

Abstract
The copolymerization behavior and the dark polymerization kinetics of highly reactive novel acrylic monomers were compared to traditional acrylate monomers. Copolymerization of thiol functionalities with novel acrylic monomers was characterized, and it was observed that the inclusion of secondary functionalities such as carbamates, carbonates, and cyclic carbonates, in acrylic monomers significantly alters the relative reactivity of the novel acrylates with thiols. While traditional aliphatic acrylates exhibited propagation to chain transfer ratios ranging between 0.8 (± 0.1)–1.5(± 0.2), the novel acrylates characterized by secondary functionalities exhibited much higher propagation to chain transfer ratios ranging from 2.8(± 0.2)–4(± 0.2). In the dark polymerization studies, the kinetics of the novel acrylates were evaluated following cessation of the UV light. The novel acrylates exhibited extensive polymerization in the dark compared to most traditional acrylates and diacrylates. For instance, cyclic carbonate acrylate was observed to attain 35 % additional conversion in the dark when the UV light was extinguished at 35 % conversion, whereas traditional acrylates such as hexyl acrylate attained only 3 % additional conversion when the UV light was extinguished at 35 %, and a diacrylate such as HDDA attained 15 % additional conversion when the UV light was extinguished at 40 % conversion. Also, through choice of appropriate monomers, the dark polymerization studies were performed such that the polymerization rate was approximately the same at the point the light was extinguished for all these monomers. The copolymerization and dark polymerization studies support the hypothesis that the nature of the propagating species in the novel acrylates is altered as compared to traditional acrylic monomers and polymerizations.
Introduction
Photopolymerization offers multiple advantages which include spatial and temporal control, solventless polymerizations, as well as compatibility with multiple solvents. Due to the multitude of advantages it offers, photopolymerization of (meth)acrylates has been used for a variety of applications including dental materials, biomaterials, coatings, polymeric membranes, microfluidic devices, stereolithography, contact lenses and adhesives1–9. However, currently used (meth)acrylates are subject to several limitations including polymerization shrinkage, oxygen inhibition and residual unsaturation which have deleterious effects on polymer performance and lifetime. Hence, development of new (meth)acrylic systems to overcome these limitations is a focus of great interest.
Decker et al.10–12 in the early 1990s and more recently, Jansen13,14, Hoyle15, and Bowman and coworkers16,17 developed and evaluated a novel class of monovinyl (meth)acrylates that exhibit polymerization kinetics rivaling or surpassing multivinyl acrylates, despite their having only a single vinyl group. These monomers also form crosslinked, insoluble polymer early in the polymerization. Owing to their faster cure kinetics, near-quantitative conversions and superior polymer properties, these materials are very attractive for use as reactive diluents. Therefore, it is of immense significance to understand the factors leading to their higher reactivity which would enable a rational design of a new generation of acrylic monomers with enhanced reactivity and superior polymer properties.
Several theories have been proposed to explain the anomalous behavior of the highly reactive monomers. Decker et al.18,19 first proposed a highly efficient hydrogen abstraction mechanism, implicating the presence of labile hydrogens in the novel acrylic monomers. Hydrogen abstraction would lead to crosslinking and subsequently suppressed termination.
Alternatively, Jansen and coworkers13,14 have theorized that for molecules with a high dipole moment, the more polar medium reduces the activation energy for propagation, leading to an acceleration in the propagation reaction rate. However, various systems characterized by relatively low values of dipole moments that have high reactivities are known. Further, recent studies have depicted that even for systems with high dipole moment, molecular dipole is not a single, decisive factor causing enhanced reactivity17.
Other investigators have implicated hydrogen bonding as a factor leading to enhanced reactivity15,20. Hydrogen bonding increases the relative viscosity of the system, particularly during early stages of polymerization. The resulting mobility restrictions inhibit termination reactions, which lead to a higher radical concentration, and hence higher reactivity. While it is true that hydrogen bonding undoubtedly contributes to enhanced reactivity, it has been shown that internal hydrogen bonding is not the single decisive factor influencing monomer reactivity16. Further, several monomer systems incapable of intermolecular hydrogen bonding are known to be extremely reactive, including several in Decker’s original work21.
Recently, acid inhibition studies were performed to improve the fundamental understanding of these novel systems and they revealed the contribution of a reactive intermediate that has at least a anionic character as a part of the novel acrylic polymerization behavior22. To enhance this fundamental understanding of the nature of the novel acrylic monomer’s double bond/propagating radical, this work focuses on examining specific features of the polymerization kinetics. Specifically, this work investigates the copolymerization behavior of the novel acrylates with thiol monomers as well as the partial cure polymerization behavior of these systems in the absence of the initiating UV light.
