator of Swe1p [23]. Hsl1p-mediated Swe1p degradation is blocked by stresses that block bud emergence [24, 25] and hsl1Δ mutants exhibit elevated levels of Cdc28p Y19 phosphorylation (Fig. 2A). Nevertheless, these cells also showed selective phosphorylation of Clb2p/Cdc28p (Fig. 2D). A lower degree of phosphorylation was detected on Clb3,4p complexes, while no appreciable phosphorylation was detected on Clb5p or Cln1,2p complexes.
The cyclin-selective phosphorylation we observed could be due to a preference of Swe1p for Clb2p complexes, or a preference of Mih1p for other complexes, or both. To assess the contribution of Mih1p, we examined Cdc28p phosphorylation in mih1Δ cells (Fig. 2E). Strikingly, these cells exhibited comparable levels of Y19 phosphorylation in Clb5p and Clb2p complexes, indicating that Clb5p/Cdc28p is normally maintained in an unphosphorylated state by Mih1p. There was also significant phosphorylation of Clb3,4p complexes in mih1Δ cells, though reproducibly at somewhat lower levels than those of Clb2p and Clb5p complexes. In aggregate, these data indicate that the Swe1p/Mih1p system is “tuned” to selectively promote phosphorylation of Clb2p-associated Cdc28p in vivo, providing an explanation for why LAT blocks nuclear division but not DNA replication or spindle assembly.
LAT treatment of mih1Δ cells does not block DNA replication
As Cdc28p associated with Clb5p became phosphorylated at a stoichiometry comparable to that of Cdc28p associated with Clb2p in mih1Δ cells (Fig. 2E), we expected that LAT treatment of mih1Δ cells would inhibit DNA replication. However, LAT treatment had absolutely no effect on the timing of DNA replication in mih1Δ cells (Fig. 3). Similar results were obtained using mih1Δ cells synchronized by centrifugal elutriation (Fig. S2). Thus, even without the protection conferred by Mih1p, Clb5p complexes retain the ability to promote DNA replication following actin stress.
Figure 3. LAT does not affect DNA replication even in the absence of Mih1p.

Cells were synchronized as in Fig. 1. A) Budding and nuclear division in clb6Δ (DLY8245 ○), clb6Δ swe1Δ (DLY8261, ), and clb6Δ mih1Δ (DLY8441, ) cells were monitored by fluorescence microscopy (n >200). B) DNA replication was monitored by flow cytometry. C) FACS profiles for untreated (black) and LAT treated (red) clb6Δ mih1Δ cells 30 min and 40 min after release from G1 arrest.
Clb5p/Cdc28p kinase activity is inhibited by Cdc28p Y19 phosphorylation
Why does Clb5p-induced DNA replication proceed unimpeded (Fig. 3) despite the Y19 phosphorylation of Clb5p-associated Cdc28p (Fig. 2) in mih1Δ cells? The simplest way to explain this observation would be that in contrast to Clb2p/Cdc28p, Clb5p/Cdc28p activity is not inhibited by Y19 phosphorylation. To test this possibility, we compared the kinase activities of Y19-phosphorylated and unphosphorylated Clb5p/Cdc28p and Clb2p/Cdc28p. We initially assumed that stoichiometric phosphorylation would best be achieved using overexpression of Swe1p, but found that such overexpression resulted in an aberrant failure to phosphorylate Clb2p/Cdc28p (Fig. S3 and supplemental results/discussion). To induce a cell cycle arrest mediated by endogenous Swe1p, we generated strains lacking Mih1p and expressing Hsl7p (which acts together with Hsl1p to promote Swe1p degradation [23]) from the regulatable GAL1 promoter. Upon transfer to dextrose medium (mih1Δ hsl7-OFF), Hsl7p expression is shut off, the protein is depleted, and the cells arrest with elongated buds (due to the inability of Clb1,2p/Cdc28p to trigger the apical-to-isotropic growth switch [26]). Under these conditions, Clb2p and Clb5p complexes are highly phosphorylated (Fig. 4A; >80% by 2D gel analysis: Fig. S3D) . We treated complexes purified from arrested cells with recombinant human Cdc25 to dephosphorylate Y19. Histone H1 kinase activities were then determined for equal amounts of phosphorylated (mock treated) and dephosphorylated (hCdc25 treated) Clb5p and Clb2p complexes. Y19 dephosphorylation clearly stimulated both Clb2p/Cdc28p and Clb5p/Cdc28p histone H1 kinase activity (Fig. 4B). Based on the degree of stimulation, Y19 phosphorylation had a greater than six-fold inhibitory effect on Clb2p/Cdc28p. Western blotting confirmed that hCdc25 treatment promoted efficient Cdc28p Y19 dephosphorylation without affecting the total amount of Cdc28p present (Fig. 4A). In addition, control experiments showed that hCdc25 treatment had no effect on unphosphorylated Cdc28p complexes from swe1Δ cells, so any effects can be ascribed to Y19 dephosphorylation (unpublished results). Thus, Y19 phosphorylation clearly inhibits Clb5p/Cdc28p as well as Clb2p/Cdc28p kinase activity in budding yeast.
