

Abstract
The reaction of prenylated carbene complexes and 2-alkynylbenzoyl derivatives has been investigated. Phenanthrene derivatives are produced if iodine is added prior to product isolation. Under these conditions alkyl migration reactions occur to form the observed products. The product yields are considerably higher using bis(prenylated) species owing to an increase in the effective molarity of dienophilic entities.
Introduction
In a series of recent manuscripts, the coupling of γ,δ-unsaturated carbene complexes (A, Scheme 1) with o-alkynylbenzoyl derivatives (B) has been demonstrated.1 In this reaction, an isobenzofuran (E) is formed, which undergoes an exo selective2 intramolecular Diels-Alder reaction3 followed by ring opening to afford hydrophenanthrenone derivatives (G) in an overall net [5+5]-cycloaddition reaction. The reaction shows a remarkable degree of versatility with respect to the R1 and R2 substituents, however the yield of the hydrophenanthrene products is considerably lower when the dienophile alkene is hindered.4 In the published examples employing unactivated trisubstituted alkenes as dienophiles (see Scheme 2) the yield was approximately 35%. Both of the examples from reference 4 featured trisubstituted alkene dienophiles contained within six-membered rings. The result in Scheme 2 is not an adequate test of the efficacy of trisubstituted alkenes in the [5+5]-cycloaddition reaction since cyclohexene dienophiles are typically less reactive relative to acyclic and smallerring analogs.5 In this manuscript, the coupling of 2-alkynylbenzoyl compounds with readily available prenylated carbene complexes, which feature an acyclic trisubstituted alkene unit, has been examined under a variety of conditions to better assess the efficacy of trisubstituted alkenes in this reaction.
Scheme 1.

Scheme 2.

Results and Discussion
The carbene complexes for this investigation are depicted in Scheme 3. The monoprenylated complex 2a was obtained through reaction of the acid chloride derived from carboxylic acid 5 with pentacarbonylchromate dianion, commonly known as the Hegedus procedure.6 Johnson orthoester Claisen rearrangement of 2-methyl-3-buten-2-ol and triethyl orthoacetate7 followed by ester hydrolysis afforded acid 5, which was transformed to the acid chloride using oxalyl chloride and then to carbene complex 2a.
Scheme 3.
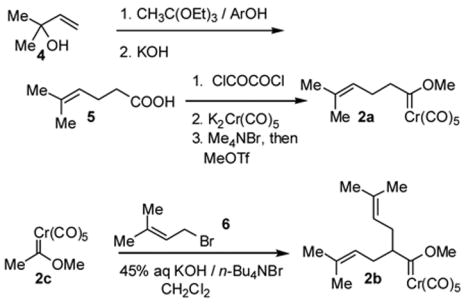
The bis(prenylated) species 2b was obtained through double alkylation of the methyl(methoxy)carbene complex 2c with allylic bromide 6 (prenyl bromide) under phase transfer conditions.8 Attempted optimization of the prenylation reaction for monoprenyl product 2a was never successful, however mixtures favoring monoprenylation could be produced if the reaction was performed under more dilute conditions.9 Alkynylbenzoyl derivatives were acquired through Sonogashira coupling involving 2-iodo or 2-bromobenzaldehyde or the corresponding acetophenone derivatives.
The first example tested was the coupling of carbene complex 2a with 2-ethynylbenzaldehyde (1a) (Scheme 4). In this reaction the keto-alcohol 7a was obtained in 41% yield as a single diastereomer arising from exo Diels-Alder reaction followed by ring opening (as depicted in Scheme 1). This reaction was successful only under high dilution conditions, perhaps due to competing polymerization of the alkyne induced by the carbene complex.10 In this reaction, the crude reaction product showed the appearance of peaks at δ 5.2–5.4. This signal is indicative of unreacted starting alkene and thus suggests that the Diels-Alder step of the reaction was not efficient.
Scheme 4.

