

Abstract
The short arm of chromosome 3 contains multiple regions that are frequently affected by loss of heterozygosity in human cancers. The TMEM7 gene, which encodes a transmembrane protein, is among the candidate tumor suppressor genes located at one of these regions, 3p21.3. This gene is expressed specifically in the liver, and the encoded protein shares substantial sequence homology with human and mouse 28-kDa interferon-alpha (IFN-α) responsive protein. On the basis of these observations, we have now investigated the possible role of TMEM7 in the development of hepatocellular carcinoma (HCC). We examined TMEM7 expression in 20 primary HCC and 18 HCC cell lines and found recurrent functional alterations. While TMEM7 mRNA was expressed in normal hepatic cells, down-regulation or inactivation of the gene was detected in 85% and 33% of primary HCC and HCC cell lines, respectively. To identify the mechanisms responsible for down-regulation or silencing of TMEM7, we examined genomic deletion and mutation as well as the effect of inhibitors of DNA methyltransferase and histone deacetylase on cells with low levels or lacking endogenous TMEM7 expression. Homozygous deletion of TMEM7 was not detected in 17 pairs of human HCC and the corresponding non-cancerous liver tissues or in any of 18 HCC cell lines studied. Likewise, mutation of the gene was not detected in 18 HCC cell lines with low or normal TMEM7 expression. Treatment of two out of six cell lines exhibiting down-regulation or loss of TMEM7 with 5-aza-2’-deoxycytidine and trichostatin A resulted in a additive increase in TMEM7 expression, implicating aberrant DNA methylation and histone deacetylation in the transcriptional silencing of this gene. Ectopic expression of TMEM7 in two TMEM7 deficient HCC lines suppressed cell proliferation, colony formation, and cell migration in vitro and reduced tumor formation in nude mice. Treatment of two highly invasive HCC cell lines with IFN-α for 7 days significantly increased TMEM7 expression and in inhibited cell migration. These observations implicate loss of TMEM7 expression in hepatocarcinogenesis and suggest that modification of TMEM7 expression by IFN-α may have potential therapeutic relevance in a subset of HCC.
Keywords: TMEM7, cell growth inhibition, cell migration inhibition, interferon alpha, reduction of tumorigenicity
Introduction
Hepatocellular carcinoma (HCC) is one of the most common human cancers worldwide and accounts for 90% of all liver carcinomas. It is estimated that more than 560,000 new cases of HCC are diagnosed annually, most of them having an unfavorable prognosis. The mortality rate of HCC is high because many tumors are asymptomatic until late stages of development. Various factors, including infection with hepatitis B or C viruses, dietary aflatoxin, alcohol consumption, and exposure to chemical carcinogens, have been implicated in the etiology of HCC [1]. In Western countries, the incidence of HCC has increased markedly in the last several years as a result of an increased incidence of hepatitis virus infection [2,3]. Both chemical carcinogens and oncogenic viruses induce DNA damage in liver cells that manifests at the chromosome level as deletions, duplications, or translocations. The progression to HCC is a slow process that includes several distinct stages that are associated with the sequential accumulation of genomic alterations [1]. In addition, epigenetic changes are thought to play an important role in hepatocarcinogenesis [4].
Chromosomal regions that are frequently deleted in cancer are thought to be the loci of tumor suppressor genes, whose loss or inactivation contribute to unrestricted cell proliferation. Tumor suppressor genes that are silenced by promoter methylation are thus often located at chromosomal regions that are deleted in cancer cells [5,6]. Deletions of the short arm of chromosome 3 are common in various types of cancer. Although region 3p21.3 is most frequently affected by homozygous and heterozygous deletions in many cancers, the identification of tumor suppressor genes at this site has been difficult [7]. Among bona fide and candidate tumor suppressor genes localized to this region of 3p, the gene for TMEM7 (transmembrane protein 7) was identified by combined deletion mapping and by the “elimination test” i.e. SCID passage of chromosome 3 monochromosomal hybrids and mapping of the non-randomly lost regions [8,9].
We found that TMEM7 shares 38% sequence identity and 45% sequence similarity with human and mouse 28-kDa interferon-α (IFN-α) responsive protein [10,11]. The antiviral, antiproliferative, apoptosis-inducing, anti-angiogenic, and immunologic effects of IFNs are mediated by transcriptional induction of various genes [12]. These observations, together with the TMEM7 expression adult and fetal liver and its chromosomal location, prompted us to investigate whether this gene may contribute to the pathogenesis of HCC. We have now examined various HCC cell lines and primary tumors for alterations of TMEM7. Our results show that expression of TMEM7 was down-regulated or silenced in both primary tumors and HCC cell lines, and that either ectopic expression of TMEM7 or IFN-α induced up-regulation of endogenous TMEM7 suppressed tumor cell proliferation or invasion.