Figure 4. Tyrosine phosphorylation inhibits both Clb5p- and Clb2p-associated kinase activity.

A) mih1Δ GAL1-HSL7 cells (DLY8197 and DLY8199) were grown in dextrose-containing medium for 22 h to deplete Hsl7p (mih1Δ hsl7-OFF). Clb2p/Cdc28p and Clb5p/Cdc28p complexes were isolated, treated with or without recombinant hCdc25, and the ratio of pY19-to-total Cdc28p was assessed. B) Phosphorylated (mock treated, □) and dephosphorylated (hCDC25 treated, ) Cdc28p complexes were assayed for their ability to phosphorylate histone H1 in vitro. The amount of 32P incorporation was normalized to the amount of Cdc28p present in the reaction and is plotted in arbitrary units (mean of two replicates).
The CKI Sic1p protects Clb5p/Cdc28p complexes from Y19 phosphorylation
If Clb5p/Cdc28p complexes can become phosphorylated at high stoichiometry in mih1Δ cells, and this phosphorylation inhibits their kinase activity, then how is it that they can still promote timely DNA replication? One possibility is that Clb5p/Cdc28p action is more rapid than Clb5p/Cdc28p inhibition by Swe1p, providing a window of opportunity for DNA replication before Clb5p complexes are inactivated. Slow inhibition by Swe1p could be due to intrinsically slow phosphorylation of Clb5p/Cdc28p (see below) and/or to initial protection of Clb5p/Cdc28p from Swe1p by the CKI Sic1p [13]. The sudden, switch-like degradation of Sic1p at G1/S [27, 28] could then unleash a stockpile of unphosphorylated Clb5p/Cdc28p.
To test whether Sic1p can protect Clb5p/Cdc28p complexes from Y19 phosphorylation, we examined the phosphorylation state of Clb5p-associated Cdc28p in mih1Δ cells that were released from pheromone arrest in the presence or absence of a stabilized form of Sic1p (Sic1Δ4p) [27]. As shown in Fig. 5B, Clb5p-associated Cdc28p remained completely unphosphorylated when stabilized Sic1p was present, suggesting that Sic1p degradation must normally precede Swe1p-mediated Clb5p/Cdc28p phosphorylation.
Figure 5. CKIs protect bound CDK from tyrosine phosphorylation.

A) Representative DIC images of mih1Δ (DLY8700) and GAL1-SIC1Δ4p mih1Δ (DLY8701) cells grown in YEPS (SUC) and induced to express Sic1p by addition of galactose for 5 h (GAL). B) Cells from the same strains were grown in YEPS and arrested in G1 with pheromone. Sic1pΔ4p expression was induced with galactose 30 min prior to release from the G1 arrest and maintained following release into YEPG. Cdc28p Y19 phosphorylation was assessed for Clb5p/Cdc28p complexes isolated from cells collected 75 min after release when the majority of the cells had budded. C) Purified Cyclin A/Cdk2 was incubated with GST-Wee1 and γ-32P-ATP in the presence or absence of p27Kip1 as described in the Materials and Methods. Phosphorylated Cdk2 was visualized using a phosphoimager.