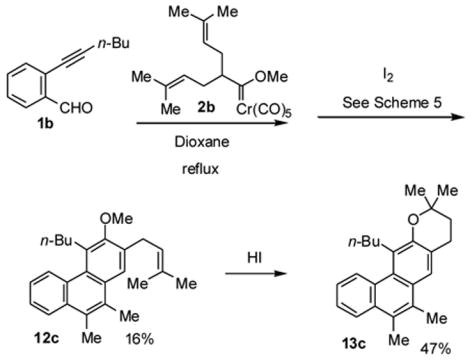
In order to better evaluate the efficiency of later steps in the reaction, internal alkynes were employed as substrates, which are less susceptible to polymerization side reactions. Although the hydrophenanthrenone alcohol analogous to 7 could be produced from the coupling of carbene complex 2a with o-hexynylbenzaldehyde (1b), the butyl group introduces a complexity into the 1H NMR spectrum that makes it difficult to evaluate the reaction prior to final purification. An alternative workup procedure was developed that removes all of the chiral centers and thus simplifies the analysis. After the carbene complex alkyne-coupling reaction had been refluxed for 24 h in dioxane, iodine was added to the reaction and the reflux further continued. Under these conditions the major product was the phenanthrene derivative 12b (Scheme 5) in 36% yield. This reaction was not clean, and the crude 1H NMR showed numerous signals in the vicinity of δ 5.3, indicating an incomplete Diels-Alder reaction. The phenanthrene structure is supported by the existence of a bay region aromatic proton at δ 8.51, the increase in chemical shift for the benzylic protons now located in the bay region (t at δ 3.22), and the increase in chemical shift for the methyl groups (accidentally isochronous singlet integrating for 6H at δ 2.65). The mechanistic process in Scheme 5 can account for phenanthrene formation. The likely product before iodine addition is diene-alcohol 8b. Reaction with iodine affords the dihydrophenanthrene (9b) and HI,11 which induces formation of a carbocation (10b). The migration of the methyl group leads to a highly-stabilized p-quinonemethide cation (11b).12 Exposure of the bis(prenylated) carbene complex 2b (Scheme 6) to the same conditions led to predominantly the phenanthrene-coumarin derivative 13c, accompanied by the uncyclized compound 12c as a minor byproduct in some experimental runs. Presumably in this reaction the HI byproduct induces the formation of the cyclic ether. The total yield of adducts is considerably higher in this case (combined yield 63%), likely due to the availability of two identical dienophiles for the Diels-Alder reaction, which doubles the effective concentration of dienophiles in the vicinity of the isobenzofuran. The spectator prenyl group can also introduce a conformational bias favoring the Diels-Alder reaction.13
Scheme 5.

Scheme 6.
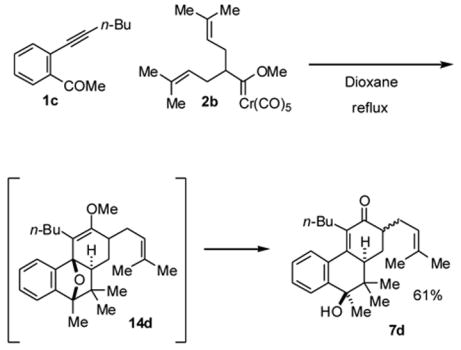
The reaction of the bis(prenylated) species 2b with o-alkynylacetophenone derivative 1c was attempted (Scheme 7). This reaction afforded initially the moderately stable oxanorbornene derivative 14d, which upon hydrolysis afforded keto-alcohol 7d as a 1:1 mixture of two (out of 4 possible) diastereomers. The oxanorbornene structure was assigned to the intermediate based on the unusually low chemical shift of the endo methyl group (δ 0.4) which would arise from anisotropic shielding expected for the bridged systems.14 The iodine workup described in Scheme 6 afforded a complex mixture of products. The reaction was not diastereoselective with respect to the spectator prenyl group. This lack of stereoinduction is contrary to previous studies where a high degree of relative asymmetric induction was observed for carbene complex-alkynylbenzaldehyde couplings when using carbene complexes that feature a chiral center at the β-position. Since the conversion of the oxanorbornene 14d to 7d likely involves an oxonium ion adjacent to the prenyl group, the stereochemistry more than likely arises from thermodynamic control and does not reflect any relative asymmetric induction in the carbene addition step.
Scheme 7.
