

Abstract
Vascular endothelial growth factor (VEGF) signaling is critical for both normal and disease-associated vascular development. Dysregulated VEGF signaling has been implicated in ischemic stroke, tumor angiogenesis, and many other vascular diseases. VEGF signals through several effectors, including the Rho family of small GTPases. As a member of this family, Rac1 promotes VEGF-induced endothelial cell migration by stimulating the formation of lamellipodia and membrane ruffles. To form these membrane protrusions, Rac1 is activated by guanine nucleotide exchange factors (GEFs) that catalyze the exchange of GDP for GTP. The goal of this study was to identify the GEF responsible for activating Rac1 in response to VEGF stimulation. We have found that VEGF stimulates biphasic activation of Rac1 and for these studies we focused on the peak of activation that occurs at 30 min. Inhibition of VEGFR-2 signaling blocks VEGF-induced Rac1 activation. Using a Rac1 nucleotide-free mutant (G15ARac1), which has a high affinity for binding activated GEFs, we show that the Rac GEF Vav2 associates with G15ARac1 after VEGF stimulation. Additionally, we show that depleting endothelial cells of endogenous Vav2 with siRNA prevents VEGF-induced Rac1 activation. Moreover, Vav2 is tyrosine phosphorylated upon VEGF treatment, which temporally correlates with Rac1 activation and requires VEGFR-2 signaling and Src kinase activity. Finally, we show that depressing Vav2 expression by siRNA impairs VEGF-induced endothelial cell migration. Taken together, our results provide evidence that Vav2 acts downstream of VEGF to activate Rac1.
Keywords: VEGF, Vav2, Rac, cell migration, angiogenesis
INTRODUCTION
Angiogenesis, the sprouting of new blood vessels from a pre-existing vascular network, occurs in many physiological and pathological conditions. This process is initiated by the migration of endothelial cells in response to chemotactic agents such as vascular endothelial growth factor (VEGF). VEGF is a soluble glycoprotein that promotes wound healing and blood vessel formation during specific biological processes such as pregnancy [1]. Though VEGF signaling is required for normal vascularization, aberrant signaling is linked to many diseases because of its ability to stimulate inappropriate cell proliferation, motility, and permeability. For example, in tumor angiogenesis, VEGF enhances tumor growth by inducing the migration and organization of surrounding endothelial cells into nascent blood vessels [1,2].
The formation of lamellipodia, protrusive structures at the leading edge of migrating cells, is critical for cell migration [3]. Previous work has shown that VEGF signaling induces lamellipodia formation and increases the migration of endothelial cells [4–6]. During this process, VEGF elicits its biological effects such as migration through VEGF receptor-2 (VEGFR-2), a member of the VEGF family of receptor tyrosine kinases found in endothelial cells [7].
VEGF-induced endothelial cell migration is mediated by many signaling molecules including the Rho family of small GTPases. Rho GTPases transduce signals from extracellular stimuli to cause changes in cell behavior [8]. The best characterized members of the family, RhoA, Cdc42, and Rac1 affect many cellular processes required for growth and survival including cytoskeletal organization, cell morphology, and adhesion. Of these GTPases, active Rac1 has been shown to stimulate cell migration by inducing lamellipodia formation. The effects of VEGF on endothelial cell motility are inhibited when Rac1 activity is perturbed [4], suggesting that VEGF mediates actin remodeling through Rac1.
Like other Rho GTPases, Rac1 acts as a molecular switch that cycles from an inactive GDP-bound state to an active GTP-bound state. Since Rac1 has an intrinsically low rate of GDP-GTP cycling, this process is promoted by guanine nucleotide exchange factors (GEFs) [9–12]. GEFs often couple signaling pathways that involve the activation of cell surface receptors such as integrins and growth factor receptors to Rho GTPases. In this study, we asked which exchange factor mediates Rac1 activity, downstream of VEGF signaling in endothelial cells and how its activity is regulated by VEGF.
MATERIALS AND METHODS
Cell culture
Pooled human umbilical vein endothelial cells (HUVECs) and human dermal microvascular endothelial cells (HMVEC-d) were obtained from Cambrex (East Rutherford, NJ) and grown in Clonetics EGM-2 or EGM-2-MV (for HMVEC-d) media according to manufacturer’s instructions at 37°C and 10% CO2. Cells were used between passages 3–6. COS7 cells were grown in Dulbecco’s modified Eagles medium (Sigma, St. Louis, MO) containing 10% fetal bovine serum (Sigma) and penicillin/streptomycin/fungizone (Invitrogen, Carlsbad, CA). Jurkat T cells were grown in RPMI 1640 media supplemented with 10% fetal calf serum (BioWhittaker, Rockland, ME).
Antibodies and pharmacological reagents
Vav2 rabbit antiserum was generated previously and used as previously described [13]. Vav2 rabbit polyclonal antibody (pAb) used for GST-G15ARac1 pulldown was obtained from Zymed (San Francisco, CA). Vav2 monoclonal antibody (mAb) was purchased from Babraham Bioscience Technologies (London, England). Anti-phosphotyrosine (pY20), Rac1, Cdc42, and Sos1 mAbs were purchased from BD Transduction Laboratories (San Jose, CA). Anti-phosphotyrosine (pY99) mAb, VEGF-1 pAb, RhoA mAb, β-PIX goat-pAb, and phospho-Vav2 (pY172) rabbit-pAb were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Vav3 pAb, Src mAb and cortactin (clone 4F11) mAb was obtained from Upstate Biotechnology (Lake Placid, NY). Vav1, VEGF-2 and VEGF-3 pAbs were purchased from Cell Signaling Technology. Phospho-Src (pY418) rabbit-pAb was purchased from Biosource (Camarillo, CA). Recombinant human VEGF165 was purchased from R&D Systems (Minneapolis, MN). VEGFR-2 inhibitors SU1498 and ZM323881, human α-thrombin, Src inhibitors PP2 and SU6656, and inactive Src compound PP3 were purchased from Calbiochem (San Diego, CA). Glutathione-Sepharose was purchased from Amersham Biosciences (Uppsala, Sweden).
siRNA transfection
HUVECs were transfected at 90–100% confluency with the siRNA oligos indicated in each experiment according to the manufacturer’s protocol using RNAifect Transfection Reagent (Qiagen, Valencia, CA). After the delivery of the 2.5 μg for HUVECs and 5.0 μg for HMVEC-d siRNA oligos for 4 h, the transfection medium was replaced with fresh EGM-2 media for 24–36 h. Vav2-specific siRNA oligos against the human sequence were as follows: 5′-UCACAGAGGCCAAGAAAUUUU-3′ and 5′-AAUUUCUUGGCCUCUGUGAUU -3′. VEGFR-2 (KDR)-specific siRNA oligos against the human sequence were as follows: 5′-GGAAAUCUCUUGCAAGCUAUU - 3′ and 5′ – UAGCUUGCAAGAGAUUUCCUU -3′. Rac1-specific siRNA oligos against the human sequence were as follows: 5′-GUUCUUAAUUUGCUUUUCC-3′ and 5′-GGAAAAGCAAUUAAGAAC -3′ and were used at 20 nM as described by Deroanne et al [14]. siGLO, a non-specific siRNA oligo with a fluorescent rhodamine tag, was used as a control (Dharmacon, Chicago, IL).