Experimental
Materials
Cyclic carbonate acrylate (CCA) was prepared by the reaction of 4-hydroxy methyl-1,3-dioxolan-one (Huntsman, Salt Lake City, Utah), with acryloyl chloride (Aldrich Chemicals, Milwaukee, WI) in the presence of triethylamine17. Ethyl linear carbonate acrylate (LCA) was prepared by reaction of ethyl chloroformate with hydroxyethyl acrylate (Aldrich Chemicals, Milwaukee, WI)16,23. The monomer phenyl carbonate ethyl acrylate was prepared by the reaction of phenyl chloroformate and hydroxy ethyl acrylate (Aldrich Chemicals, Milwaukee, WI)16,23. Phenyl carbamate ethyl acrylate was prepared by a reaction of phenyl isocyanate (Aldrich Chemicals, Milwaukee, WI) with hydroxyethyl acrylate (Aldrich Chemicals, Milwaukee, WI)17. The synthesized monomers did not contain any added inhibitors. The 1H NMR (400 MHz, CDCl3) spectra of the synthesized monomers collected on a Varian Innova DSX-400 spectrometer are; cyclic carbonate acrylate(CCA):(400 MHz, CDCl3) δ6.42 (d, 1H), δ6.15 (dd, 1H), δ5.9 (d, 1H), δ4.95 (m, 1H), δ4.58 (t, 1H), δ4.4 (q, 1H), δ4.3 (m, 2H); ethyl linear carbonate acrylate (LCA): δ6.29 (d, 1H), δ5.96 (m, 1H), δ5.72 (d, 1H), δ4.25 (m, 4H), δ4.05 (m. 2H), δ1.15 (m, 3H); phenyl carbonate ethyl acrylate: δ 7.1–7.4 (m, 5H), δ 6.4 (d, 1H), δ 6.05 (q, 1H), δ 5.85 (d, 1H), δ 4.4 (m, 4H); phenyl carbamate ethyl acrylate: (400 MHz, CDCl3) δ7.4 (m, 5H), δ7.05 (m, 1H), δ6.7 (s, 1H), δ6.42 (d, 1H), δ6.05 (dd, 1H), δ5.82 (d, 1H), δ4.4 (s, 4H). The monomers hexyl acrylate, ethoxyethyl acrylate, tetrahydrofurfuryl acrylate, lauryl acrylate, and butyl mercaptopropionate were purchased from Aldrich Chemicals (Milwaukee, WI) and used as received. 1,6-hexanediol diacrylate (HDDA) was purchased from Aldrich Chemicals (Milwaukee, WI) deinhibited through vacuum distillation prior to use. The photoinitiator 2,2-dimethoxy-2-phenylacetophenone(DMPA) was purchased from Ciba-Geigy ( Hawthorne, NY). The structures of all the monomers used in the study are depicted in Figure 1.
Figure 1.
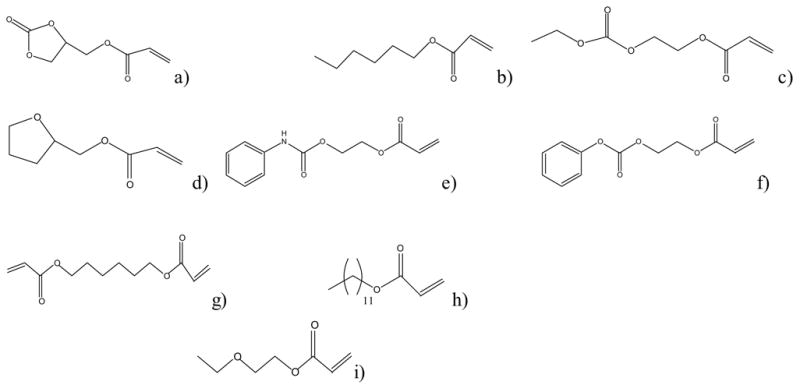
The structures of the monomers used in the study a) cyclic carbonate acrylate (CCA) b) hexyl acrylate c) ethyl linear carbonate acrylate (LCA) d) tetrahydrofurfuryl acrylate e) phenyl carbamate ethyl acrylate f) phenyl carbonate ethyl acrylate g) hexanediol diacrylate (HDDA) h) lauryl acrylate i) ethoxyethyl acrylate
Methods
Fourier Transform Infrared Spectroscopy
FTIR studies were conducted with a Nicolet 760 Magna FTIR spectrometer (Nicolet, Madison, WI). Samples were laminated by placing between two NaCl crystals with approximate film thicknesses of 15–20 μm and placed in a horizontal transmission apparatus16. Samples were irradiated with an ultraviolet light source (Ultracure 100SS 100W high pressure mercury vapor short-arc lamp, EXFO, Mississaugua, Ontario, Canada) filtered and centered at 365 nm for a duration of 5 minutes. Irradiation intensity was monitored using a Cole-Parmer Instruments Co. Series 9811 radiometer (Vernon Hills, IL). The initiator used was 0.1 wt % of 2,2-dimethoxy-2-phenylacetophenone (DMPA) for all samples. The thiol conversion was monitored by measuring the SH absorption peak24–26 at 2570 cm−1, and the acrylate conversion was monitored by measuring the C=C stretching vibration at 1630 cm−1 or the C=C twisting vibration around 810 cm−1.
Dynamic Mechanical Analysis
Dynamic mechanical analysis in extension mode (Perkin-Elmer DMA-7, Perkin-Elmer, Norwalk, CT) was utilized for determining the modulus, glass transition temperature and crosslinking density. Loss tangent and storage modulus were determined as a function of temperature, applying a sinusoidal stress at a frequency of 1 Hz. The temperature of the sample was increased from −30 to 170 °C at a rate of 5 °C per minute. The crosslink density was determined from the equation (1)
| (1) |
where E′ storage modulus in the rubbery plateau, R is the universal gas constant, and T is the absolute temperature at which the storage modulus is measured in the rubbery plateau.