Conclusions
In conclusion, simple unactivated trisubstituted alkenes can serve as dienophiles in the net [5+5]-cycloaddition reaction that occurs in the coupling of alkynylbenzoyl derivatives with γ,δ-unsaturated carbene complexes. The most efficient substrates are bis(prenylated) carbene complexes, which increase the effective dienophile concentration in the vicinity of the isobenzofuran intermediate and can induce a conformational bias favoring the Diels-Alder step. When iodine is added to the reaction, aromatization occurs, affording phenanthrene derivatives through an alkyl migration process. Under these conditions the spectator prenyl groups cyclized to afford dimethylcoumarin derivatives, which have been identified as important synthetic targets for drug discovery.15
Experimental:16
Synthesis of 5-methyl-4-hexenoic acid (5). A solution of 2-methyl-3-buten-2-ol (1.50 g, 17.4 mmol) and 2-nitrophenol (0.160 g, 1.15 mmol) in triethyl orthoacetate (26.40 g, 162.4 mmol) was heated to reflux for 36 h. The resulting solution was allowed to cool to room temperature and a 1:1 mixture of methanol and water (25 mL) was added. Two drops of concentrated hydrochloric acid were added and the solution was stirred for 12 h at room temperature. Diethyl ether (100 mL) and saturated aqueous sodium chloride solution (100 mL) were added and the aqueous layer was drained off. The ether layer was washed once more with saturated aqueous sodium chloride solution and dried over sodium sulfate. The solvent was removed on a rotary evaporator, and to the residue was added a solution of potassium hydroxide (1.25 g, 22.0 mmol) in ethanol (30 mL) and water (5.5 mL). This solution was heated to reflux for a 24h period. The mixture was acidified to pH 3 and extracted with ether. The ether layer was washed once with water and once with saturated aqueous sodium chloride solution and dried over sodium sulfate. The solvent was removed on a rotary evaporator and the residue was purified by flash chromatography on silica using 9:1 hexane: ethyl acetate as eluent. A colorless oil (1.49 g, 67% yield) identified as carboxylic acid 5 was obtained. 1H NMR (CDCl3): δ 5.10 (t, 1H, J = 4.0 Hz), 2.42-2.25 (m, 4H), 1.69 (s, 3H), 1.62 (s, 3H). The COOH peak was not detected. The spectral data are in agreement with the reported values.17
Synthesis of carbene complex 2a
a. Preparation of dipotassium pentacarbonylchromate
Graphite (2.860 g, 238.5 mmol) was heated at 150°C – 160 °C and stirred under argon for 2 h. The graphite was cooled to room temperature and potassium (1.160 g, 29.7 mmol) was added as tiny pieces under a positive flow of argon. The mixture was heated to 160 °C and stirred for 1 h. THF was added to the bronze colored material and the mixture was cooled to −78 °C. Chromium hexacarbonyl (2.970 g, 13.5 mmol) was added under a positive flow of argon. The reaction mixture was stirred for 30 min at −78 °C and placed in an ice bath until a thick slurry was formed. This mixture was directly used in the next step. b. Preparation of tetramethylammonium salt. To the solution of dipotassium pentacarbonylchromate in THF at −78 °C obtained from step a, the acid chloride (0.375 g, 2.56 mmol)18 in THF (10 mL) was added dropwise. The resultant mixture was stirred at −78 °C for 15 min, at 0 °C for 1.5 h, and at room temperature for 1 h. The solvent was removed under reduced pressure. Cold water was added to the residue, which was then filtered through Celite. To the filtrate was added a saturated aqueous solution of tetramethylammonium bromide (25 mL), leading to immediate precipitation of the ammonium salt. The precipitate was removed by extracting the aqueous layer with dichloromethane. The organic layer was then dried over anhydrous sodium sulfate and the solvent was removed on a rotary evaporator to afford a pale yellow solid. c. Alkylation of tetramethylammonium salt. To a solution of the ammonium salt in dichloromethane obtained in step b, methyl triflate (0.755 g, 4.60 mmol) was added at 0 °C. The reaction mixture was then stirred at 0 °C for 0.5 h and at room temperature for 1 h. The mixture was filtered through Celite, washed with saturated aqueous sodium bicarbonate solution, water, saturated aqueous sodium chloride solution and dried over sodium sulfate. The solvent was removed on a rotary evaporator to yield the carbene complex. Final purification by flash column chromatography using hexane as the eluent afforded a yellow liquid identified as carbene complex 2a (0.452 g 68 % yield). 1H NMR (CDCl3): δ 5.06 (t, 1H, J = 7.0 Hz), 4.79 (s, 3H), 3.33 (t, 2H, J = 7.5 Hz), 2.27-2.08 (m, 2H) 1.69 (s, 3H), 1.60 (s, 3H); 13C NMR (CDCl3): δ 363.6, 223.2, 216.4, 133.2, 122.1, 67.6, 62.7, 25.6, 25.1, 17.5; IR (neat): 2059, 1945 cm−1.