Immunoprecipitation and immunoblotting
For immunoprecipitation, HUVECs were starved in serum-free Optimem media for 1 h at 37°C under 10% CO2. Cells were then pretreated with specific VEGFR-2 inhibitors (50 μM SU1498 and 10 nM ZM323881) or Src inhibitors (30 μM PP2 or PP3, the latter being an inactive control) for 30 min at 37°C. Subsequently, cells were rinsed with PBS and treated with 10 ng/mL VEGF165 for the time specified. Cells were washed in cold PBS prior to scraping into modified ice-cold RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.25% deoxycholate (DOC), 1.5 mM MgCl2, 1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM NaF, 10 mM pervanadate, 10 μg/mL leupeptin, 10 μg/mL aprotinin) on ice. Lysates were clarified by centrifugation at 16,000 × g for 10 min, precleared with protein A- or protein G-Sepharose beads for 30 min, and then incubated with antibody and beads for 2 h at 4°C. The beads were washed three times in RIPA buffer and bound proteins were eluted with SDS sample buffer.
For immunoblotting, cells were washed in cold PBS and scraped into general lysis buffer (50 mM Tris-HCL, pH 7.6, 150 mM NaCl, 1% Triton X-100, 0.25% DOC, 2 mM EGTA, 1 mM PMSF, 10 μg/mL leupeptin, 10 μg/mL aprotinin). Cells were lysed on ice and clarified by centrifugation. For both techniques, protein concentrations were determined using detergent-compatible (DC) protein assay kit (Bio-Rad, Hercules, CA) according to the manufacturer’s instructions. Equal amounts of total protein were denatured in SDS sample buffer, run on 10% SDS-polyacrylamide gels and transferred to Immobilon-P membrane. Blots were probed with primary antibody and then horseradish peroxidase-conjugated secondary antibody, visualized by enhanced chemiluminescence according to the manufacturer’s instructions.
Chemotaxis assay
To assess the migratory behavior of endothelial cells transfected with Vav2 siRNA, chemotaxis assays were performed using Transwell polycarbonate filters (6.5-mm diameter, 8-μm pore size) (Costar, Acton, MA). The top and bottom surfaces of the filters were coated with 10 μg/mL fibronectin in PBS overnight at 4°C. After overnight incubation, 10 ng/mL VEGF165 in serum-free Optimem media was added to the lower chamber where indicated. Equal amounts of Vav2 siRNA transfected cells (105) in serum-free Optimem media were added to the upper chamber and allowed to migrate for 4 h at 37°C. After 4 h, migrated cells were rinsed, fixed, stained with Hoechst 33342 (Molecular Probes, Eugene, OR) and the number of cells that migrated to the other side of the filter were counted. At least three fields per filter were counted using fluorescent microscopy.
GTPase activation assays
The GTPase activity of RhoA, Rac1, and Cdc42 was measured as previously described [15] with some modifications. Cells were rinsed with cold PBS and lysed in RIPA buffer as described. Lysates were cleared by centrifugation and protein concentration was measured. Equal amounts of protein (400–600 μg) were incubated with 50 μg GST fusion protein of Rho-binding domain of Rhotekin (GST-RBD) or GST fusion protein of CRIB-binding domain of PAK (GST-PBD) bound to glutathione-Sepharose beads for 30 min at 4°C. Active RhoA was precipitated with GST-RBD and active Rac1 and Cdc42 were precipitated with GST-PBD. Beads were washed three times in buffer (50 mM Tris-HCl, pH 7.6, 500 mM NaCl, 1% Triton X-100, 0.5 mM MgCl2, 1 mM PMSF, 10 μg/mL leupeptin, 10 μg/mL aprotinin) and associated proteins were eluted with sample buffer. The amount of active RhoA, Rac1, and Cdc42 was then analyzed by immunoblotting and quantified by densitometric analysis using ImageJ software (NIH).
Precipitation of Activated GEFs with Recombinant Rac1 Protein
To perform the pulldown assay with nucleotide-free Rac1 mutant (G15ARac1), HUVECs were lysed and processed as described in Garcia-Mata et al. [16]. Briefly, cells were lysed in a buffer consisting of 1% Triton X-100, 20 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM MgCl2, and protease inhibitors. Lysates were cleared by centrifugation at 16,000 × g for 10 min. Equal amounts of protein (400–600 μg) were incubated at 4 °C for 60 min with 20 μg of GST or GST fusion proteins containing nucleotide-free Rac1 protein (G15ARac1) bound to glutathione-Sepharose. After 1 h of incubation, the beads were washed 4 times with lysis buffer. Samples were immunoblotted using antibodies against Vav2, Sos1 and β-PIX. Membranes were also stained with Ponceau S prior to immunoblotting to confirm that similar amounts of fusion proteins were used in all experiments.
Scratch wound migration assays
HUVEC monolayers were grown on growth factor-reduced Matrigel (BD Biosciences, San Jose, CA) for 48 h in glass-bottomed dishes (MatTek Corporation, Ashland, MA). Cells were starved for 1 h in serum-free Optimem media prior to the experiment. The monolayer was scratch-wounded with a pipet tip (Fisher Scientific, Suwanee, GA) and then washed to remove debris. Cells were allowed to migrate for 48 h with images collected at desired time points. In both assays, images were obtained using a 10× objective on a Zeiss Axiovert microscope (Zeiss, Thornwood, NY) and analyzed using MetaMorph Imaging software (Universal Imaging Corp., West Chester, PA).
Statistical Analysis
Statistical analysis was calculated using either Student’s t-test or 2-way analysis of variance (ANOVA) where indicated.
RESULTS
VEGF activates Rac1 through VEGFR-2 in endothelial cells
We measured Rac1 activity by using GST-PBD to pulldown GTP-Rac. Work by Zeng et al. showed a rapid and transient increase in Rac1 activity around 5 min of VEGF treatment [17]. In preliminary work, we confirmed these results and found that there is a peak of Rac activity at 1–5 min which then declines. Extending the time course, we have found that there is a second peak of Rac activity that reaches a maximum 30 min after VEGF stimulation in both HUVECs and HMVEC-d (Fig. 1A). VEGF has also been shown to activate other small GTPases [17]. We found that RhoA transiently becomes activated 5–15 min after VEGF treatment. This data confirmed work by other groups [18,19] that demonstrated a rapid and transient increase of RhoA activity in the first few minutes after VEGF treatment. Cdc42 was also activated 5–15 min after stimulation with VEGF; this activity declined and elevated again around 60 min. The activation of RhoA and Cdc42 decreased at 30 min and thus, do not correlate with the second peak of Rac activity (Fig. 1B). In this study, we have focused on this second peak of Rac activity.
Figure 1. VEGF activates Rac1 in endothelial cells through VEGFR-2.