Results and Discussion
Copolymerization with Thiol Monomers
Traditional acrylic monomers copolymerize with thiol functionalities through a mixed step-chain growth polymerization. In this polymerization mechanism, depicted below, while acrylates readily homopolymerize through a chain growth mechanism, they are also capable of abstracting a hydrogen from the thiol monomers (chain transfer). Therefore, when a mixture of acrylate and thiol monomers is polymerized, there are two possible reactions in which the acrylic radical participates: propagation through another acrylate functional group (step 1) and chain transfer to thiol (step 2). The thiyl radical, formed through the chain transfer step, further consumes the acrylic functional groups by propagating through the carbon-carbon double bond (step 3).
To characterize the polymerization behavior of traditional acrylic monomers with thiol functionalities, copolymerization studies of hexyl acrylate were conducted with butyl mercaptopropionate as a chain transfer agent. Figure 2a presents the copolymerization kinetics of these monomers at various thiol:acrylate stoichiometric conditions. These experiments reveal that the addition of thiol to hexyl acrylate results in considerable enhancement in acrylate polymerization kinetics. To understand this phenomenon better, presented below are polymerization rates of acrylic monomers in both the absence (equation 2) and presence (equation 3) of thiol25.
Figure 2.
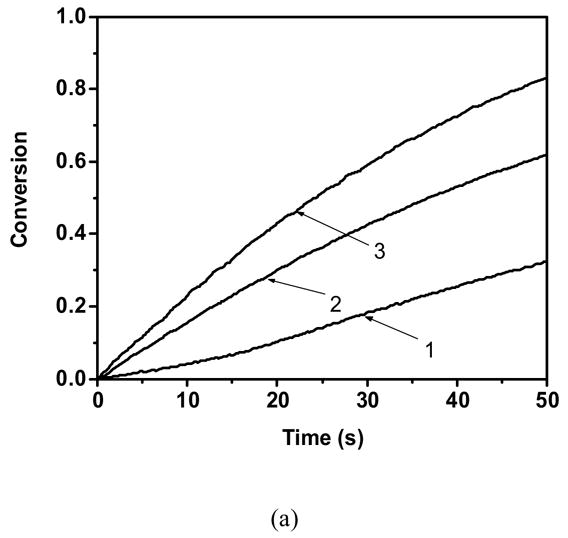
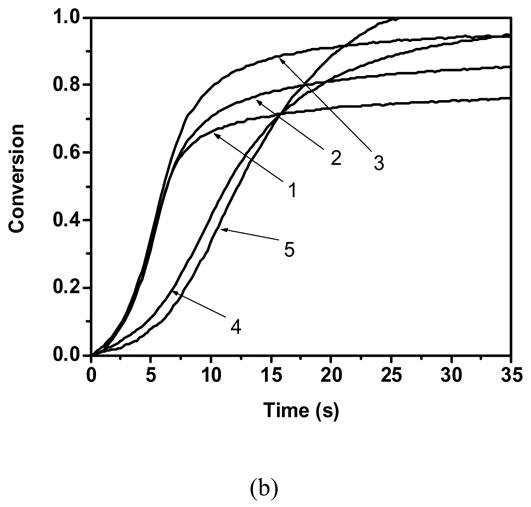
Acrylate conversion as a function of time for a). hexyl acrylate and butyl mercaptopropionate 1) 100:0 2)70:30 3) 50:50 b). Cyclic carbonate acrylate and butyl mercaptopropionate 1)100:0 2) 95:05 3) 90:10 4)75:25 5)50:50 Polymerization conditions: light intensity = 5 mW/cm2, initiator concentration = 0.1 wt %, room temperature.
| (2) |
| (3) |
where, kpCC is the homopolymerization kinetic constant, kpSC is the thiyl radical propagation kinetic constant, kCT is the chain-transfer kinetic constant of the acrylate to thiol, kt is the termination kinetic constant and Ri is the initiation rate, , and
Therefore, the enhancement in the polymerization rate of acrylate associated with the addition of thiol is given by
| (4) |
From equation 4, it is clear that the increase in acrylic monomer reactivity upon thiol functional group addition occurs when parameter b is greater than parameter a, i.e., when kpSC > kpCC. Further, previous work in our group has demonstrated that the ratio of the thiyl radical propagation (kpSC) to homopolymerization (kpCC) kinetic constant is approximately 13 for the hexyl acrylatethiol reaction presented here25. Therefore, the increase in acrylic monomer reactivity upon addition of thiol is not unexpected, and it is due to the higher propensity of the thiyl radical to propagate through the acrylic double bond as contrasted with the propagation of an acrylic radical through the acrylic double bond.
However, addition of thiol does not lead to a rate enhancement in the polymerization kinetics of the novel acrylic monomers, as shown for CCA in Figure 2 b. In contrast, the addition of thiol in similar quantities as used with hexyl acrylate slowed the novel acrylate’s polymerization kinetics (Figure 2 b). The negligible impact of the low thiol concentration on cyclic carbonate acrylate clearly suggests that the homopolymerization kinetic parameter is greater than the thiyl radical propagation kinetic parameter. Further, the suppression in the polymerization kinetics upon addition of large amounts of monothiol reflects the chain transfer ability of thiols24,27,28. While kinetically the thiol is not increasing the polymerization rate as in the case with traditional acrylic systems, the thiol monomer here further functions as a chain-transfer agent, suppressing autoacceleration effects by reducing the molecular weight of the growing kinetic chains27–29. Further, the reduction in the molecular weight of the growing chains also leads to an increase in the observed overall conversion.