Materials and Methods
Primary HCC specimens and PCR analysis of TMEM7
Primary tumor specimens were obtained by surgical resection of HCC from 17 patients in Qidong, China. Each tumor sample was matched with its surrounding noncancerous liver tissue. The tumors were positive for hepatitis B virus (HBV) surface antigen and /or PCR detection of the HBVx gene. DNA was extracted from tumor tissue samples, and portions of TMEM7 and of the β-globin gene (internal control) were amplified by a multiplex polymerase chain reaction (PCR) with the primers 5’-GATCCTGAAAAACCTGGTGT-3’ and 5’-TCCAAGGCTCATATAGCAGT-3’ for TMEM7 (sense and antisense, respectively, yielding a product of 369 bp) and 5’-GAAGAGCCAAGGACAGGTAC-3’ and 5’-CAACTTCATCCACGT TCACC-3’ for the β-globin gene (sense and antisense, respectively, yielding a product of 268 bp). PCR was performed with a Peltier Thermal Cycler (MJ Research, Watertown, MA) according to standard protocols.
HCC cell culture, mutation screening, and RT-PCR analysis of TMEM7 expression
Eighteen HCC cell lines used in the present study were previously described and analyzed by comparative genomic hybridization (CGH) [13]. Cells were cultured under a humidified atmosphere of 5% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DMEM)–F12 supplemented with 10% fetal bovine serum, 2 mM glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). They were passaged by exposure to trypsin. Total RNA was isolated from the cells with the use of an RNeasy kit (Qiagen, Valencia, CA) and was subjected to reverse transcription (RT) and multiplex PCR analysis with the TMEM7-specifc primers described above and with the primers 5’-CTCGCGAACAAGGGATTATG-3’ and 5’-ACACAGCCCAGCCACATTA-3’ (sense and antisense, respectively, yielding a product of 507 bp) specific for DLC1 (internal control). TMEM7 PCR products amplified from the HCC cell lines were also purified and sequenced for detection of mutations. Total RNA from HCC cell lines and from 20 primary HCC samples were analyzed by quantitative real time PCR for expression of TMEM7. DNA from these primary HCC samples was not available. The TMEM7 primer sets, probes, and an endogenous control GAPDH were purchased from Applied Biosystems (Applied Biosystems, Foster City, CA). The ABI PRISM 7900HT Sequence Detection System (PE Applied Biosystems, Foster City, CA) was used with a universal cycling condition. The 2–{Delta}{Delta}Ct method was used to calculate the relative fold difference of TMEM7 mRNA expression in all samples. Two-fold increased or decreased expression was considered significant.
Tissue distribution of TMEM7 mRNA
The abundance of TMEM7 mRNA in human organs and tissues was determined with the use of Rapid-Scan Gene Expression Panels (OriGene Technologies, Rockville, MD). TMEM7 primers used as above and 5’-GTGGGGCGCCCCAGGCACCA-3’ and 5’-CTCCTTAATGTCACGCACGATTTC-3’ (sense and antisense, respectively, yielding a product of 540 bp) for the actin gene (internal control). PCR was performed with a Peltier Thermal Cycler, and 10 μl of each reaction mixture were then subjected to electrophoresis.
Plasmid construction and cell transfection
A full-length TMEM7 cDNA was subcloned into pcDNA-DEST40 and pcDNA-DEST47-GFP (Invitrogen, Carlsbad, CA), vectors designed for high-level stable or transient expression in mammalian cells. The sequence and orientation of the constructs were confirmed by DNA sequencing. Cells were transfected with the vectors using Lipofectamine 2000 (Invitrogen) and were subjected to selection 48 h after transfection by incubation in complete medium containing G418 (Invitrogen) at 200 or 250 μg/ml. Expression of TMEM7 cDNA in the transfected cell lines was assessed by RT-PCR analysis. The efficiency of transient transfection was evaluated under the same experimental conditions with pcDNA-DEST47-GFP vectors. Selected stable transfectants were expanded for analysis of tumorigenicity.
Assay of cell growth, colony formation, and cell migration
Exponentially growing cells were transfected with either pcDNA-DEST40 or pcDNA-DEST40-TMEM7, harvested 2 days later, and transferred to six-well plates (3 × 104 cells per well) in G418 selection medium. The cells were harvested with trypsin at 48-h intervals during incubation for up to 10 days and the number of viable cells was determined by staining with trypan blue. Colony formation was assayed with a cell transformation detection kit (Chemicon, Temecula, CA). The transfected cells were thus suspended in 0.4% agar, layered onto a 0.8% agar base, and incubated at 37°C until colonies formed. The colonies were counted after staining with the solution included with the kit. For assay of cell migration in vitro, transiently transfected cells (2 × 105) were suspended in serum-free medium and seeded into the upper compartment of BIO-COAT invasion chambers containing a membrane (pore size, 8 μm) that had been coated with Matrigel (Becton Dickinson, Bedford, MA). The lower compartment of the chambers was filled with complete medium. After incubation for 24 h at 37°C, cells that had migrated to the bottom surface of the membrane were fixed, stained with Diff-Quick (Dade Behring, Newark, DE), and counted.