(A) HUVECs (top panels) or HMVEC-d (bottom panels) were serum starved for 1 h and treated with 10 ng/mL VEGF for the specified times and subsequently assayed for Rac1 activity as described in Experimental Procedures. The upper left panel shows Rac-GTP levels after VEGF treatment. The lower left panel shows total Rac1 levels. The histogram on the right shows quantification of the increase in Rac1 activity after VEGF stimulation in four independent experiments. (B) VEGF activates RhoA and Cdc42. HUVECs were serum starved for 1 h and treated with 10 ng/mL VEGF for the specified times and subsequently assayed for RhoA or Cdc42 activity as described in Experimental Procedures. The upper left panel shows GTP levels of RhoA or Cdc42 after VEGF treatment. The lower left panel shows total levels of RhoA or Cdc42 expression. The histogram on the right shows quantification of the Rho-GTP or Cdc42-GTP levels after VEGF stimulation in four independent experiments. (C) VEGF receptor-2 inhibitors block VEGF-induced Rac1 activation. Cells were pretreated with either 50 μM SU1498 or 10 nM ZM323881 (VEGFR-2 inhibitors) for 30 min at 37°C, followed by VEGF treatment for 30 min. Untreated (un) denotes cells not treated with VEGF. The upper left panel shows that both inhibitors blocked Rac1 activity at 30 min. The lower left panel shows equal protein loading of total Rac1. The histogram on the right shows quantification of Rac1 activation. (D) Knockdown of VEGFR-2 protein expression by siRNA. HUVECs were transfected with either siGLO, a non-specific control siRNA oligo or VEGFR-2-specific siRNA oligos as described in Experimental Procedures. After 24 h, the cells were serum-starved for 1 h, treated with VEGF for 30 min and assayed for active Rac1. Quantification of protein expression is shown in the histogram on the right (n = 3; **p < 0.01). (E) VEGFR-2-specific siRNA blocks Rac1 activity after VEGF stimulation. HUVECs were assayed 24 h after transfection with VEGFR-2-specific siRNA. The upper left panel shows VEGF-induced Rac1 activity is decreased in VEGFR-2 siRNA-transfected cells compared to control cells. The middle left panel confirms equal levels of total Rac1, and the lower left panel shows levels of VEGFR-2 expression following siRNA treatment. The histogram on the right represents the average Rac1 activation relative to total Rac1 levels in four independent experiments (n = 4; *p < 0.05 by 2-way ANOVA).
VEGF functions are mediated by three VEGF receptors (VEGFR-1, VEGFR-2, and VEGFR-3) expressed in HUVECs, but previous work has shown that the VEGFR-2 is the main receptor responsible for processes such as cell migration [7,20–22]. To examine the role of this receptor in VEGF signaling to Rac1, we first used two structurally different but selective VEGFR-2 inhibitors, SU1498 and ZM323881. Both inhibitors blocked VEGF-induced Rac1 activation at 30 min (Fig. 1C). Using siRNA against VEGFR-2, we successfully depleted VEGFR-2 expression in HUVECs, whereas VEGFR-1, VEGFR-3, and β-actin protein expression were not affected (Fig. 1D). VEGF-induced Rac1 activation was inhibited when VEGFR-2 expression was depleted by siRNA targeting (Fig. 1E). Taken together, these results indicate that expression and activation of VEGFR-2 is required to mediate VEGF-induced Rac1 activation at 30 min.
Exchange factor Vav2 couples VEGF signaling to Rac1
It has been shown that several GEFs activate Rac1 in response to EGF and PDGF signaling [13,23–25]. To determine which exchange factors activate Rac1 downstream of VEGF, we first investigated the endogenous expression of candidate Rac1 GEFs in endothelial cells. We identified Vav2, β-PIX and Sos-1 in HUVECs by western blotting (Fig. 2A). Vav2 is a member of a family of closely related GEFs and we looked for the presence of Vav1 and Vav3 as well. Vav1 was not detected, consistent with its reported distribution being restricted to hematopoietic cells. With Vav3, a weakly cross-reacting band was detected migrating slightly faster than the corresponding band detected in COS7 cells. However, we did not detect the presence of Vav3 mRNA by RT-PCR in HUVECs (data not shown). Interestingly, in HMVEC-d cells, we did detect low levels of Vav3 both by western blotting (Fig. 2A) and by RT-PCR (data not shown).
Figure 2. Vav2 couples VEGF signaling to Rac1.


(A) Endogenous expression of candidate Rac1 GEFs in endothelial cells. Equivalent total protein from HMVEC-d, HUVECs or COS7 cells was probed with antibodies against the specific Rac1 GEFs Vav2 and Vav3. Similarly, expression of Vav1 was probed in HMVEC-d, HUVECs, or Jurkat T cells. Equivalent levels of protein from HUVECs or COS7 cells were also probed with antibodies against other Rac1 GEFs, Sos1 and β-PIX. β-actin (bottom panel) indicated equal amount of protein was loaded. (B) VEGF stimulates Vav2-Rac1 complex formation. HUVECs were serum-starved and then pretreated in the presence or absence of 50 μM SU1498. Cells were treated with 10 ng/mL VEGF for 30 min, lysed, and lysates were incubated with the nucleotide-free Rac1 mutant fused to GST (GST-G15ARac1) on glutathione-Sepharose beads for 60 min at 4°C. Antibodies against Vav2 (upper panel), Sos1 (middle panel), and β-Pix (lower panel) were used to detect binding of these proteins to G15ARac1. The histogram shows the % GEF association to G15ARac1 in four independent experiments (n = 4; *p < 0.05). (C) Knockdown of Vav2 protein expression with Vav2-specific siRNA. HUVECs were transfected with either non-specific control siRNA (siGLO) or Vav2-specific siRNA oligos as described in Experimental Procedures. The upper left panel shows that Vav2 expression was significantly decreased by siRNA. The marked expression levels of Vav3, Sos1, and β-actin are indicated in the panels and were unaffected by Vav2 siRNA. The histogram on the right illustrates the fold change in Vav2 protein expression after siRNA, but no change in Vav3 or Sos1 expression (n = 3; **p < 0.01). (D) Depleting Vav2 by siRNA perturbs Rac1 activation. HUVECs (top panels) or HMVEC-d (bottom panels) were transfected with Vav2-specific siRNA as described in Experimental Procedures. After 24 h, the cells were serum-starved for 1 h, treated with VEGF for 30 min and assayed for active Rac1. The upper left panel shows that Vav2 siRNA cells prevented VEGF-induced Rac1 activity compared to control cells. The middle left panel shows equal levels of total Rac1. The lower left panel shows knockdown of Vav2 expression by siRNA. The histogram on the right shows the average Rac1 activation relative to total Rac1 levels in three independent experiments (n = 3; *p < 0.05 by 2-way ANOVA).
In order to determine which GEF might be involved in Rac1 activation downstream from VEGF stimulation of HUVECs, we used nucleotide-free Rac1 in a pulldown strategy. This technique is based on the concept that GEFs bind small GTPases with high affinity when the GTPase is in a transitional nucleotide-free state [16,26]. As such, the nucleotide-free Rac1 mutant G15ARac1 has higher affinity for binding active GEFs, and our laboratory has used this mutant previously to bind and identify active GEFs [16]. Using a GST fusion protein of the G15ARac1 mutant, we found that 30 min VEGF treatment increased the association of Vav2 with G15ARac1. Conversely, when we inhibited VEGFR-2 signaling with SU1498, VEGF treatment did not stimulate the interaction between Vav2 and G15ARac1 (Fig. 2B). G15ARac1 failed to pull down Sos1 after VEGF treatment, suggesting that Sos1 is not involved in VEGF-induced signaling at this time point in endothelial cells. Low levels of β-PIX were detected binding to the G15ARac1 mutant, but the level of binding was not increased by VEGF treatment. Control experiments using GST showed no evidence of non-specific GEF binding (data not shown). We have also observed that silencing of VEGFR-2 by RNAi perturbs VEGF-stimulated Vav2 binding to G15ARac1 (data not shown). These results indicated that of the GEFs examined only Vav2 increased its association with G15ARac1 after VEGF stimulation through the VEGFR-2 signaling.