It was expected that the propagation to chain transfer ratios for the thiol and acrylate would also be different compared to traditional acrylic systems. Hence, we attempted to obtain the propagation to chain transfer kinetic constant ratio (kp/kct) for several thiol-acrylate systems. Based on steps 1–3 from Scheme 1, of the thiol-acrylate reaction, the relative rates of thiol and acrylate consumption are given by21:
Scheme 1.
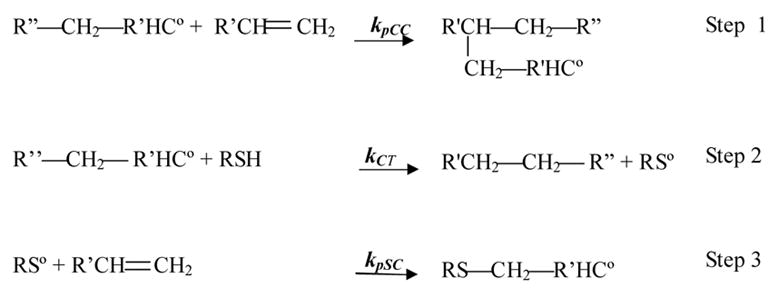
The polymerization sequence of thiol-acrylate systems.
| (5) |
These ratios are obtained by optimizing the kp/kct parameter for a least squares fit of the thiol functional group conversion as a function of the acrylate functional group conversion over a range of 0–80 % acrylate conversion. The kp/kct parameter was determined for two different thiol/acrylate initial ratios of 50/50 wt% and 30/70 wt%. Since the kp/kct parameter was fit over a range of conversion from 0–80 %, where the stoichiometric ratios of thiol to acrylate change as the reaction progresses, two initial thiol:acrylate mixtures of different ratios were adequate for the estimation of kp/kct.
Studies previously revealed that most traditional acrylates have kp/kct ratios ranging from 0.8–1.524,30 (Table 1). However, the novel acrylates functionalized by carbamate, carbonate and cyclic carbonate were found to have much higher kp/kct ratios ranging from 2.8 (± 0.2)–4 (± 0.2) (Figure 3 and Table 1). Acrylates such as hexyl acrylate, lauryl acrylate, and HDDA which are very structurally similar, showed propagation to chain transfer ratios around 1.0 ( ± 0.1) – 1.4 ( ± 0.1)whereas the ethoxyethyl acrylate depicted a slightly higher kp/kct ratio of 1.5 ± 0.2. The novel acrylates exhibit much higher kp/kct ratios as compared to any of the aliphatic acrylates. As the propagation kinetic constants for CCA and HDDA are very similar31, 5 × 104 ± 0.4 × 104 lmol−1s, under identical polymerization conditions and measured with similar analytical techniques the unusually high kp/kct ratio of CCA is due to the decreased chain transfer kinetic constant associated with the reaction of the propagating radical in chain transfer to the thiol. This difference in thiol chain transfer ability of CCA suggests an inherent difference in the nature of the cyclic carbonate acrylate’s propagating acrylic radical as compared to that of traditional acrylates. This hypothesis, if true, could potentially explain many unusual properties of the novel acrylic systems including their unusually high reactivity. This hypothesis is also supported by the acid inhibition studies which suggested that the propagating acrylic radical might possess a greater ionic character22. The kp/kct ratios of various novel acrylates as well as traditional acrylates are presented in Table 1.
Table 1.
The ratio of propagation to chain transfer kinetic constants (kp/kct) ratios for various acrylates reacting with butyl mercaptopropionate.
| Monomer | Copolymerization Ratio (Propagation to Acrylate/Chain transfer to thiol) (kp/kct) |
|---|---|
| Lauryl Acrylate | 0.9 ± 0.1 |
| Hexyl Acrylate | 1.0 ± 0.1 |
| Ethoxyethyl Acrylate | 1.5 ± 0.2 |
| Hexanediol Diacrylate | 1.4 ± 0.1 |
| Cyclic Carbonate Acrylate | 4.0 ± 0.2 |
| Ethyl Linear Carbonate Ethyl Acrylate | 3.0 ± 0.2 |
| Phenyl Carbonate Ethyl Acrylate | 2.8 ± 0.2 |
| Phenyl Carbamate Ethyl Acrylate | 3.4 ± 0.6 |
Polymerization conditions: light intensity = 5 mW/cm2, initiator concentration = 0.1 wt % DMPA, ambient temperature.
Figure 3.