Synthesis of carbene complex 2b
To a 25mL round bottom flask containing methylcarbene complex 2c19 (1.60 g, 6.40 mmol) and tetrabutylammonium bromide (0.294 g, 0.90 mmol) in dichloromethane (10 mL) was added 45% aqueous potassium hydroxide solution (15 mL). To this suspension 3-methyl-1-bromo-2-butene (6) (2.86 g, 19.2 mmol) was added and the mixture was stirred at room temperature until TLC analysis indicated the absence of the reactant carbene complex. The reaction mixture was diluted with water and extracted with dichloromethane. The extracts were dried over sodium sulfate, concentrated, and purified by flash column chromatography using hexanes: ethyl acetate (50:1) as eluent. A yellow oil identified as carbene complex 2b was obtained (2.17 g, 88% yield). 1H NMR (CDCl3): δ 5.09 (t, 2H, J = 7.6 Hz), 4.79 (s, 3H), 4.11(quintet, 1H, J = 7.0 Hz), 2.17 (m, 2H), 1.95 (m, 2H), 1.66 (s, 6H), 1.56 (s, 6H); 13C NMR (CDCl3): δ 370.3, 233.3, 216.3, 133.2, 121.5, 71.7, 67.5, 30.7, 25.6, 17.5; IR (neat): 2079, 1945 cm−1. The spectral data are in agreement with those previously reported for this compound.8 In an experimental run where too much dichloromethane was employed (ca 50 mL) the major product was monoprenylated compound 2a.
General Procedure 1
Reaction of γ,δ-unsaturated carbene complexes with o-hexynylbenzaldehyde (1b) or o-hexynylacetophenone (1c). A solution of 1.0 eq of hexynylbenzaldehyde 1b (or hexynylacetophenone 1c) in dioxane (10 mL) under nitrogen was placed in a two necked flask fitted with a reflux condenser, addition funnel and stirrer. This solution was brought to a gentle reflux, and a 1M solution of carbene complex (1.0–1.2 eq) in dioxane was added dropwise to the refluxing solution over a period of 2 h. After the addition was complete, the mixture was allowed to reflux for a period of 24–36 h. A solution of iodine (1 eq) in dioxane (5 mL) was then added dropwise to the refluxing mixture. The reaction mixture was refluxed for 24 h. The reaction mixture was cooled to room temperature and concentrated on a rotary evaporator. Ethyl acetate (25mL) was added and the residue was filtered through a bed of Celite. The filtrate was poured into aqueous sodium thiosulfate solution and the aqueous layer was extracted with a 1:1 mixture of hexane: ethyl acetate. The combined organic layers were washed with brine and dried over sodium sulfate. The solvent was removed on a rotary evaporator and the crude residue was purified using flash chromatography on silica gel.
Reaction of monoprenylated carbene complex 2a with o-ethynylbenzaldehyde (1a). A solution of o-ethynylbenzaldehyde20 (0.120 g, 0.92 mmol) and carbene complex 2a (0.131 g, 0.41 mmol) in dioxane (15 mL) in an addition funnel was added to refluxing dioxane (10 mL) over a 3h period. After the addition was complete, the mixture was refluxed for a period of 24 h. The reaction mixture was cooled to room temperature and concentrated on a rotary evaporator. Ethyl acetate (25 mL) was added and the residue was filtered through Celite. The solvent was removed on a rotary evaporator and the crude products were purified by flash column chromatography using 3:1 hexanes: ethyl acetate as eluent to afford compound 7a (0.042 g, 41 % yield). 1H NMR (CDCl3): δ 7.78 (d, 1H, J = 7.6 Hz), 7.75 (d, 1H, J = 7.6 Hz), 7.47 (t, 1H, J = 7.6 Hz), 7.32 (t, IH, J = 7.6 Hz), 6.74 (d, 1H, J = 2.0 Hz), 4.58 (s, 1H), 2.62 (ddd, 1H, J = 16.8, 4.0, 2.6 Hz), 2.54 (ddd, 1 H, J = 11.2, 4.4. 2.0 Hz), 2.40 (ddd, 1H, J = 16.8, 14.8, 4.8 Hz), 2.26 (dddd, 1H, J = 13.2, 4.8, 4.4, 2.6 Hz), 1.89 (dddd, 1H, J = 14.6, 13.2, 11.2, 4.0 Hz), 1.26 (s, 3H), 0.65 (s, 3H); Irradiate at δ 6.74: δ 2.44 (dd, J = 12.0, 4.2 Hz); 13C NMR (CDCl3): δ 199.6, 155.7, 140.7, 131.2, 130.8, 127.6, 126.6, 124.2, 122.1, 45.6, 40.0, 37.1, 29.7, 24.3, 22.6, 13.5; IR (neat): 3417 (s), 1652 (s), 1587 (m) cm−1; MS (CI): (m/e) 243 (M+1, 89), 242 (M, 100), 224 (M – H2O, 16), 209 (39), 199 (54), 181 (41), 171 (45), 157 (36), 141 (34), 128 (43), 115 (46), 91(15), 77(23); HRMS: Calcd for C16H18O2 242.13068, found 242. 12992.