Detection of IFN-α responses
Two highly invasive HCC cell lines, HLF and MHCC97, that express moderate levels of TMEM7 were cultured in 100-mm dishes and incubated with native IFN-α (Schering, Kenilworth, NJ) at 100 or 1000 IU/ml or with vehicle for 7 days, with a change of medium every 2 days. The cells were subjected to RT and real-time PCR analysis for measurement of TMEM7 mRNA or were assayed for cell migration as described above. For quantitative RT-PCR analysis, total RNA was isolated from the cells and converted to cDNA, 200 ng of which were then subjected to real-time PCR with TaqMan Universal PCR Master Mix and an ABI Prism Sequence Detection Instrument 7900 (Applied Biosystems, Foster City, CA). Primers and TaqMan probes for TMEM7 (Assay ID: Hs00230189-m1) and for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene (internal control) were obtained from Applied Biosystems.
Tumorigenicity assay
HCC cells (PLC/PRF/5 and SNU 398) stably transfected with pcDNA-DEST40-TMEM7 or with the empty vector were harvested. The cell viability was assessed by staining with trypan. Viable cells (2 × 106) were injected subcutaneously at the proximal dorsal midline of 6-week-old male athymic nu/nu mice (Harlan). The size of the resulting tumors was measured in two dimensions twice a week for up to 6 weeks.
Effects of DNA methylation and histone deacetylation inhibitors on TMEM7 expression
HCC cell lines with undetectable or low levels of TMEM7 expression were cultured in six-well plates to 30 to 40% confluence and were then incubated with 1 μM 5-aza-2’-deoxycytidine (Sigma-Aldrich, St. Louis, MO), a DNA methyltransferase inhibitor, for 72 h; with 500 nM trichostatin A (Sigma-Aldrich), a histone deacetylase inhibitor, for 12 h; or with the combination of 1 μM 5-aza-2’-deoxycytidine (for 72 h) and 500 nM trichostatin A (added only during the last 12 h). Total RNA was then extracted from the cells and subjected to RT and real-time PCR analysis for quantitation of TMEM7 mRNA as described above.
Statistical analysis
All data are representative of at least three independent experiments. Quantitative data are presented as means ± SD, and the significance of differences between means was determined by Student’s t test. A P value of <0.05 was considered statistically significant.
Results
Structure of TMEM7
Human TMEM7 encompasses ~ 3 kbp of genomic DNA, comprises two exons consisting of a total of 791 bp, and encodes a protein of 232 amino acids with a predicted molecular size of 27 kDa (Figure 1A). The SMART program predicts that the TMEM7 protein contains a single transmembrane domain that comprises amino acids 211 to 228 (LSIFCCCVILIVIVVIVV). Sequence alignment with the use of GCG Gap-Global revealed that human TMEM7 shares 38% sequence identity and 45% sequence similarity with human or mouse 28-kDa IFN-α responsive protein (Figure 1B). No conserved domains were detected in TMEM7 by the NCBI Conserved Domain Search program.
Figure 1.

Structure of human TMEM7 (A) and sequence alignment of human TMEM7 with human (HSA) or mouse (Mu) 28-kDa IFN-α responsive proteins (B). Identical residues in the sequence alignment are shaded, and dashes represent gaps introduced to optimize the alignment.
Expression and mutation screening of TMEM7 in HCC cell lines
The expression of TMEM7 in 18 HCC cell lines was examined by RT-PCR analysis. The loss or near loss of TMEM7 expression in the absence of homozygous deletion of the gene was apparent in six (33%) of the eighteen cell lines (Figure 2A). Sequencing of TMEM7 exon 2 in these HCC cell lines did not detect any mutations (data not shown).
Figure 2.