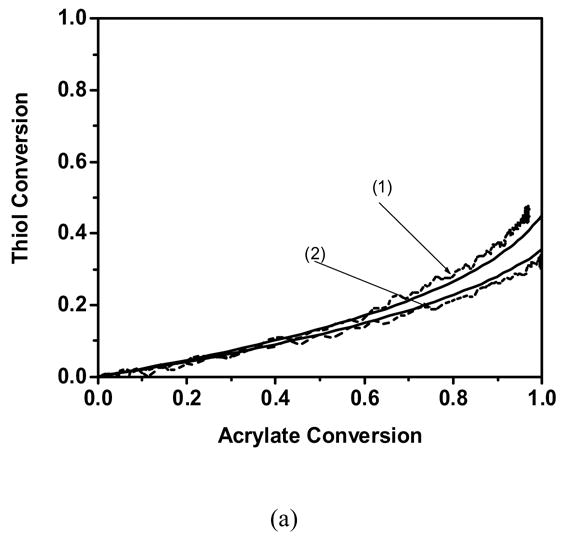
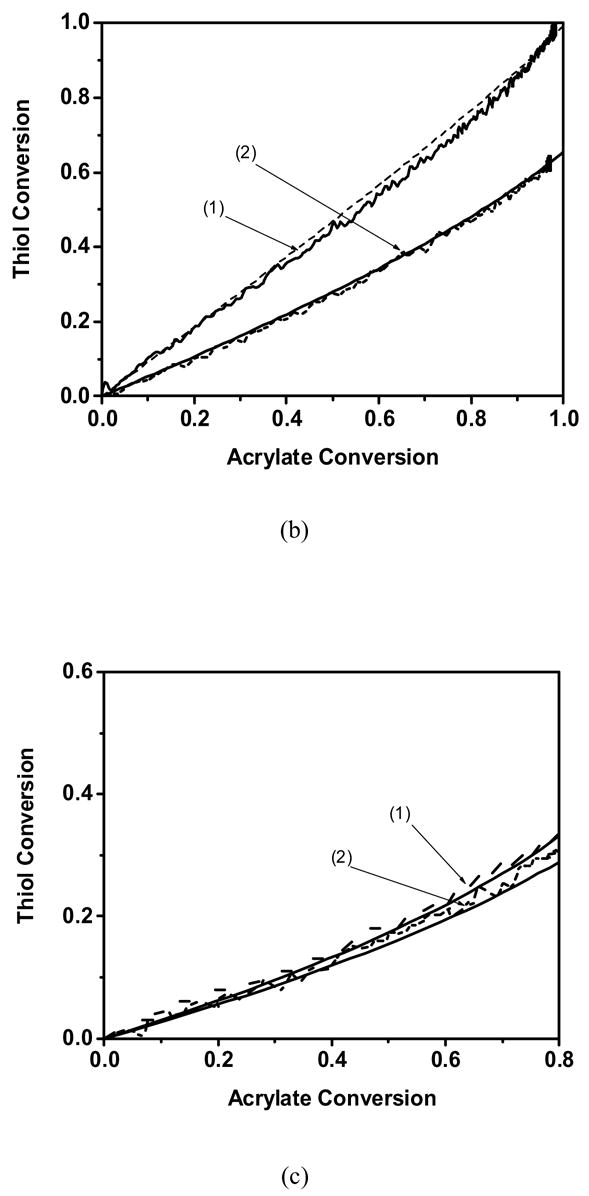
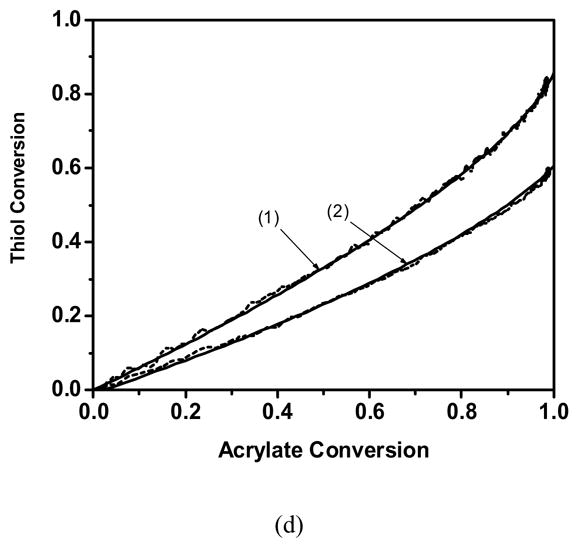
The thiol conversion as a function of the acrylate conversion for a variety of monomers and conditions. (— ) Theoretical fit and ( – – ) Experimental profile of(1) 50/50 wt % acrylate/butyl mercaptopropionate, (2) 70/30 wt % acrylate/butyl mercaptopropionate. Experimentally observed profile of a) Cyclic carbonate acrylate b) Hexyl acrylate c) Phenyl carbonate ethyl acrylate d) Ethoxyethyl acrylate. Polymerization conditions: light intensity = 5 mW/cm2, initiator concentration = 0.1 wt % DMPA, temperature = 25 °C.