To study further the role of Vav2 in VEGF-induced Rac1 activation, we used siRNA against Vav2 to decrease Vav2 expression in endothelial cells. Silencing Vav2 resulted in a decrease of Vav2 protein expression, but no changes in Vav3, Sos1 or β-actin (Fig. 2C). Following knockdown of Vav2 expression there was impaired Rac1 activation in response to VEGF in both HUVECs and HMVEC-d (Fig. 2D). In the HUVECs, we used two different siRNA oligos against Vav2 and observed comparable results (data not shown). Silencing Vav2 with siRNA did not affect expression of Vav3 in HMVEC-d (data not shown). Collectively, these experiments suggest that Vav2 expression is required for VEGF-induced Rac1 activation at the 30 min time point in these two endothelial cell types.
Vav2 is tyrosine phosphorylated in response to VEGF
The pulldown assay with G15ARac1 suggested that Vav2 is activated downstream of VEGF treatment. Given that Vav2 is tyrosine phosphorylated in response to EGF and PDGF signaling, and that tyrosine phosphorylation has been associated with Vav2 activation [13,27], we examined the tyrosine phosphorylation of Vav2 following VEGF treatment. We found that Vav2 tyrosine phosphorylation was stimulated by VEGF and peaked at 30 min of VEGF stimulation (Fig. 3A). VEGF did not induce detectable tyrosine phosphorylation of Sos1 (data not shown). To determine whether tyrosine phosphorylation of Vav2 was downstream of VEGFR-2, we applied the pharmacological inhibitors of VEGFR-2 used earlier to inhibit Rac1 activation. Both SU1498 and ZM323881 prevented Vav2 tyrosine phosphorylation in response to VEGF (Fig. 3B). In addition, silencing VEGFR-2 with siRNA decreased VEGF-induced Vav2 tyrosine phosphorylation (Fig. 3C). These results suggest that VEGF-induced Vav2 tyrosine phosphorylation requires VEGFR-2 signaling.
Figure 3. Vav2 is tyrosine phosphorylated in response to VEGF treatment.
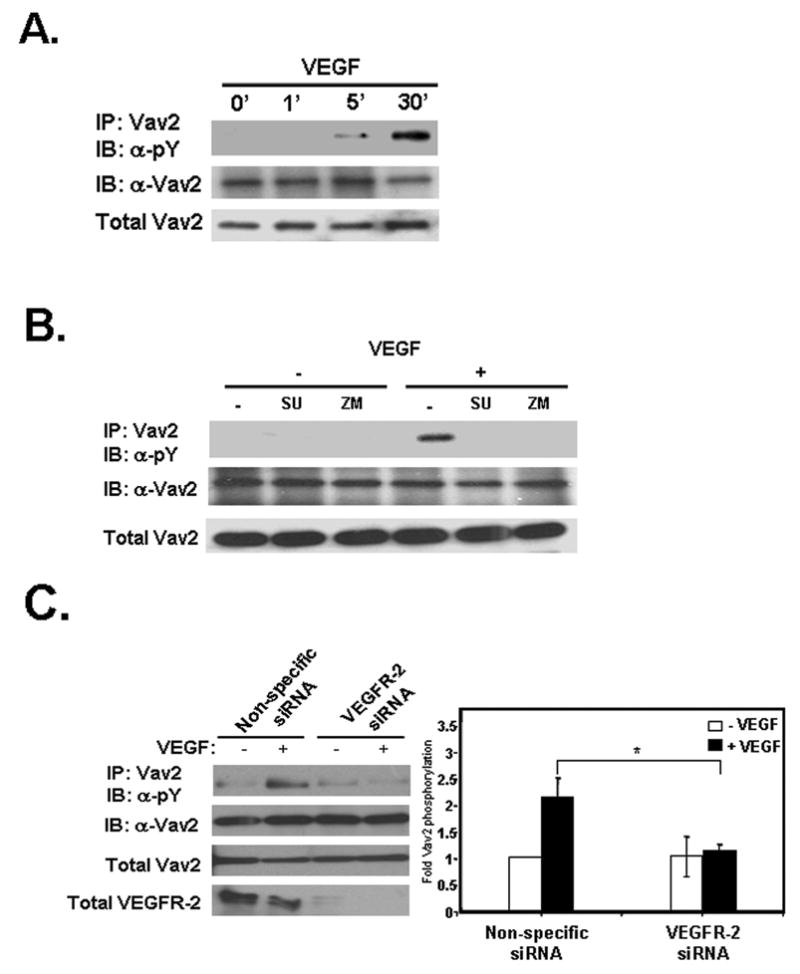
(A) Serum-starved HUVECs were stimulated with 10 ng/mL VEGF, endogenous Vav2 immunoprecipitated, and then probed for phosphotyrosine. Vav2 is tyrosine phosphorylated after VEGF stimulation and peaked at 30 min (upper panel). The middle panel shows equal Vav2 levels in every immunoprecipitation, whereas the lower panel shows equal protein loading in cell lysates. (B) Vav2 tyrosine phosphorylation is blocked with VEGFR-2 inhibitors. Cells were pretreated with 50 μM SU1498 or 10 nM ZM323881 for 30 min at 37°C, followed by VEGF addition for 30 min. In the upper panel, immunoprecipitates of Vav2 were probed with anti-phosphotyrosine antibody, showing that inhibiting VEGFR-2 signaling prevents Vav2 tyrosine phosphorylation. The middle panel shows equal Vav2 levels in every immunoprecipitation, and the lower panel shows equal protein loading in cell lysates. Blots are representative of three independent experiments. (C) Vav2 tyrosine phosphorylation is blocked with VEGFR-2 siRNA. HUVECs were transfected with either non-specific control siRNA (siGLO) or VEGFR-2-specific siRNA oligos as described in Experimental Procedures. Cells were assayed 24 h after transfection with VEGFR-2-specific siRNA. Serum-starved cells were stimulated with 10 ng/mL VEGF, endogenous Vav2 immunoprecipitated, and then probed for phosphotyrosine. The upper left panel shows that VEGFR-2 siRNA cells prevented Vav2 tyrosine phosphorylation 30 min after VEGF stimulation compared with control cells. The middle panels confirm equal Vav2 levels in every immunoprecipitation and equal protein loading in cell lysates. The lower panel shows levels of VEGFR-2 expression after siRNA treatment. The histogram on the right shows the average Vav2 tyrosine phosphorylation relative to total Vav2 levels in four independent experiments (n = 4; *p < 0.05 by 2-way ANOVA).