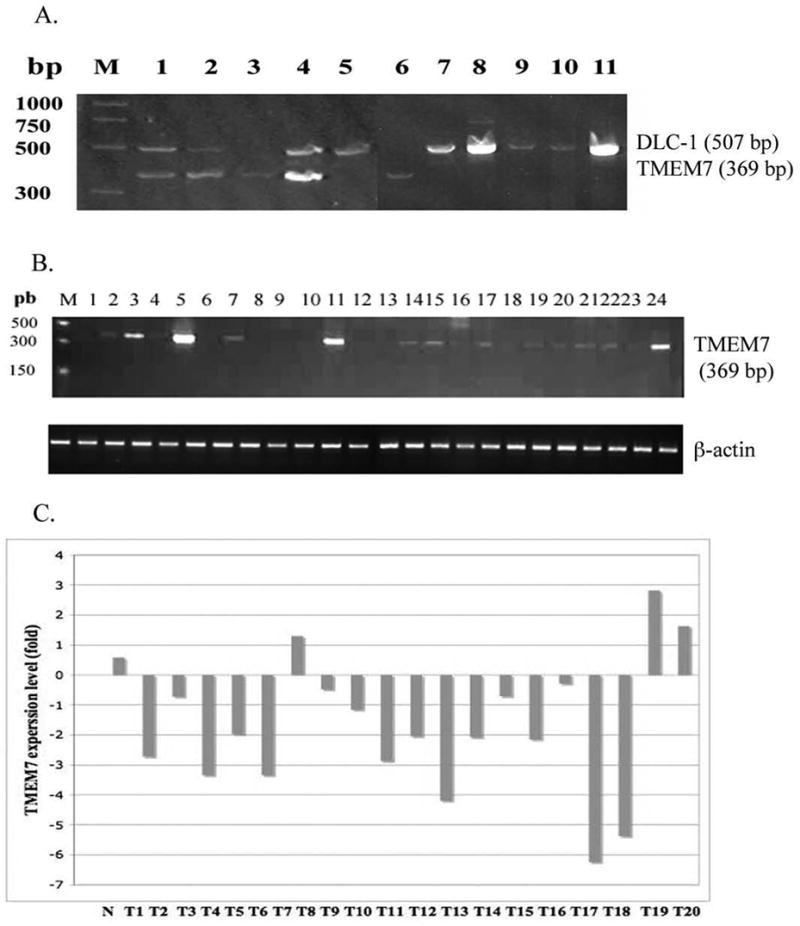
RT-PCR analysis of TMEM7 expression in human organs or tissues, primary HCC samples, and in HCC cell lines. (A) Expression of TMEM7 in HCC cell lines. Total RNA was isolated from the cell lines and subjected to RT-PCR analysis with primers specific for TMEM7 and for DLC1 (internal control). The cell lines Huh 6, HuH7, Chang, SK-Hep-1, and Focus (lanes 1 to 4 and lane 6, respectively) manifested a moderate level of TMEM7 expression, whereas TMEM7 mRNA was undetectable or present at low levels in SNU449, PLC/PRF/5, HLE, SNU 387, SNU 398, and SNU 475 (lane 5 and lanes 7 to 11, respectively). A moderate level of TMEM7 expression was also detected in the HCC cell lines HLF, MHCC97, 7703K, HepG2, Hep3B, Hep40, and SNU 182 (data not shown). (B) Expression of TMEM7 in human organs and tissues measured with a panel of cDNAs prepared from brain, heart, kidney, spleen, liver, colon, lung, small intestine, skeletal muscle, stomach, testis, placenta, salivary gland, thyroid, adrenal gland, pancreas, ovary, uterus, prostate, skin, plasma leukocytes, bone marrow, fetal brain, and fetal liver (lanes 1 to 24, respectively). PCR was performed with primers specific for TMEM7 and for the β-actin gene (internal control). Lane M, molecular size standards. (C). Expression of TMEM7 in primary HCC samples. N; normal liver sample and T1–T20, primary HCC tumor samples. Among 20 tumors examined, 17 displayed down regulation of TMEM7 expression.
Expression of TMEM7 in human tissues and primary HCC
Expression of TMEM7 in 24 human organs or tissues was examined with an RT-PCR–based system. TMEM7 mRNA was most abundant in adult liver but was also present in smaller amounts in kidney, testis, and fetal liver (Figure 2B). It was not detected or was present at only low levels in the other organs and tissues examined. Quantitative real time PCR analysis of 20 primary HCC tumors showed down regulation of TMEM7 expression in 17 tumors (85%) as compared normal liver tissue (Figure 2C). Homozygous deletion of TMEM7 was not detected by Southern blotting in 17 primary HCC specimens and their matched normal tissue samples (data not shown).
Effects of TMEM7 on cell growth, colony formation, and cell migration
We examined the effects of TMEM7 on various cellular activities by transient transfection of two HCC cell lines, SNU 398 and PLC/PRF/5, which contain only a small amount of endogenous TMEM7 mRNA with either pcDNA-DEST40-TMEM7 and pcDNA-DEST47-GFP-TMEM7 or the corresponding empty vectors. Using fluorescent microscopy, we detected approximately 65 to 75% transfection efficiency in SNU 398 and PLC/PRF/5 cells transfected with pcDNA-DEST47-GFP-TMEM7. Transfection of each of the two HCC cell lines with the TMEM7 vector resulted in suppression of cell growth that was first apparent 4 days after transfection compared with the rate of increase in cell number observed in cells transfected with the empty vector (Figure 3A). Transfection with the TMEM7 vector also resulted in a marked reduction in both the number and size of colonies formed by each cell line compared with that apparent with cells transfected with the empty vector (Figure 3B). We also examined the effect of TMEM7 expression on the invasiveness of SNU 398 and PLC/PRF/5 cells in vitro. Although the proportions of PLC/PRF/5 and SNU 398 cells transfected with the empty vector that migrated through a Matrigel-coated membrane differed by a factor of >2, ectopic expression of TMEM7 inhibited cell migration by >50% in both HCC cell lines (Figure 3C).
Figure 3.