Hence, the nature of the propagating acrylic species is hypothesized to affect the nature of both kp/kct ratios of these monomers with thiols as well as their enhanced reactivity towards photopolymerization. Thus, the kp/kct ratios of these acrylate monomers are plotted against the average polymerization rates to examine possible correlations between reactivity to photopolymerization and a inclination to react with thiols (Figure 4). It can be inferred from Figure 4 that the novel acrylates with higher kp/kct ratios also exhibited enhanced average polymerization rates. However, no monotonic correlation between average polymerization rates and copolymerization ratios was found, since polymerization rates are also affected by factors including increased viscosity effects of hydrogen bonding, and autoacceleration effects7,15,32. For instance, phenyl carbamate ethyl acrylate which exhibited a kp/kct ratio of 3.4 ± 0.6 is observed to be approximately 3-fold more reactive as compared to CCA because of increased viscosity effects due to hydrogen bonding32. Similarly, HDDA with a lower kp/kct ratio of 1.4 ± 0.1 is also observed to exhibit reactivity equivalent to the linear carbonate ethyl acrylate due to the contribution of typical autoacceleration effects associated with the polymerization of diacrylates6. Though both hydrogen bonding interactions and crosslinking affect the polymerization rate, neither of these interactions are expected to have a primary effect on the reaction with the thiol.
Figure 4.
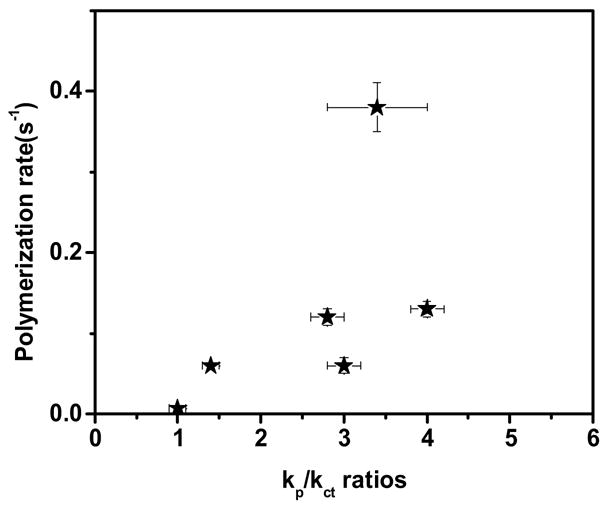
Average bulk polymerization rates of various acrylates (10–50 % conversion) versus their kp/kct ratios with butyl mercaptopropionate. The polymerization rates have been normalized with respect to initial double bond concentration of acrylates. Polymerization conditions: light intensity = 5 mW/cm2, initiator concentration = 0.1 wt% DMPA, room temperature, weight ratios of acrylate/thiol ranging from 70/30 to 50/50.
Dark Polymerization Studies
Once the photopolymerization of the novel acrylates is initiated and, subsequently, the UV light is extinguished, the novel acrylates are observed to polymerize and achieve extensive conversions in the dark. Table 2 presents the extent of conversion in the dark for various monomers under similar initiation conditions. The above behavior is also very atypical of traditional acrylic systems. Cyclic carbonate ethyl acrylate and phenyl carbamate ethyl acrylate are observed to polymerize and attain ~ 30–40% conversions after the light is extinguished, whereas traditional acrylates such as hexyl acrylate and tetrahydrofurfuryl acrylates attain only ~ 2–3 % conversion in the dark. A certain amount of increased dark conversion in novel acrylates is attributed to the reduction in termination kinetics due to the presence of intermolecular interactions such as hydrogen bonding and mobility restrictions generated by the small amount of crosslinking that occurs. Increased mobility restrictions reduce the termination kinetics, increasing the radical lifetimes and the amount of dark conversion. Hence, the novel acrylates would be expected to exhibit increased amounts of dark polymerization compared to traditional monoacrylates. However, as discussed in the following section, the novel acrylates were observed to exhibit significantly enhanced dark polymerization, even compared to traditional diacrylates such as HDDA.
Table 2.
Dark conversion data for various acrylate monomers is presented.
| Monomer | Conversion at which the UV light was extinguished (%) | Dark Polymerization (% conversion after the UV light was extinguished) | Rp at the conversion where the light was extinguished (s−1) |
|---|---|---|---|
| Hexyl Acrylate | 35 ± 1 | 3 ± 1 | 0.007 ± 0.002 |
| Tetrahydrofurfuryl Acrylate | 35 ± 1 | 7 ± 2 | 0.02 ± 0.03 |
| Hexanediol Diacrylate | 10 ± 2 | 7 ± 2 | 0.05 ± 0.02 |
| Hexanediol Diacrylate | 40 ± 3 | 15 ± 3 | 0.08 ± 0.02 |
| Cyclic Carbonate Acrylate | 35 ± 5 | 35 ± 3 | 0.16 ± 0.02 |
| Phenyl Carbamate Ethyl Acrylate | 36 ± 8 | 40 ± 3 | 0.38 ± 0.03 |
| Ethyl Linear Carbonate Ethyl Acrylate | 34 ± 3 | 25 ±3 | 0.09 ± 0.02 |
Polymerization conditions: light intensity = 5 mW/cm2, initiator concentration = 0.1 wt% DMPA, room temperature.
HDDA was observed to achieve a higher dark conversion of around 7 %, when the UV light source was extinguished at 10 % conversion and 15 % conversion when the UV light source was extinguished at 40 % conversion. HDDA is expected to polymerize to a greater extent in the dark since it is crosslinked to a greater extent, leading to higher mobility restrictions and greater radical life-times. However, CCA reacted an additional 35 % conversion following extinguishing of the light source compared to the 12 % additional conversion exhibited by HDDA under the same initiation conditions.