Tyrosine phosphorylation of Vav2 is mediated by Src
The non-receptor tyrosine kinase Src is a known effector of VEGF signaling and is important for endothelial cell migration [28–30]. Previous work has demonstrated that Src phosphorylates exchange factors that activate Rho proteins [31–33]. To confirm that Src is activated by VEGF, we used phosphospecific antibodies that bind to phosphorylated tyrosine 418 of Src. This tyrosine residue is found in the activation loop of Src and its phosphorylation leads to Src activation [34]. VEGF treatment of HUVECs stimulated Src phosphorylation at tyrosine 418 and this was blocked by the Src inhibitor PP2 (Fig. 4A). To investigate the role of Src in Vav2 tyrosine phosphorylation, Src kinase activity was blocked with PP2. Immunoprecipitations with Vav2 revealed that, in response to VEGF, Vav2 tyrosine phosphorylation was decreased in cells treated with PP2, whereas the inactive analog PP3 did not affect Vav2 tyrosine phosphorylation (Fig. 4B). It has been shown previously that phosphorylation of tyrosine 172 in Vav2 relieves an intramolecular inhibition, potentially resulting in increased activity of Vav2 by allowing access to the DH domain [27,35]. Using a phosphospecific antibody, we explored whether Vav2 was specifically phosphorylated at tyrosine 172 upon VEGF treatment. VEGF treatment induced phosphorylation of Vav2 on tyrosine 172 (Fig. 4C). This phosphorylation was blocked by PP2, but not PP3, suggesting that this phosphorylation depended on Src kinase activity. Lastly, to determine if Src kinase activity in turn perturbs Rac1 activation, we treated cells with the Src inhibitors PP2 or SU6656. Both inhibitors, but not PP3, abolished VEGF-induced Rac1 activation (Fig. 4D). These results were also seen in HMVEC-d (data not shown). Together, these data indicate that VEGF-induced tyrosine phosphorylation of Vav2, in particular on tyrosine 172, is regulated by Src and that Src kinase activity is required for activation of Rac1 in response to VEGF.
Figure 4. VEGF-induced Vav2 tyrosine phosphorylation is regulated by Src.

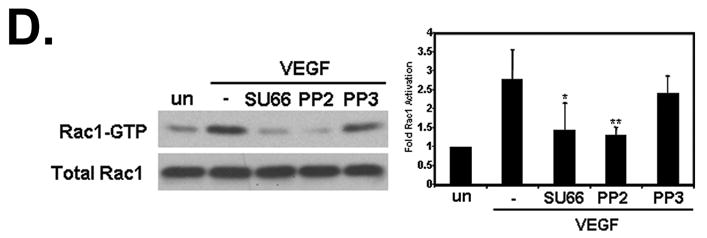
(A) Serum-starved HUVECs were pretreated with either 30 μM PP2 (Src inhibitor) or PP3 (the inactive analog) for 30 min at 37°C and then treated with VEGF. Untreated (un) denotes cells not treated with 10 ng/mL VEGF for 30 min. Lysates were immunoblotted for phosphorylation of tyrosine 418 on Src and showed that Src is catalytically activated after VEGF treatment (upper panel). PP2, but not the control compound PP3, blocks Src tyrosine phosphorylation. The middle panel shows equal Src levels in every immunoprecipitation, whereas the lower panel shows equal protein loading in cell lysates. The histogram on the right shows the average Src tyrosine phosphorylation relative to total Src levels in three independent experiments (n = 3; *p < 0.05). (B) Src inhibition blocks Vav2 tyrosine phosphorylation. HUVECs were serum starved and pretreated as described under “A”. Cells were lysed and Vav2 was immunoprecipitated followed by immunoblotting for phosphotyrosine. Inhibiting Src with PP2 prevents Vav2 tyrosine phosphorylation, whereas Vav2 phosphorylation was unaffected by PP3 (upper panel). The middle panel shows equal Vav2 levels in every immunoprecipitation, whereas the lower panel shows equal protein loading in cell lysates. The histogram on the right shows the average Vav2 tyrosine phosphorylation relative to immunoprecipitated Vav2 levels in three independent experiments (n = 3; *p < 0.05). (C) The Src inhibitor PP2 blocks Vav2 phosphorylation specifically on tyrosine 172. Serum-starved HUVECs were pretreated as described under “A”. Phosphorylation of Vav2 on Y172 was analyzed by western blotting using a phospho-epitope-specific antibody. The upper panel shows that inhibition of Src with PP2 prevents phosphorylation at this residue. The middle panel shows equal Vav2 levels in every immunoprecipitation, whereas the lower panel shows equal protein loading in cell lysates. The histogram on the right shows the average Vav2 phosphorylation on tyrosine 172 activation relative to total Vav2 expression in four independent experiments (n = 4; *p < 0.05). (D) VEGF-induced Rac1 activation is regulated by Src activity. HUVECs were serum starved for 1 h, pretreated with either 30 μM PP2 or 5 μM SU6656 (Src inhibitor) or 30 μM PP3 for 30 min at 37°C. Cells were then treated with 10 ng/mL VEGF and subsequently assayed for Rac1 activity as described in Experimental Procedures. The upper left panel shows Rac-GTP levels after VEGF treatment. The lower left panel shows total Rac1 levels. The histogram on the right shows quantification of the increase in Rac1 activity after VEGF stimulation. Data is representative of five independent experiments (n = 5; *p < 0.05, **p < 0.01).
Vav2 is necessary for VEGF-induced endothelial cell migration
Previous work has shown that VEGF stimulates HUVEC migration [4,5,36]. Since our findings suggested that Vav2 specifically activates Rac1 during VEGF signaling, we investigated whether Vav2 was required for the migration of HUVECs and HMVEC-d towards VEGF. We knocked down expression of Vav2 in both cell types using siRNA and assayed for chemotactic migration across Transwell filters. As expected, we found that migration of endothelial cells was increased when a VEGF gradient was present (Fig. 5A, closed bars). However, migration was significantly decreased in both endothelial cell types when Vav2 was depleted using siRNA (Fig. 5A). Studies performed in HUVECs with two different siRNAs against Vav2 showed similar results (data not shown). Since Vav2 is known to activate not only Rac1 but also RhoA and Cdc42 in vitro, we determined if silencing of Rac1 conferred similar migratory defects as seen with Vav2 siRNA. Using siRNA against Rac1 to knockdown Rac1 expression, we found that HUVEC migration towards VEGF was significantly decreased as was seen with the knockdown of Vav2 (Fig. 5B). These observations show that Vav2 expression contributes to chemotactic migration of endothelial cells towards VEGF and that Rac1 is important for this migration. To investigate further the role of Vav2 in HUVEC migration, we performed a “scratch wound” assay comparing migration of HUVECs in which Vav2 levels had been depressed by siRNA with cells expressing control siRNA. After being stimulated with VEGF, endothelial cells depleted of Vav2 by siRNA revealed decreased wound closure compared to control cells (Fig. 5C).
Figure 5. Vav2 silencing by siRNA reduces HUVEC chemotaxis and wound closure.