Inhibition of HCC cell growth, colony formation, and cell migration by ectopic expression of TMEM7. (A) Cell growth. PLC/PRF/5 or SNU 398 cells were transiently transfected with pcDNA-DEST40-TMEM7 or pcDNA-DEST40, harvested after 48 h, and transferred to six-well plates (3 × 104 cells per well). The number of viable cells was determined at various times thereafter (upper panel) and the wells were photographed after 10 days (lower panel). (B) Colony formation. Cells transfected as in (A) were harvested and assayed for colony formation. Colonies formed after 12 days were stained, counted (upper panel), and photographed (lower panel). Data are expressed as colony formation efficiency relative to that of each cell line transfected with the empty vector. (C) Cell migration. Cells transfected as in (A) were harvested and assayed for cell invasiveness with a Matrigel-coated membrane. Cells that had migrated through the membrane after incubation for 24 h were stained and counted . Data are expressed as the average number of invading cells per microscopic field (400×) determined from 10 fields per membrane. Quantitative data in (A) through (C) are means ± SD of values from three independent experiments.
Effects of IFN-α on TMEM7 expression in and migration of HCC cells
To determine whether TMEM7 is an IFN-α responsive gene, we examined the effect of IFN-α on the abundance of TMEM7 mRNA in HLF and MHCC97 cells, both of which are highly invasive and express TMEM7 at a moderate level. Quantitative RT-PCR analysis revealed that incubation of cells with IFN-α for 7 days resulted in a concentration-dependent increase in the amount of TMEM7 mRNA in each cell line (Figure 4A). We also examined the effect of IFN-α on cell migration in vitro. Although incubation of cells with IFN-α at 100 IU/ml for 7 days had no effect on cell migration (data not shown), incubation with IFN-α at 1000 IU/ml resulted in >50% inhibition of cell invasiveness (Figure 4B).
Figure 4.

Induction of TMEM7 expression in and inhibition of migration of HCC cells by IFN-α. (A) TMEM7 expression. HLF and MHCC97 cells were incubated in the absence or presence of IFN-α (100 or 1000 IU/ml) for 7 days, after which the abundance of TMEM7 mRNA was determined by quantitative RT-PCR analysis. (B) Cell migration. HLF and MHCC97 cells were exposed to IFN-α (1000 IU/ml) for 7 days and were assayed for cell invasiveness with a Matrigel-coated membrane during the final 24 h of exposure. Cells that had migrated through the membrane were stained, photographed (upper panel), and counted (lower panel). Data are expressed as the average number of invading cells per microscopic field (400×) determined from 10 fields per membrane. Quantitative data in (A) and (B) are means ± SD of values from three independent experiments.
Effect of TMEM7 on tumorigenicity
To examine the effect of TMEM7 on tumorigenicity in vivo, we injected HCC cells stably transfected with pcDNA-DEST40-TMEM7, which showed high levels of TMEM7 expression as quantified by real time PCR, or with the empty vector into athymic nude mice. Restoration of TMEM7 expression reduced the ability of SNU 398 and PLC/PRF/5 cells to form tumors in nude mice. Compared with cells transfected with the empty vector, those transfected with the TMEM7 vector showed an increased latency to tumor formation and formed markedly smaller and fewer tumors (Table 1).
Table 1.
Inhibition of in vivo tumorigenicity of HCC cells by restoration of TMEM7 expression.
| Cell Line | number of cells inoculated | Latency (days) | Tumor size (W×L mm) | # of tumors/# of inoculations |
|---|---|---|---|---|
| SNU 398 Vector | 2×106 | 30 | 12×14 | 20/20 |
| SNU 398 TMEM7 | 2×106 | 45 | 6×8 | 10/20 |
| PCL/PRF/5 Vector | 2×106 | 45 | 13×16 | 19/20 |
| PCL/PRF/5 TMEM7 | 2×106 | 60 | 8×10 | 13/20 |
Inactivation of TMEM7 by DNA methylation and histone deacetylation in HCC cells
Given that homozygous deletion and mutation were not responsible for down regulation or loss of TMEM7 expression in the HCC cell lines studied, we examined the possible role of DNA methylation or histone deacetylation in TMEM7 silencing by treating four of the affected cell lines with 5-aza-2’-deoxycytidine or trichostatin A, respectively. Whereas these agents alone or together had no effect on TMEM7 expression in PLC/PRF/5 or HLE cells, 5-aza-2’-deoxycytidine induced 6.4- and 4.5-fold increases in the amount of TMEM7 mRNA in SNU 398 and SNU449 cells, respectively (Figure 5). Although trichostatin A alone had no effect on TMEM7 expression in the latter two cell lines, it potentiated the effect of 5-aza-2’-deoxycytidine about 27 to 34% more.
Figure 5.