In principle, the protection conferred by Sic1p could be direct (Sic1p binding occludes Y19 so that Swe1p cannot phosphorylate it) or indirect (stabilized Sic1p somehow leads to Swe1p inactivation). We have not been able to generate highly active Swe1p for in vitro studies to test the direct protection model. However, Sic1p is functionally analogous to the p21/p27 family of mammalian CKIs and is thought to share a similar structure in the CDK inhibitory domain [29]. Structural considerations suggest that p27Kip1 binding would prevent access of Wee1 to the target CDK tyrosine residue [30]. Indeed, the addition of p27Kip1 completely inhibited all observable phosphorylation of Cdk2 by Wee1 in vitro (Fig. 5C). Taken together, these findings suggest that CKI binding directly protects the bound cyclin/CDK complex from phosphorylation by Wee1 in both yeast and animal cells.
Swe1p phosphorylates Clb2p and Clb5p complexes at different rates in vivo
Slow phosphorylation of Clb5p/Cdc28p by Swe1p following Sic1p degradation may contribute to a “window of opportunity”. To assess how rapidly Clb5p/Cdc28p becomes phosphorylated following Sic1p degradation, we devised an in vivo kinase assay. The assay involves monitoring the phosphorylation of Clb5p/Cdc28p (and for comparison, Clb2p/Cdc28p) following activation of a conditional Swe1p in cells lacking Mih1p. Although the absolute rate of phosphorylation is influenced by the kinetics of activation of the conditional Swe1p, this assay has the advantage over standard in vitro assays that the kinase, substrate, and ATP concentrations are all physiological, and that the reaction occurs in its natural milieu.
To isolate a conditional allele of SWE1, we screened for swe1 mutants that would arrest the cell cycle of hsl1Δ mih1Δ cells at 24°C but not 37°C (see Materials and Methods). We isolated swe1-12, which contains a single point mutation changing E792 to Q. Cells harboring this allele and lacking both Hsl1p and Mih1p proliferate well at 37°C (when Swe1-12p is inactive), but arrest upon shift to 24°C (Fig. 6A). In this context, there is very little Y19 phosphorylation at 37°C, and activation of Swe1-12p following shift-down leads to a progressive increase in Cdc28p phosphorylation (Fig. 6B). By isolating Clb2p-TAP or Clb5p-TAP complexes from hsl1Δ mih1Δ swe1-12 cells, the rate of phosphorylation of the associated Cdc28p following shift-down could be assessed. To ensure that Swe1p, Clb5p, and Clb2p levels were all high and that Sic1p was absent, we arrested the cells in S phase using hydroxyurea (HU) prior to temperature shift. As shown in Fig. 6C, Clb5p complexes were phosphorylated much more slowly than Clb2p complexes after shift to 24°C. Thus, slow phosphorylation may combine with the initial Sic1p-mediated protection of Clb5p/Cdc28p complexes to allow DNA replication even in mih1Δ cells treated with LAT.
Figure 6. Swe1p phosphorylates Clb2p/Cdc28p more rapidly than Clb5p/Cdc28p.

A) swe1-12 hsl1Δ mih1Δ cells containing Clb2p-TAP (DLY8726) or Clb5p-TAP (DLY8727) were grown at 37°C and shifted to 24°C for 5 h and processed for DIC imaging. B) Cells from those strains were grown in 0.5 M HU at 37°C for 5 h and shifted to 24°C in the continued presence of HU (>90% large-budded arrest). Cdc28p phosphorylation was assessed by Western blot analysis of TCA-precipitated total cellular protein. C) Clb2p and Clb5p complexes were isolated from cells in B at the indicated times after shift to 24°C, and Cdc28p Y19 phosphorylation was assessed. D) The ratio of pY19-to-total Cdc28p was determined for the purified Cdc28p complexes shown in C.