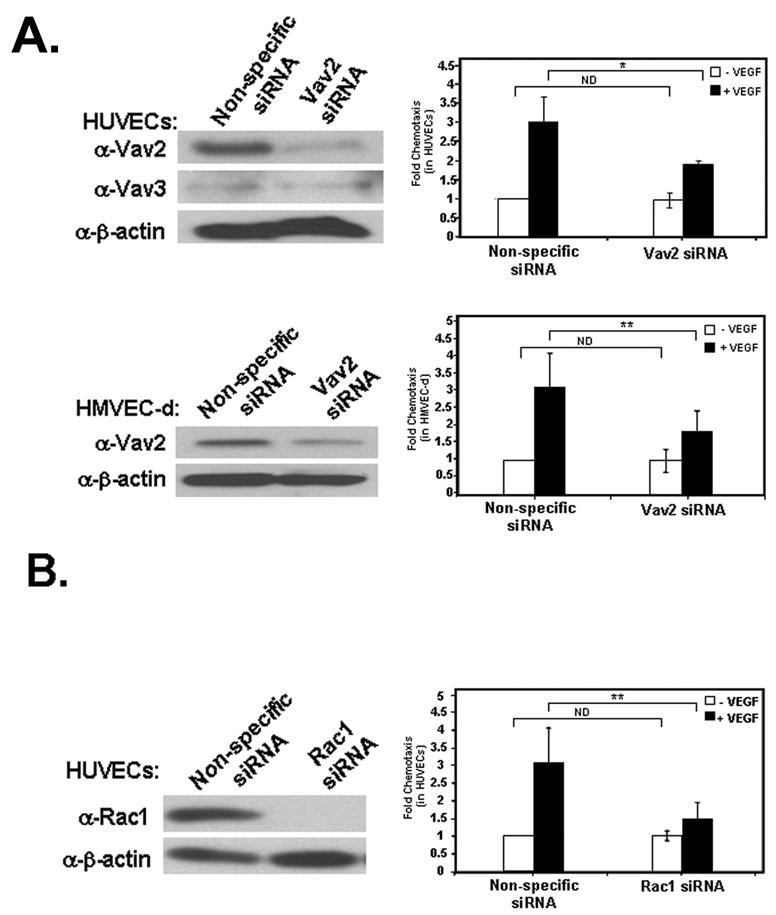

(A) HUVECs (top panels) or HMVEC-d (bottom panels) were transfected with either non-specific control (siGLO) or Vav2-specific siRNA. Blots indicate efficient knockdown of Vav2 protein levels. Cells were allowed to migrate for 4 h towards non-specific control media (open bars) or media containing 10 ng/mL VEGF (closed bars) using Transwell filters. Chemotaxis was quantified by calculating the mean number of cells that had migrated on the underside of the Transwell filter in three separate fields per filter ± standard deviation of triplicate independent experiments (n = 3; ** p < 0.01; *p < 0.05; ND, not significantly different by 2-way ANOVA). (B) Silencing of Rac1 decreases HUVEC migration towards VEGF. HUVECs were transfected with either non-specific control (siGLO) or Rac1-specific siRNA. Blots indicate efficient knockdown of Rac1 protein levels. Cells were allowed to migrate for 4 h towards non-specific control media (open bars) or media containing 10 ng/mL VEGF (closed bars) using Transwell filters. Chemotaxis was quantified by calculating the mean number of cells that had migrated onto the underside of the Transwell filter in three separate fields per filter ± standard deviation of triplicate independent experiments (n = 3; ** p < 0.01; ND, not significantly different by 2-way ANOVA). (C) Knockdown of Vav2 by siRNA prevents VEGF-induced wound closure. Cells transfected with either control siRNA or Vav2-targeting siRNA were wounded with a pipette tip and allowed to invade the wound for 48 h in the presence of 10 ng/mL VEGF. Wound closure was visualized by phase microscopy. The distance of wound closure is the average wound closure on three distinct points on each image over time and divided by the average initial wound size. At least five images were collected at each time point. Bar, 100 μm. The graph shows the mean distance of wound closure in three independent experiments (n = 3; *p < 0.05; ** p < 0.01).
DISCUSSION
VEGF signaling is well-characterized in the context of endothelial cell behavior and function, but relatively little is known about the downstream proteins that directly facilitate biological responses. Our finding that VEGF causes both early (1–5 min) and late (30 min) activation of Rac1 demonstrates that VEGF signaling controls Rac1 activity in a time-dependent manner. This finding is similar to recent data shown by Aoki et al. [37] in PC12 cells stimulated with nerve growth factor (NGF), suggesting that biphasic Rac1 activation occurs in different cell types in response to cell-specific stimuli. Our work has focused on the late phase of Rac1 activation in response to VEGF stimulation since Rac1 activation was most pronounced at these later stages.
Currently, it is unclear how specific exchange factors couple growth factor signaling to Rho proteins, thereby allowing these proteins to coordinate finely-orchestrated biological responses. Though several studies showed that VEGF causes endothelial cell migration by inducing Rac1-stimulated lamellipodia formation, the pathway between VEGF signaling and Rac1 activation was not explored [4]. In this study, we identified the exchange factor Vav2 as mediating VEGF signaling to Rac1. Using a nucleotide-free G15ARac1 mutant, we showed that Vav2 interacts with Rac1 after VEGF stimulation and this association correlated with a robust increase in Rac1 activity. Additionally, we observed that VEGF stimulated tyrosine phosphorylation of Vav2. This tyrosine phosphorylation was prevented by inhibiting either VEGFR-2 signaling or Src kinase activity. Moreover, knockdown of Vav2 not only inhibited the increase in Rac1 activity in response to VEGF stimulation, but also inhibited VEGF-stimulated migration. Together these results indicate that Vav2 plays a critical role in the response to VEGF. It is interesting that the related GEF, Vav3, was also detected at low levels in HMVEC-d. The similarity of this GEF to Vav2 raises the possibility that it too may contribute to Rac1 activation downstream from VEGF stimulation. However, because the knockdown of Vav2 significantly decreases VEGF-induced Rac1 activation and cell migration, the contribution of Vav3 to this response must be small at best.
Vav2 is one of the better characterized Rac1 GEFs and in several cell types is necessary for processes that require rearrangement of the actin cytoskeleton, including spreading and migration [31,38,39]. Recent data have shown that Vav2 is critical for Rac1 activity downstream of both adhesion molecules and growth factor receptors in diverse cell types. Two recent studies have shown that Vav2 is involved in nectin- and E-cadherin-induced activation of Rac1 in MDCK cells [40,41]. Similarly, Vav2 has been implicated in neuronal cell migration and neurite outgrowth mediated by adhesion molecule L1 [42], and Rac1-dependent Schwann cell migration [43]. Vav2 has also been shown to promote cell migration in response to EGF and UTP nucleotide signaling [13,44,45]. We conclude that Vav2 is an exchange factor commonly used by cells to activate Rac1 and promote migration in response to diverse signaling pathways.
In immunoprecipitation studies, we observed that Vav2 may not directly associate with VEGFR-2 (data not shown), thus other intermediate proteins may link VEGF signaling to Vav2 activity. Vav2 is regulated by tyrosine phosphorylation and/or membrane targeting, but the exact regulatory mechanism is controversial. Vav2 is tyrosine phosphorylated in response to EGF and PDGF signaling and this phosphorylation correlates with enhanced migration of fibroblasts [13,27,46]. However, it is still unclear whether this tyrosine phosphorylation directly regulates Vav2 exchange activity.