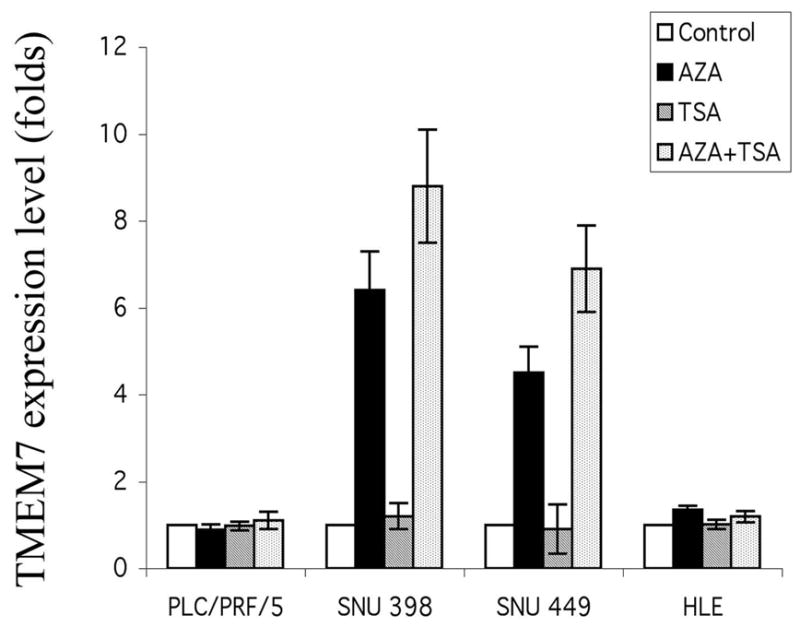
Effects of DNA methyltransferase and histone deacetylase inhibitors on TMEM7 expression in HCC cells. The indicated cell lines were incubated with 5-aza-2’-deoxycytidine (AZA), with trichostatin A (TSA), or with both agents as described in Materials and Methods, after which the abundance of TMEM7 mRNA was determined by quantitative RT-PCR analysis.
Discussion
The identification of cancer-related genes, which constitute only a small fraction of the human genome, is fundamental to the development of new therapeutics. We have now shown that expression of TMEM7 is down-regulated in 85% of the primary HCC and in 33% of the HCC cell lines studied. Restoration of the expression of this gene in such cell lines by vector transfection results in inhibition of tumorigenic properties in vitro and in vivo. Ectopic expression of TMEM7 thus suppressed cell growth and migration in vitro as well as tumor formation in vivo, showing that TMEM7 acts as a tumor suppressor gene in HCC cells.
About 30% of HCC patients exhibit loss of heterozygosity on chromosome 3p, and loss of DNA copy number at 3p has been detected by CGH in the HCC cell lines used in the present study [13]. Current analysis showed that homozygous deletion and mutation of TMEM7 were not responsible for down-regulation of the expression of this gene in the affected cell lines and were also not detected in the primary HCC tumor specimens. These results are not surprising given the fact homozygous deletions are rare in primary HCC tumors and HCC cell lines [14,15] and that none of the other 3p21.3 genes appear to be mutational targets in cancers. However, as in other types of cancer, aberrant DNA methylation and histone deacetylation also contribute to the inactivation of tumor suppressor genes in HCC [4,16]. Down-regulation or complete silencing of the expression of RASSF1A, BLU, SEMA3B, FHIT, RIZ, and VHL, all of which are candidate or bona fide tumor suppressor genes on chromosome 3p, has been detected in HCC and shown to be the result of genetic or epigenetic mechanisms [17,18–21]. In the present study, treatment of TMEM7-deficient HCC cell lines with trichostatin A alone had no effect on TMEM7 expression. However, this histone deacetylase inhibitor potentiated the stimulatory effect of 5-aza-2-deoxycytidine on TMEM7 expression in two HCC cell lines. A similar effect of these two agents was recently observed in prostate cancer cell lines and is consistent with the notion that optimal reexpression of genes silenced by promoter methylation and histone deacetylation can be achieved by treatment with inhibitors of both processes [22]. Most recently the molecular mechanisms responsible for such optimal response by sequential treatment with methyltransferase inhibitors and histone deacetylase inhibitors have been reported [23]. Drugs that bring about DNA demethylation or histone acetylation have been shown to have therapeutic potential in cancer and the responses to their combination justify the testing in randomized clinical trials [6,23,24]. Other mechanisms responsible for TMEM7 deregulation, such as phosphorylation and ubiquitination remain to be examined .
Ectopic expression of TMEM7 in TMEM7-deficient HCC cells resulted in inhibition of cell proliferation, colony formation, and cell migration in vitro as well as of tumor formation in nude mice. Although restoration of TMEM7 expression in HCC cells did not prevent tumor formation in most injected mice, it reduced both the number of mice that developed tumors and tumor size as well as increased the latency to tumor formation. We previously showed that ectopic expression of DLC-1, which functions as tumor suppressor gene, resulted in a similar reduction in the frequency and size of tumors formed by HCC cells in this model system [25]. In breast and lung cancer, DLC-1 abolished the in vivo tumorigenicity [26,27].