Discussion
Yeast cells live in a fluctuating environment in which temperature, osmolarity, nutrients, and toxins can change rapidly as a result of the day/night cycle, weather patterns, and other organisms. Many such changes provoke a transient stress response that depolarizes the actin cytoskeleton, halts bud growth, and engages the morphogenesis checkpoint [4]. Under these circumstances it is clearly advantageous to delay nuclear division until the bud is ready to receive a daughter nucleus. In contrast, there is no evident benefit in delaying DNA replication once cells have committed to enter the cell cycle; indeed, lengthening S phase might render cells vulnerable to DNA damaging agents. Consistent with these suppositions, the morphogenesis checkpoint delays nuclear division without affecting S phase ([12, 32] and this work).
Specificity in the function and phosphorylation of different B cyclin/Cdc28p complexes
By deleting different cyclins and subjecting cells to actin stress, we found that, unlike in lab-grown cells where Clb1-6p have overlapping roles [17-20], in actin-stressed cells only Clb5,6p promote DNA replication and only Clb3,4p promote spindle assembly. Thus, Clb3-6p retain their biological function in stressed conditions, presumably due to protection of the associated Cdc28p from tyrosine phosphorylation. Indeed, in hsl1Δ cells, which mimic partial activation of the checkpoint, only a low level of inhibitory phosphorylation was observed on Clb3-6p/Cdc28p. Using mih1Δ and swe1-Ts mutant strains and assaying phosphorylation stoichiometry on different Cdc28p complexes in vivo, we found that the low phosphorylation of Clb5p/Cdc28p relative to Clb2p/Cdc28p involved both reduced Swe1p-mediated phosphorylation and enhanced Mih1p-mediated dephosphorylation of Clb5p/Cdc28p complexes.
The differences in the extent of phosphorylation we detected could arise from intrinsic biochemical preferences on the part of Swe1p and Mih1p for different cyclin/Cdc28p complexes, the presence of binding proteins that affect access of Swe1p/Mih1p to specific substrates, and/or the degree to which Swe1p/Mih1p and substrate complexes are co-expressed in time and space. The latter hypothesis appears unlikely to account for our results. First, Swe1p and Clb5p both accumulate in late G1/S phase, while Clb2p accumulates in G2/M and Mih1p is present constitutively ([13], unpublished results). This expression pattern would favor phosphorylation of Clb5p/Cdc28p rather than Clb2p/Cdc28p. Second, Swe1p, Clb5p, and Clb2p are all concentrated in the nucleus [33-35], while Mih1p is predominantly cytoplasmic for most of the cell cycle (except late anaphase: unpublished results). Swe1p and Clb2p are also localized at the mother-bud neck [33, 35, 36]. Thus, Mih1p would have greater access to Clb2p/Cdc28p than Clb5p/Cdc28p, yet we observed that Mih1p specifically keeps Clb5p/Cdc28p dephosphorylated despite the reduced access. These considerations imply that substrate preference and/or other regulators account for the specificities we have documented.
One regulator that we identified as influencing Cdc28p tyrosine phosphorylation is the CKI Sic1p. Sic1p can bind to various Clb/Cdc28p complexes in vitro [37], but in vivo it binds primarily to Clb5p and Clb6p complexes, because it is degraded before the other B cyclins accumulate [38]. We found that stabilized Sic1p protected Clb5p/Cdc28p from Swe1p-mediated phosphorylation in vivo. The structural basis for inhibition of Clb5p/Cdc28p by Sic1p is thought to be similar to that of the mammalian CKI p27Kip1 [29], whose crystal structure has been solved in complex with Cyclin A/Cdk2 [30]. In this structure, p27Kip1 exists in an extended conformation and makes multiple interactions with both the kinase and cyclin subunits. The region surrounding the site of phosphorylation (Tyr15 of Cdk2) by Wee1 is particularly distorted compared to the free Cyclin A/Cdk2 complex. We found that stoichiometric amounts of p27Kip1 completely abrogated the ability of Wee1 to phosphorylate Cyclin A/Cdk2 in vitro. This suggests that CKI binding protects cyclin/CDK complexes from inhibitory phosphorylation by perturbing the recognition site for the inhibitory kinases, in both yeast and mammalian cells.