Work from other labs has indicated that the non-receptor tyrosine kinase Src may phosphorylate Vav2 downstream of VEGF signaling [33,47]. Eliceiri et al. [47] showed that endogenous Src kinase activity is required for VEGF-induced angiogenesis. Another study showed that Vav2 is tyrosine phosphorylated in cells transfected with an active form of c-Src (Src Y529F) [33]. Overexpression of wild-type Vav2 fails to activate Rac1 or induce lamellipodia formation in cells that are expressing a dominant negative Src or cells that are treated with Src inhibitor PP2 [31]. These data support a role for Src in Vav2 activation. Gavard and Gutkind [48] published that Src-dependent phosphorylation of Vav2 upon VEGF stimulation promotes VE-Cadherin endocytosis, contributing to endothelial cell permeability. To complement those studies, we show here that VEGF-induced Vav2 tyrosine phosphorylation and downstream activation of Rac1 depends on Src kinase activity in endothelial cells and is responsible for migration and wound closure. Collectively, these data illustrate an important role for Vav2 in endothelial cells responding to VEGF. Work by Sauzeau et al. [49] has shown that Vav2 knockout mice have defects in cardiovascular function characterized by an increase in hypertension and tachycardia, suggesting that Vav2 is not only essential for the regulation of endothelial cells, but other vascular cells as well.
The stimulation of endothelial migration by VEGF is important for normal angiogenesis. However, there are several situations where angiogenesis can have pathological consequences. For example, much attention has been paid to the vascularization of tumors, which facilitates tumor growth. Similarly, inappropriate blood vessel formation underlies several pathological situations, such as age-related macular degeneration. Understanding the signaling pathways by which VEGF stimulates Rac1 activation and cell migration may reveal new therapeutics targets. Our identification of Vav2 as an exchange factor activated in response to VEGF stimulation suggests Vav2 as a potential target for drug development.
Acknowledgments
The authors thank Drs. Erika Wittchen and Etienne Boulter for helpful comments and experimental assistance during the preparation of the manuscript. The authors acknowledge Lisa Sharek for outstanding technical assistance and thank Matthew Darrow, Dr. David Riddle, Peter Miller, Jed Ferguson, and Dr. Cercina Onesto for providing reagents in certain experiments. This work was supported in part by National Institutes of Health Grant HL45100 and a Kenan Professorship (K.B.). TAG is supported by a predoctoral fellowship from the American Heart Association. JDvB is supported by the Ter Meulen Fund, Royal Netherlands Academy of Arts and Sciences.
ABBREVIATIONS
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
- GEF
guanine nucleotide exchange factor
- GAP
GTPase activating protein
- HUVECs
human umbilical vein endothelial cells
- HMVEC-d
human microvascular endothelial cells-dermal
- EGF
epidermal growth factor
- PDGF
platelet-derived growth factor
- RT-PCR
reverse transcriptase-polymerase chain reaction
- siRNA
short interfering ribonucleic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ferrara N. Role of vascular endothelial growth factor in physiologic and pathologic angiogenesis: therapeutic implications. Semin Oncol. 2002;29:10–14. doi: 10.1053/sonc.2002.37264. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 3.Small JV, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol. 2002;12:112–120. doi: 10.1016/s0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- 4.Soga N, Connolly JO, Chellaiah M, Kawamura J, Hruska KA. Rac regulates vascular endothelial growth factor stimulated motility. Cell Commun Adhes. 2001;8:1–13. doi: 10.3109/15419060109080703. [DOI] [PubMed] [Google Scholar]
- 5.Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15:2169–2177. doi: 10.1038/sj.onc.1201380. [DOI] [PubMed] [Google Scholar]
- 6.Yamazaki D, Suetsugu S, Miki H, Kataoka Y, Nishikawa S, Fujiwara T, Yoshida N, Takenawa T. WAVE2 is required for directed cell migration and cardiovascular development. Nature. 2003;424:452–456. doi: 10.1038/nature01770. [DOI] [PubMed] [Google Scholar]
- 7.Reynolds AR, Reynolds LE, Nagel TE, Lively JC, Robinson SD, Hicklin DJ, Bodary SC, Hodivala-Dilke KM. Elevated Flk1 (vascular endothelial growth factor receptor 2) signaling mediates enhanced angiogenesis in beta3-integrin-deficient mice. Cancer Res. 2004;64:8643–8650. doi: 10.1158/0008-5472.CAN-04-2760. [DOI] [PubMed] [Google Scholar]
- 8.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 9.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 10.Cherfils J, Chardin P. GEFs: structural basis for their activation of small GTP-binding proteins. Trends Biochem Sci. 1999;24:306–311. doi: 10.1016/s0968-0004(99)01429-2. [DOI] [PubMed] [Google Scholar]
- 11.Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 12.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 13.Liu BP, Burridge K. Vav2 activates Rac1, Cdc42, and RhoA downstream from growth factor receptors but not beta1 integrins. Mol Cell Biol. 2000;20:7160–7169. doi: 10.1128/mcb.20.19.7160-7169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deroanne C, Vouret-Craviari V, Wang B, Pouyssegur J. EphrinA1 inactivates integrin-mediated vascular smooth muscle cell spreading via the Rac/PAK pathway. J Cell Sci. 2003;116:1367–1376. doi: 10.1242/jcs.00308. [DOI] [PubMed] [Google Scholar]
- 15.Ren XD, Kiosses WB, Schwartz MA. Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. Embo J. 1999;18:578–585. doi: 10.1093/emboj/18.3.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 2006;406:425–437. doi: 10.1016/S0076-6879(06)06031-9. [DOI] [PubMed] [Google Scholar]
- 17.Zeng H, Zhao D, Mukhopadhyay D. Flt-1-mediated down-regulation of endothelial cell proliferation through pertussis toxin-sensitive G proteins, beta gamma subunits, small GTPase CDC42, and partly by Rac-1. J Biol Chem. 2002;277:4003–4009. doi: 10.1074/jbc.M110842200. [DOI] [PubMed] [Google Scholar]
- 18.Zeng H, Zhao D, Mukhopadhyay D. KDR stimulates endothelial cell migration through heterotrimeric G protein Gq/11-mediated activation of a small GTPase RhoA. J Biol Chem. 2002;277:46791–46798. doi: 10.1074/jbc.M206133200. [DOI] [PubMed] [Google Scholar]
- 19.van Nieuw Amerongen GP, Koolwijk P, Versteilen A, van Hinsbergh VW. Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler Thromb Vasc Biol. 2003;23:211–217. doi: 10.1161/01.atv.0000054198.68894.88. [DOI] [PubMed] [Google Scholar]