Based on the 38% sequence identity and 45% sequence similarity of the 28-kDa IFN-α-responsive protein with TMEM7 we addressed the question whether there are functional similarities and/or cooperation between them. IFN-α manifests antiviral as well various antitumor activities that include immunomodulatory, antiproliferative, and anti-angiogenic effects [28,29]. Chemoadjuvant therapy with IFN-α has been shown to be highly effective, inhibiting dissemination to secondary sites, in individuals with advanced and metastatic HCC. We have shown that exposure of highly invasive HCC cell lines to IFN-α resulted in a marked increase in the abundance of TMEM7 mRNA as well as inhibition of cell migration in vitro. A similar effect of IFN-α on cell invasiveness has been observed in HLF cells [29]. IFN-α also exerts antiproliferative and antimetastatic effects on HCC through inhibition of matrix metalloproteinase expression. The migration of and invasion by HCC cells require matrix metalloproteinase activity [29,30]. Lactoferrin, which is encoded by another candidate tumor suppressor gene located in the region of deletion at 3p21.3 in cancer cells, exerts an immunomodulatory effect through up-regulation of IFN-α expression in healthy individuals [31,32].
In summary, we have shown that TMEM7 appears to be an IFN-α responsive gene and manifests tumor suppressor activity that may be important in the pathogenesis of HCC. Our observations suggest that TMEM7 has potential therapeutic applications not only for cancer but also for inflammatory or infectious diseas
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NCI.
Nonstandard abbreviations
- TMEM7
transmembrane protein 7
- HCC
hepatocellular carcinoma
- IFN-α
interferon alpha
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002;31:339–46. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. N Engl J Med. 1999;340:745–50. doi: 10.1056/NEJM199903113401001. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka Y, Hanada K, Mizokami M, Yeo AE, Shih JW, Gojobori T, Alter HJ. Inaugural Article: A comparison of the molecular clock of hepatitis C virus in the United States and Japan predicts that hepatocellular carcinoma incidence in the United States will increase over the next two decades. Proc Natl Acad Sci U S A. 2002;99:15584–9. doi: 10.1073/pnas.242608099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katoh H, Shibata T, Kokubu A, Ojima H, Fukayama M, Kanai Y, Hirohasi S. Epigenetic instability and chromosomal instability in hepatocellular carcinoma. Am J Pathol. 2006;168:1375–84. doi: 10.2353/ajpath.2006.050989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–28. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 6.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 7.Huebner K. Tumor suppressors on 3p: a neoclassic quartet. Proc Natl Acad Sci U S A. 2001;98:14763–5. doi: 10.1073/pnas.261586598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiss H, Yang Y, Kiss C, Andersson K, Klein G, Imreh S, Dumansky JP. The transcriptional map of the common eliminated region 1 (C3CER1) in 3p21.3. Eur J Hum Genet. 2002;10:52–61. doi: 10.1038/sj.ejhg.5200758. [DOI] [PubMed] [Google Scholar]
- 9.Imreh S, Klein G, Zabarovsky ER. Search for unknown tumor-antagonizing genes. Genes Chromosomes Cancer. 2003;38:307–21. doi: 10.1002/gcc.10271. [DOI] [PubMed] [Google Scholar]
- 10.Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmem CM, Schuler DG, Altschul SF, Zeeberg B, Buetow KH, et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci U S A. 2002;99:16899–903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–73. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- 12.Leaman DW, Chawla-Sarkar M, Jacobs B, Vyas K, Sun Y, Ozdemir A, Taolin Yi, Williams BR, Borden EC. Novel growth and death related interferon-stimulated genes (ISGs) in melanoma: greater potency of IFN-beta compared with IFN-alpha2. J Interferon Cytokine Res. 2003;23:745–56. doi: 10.1089/107999003772084860. [DOI] [PubMed] [Google Scholar]
- 13.Zimonjic DB, Keck CL, Thorgeirsson SS, Popescu NC. Novel recurrent genetic imbalances in human hepatocellular carcinoma cell lines identified by comparative genomic hybridization. Hepatology. 1999;29:1208–14. doi: 10.1002/hep.510290410. [DOI] [PubMed] [Google Scholar]
- 14.Pineau P, Marchio A, Nagamori S, Seki S, Tiollais P, Dejean A. Homozygous deletion scanning in hepatobiliary tumor cell lines reveals alternative pathways for liver carcinogenesis. Hepatology. 2003;37:852–61. doi: 10.1053/jhep.2003.50138. [DOI] [PubMed] [Google Scholar]