Coupling of monoprenylated carbene complex 2a with 2-(1-hexynyl)benzaldehyde (1b) followed by iodine. General Procedure 1 was followed using carbene complex 2a (0.140 g, 0.44 mmol) and 2-(1-hexynyl)benza1dehyde (1b)21 (0.101 g, 0.54 mmol). After a 24h reflux period, a solution of iodine (0.142 g, 0.55 mmol) in 10 mL of dioxane was added and the reflux was continued for 24 h. Purification using flash column chromatography using 19:1 hexanes: ethyl acetate yielded a compound assigned as phenanthrene 12b (0.046 g, 36 %). 1H NMR (CDCl3): δ 8.51 (d, 1H, J = 8.2 Hz), 8.07 (d, 1H, J = 6.7 Hz), 7.97 (d, 1H, J = 9.2 Hz), 7.65-7.43 (m, 2H), 7.29 (d, 1H, J = 9.2 Hz), 3.99 (s, 3H), 3.22 (t, 2H, J = 8.5 Hz), 2.65 (s, 6H), 2.15-1.95 (m, 2H),1.75-1.55 (m, 2H), 1.09 (t, 3H, J = 7.9 Hz); 13C NMR (CDCl3): δ 156.7, 133.9, 131.0, 129.7, 129.3, 128.1, 128.0, 127.1, 126.6, 126.1, 123.8, 123.4, 122.9, 111.1, 56.2, 31.4, 29.9, 23.3, 16.3, 15.9, 14.0; MS (EI): (m/e) 293 (M +1, 30), 292 (M, 100), 249 (78), 234 (70), 219 (35), 203(13), 189 (20). HRMS: Calcd for C21H24O 292.18271, found 292.18373.
Coupling of bis(prenylated) carbene complex 2b with 2-(1-hexynyl)benzaldehyde (1b). General Procedure 1 was followed using carbene complex 2b (0.504 g, 1.30 mmol), and 2-(1-hexynyl)benzaldehyde (1b)21 (0.240 g, 1.29 mmol). After a 24h reflux period, a solution of iodine (0.280 g, 1.10 mmol) in 10 mL of dioxane was added and the reflux continued for 24 h. The product obtained was purified by flash chromatography on silica gel using hexanes: ethyl acetate (20:1) as eluent. Two compounds were isolated. The compound in the first fraction was identified as phenanthrene 13c (0.210 g, 47%). 1H NMR (CDCl3): δ 8.47 (d, 1H, J = 8.0 Hz), 8.02 (dd, 1H, J = 7.8, 1.0 Hz), 7.68 (s, lH), 7.59-7.40 (m, 2H), 3.17 (t, 2H, J = 8.0 Hz), 3.04 (t, 2H, J = 6.6 Hz), 2.64 (s, 6H), 2.14-1.98 (m, 2H), 1.91 (t, 2H, J = 6.7 Hz), 1.72-1.54 (m, 2H, J = 7.4 Hz), 1.41 (s, 6H), 1.10 (t, 3H, J = 7.4 Hz); 13CNMR(CDCl3): δ 151.5, 133.5, 129.7, 129.4, 127.7, 127.0, 126.3, 126.0, 125.6, 123.7, 123.2, 122.4, 120.7, 74.3, 33.0, 31.2, 29.8, 27.2, 23.4, 23.3, 16.3, 15.9, 14.0; MS (EI): (m/e) 347 (M +1, 28), 346 (M, 100), 247 (43), 219 (21); HRMS: Calcd for C25H30O 346.22968, found 346.23014. The minor product in the second fraction was tentatively identified as uncyclized phenanthrene 12c (0.072 g, 16% yield). Compound 12c: 1H NMR (CDCl3): δ 8.55 (d, 1H, J = 8 Hz), 8.07 (d, 1H, J = 8 Hz), 7.80 (s, 1H), 7.58 (t, 1H, J = 7.0 Hz), 7.48 (t, 1H, J = 7.0 Hz), 5.44 (t, 1H, J = 7.0 Hz), 3.84 (s, 3H), 3.60 (d, 2H, J = 7.0 Hz), 3.32 (t, 2H, J = 8.0 Hz), 2.65 (s, 3H), 2.68 (s, 3H), 1.95-1.73 (m, 2H) overlapping with 1.80 (s, 3H), 1.62-1.45 (m, 2H) overlapping with 1.55 (s, 3H); 1.09 (t, 3H, J = 7.0 Hz).