In aggregate, our results highlight several differences in the regulation of individual B cyclin/CDK complexes that can explain why the cell cycle arrest by the morphogenesis checkpoint affects nuclear division but not DNA replication. Clb5p is synthesized in late G1 and associates with abundant free Cdc28p and Sic1p [13]. Sic1p protects this complex from Swe1p until the Sic1p is degraded at G1/S. Clb5p/Cdc28p complexes are now susceptible to phosphorylation by Swe1p, but are rapidly dephosphorylated by Mih1p so that net phosphorylation remains low. In the absence of Mih1p (or if Mih1p is inhibited by the checkpoint), Clb5p/Cdc28p becomes heavily phosphorylated, but this takes a considerable amount of time, leaving a window between Sic1p degradation and inhibitory phosphorylation that suffices to allow Clb5p/Cdc28p to trigger DNA replication. In contrast to Clb5p, Clb2p is synthesized in late S/G2 when Sic1p is absent, and is phosphorylated by Swe1p more rapidly than is Clb5p/Cdc28p. Thus, in stressful conditions that trigger the morphogenesis checkpoint Clb2p/Cdc28p complexes are rapidly inhibited, thereby preventing nuclear division.
To what degree does tyrosine phosphorylation inhibit B cyclin/CDK activity?
In the model yeasts S. cerevisiae and Schizosaccharomyces pombe, both DNA replication and mitosis are triggered by members of the B cyclin family in complex with a single CDK: S. pombe has three such cyclins and S. cerevisiae has six [13, 39]. In both yeasts, a single B cyclin can drive both S and M phase in appropriately engineered mutants [40, 41], which raises the question of why these events don't happen simultaneously. An appealing answer, suggested by Stern and Nurse, is that while low levels of B cyclin/CDK activity suffice to trigger S phase, higher levels are required to trigger mitosis [42]. In S. pombe, mitosis is restrained by Wee1-mediated inhibitory phosphorylation of B cyclin/CDK complexes, but residual kinase activity can be detected and suffices to trigger DNA replication. It is unclear whether the residual activity in that case is intrinsic to tyrosine-phosphorylated cyclin/CDK complexes, or is due to a subpopulation of unphosphorylated complexes.
The phospho-mimetic allele, cdc28-Y19E, retains detectable kinase activity in vitro and can support DNA replication in S. cerevisiae [16], suggesting that even fully phosphorylated Cdc28p could initiate DNA replication, with the important caveat that the Y19E mutation may not accurately mimic phosphorylated Cdc28p. In our experiments, we found that under conditions that promote Swe1p-mediated Cdc28p phosphorylation, Clb1,2p/Cdc28p no longer promoted S phase, spindle assembly, or nuclear division. Consistent with this result, Aparicio and colleagues reported that DNA replication driven by a prematurely expressed Clb2p (instead of the normal Clb5p) was delayed by Swe1p [31]. Thus, it would appear that at least Clb1,2p complexes do not retain a biologically relevant kinase activity in the phosphorylated state.
We found that, like Clb2p/Cdc28p, Clb5p/Cdc28p in vitro kinase activity was inhibited by Y19 phosphorylation. Given the diversity of cyclin/CDK complexes that are clearly inhibited by tyrosine phosphorylation in metazoans and structural considerations suggesting that phosphorylation acts by reducing ATP binding and/or catalysis [43, 44], it seems likely that tyrosine phosphorylation universally inhibits CDK activity.
Conclusions
A number of studies have suggested that the various B cyclins in S. cerevisiae differ both in the effectiveness with which they promote particular events and in the manner in which they are regulated [45-49], providing an opportunity to fine-tune the cell cycle under different conditions. Our findings demonstrate that differences in the regulation of specific B cyclin/Cdc28p complexes by Swe1p and Mih1p tailor the cell cycle response of yeast cells to actin stress, permitting DNA replication and spindle assembly while blocking nuclear division.