Further, the increased propagation rate exhibited by the novel acrylates also translates to a greater extent of conversion in the dark for a given period of time. Hence, the kinetics in the absence of UV light, for LCA, a novel (mono)acrylate were compared to HDDA, since both monomers exhibit comparable polymerization rates of 0.09s−1 and 0.08 s−1, respectively, at the conversion at which the light is extinguished. LCA reacted an additional 25 % conversion following extinguishing of the light source compared to the 12 % additional conversion exhibited by HDDA when the UV light was extinguished at approximately 35 % conversion for both monomers (Figure 5). Increased amounts of dark conversion, at similar polymerization rates can be inferred to arise specifically from suppressed termination of radicals. Also, it can be inferred from Table 3 that the novel acrylates are crosslinked to a lesser extent as compared to diacrylates. Hence, mobility restrictions due to crosslinking leading to suppressed termination do not account for the increased amounts of dark reaction. Another reason for the altered dark polymerization behavior could be the propagating acrylic radical is inherently altered for the novel acrylates characterized by the secondary functionalities. Specifically, the acid inhibition studies conducted previously suggest that the propagating species appears to have an increased partial anionic character22. Greater partial charge on the propagating acrylic species would not only impact monomer reactivity but also would suppress termination due to increased electrostatic repulsion between partially charged radicals13 and hence, account for increased dark polymerization.
Figure 5.
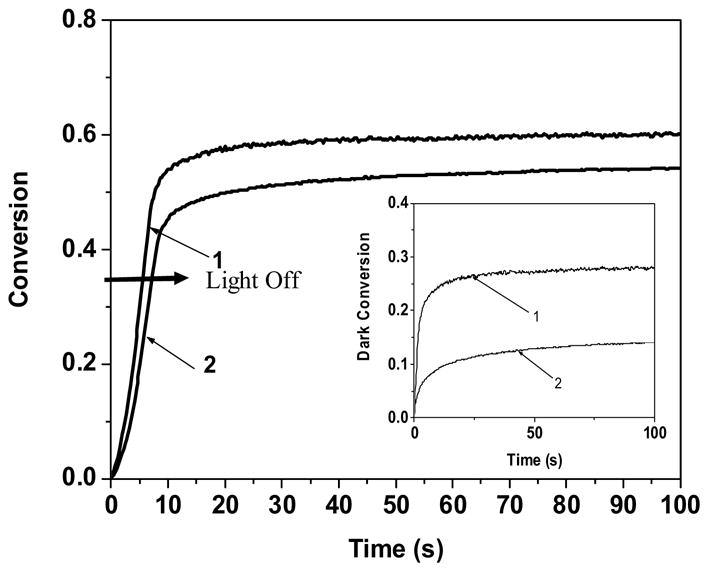
Acrylate conversion as a function of time. 1) Ethyl linear carbonate ethyl acrylate 2) Hexanediol diacrylate. The UV light was extinguished at 35 % conversion for both monomers, and conversion upon extinguishing the UV light is depicted in the inset. Polymerization conditions: light intensity prior to extinguishing the UV light = 5 mW/cm2, initiator concentration = 0.1 wt %, room temperature.
Table 3.
Crosslinking densities of various acrylate monomers are presented.
| Monomer | Crosslink Density(mol/l) |
|---|---|
| Hexanediol Diacrylate | 10.0 ± 0.4 |
| Cyclic Carbonate Acrylate | 0.04 ± 0.003 |
| Phenyl Carbamate Ethyl Acrylate | Polymer is soluble in chloroform |
| Ethyl Linear Carbonate Ethyl Acrylate | Polymer soluble in methylene chloride |
Polymerization Conditions: Light intensity = 5 mW/cm2, initiator concentration = 0.1 wt% DMPA, room temperature. Crosslink densities are calculated from the storage modulus in the rubbery region as presented in Equation 1.
Conclusions
Copolymerization of thiol functionalities with novel acrylic monomers was characterized, and it was observed that functionalizing the acrylic monomers with secondary functionalities significantly alters the relative reactivity of the novel acrylates with thiols. The novel acrylates characterized by the secondary functionalities were observed to exhibit reduced inclination to copolymerize with the thiols, with kp/kct ratios ranging from 2.8 (± 0.2) −4 (± 0.2), as compared to the traditional acrylates which depicted kp/kct ratios ranging from 0.9(± 0.1) −1.5(± 0.2). Further, it was shown that the incorporation of the secondary functionalities into the acrylate monomer alters the chain transfer ability to thiol, signifying that the nature of the propagating acrylic radical is altered.
Further, the polymerization kinetics of the novel (meth)acrylates were measured in the absence of UV light, and the novel acrylates were observed to exhibit extensive polymerization in the dark compared to traditional acrylates and diacrylates. Previously conducted acid inhibition studies22,33 have also suggested that the nature of the propagating species could be partially ionic. A greater partial charge on the propagating acrylic radical would suppress termination and lead to greater extent of polymerization in the dark. Thus, these results support the hypothesis that the nature of the propagating species in the novel acrylates is different from that in traditional acrylic systems.