Coupling of bis(prenylated)carbene complex 2b with 2-(1-hexynyl)acetophenone (1c). General Procedure 1 was followed using carbene complex 2b (0.125 g, 0.320 mmol) and 2-(1-hexynyl)acetophenone (1c)22 (0.054 g, 0.27 mmol). After a 40h reflux period, the reaction mixture was cooled to room temperature and the solvent was removed on a rotary evaporator. The residue was dissolved in ether (5 mL), 3.0M aqueous hydrochloric acid (3 mL) was added, and the mixture was stirred vigorously for 4 h. The mixture was neutralized by addition of saturated aqueous sodium bicarbonate solution. The aqueous layer was extracted three times with ether and the combined ether layers were dried over sodium sulfate. After removal of the solvent on a rotary evaporator, final purification was achieved by Flash chromatography on silica gel using 13:1 hexane: ethyl acetate as eluent. A single fraction was isolated and determined to be an approximately 1:1 mixture of stereoisomers of 7d (0.062 g, 61% yield). A second chromatographic separation generated enough pure material for characterization of one of the isomers. 1H NMR (CDCl3): δ 7.66 (dd, 1H, J = 8.0, 1.8 Hz), 7.44-7.40 (m, 2H), 7.32 (dd, 1H, J = 7.1, 1.5 Hz), 5.19 (br t, 1H, J = 7.2 Hz), 2.83 (dd, 1H, J = 11.7, 4.7 Hz), 2.67-2.55 (m, 2H), 2.43 (dd, 1H, J = 11.2, 3.2 Hz), 2.37-2.17 (m, 2H), 2.07 (ddd, 1H, J = 15.7, 8.0, 8.0 Hz), 1.74 (s, 3H), 1.66 (s, 3H), 1.49 (s, 3H), 1.42-1.31 (m, 4H), 1.25 (d, 2H, J = 12.0 Hz), 0.94 (s, 3H), 0.89 (t, 3H, J = 7.1 Hz), 0.86 (s, 3H); 13C NMR (CDCl3): δ 201.2, 151.9, 144.7, 135.9, 133.2, 132.8, 129.7, 128.7, 126.5, 124.8, 122.1, 75.4, 46.8, 44.7, 42.0, 31.6, 30.1, 28.1, 25.9, 25.8, 24.5, 24.0, 23.1, 18.8, 17.9, 13.8; IR (neat): 3500 (s), 1660 (s) cm−1; HRMS (CI): calcd for C26H37O2 (M+H)+ 381.2793, found 381.2793.
Supplementary Material
Supplementary Material Available: General Experimental and photocopies of 1H NMR and 13C NMR spectra for compounds carbene complex 2a and coupling products 7a, 12b, 13c, and 7d.
Acknowledgments
This research was supported by the SCORE Program of NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For the most recent example, see: Li R, Zhang L, Camacho-Davila A, Herndon JW. Tetrahedron Lett. 2005;46:5117–5120.