In contrast to yeasts, metazoans employ different classes of cyclin acting with different CDKs to promote S phase and mitosis. Moreover, metazoans are known to employ Wee1-mediated CDK phosphorylation to delay DNA replication as well as mitosis [3], suggesting that there is less target specificity on the part of metazoan Wee1/Cdc25 than that exhibited by yeast Swe1p/Mih1p. These differences raise interesting questions regarding the evolution of cell cycle regulators: When was specificity in the action of Wee1/Cdc25 acquired (or lost) during evolution? Why did different classes of cyclins evolve? Did changes in cyclin functional specialization co-evolve with changes in their regulation by tyrosine phosphorylation? Further investigation of these issues should shed light on the evolution of cell cycle control strategies.
Supplementary Material
Acknowledgments
We thank Sally Kornbluth, Steve Haase, Dave MacAlpine, Audrey Howell, Lukasz Kozubowski, and Matt Cohen for critical comments on the manuscript. Thanks to Steve Haase and members of the Lew lab for many stimulating discussions. We also thank Steve Haase for providing the SPC42-GFP and GAL1-SIC1Δ4P constructs. This work was supported by NIH/NIGMS grant GM53050 to DJL.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 2.Humphrey T. DNA damage and cell cycle control in Schizosaccharomyces pombe. Mutat. Res. 2000;451:211–226. doi: 10.1016/s0027-5107(00)00051-8. [DOI] [PubMed] [Google Scholar]
- 3.Donzelli M, Draetta GF. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003;4:671–677. doi: 10.1038/sj.embor.embor887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lew DJ. The morphogenesis checkpoint: how yeast cells watch their figures. Curr. Opin. Cell Biol. 2003;15:648–653. doi: 10.1016/j.ceb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Falck J, Mailand N, Syljuasen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 6.Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J. Rapid destruction of human Cdc25A in response to DNA damage. Science. 2000;288:1425–1429. doi: 10.1126/science.288.5470.1425. [DOI] [PubMed] [Google Scholar]
- 7.Costanzo V, Robertson K, Ying CY, Kim E, Avvedimento E, Gottesman M, Grieco D, Gautier J. Reconstitution of an ATM-dependent checkpoint that inhibits chromosomal DNA replication following DNA damage. Mol. Cell. 2000;6:649–659. doi: 10.1016/s1097-2765(00)00063-0. [DOI] [PubMed] [Google Scholar]
- 8.Jin P, Gu Y, Morgan DO. Role of inhibitory CDC2 phosphorylation in radiation-induced G2 arrest in human cells. J. Cell. Biol. 1996;134:963–970. doi: 10.1083/jcb.134.4.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poon RY, Chau MS, Yamashita K, Hunter T. The role of Cdc2 feedback loop control in the DNA damage checkpoint in mammalian cells. Cancer. Res. 1997;57:5168–5178. [PubMed] [Google Scholar]
- 10.Rhind N, Furnari B, Russell P. Cdc2 tyrosine phosphorylation is required for the DNA damage checkpoint in fission yeast. Genes Dev. 1997;11:504–511. doi: 10.1101/gad.11.4.504. [DOI] [PubMed] [Google Scholar]
- 11.Booher RN, Deshaies RJ, Kirschner MW. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993;12:3417–3426. doi: 10.1002/j.1460-2075.1993.tb06016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lew DJ, Reed SI. A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J. Cell Biol. 1995;129:739–749. doi: 10.1083/jcb.129.3.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lew DJ, Weinert T, Pringle JR. Cell cycle control in Saccharomyces cerevisiae. Vol. 3. Cole Spring Harbor Laboratory Press; Plainview, N.Y.: 1997. [Google Scholar]
- 14.Russell P, Moreno S, Reed SI. Conservation of mitotic controls in fission and budding yeasts. Cell. 1989;57:295–303. doi: 10.1016/0092-8674(89)90967-7. [DOI] [PubMed] [Google Scholar]