- 15.Thorgeirsson SS. Hunting for tumor suppressor genes in liver cancer. Hepatology. 2003;37:739–41. doi: 10.1053/jhep.2003.50174. [DOI] [PubMed] [Google Scholar]
- 16.Yang B, Guo M, Herman JG, Clark DP. Aberrant promoter methylation profiles of tumor suppressor genes in hepatocellular carcinoma. Am J Pathol. 2003;163:1101–7. doi: 10.1016/S0002-9440(10)63469-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du Y, Carling T, Fang W, Piao Z, Sheu JC, Huang S. Hypermethylation in human cancers of the RIZ1 tumor suppressor gene, a member of a histone/protein methyltransferase superfamily. Cancer Res. 2001;61:8094–9. [PubMed] [Google Scholar]
- 18.Schagdarsurengin U, Wilkens L, Steinemann D, Flemming P, Kreipe HH, Pfeifer GP, Schlegelberger B, Damman R. Frequent epigenetic inactivation of the RASSF1A gene in hepatocellular carcinoma. Oncogene. 2003;22:1866–71. doi: 10.1038/sj.onc.1206338. [DOI] [PubMed] [Google Scholar]
- 19.Tischoff I, Markwarth A, Witzigmann H, Uhlmann D, Hauss J, Mirmohammadsadegh A, Wittekind C, Hengge UR, Tannapfel A. Allele loss and epigenetic inactivation of 3p21.3 in malignant liver tumors. Int J Cancer. 2005;115:684–9. doi: 10.1002/ijc.20944. [DOI] [PubMed] [Google Scholar]
- 20.Yu J, Ni M, Xu J, Zhang H, Gao B, Gu J, Chen J, Zhang L, Wu M, Zhen S, Zhu J. Methylation profiling of twenty promoter-CpG islands of genes which may contribute to hepatocellular carcinogenesis. BMC Cancer. 2002;2:29. doi: 10.1186/1471-2407-2-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yuan BZ, Keck-Waggoner C, Zimonjic DB, Thorgeirsson SS, Popescu NC. Alterations of FHIT gene in human hepatocellular carcinoma. Cancer Res. 2000;60:1049–53. [PubMed] [Google Scholar]
- 22.Guan M, Zhou X, Soulitzis N, Spandidos DA, Popescu NC. Aberrant methylation and deacetylation of deleted in liver cancer-1 gene in prostate cancer: potential clinical applications. Clin Cancer Res. 2006;12:1412–9. doi: 10.1158/1078-0432.CCR-05-1906. [DOI] [PubMed] [Google Scholar]
- 23.Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M, Grever M, Galm O, Dauses T, Karp JE, Rudek MA, Zhao M, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66:6361–9. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 24.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 25.Zhou X, Thorgeirsson SS, Popescu NC. Restoration of DLC-1 gene expression induces apoptosis and inhibits both cell growth and tumorigenicity in human hepatocellular carcinoma cells. Oncogene. 2004;23:1308–13. doi: 10.1038/sj.onc.1207246. [DOI] [PubMed] [Google Scholar]
- 26.Yuan BZ, Zhou X, Durkin ME, Zimonjic DB, Gumundsdottir K, Eyfjord JE, Thorgeirsson SS, Popescu NC. DLC-1 gene inhibits human breast cancer cell growth and in vivo tumorigenicity. Oncogene. 2003;22:445–50. doi: 10.1038/sj.onc.1206064. [DOI] [PubMed] [Google Scholar]
- 27.Yuan BZ, Jefferson AM, Baldwin KT, Thorgeirsson SS, Popescu NC, Reynolds SH. DLC-1 operates as a tumor suppressor gene in human non-small cell lung carcinomas. Oncogene. 2004;23:1405–11. doi: 10.1038/sj.onc.1207291. [DOI] [PubMed] [Google Scholar]
- 28.Bogdan C. The function of type I interferons in antimicrobial immunity. Curr Opin Immunol. 2000;12:419–24. doi: 10.1016/s0952-7915(00)00111-4. [DOI] [PubMed] [Google Scholar]
- 29.Kaneko F, Saito H, Saito Y, Wakabayashi K, Nakamoto N, Tada S, Suzuki S, Tsunematsu S, Kumagai N, Ishii H. Down-regulation of matrix-invasive potential of human liver cancer cells by type I interferon and a histone deacetylase inhibitor sodium butyrate. Int J Oncol. 2004;24:837–45. [PubMed] [Google Scholar]
- 30.Giannelli G, Bergamini C, Fransvea E, Marinosci F, Quaranta V, Antonaci S. Human hepatocellular carcinoma (HCC) cells require both alpha3beta1 integrin and matrix metalloproteinases activity for migration and invasion. Lab Invest. 2001;81:613–27. doi: 10.1038/labinvest.3780270. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, Li J, Szeles A, Imreh MP, Kost-Alimova M, Kiss H, Kholodnyuk I, Fedorova L, Darai E, Kein G, Imreh S. Consistent downregulation of human lactoferrin gene, in the common eliminated region 1 on 3p21.3, following tumor growth in severe combined immunodeficient (SCID) mice. Cancer Lett. 2003;191:155–64. doi: 10.1016/s0304-3835(02)00677-8. [DOI] [PubMed] [Google Scholar]
- 32.Ishikado A, Imanaka H, Kotani M, Fujita A, Mitsuishi Y, Kanemitsu T, Tamura Y, Makino T. Liposomal lactoferrin induced significant increase of the interferon-alpha (IFN-alpha) producibility in healthy volunteers. Biofactors. 2004;21:69–72. doi: 10.1002/biof.552210113. [DOI] [PubMed] [Google Scholar]