- 2.Six-membered ring-forming intramolecular Diels-Alder reactions of isobenzofurans are normally exo selective. Meegalla SK, Rodrigo R. Synthesis. 1989:942–944.Yamaguchi Y, Yamada H, Hayakawa K, Kanematsu K. J Org Chem. 1987;52:2040–2046.Tobia D, Rickborn B. J Org Chem. 1987;52:2611–2615.
- 3.For a review of isobenzofurans, see: Friedrichsen W. Adv Heterocycl Chem. 1999;73:1–96.
- 4.Ghorai BK, Menon S, Johnson DL, Herndon JW. Org Lett. 2002;4:2121–2124. doi: 10.1021/ol025808y. [DOI] [PubMed] [Google Scholar]
- 5.For a systematic study, see: Fringuelli F, Pizzo F, Taticchi A, Halls TDJ, Wenkert E. J Org Chem. 1982;47:5056–5065.
- 6.Moser WH, Hegedus LS. J Am Chem Soc. 1996;118:7873–7880. [Google Scholar]
- 7.Eng HM, Myles DC. Tetrahedron Lett. 1999;40:2275–2278. [Google Scholar]
- 8.Amin SR, Sarkar A. Organometallics. 1995;14:547–550. [Google Scholar]
- 9.The analogous process involving allyl bromide is successful. Zhang Y, Herndon JW. J Org Chem. 2002;67:4177–4185. doi: 10.1021/jo011136y.
- 10.Katz ATJ, Lee SJ. J Am Chem Soc. 1980;102:422–424. [Google Scholar]; The polymerization can be suppressed if the concentration of alkyne is kept low. Wulff WD, Kaesler RW, Peterson GA, Tang P-C. J Am Chem Soc. 1985;107:1060–1062.
- 11.For a similar transformation on a less activated ring, see: Hegde SG, Kassim AM, Kennedy AI. Tetrahedron. 2001;57:1689–1698.
- 12.For a recent discussion of this stabilization, see: DiLabio GA, Ingold KU. J Org Chem. 2004;69:1620–1624. doi: 10.1021/jo035693r.
- 13.This effect is expected to be less pronounced than the gem dialkyl effect. Jung ME, Gervay J. J Am Chem Soc. 1991;113:224–232.
- 14.In a related benzonorbornene the chemical shift of the endo methyl group is δ 0.62. Hemetsberger H, Nobbe M. Tetrahedron. 1988;44:67–80.
- 15.a. Nicolaou KC, Pfefferkorn JA, Cao GQ. Angew, Chem Int Ed. 2000;39:734–739. [PubMed] [Google Scholar]; b. Nicolaou KC, Pfefferkorn JA, Schuler F, Roecker AJ, Cao GQ, Casida JE. Chem Biol. 2000;7:979–992. doi: 10.1016/s1074-5521(00)00047-8. [DOI] [PubMed] [Google Scholar]
- 16.For a recent General Experimental, see the Supplementary Material.
- 17.Mori K, Sugai T, Maeda Y, Okazaki T, Noguchi T, Naito H. Tetrahedron. 1985;41:5307. [Google Scholar]
- 18.This compound was prepared in situ from carboxylic acid 5 and oxalyl chloride (see reference 6). The crude product from evaporation of dichloromethane and excess oxalyl chloride was employed.
- 19.Hegedus LS, McGuire MA, Schultze LM. Org Synth. Coll. 1987;658:140–145. 216–221. or. [Google Scholar]
- 20.This compound was prepared from Sonogashira coupling of trimethylsilylacetylene and 2-bromobenzaldehyde followed by desilylation according to a literature procedure. Acheson RM, Lee GCM. J Chem Soc, Perkin Trans 1. 1987:2321–2332.
- 21.This compound was prepared from Sonogashira coupling of 1-hexyne and 2-bromobenzaldehyde according to a literature procedure. Sakamoto T, Kondo Y, Miura N, Hayashi K, Yamanaka H. Heterocycles. 1986;24:2311–2314.
- 22.This compound was prepared from the Sonogashira coupling of 2-bromoacetophenone and 1-hexyne according to a literature procedure. Herndon JW, Zhang Y, Wang K. J Organometal Chem. 2001;634:1–4.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material Available: General Experimental and photocopies of 1H NMR and 13C NMR spectra for compounds carbene complex 2a and coupling products 7a, 12b, 13c, and 7d.