- 15.McMillan JN, Sia RA, Lew DJ. A morphogenesis checkpoint monitors the actin cytoskeleton in yeast. J. Cell Biol. 1998;142:1487–1499. doi: 10.1083/jcb.142.6.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim HH, Goh PY, Surana U. Spindle pole body separation in Saccharomyces cerevisiae requires dephosphorylation of the tyrosine 19 residue of Cdc28. Mol. Cell. Biol. 1996;16:6385–6397. doi: 10.1128/mcb.16.11.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Epstein CB, Cross FR. CLB5: a novel B cyclin from budding yeast with a role in S phase. Genes Dev. 1992;6:1695–1706. doi: 10.1101/gad.6.9.1695. [DOI] [PubMed] [Google Scholar]
- 18.Schwob E, Nasmyth K. CLB5 and CLB6, a new pair of B cyclins involved in DNA replication in Saccharomyces cerevisiae. Genes Dev. 1993;7:1160–1175. doi: 10.1101/gad.7.7a.1160. [DOI] [PubMed] [Google Scholar]
- 19.Fitch I, Dahmann C, Surana U, Amon A, Nasmyth K, Goetsch L, Byers B, Futcher B. Characterization of four B-type cyclin genes of the budding yeast Saccharomyces cerevisiae. Mol. Biol. Cell. 1992;3:805–818. doi: 10.1091/mbc.3.7.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richardson H, Lew DJ, Henze M, Sugimoto K, Reed SI. Cyclin-B homologs in Saccharomyces cerevisiae function in S phase and in G2. Genes Dev. 1992;6:2021–2034. doi: 10.1101/gad.6.11.2021. [DOI] [PubMed] [Google Scholar]
- 21.Surana U, Robitsch H, Price C, Schuster T, Fitch I, Futcher AB, Nasmyth K. The role of CDC28 and cyclins during mitosis in the budding yeast S. cerevisiae. Cell. 1991;65:145–161. doi: 10.1016/0092-8674(91)90416-v. [DOI] [PubMed] [Google Scholar]
- 22.Hadwiger JA, Reed SI. Nucleotide sequence of the Saccharomyces cerevisiae CLN1 and CLN2 genes. Nucleic Acids Res. 1990;18:4025. doi: 10.1093/nar/18.13.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McMillan JN, Longtine MS, Sia RA, Theesfeld CL, Bardes ES, Pringle JR, Lew DJ. The morphogenesis checkpoint in Saccharomyces cerevisiae: cell cycle control of Swe1p degradation by Hsl1p and Hsl7p. Mol. Cell. Biol. 1999;19:6929–6939. doi: 10.1128/mcb.19.10.6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sia RA, Bardes ES, Lew DJ. Control of Swe1p degradation by the morphogenesis checkpoint. EMBO J. 1998;17:6678–6688. doi: 10.1093/emboj/17.22.6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theesfeld CL, Zyla TR, Bardes EG, Lew DJ. A monitor for bud emergence in the yeast morphogenesis checkpoint. Mol Biol Cell. 2003;14:3280–3291. doi: 10.1091/mbc.E03-03-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lew DJ, Reed SI. Morphogenesis in the yeast cell cycle: regulation by Cdc28 and cyclins. J. Cell Biol. 1993;120:1305–1320. doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verma R, Annan RS, Huddleston MJ, Carr SA, Reynard G, Deshaies RJ. Phosphorylation of Sic1p by G1 Cdk required for its degradation and entry into S phase. Science. 1997;278:455–460. doi: 10.1126/science.278.5337.455. [DOI] [PubMed] [Google Scholar]
- 28.Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M. Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature. 2001;414:514–521. doi: 10.1038/35107009. [DOI] [PubMed] [Google Scholar]
- 29.Barberis M, De Gioia L, Ruzzene M, Sarno S, Coccetti P, Fantucci P, Vanoni M, Alberghina L. The yeast cyclin-dependent kinase inhibitor Sic1 and mammalian p27Kip1 are functional homologues with a structurally conserved inhibitory domain. Biochem J. 2005;387:639–647. doi: 10.1042/BJ20041299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- 31.Hu F, Aparicio OM. Swe1 regulation and transcriptional control restrict the activity of mitotic cyclins toward replication proteins in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2005;102:8910–8915. doi: 10.1073/pnas.0406987102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sia RA, Herald HA, Lew DJ. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol. Biol. Cell. 1996;7:1657–1666. doi: 10.1091/mbc.7.11.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Longtine MS, Theesfeld CL, McMillan JN, Weaver E, Pringle JR, Lew DJ. Septin-dependent assembly of a cell cycle-regulatory module in Saccharomyces cerevisiae. Mol. Cell. Biol. 2000;20:4049–4061. doi: 10.1128/mcb.20.11.4049-4061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jacobson MD, Gray S, Yuste-Rojas M, Cross FR. Testing cyclin specificity in the exit from mitosis. Mol. Cell. Biol. 2000;20:4483–4493. doi: 10.1128/mcb.20.13.4483-4493.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hood JK, Hwang WW, Silver PA. The Saccharomyces cerevisiae cyclin Clb2p is targeted to multiple subcellular locations by cis- and trans-acting determinants. J. Cell Sci. 2001;114:589–597. doi: 10.1242/jcs.114.3.589. [DOI] [PubMed] [Google Scholar]
- 36.Bailly E, Cabantous S, Sondaz D, Bernadac A, Simon MN. Differential cellular localization among mitotic cyclins from Saccharomyces cerevisiae: a new role for the axial budding protein Bud3 in targeting Clb2 to the mother-bud neck. J. Cell Sci. 2003;116:4119–4130. doi: 10.1242/jcs.00706. [DOI] [PubMed] [Google Scholar]
- 37.Mendenhall MD. An inhibitor of p34CDC28 protein kinase activity from Saccharomyces cerevisiae. Science. 1993;259:216–219. doi: 10.1126/science.8421781. [DOI] [PubMed] [Google Scholar]
- 38.Schwob E, Bohm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell. 1994;79:233–244. doi: 10.1016/0092-8674(94)90193-7. [DOI] [PubMed] [Google Scholar]
- 39.Fisher D, Nurse P. Cyclins of the fission yeast Schizosaccharomyces pombe. Semin. Cell Biol. 1995;6:73–78. doi: 10.1016/1043-4682(95)90003-9. [DOI] [PubMed] [Google Scholar]
- 40.Fisher DL, Nurse P. A single fission yeast mitotic cyclin B p34cdc2 kinase promotes both S-phase and mitosis in the absence of G1 cyclins. EMBO J. 1996;15:850–860. [PMC free article] [PubMed] [Google Scholar]
- 41.Haase SB, Reed SI. Evidence that a free-running oscillator drives G1 events in the budding yeast cell cycle. Nature. 1999;401:394–397. doi: 10.1038/43927. [DOI] [PubMed] [Google Scholar]
- 42.Stern B, Nurse P. A quantitative model for the cdc2 control of S phase and mitosis in fission yeast. Trends Genet. 1996;12:345–350. [PubMed] [Google Scholar]
- 43.Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 1995;376:313–320. doi: 10.1038/376313a0. [DOI] [PubMed] [Google Scholar]
- 44.Bartova I, Otyepka M, Kriz Z, Koca J. Activation and inhibition of cyclin-dependent kinase-2 by phosphorylation; a molecular dynamics study reveals the functional importance of the glycine-rich loop. Protein Sci. 2004;13:1449–1457. doi: 10.1110/ps.03578504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grandin N, Reed SI. Differential function and expression of Saccharomyces cerevisiae B-type cyclins in mitosis and meiosis. Mol. Cell. Biol. 1993;13:2113–2125. doi: 10.1128/mcb.13.4.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cross FR, Yuste-Rojas M, Gray S, Jacobson MD. Specialization and targeting of B-type cyclins. Mol. Cell. 1999;4:11–19. doi: 10.1016/s1097-2765(00)80183-5. [DOI] [PubMed] [Google Scholar]
- 47.Cross FR, Jacobson MD. Conservation and function of a potential substrate-binding domain in the yeast Clb5 B-type cyclin. Mol. Cell. Biol. 2000;20:4782–4790. doi: 10.1128/mcb.20.13.4782-4790.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Archambault V, Chang EJ, Drapkin BJ, Cross FR, Chait BT, Rout MP. Targeted proteomic study of the cyclin-Cdk module. Mol. Cell. 2004;14:699–711. doi: 10.1016/j.molcel.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 49.Loog M, Morgan DO. Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature. 2005;434:104–108. doi: 10.1038/nature03329. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.