Acknowledgments
The authors thank the National Science Foundation Industrial University Cooperative Research Center for Fundamentals and Applications of Photopolymerizations and NIH grant DE - 10959 for funding this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Decker C, Elzaouk B. Journal of Applied Polymer Science. 1997;65:833–844. [Google Scholar]
- 2.Hutchison JB, Haraldsson KT, Good BT, Sebra RP, Luo N, Anseth KS, Bowman CN. Lab on a Chip. 2004;4:658–662. doi: 10.1039/b405985a. [DOI] [PubMed] [Google Scholar]
- 3.Finger WJ, Lee KS, Podszun W. Dental Materials. 1996;12:256–261. doi: 10.1016/s0109-5641(96)80032-7. [DOI] [PubMed] [Google Scholar]
- 4.Bernhard P, Hofmann M, Schulthess A, Steinmann B. Chimia. 1994;48:427–430. [Google Scholar]
- 5.Decker C. Nuclear Instruments & Methods in Physics Research Section B-Beam. Interactions with Materials and Atoms. 1999;151:22–28. doi: 10.1016/s0168-583x(99)00546-7. [DOI] [PubMed] [Google Scholar]
- 6.Anseth KS, Wang CM, Bowman CN. Polymer. 1994;35:3243–3250. [Google Scholar]
- 7.Lovell LG, Stansbury JW, Syrpes DC, Bowman CN. Macromolecules. 1999;32:3913–3921. [Google Scholar]
- 8.Lu H, Lovell LG, Bowman CN. Macromolecules. 2001;34:8021–8025. [Google Scholar]
- 9.Lu H, Stansbury JW, Nie J, Berchtold KA, Bowman CN. Biomaterials. 2005;26:1329–1336. doi: 10.1016/j.biomaterials.2004.04.041. [DOI] [PubMed] [Google Scholar]
- 10.Decker C, Moussa K. European Polymer Journal. 1991;27:881–889. [Google Scholar]
- 11.Decker C, Moussa K. European Polymer Journal. 1991;27:403–411. [Google Scholar]
- 12.Decker C, Moussa K. Die Macromolekulare Chemie. 1991;192:507–522. [Google Scholar]
- 13.Jansen JFGA, Dias AA, Dorschu M, Coussens B. Macromolecules. 2003;36:3861–3873. [Google Scholar]
- 14.Jansen JFGA, Dias AA, Dorschu M, Coussens B. Macromolecules. 2002;35:7529–7531. [Google Scholar]
- 15.Lee TY, Roper TM, Jonsson ES, Guymon CA, Hoyle CE. Macromolecules. 2004;37:3659–3665. [Google Scholar]
- 16.Berchtold KA, Nie J, Stansbury JW, Hacioglu B, Beckel ER, Bowman CN. Macromolecules. 2004;37:3165–3179. [Google Scholar]
- 17.Kilambi H, Beckel ER, Berchtold KA, Stansbury JW, Bowman CN. Polymer. 2005;46:4735–4742. [Google Scholar]
- 18.Decker C, Elzaouk B. European Polymer Journal. 1990;26:393–401. [Google Scholar]
- 19.Decker C, Elzaouk B, Decker D. Journal of Macromolecular Science-Pure and Applied Chemistry. 1996;A33:173–190. [Google Scholar]
- 20.Jansen J, Dias AA, Dorschu M, Coussens B. Abstracts of Papers of the American Chemical Society. 2001;222:U321–U321. [Google Scholar]
- 21.Decker C, Moussa K. Macromolekulare Chemie, Rapid Communications. 1990;11:159–167. [Google Scholar]
- 22.Beckel ER, Stansbury JW, Bowman CN. Macromolecules. 2005;38:9474–9481. [Google Scholar]
- 23.Beckel ER. PhD Thesis, University of Colorado. 2004. [Google Scholar]
- 24.Cramer NB, Bowman CN. Journal of Polymer Science Part a-Polymer Chemistry. 2001;39:3311–3319. [Google Scholar]
- 25.Reddy SK, Cramer NB, O’Brien AK, Cross T, Raj R, Bowman CN. Macromolecular Symposia. 2004;206:361–374. [Google Scholar]
- 26.Reddy SK, Sebra RP, Anseth KS, Bowman CN. Journal of Polymer Science Part a-Polymer Chemistry. 2005;43:2134–2144. [Google Scholar]
- 27.Rydholm AE, Held NL, Bowman CN, Anseth KS. Macromolecules. 2006;39:7882–7888. doi: 10.1021/ma060858u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reddy SK, Cramer NB, Bowman CN. Macromolecules. 2006;39:3681–3687. [Google Scholar]
- 29.Cramer NB, Reddy SK, O’Brien AK, Bowman CN. Macromolecules. 2003;36:7964–7969. [Google Scholar]
- 30.Brandrup JIEH, editor. Polymer Handbook. John Wiley & Sons; New York: 1989. [Google Scholar]
- 31.Berchtold KA. PhD Thesis, University of Colorado. 2001. [Google Scholar]
- 32.Kilambi H, Stansbury JW, Bowman CN. Macromolecules. 2007;40:47–54. doi: 10.1021/ma062708p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kilambi H, Konopka D, Stansbury JW, Bowman CN. Factors effecting acid inhibition in novel acrylates. Journal of Polymer Science Part A, Polymer Chemistry In Press [Google Scholar]