- 20.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 21.Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- 22.Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 23.Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, Tempst P, Hawkins PT, Stephens LR. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–821. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- 24.Shinohara M, Terada Y, Iwamatsu A, Shinohara A, Mochizuki N, Higuchi M, Gotoh Y, Ihara S, Nagata S, Itoh H, Fukui Y, Jessberger R. SWAP-70 is a guanine-nucleotide-exchange factor that mediates signalling of membrane ruffling. Nature. 2002;416:759–763. doi: 10.1038/416759a. [DOI] [PubMed] [Google Scholar]
- 25.Sini P, Cannas A, Koleske AJ, Di Fiore PP, Scita G. Abl-dependent tyrosine phosphorylation of Sos-1 mediates growth-factor-induced Rac activation. Nat Cell Biol. 2004;6:268–274. doi: 10.1038/ncb1096. [DOI] [PubMed] [Google Scholar]
- 26.Arthur WT, Ellerbroek SM, Der CJ, Burridge K, Wennerberg K. XPLN, a guanine nucleotide exchange factor for RhoA and RhoB, but not RhoC. J Biol Chem. 2002;277:42964–42972. doi: 10.1074/jbc.M207401200. [DOI] [PubMed] [Google Scholar]
- 27.Tamas P, Solti Z, Bauer P, Illes A, Sipeki S, Bauer A, Farago A, Downward J, Buday L. Mechanism of epidermal growth factor regulation of Vav2, a guanine nucleotide exchange factor for Rac. J Biol Chem. 2003;278:5163–5171. doi: 10.1074/jbc.M207555200. [DOI] [PubMed] [Google Scholar]
- 28.Schlessinger J. New roles for Src kinases in control of cell survival and angiogenesis. Cell. 2000;100:293–296. doi: 10.1016/s0092-8674(00)80664-9. [DOI] [PubMed] [Google Scholar]
- 29.Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, McSharry H, Iwakura A, Yoon YS, Himes N, Burstein D, Doukas J, Soll R, Losordo D, Cheresh D. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004;113:885–894. doi: 10.1172/JCI20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chou MT, Wang J, Fujita DJ. Src kinase becomes preferentially associated with the VEGFR, KDR/Flk-1, following VEGF stimulation of vascular endothelial cells. BMC Biochem. 2002;3:32. doi: 10.1186/1471-2091-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marignani PA, Carpenter CL. Vav2 is required for cell spreading. J Cell Biol. 2001;154:177–186. doi: 10.1083/jcb.200103134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miyamoto Y, Yamauchi J, Itoh H. Src kinase regulates the activation of a novel FGD-1-related Cdc42 guanine nucleotide exchange factor in the signaling pathway from the endothelin A receptor to JNK. J Biol Chem. 2003;278:29890–29900. doi: 10.1074/jbc.M301559200. [DOI] [PubMed] [Google Scholar]
- 33.Servitja JM, Marinissen MJ, Sodhi A, Bustelo XR, Gutkind JS. Rac1 function is required for Src-induced transformation. Evidence of a role for Tiam1 and Vav2 in Rac activation by Src. J Biol Chem. 2003;278:34339–34346. doi: 10.1074/jbc.M302960200. [DOI] [PubMed] [Google Scholar]
- 34.Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 1999;3:629–638. doi: 10.1016/s1097-2765(00)80356-1. [DOI] [PubMed] [Google Scholar]
- 35.Aghazadeh B, Lowry WE, Huang XY, Rosen MK. Structural basis for relief of autoinhibition of the Dbl homology domain of proto-oncogene Vav by tyrosine phosphorylation. Cell. 2000;102:625–633. doi: 10.1016/s0092-8674(00)00085-4. [DOI] [PubMed] [Google Scholar]
- 36.Soga N, Namba N, McAllister S, Cornelius L, Teitelbaum SL, Dowdy SF, Kawamura J, Hruska KA. Rho family GTPases regulate VEGF-stimulated endothelial cell motility. Exp Cell Res. 2001;269:73–87. doi: 10.1006/excr.2001.5295. [DOI] [PubMed] [Google Scholar]
- 37.Aoki K, Nakamura T, Matsuda M. Spatio-temporal regulation of Rac1 and Cdc42 activity during nerve growth factor-induced neurite outgrowth in PC12 cells. J Biol Chem. 2004;279:713–719. doi: 10.1074/jbc.M306382200. [DOI] [PubMed] [Google Scholar]
- 38.Bustelo XR. Regulatory and signaling properties of the Vav family. Mol Cell Biol. 2000;20:1461–1477. doi: 10.1128/mcb.20.5.1461-1477.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hornstein I, Alcover A, Katzav S. Vav proteins, masters of the world of cytoskeleton organization. Cell Signal. 2004;16:1–11. doi: 10.1016/s0898-6568(03)00110-4. [DOI] [PubMed] [Google Scholar]
- 40.Kawakatsu T, Ogita H, Fukuhara T, Fukuyama T, Minami Y, Shimizu K, Takai Y. Vav2 as a Rac-GDP/GTP exchange factor responsible for the nectin-induced, c-Src- and Cdc42-mediated activation of Rac. J Biol Chem. 2005;280:4940–4947. doi: 10.1074/jbc.M408710200. [DOI] [PubMed] [Google Scholar]
- 41.Fukuyama T, Ogita H, Kawakatsu T, Inagaki M, Takai Y. Activation of Rac by cadherin through the c-Src-Rap1-phosphatidylinositol 3-kinase-Vav2 pathway. Oncogene. 2006;25:8–19. doi: 10.1038/sj.onc.1209010. [DOI] [PubMed] [Google Scholar]
- 42.Schmid RS, Midkiff BR, Kedar VP, Maness PF. Adhesion molecule L1 stimulates neuronal migration through Vav2-Pak1 signaling. Neuroreport. 2004;15:2791–2794. [PubMed] [Google Scholar]
- 43.Yamauchi J, Chan JR, Shooter EM. Neurotrophins regulate Schwann cell migration by activating divergent signaling pathways dependent on Rho GTPases. Proc Natl Acad Sci U S A. 2004;101:8774–8779. doi: 10.1073/pnas.0402795101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bagchi S, Liao Z, Gonzalez FA, Chorna NE, Seye CI, Weisman GA, Erb L. The P2Y2 nucleotide receptor interacts with alphav integrins to activate Go and induce cell migration. J Biol Chem. 2005;280:39050–39057. doi: 10.1074/jbc.M504819200. [DOI] [PubMed] [Google Scholar]
- 45.Seye CI, Yu N, Gonzalez FA, Erb L, Weisman GA. The P2Y2 nucleotide receptor mediates vascular cell adhesion molecule-1 expression through interaction with VEGF receptor-2 (KDR/Flk-1) J Biol Chem. 2004;279:35679–35686. doi: 10.1074/jbc.M401799200. [DOI] [PubMed] [Google Scholar]
- 46.Hawkins PT, Eguinoa A, Qiu RG, Stokoe D, Cooke FT, Walters R, Wennstrom S, Claesson-Welsh L, Evans T, Symons M, et al. PDGF stimulates an increase in GTP-Rac via activation of phosphoinositide 3-kinase. Curr Biol. 1995;5:393–403. doi: 10.1016/s0960-9822(95)00080-7. [DOI] [PubMed] [Google Scholar]
- 47.Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, Cheresh DA. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol Cell. 1999;4:915–924. doi: 10.1016/s1097-2765(00)80221-x. [DOI] [PubMed] [Google Scholar]
- 48.Gavard J, Gutkind JS. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat Cell Biol. 2006;8:1223–1234. doi: 10.1038/ncb1486. [DOI] [PubMed] [Google Scholar]
- 49.Sauzeau V, Jerkic M, Lopez-Novoa JM, Bustelo XR. Loss of Vav2 proto-oncogene causes tachycardia and cardiovascular disease in mice. Mol Biol Cell. 2007;18:943–952. doi: 10.1091/mbc.E06-09-0877. [DOI] [PMC free article] [PubMed] [Google Scholar]