

Abstract
Experimental studies have shown that blockade of the angiotensin II type-1 (AT1) receptor is effective in the mitigation and treatment of radiation-induced chronic renal failure. We have shown that blockade of the angiotensin II type-2 (AT2) receptor with PD-123319 also had a modest, but reproducible, beneficial effect in experimental radiation nephropathy, and that it might also augment the efficacy of an AT1 blocker (L-158,809). Those studies could not exclude the possibility that the effects of AT2 blockade were non-specific. The current studies confirm the efficacy of AT2 blockade for mitigation of experimental radiation nephropathy, but paradoxically find no detectable level of AT2 receptor binding in renal membranes. However, a bioassay showed that the circulating levels of the AT2 blocker were orders-of-magnitude too low to block AT1 receptors. We conclude that the effect of AT2 blockade in radiation nephropathy cannot be explained by binding to the AT1 receptor, and that the efficacy of the AT1 blockade in the same model cannot be explained by unopposed overstimulation of the AT2 receptor.
Radiation-induced chronic renal failure is well-documented in subjects undergoing total body irradiation (TBI) for hematopoietic stem cell transplants,1,2 and in subjects receiving radiolabeled biologicals for cancer therapy.3,4 We and others have shown the benefit of blockade of the renin-angiotensin system in experimental5–7 and clinical8,9 radiation nephropathy. In a rat model of radiation nephropathy, the use of angiotensin II (AII) blockade,5,6 or reciprocally the use of AII infusion,10 have shown that the renin-angiotensin system is particularly important between one and three months after irradiation. Further, the efficacy of an AII type-1 (AT1) receptor blocker strongly suggests that the mechanism of injury is via the AT1 receptor.5,9 It has been suggested that the benefit of the AT1 receptor blockade might be via over-stimulation of the unblocked angiotensin II type-2 (AT2) receptor.11 This hypothesis implied that blockade of the AT2 receptor would negate or even reverse the effects of AT1 blockade. Initial studies in our model have shown that AT2 blockade has a modest, but reproducible, beneficial effect in experimental radiation nephropathy.12,13 A similar benefit of AT2 blockade was found by Cao et al14 in the remnant kidney model. However, these studies could not exclude the possibility that the effects of AT2 blockade were non-specific, possibly via binding to the AT1 receptor. We undertook studies to confirm the efficacy of AT2 receptor blockade in experimental radiation nephropathy, and to elucidate the pharmacologic basis for this effect.
MATERIALS AND METHODS
Rat radiation nephropathy model
A fractionated TBI regimen followed by bone marrow transplantation (BMT) was used to cause radiation nephropathy.15,16 This radiation nephropathy is characterized by proteinuria, azotemia and progressive hypertension that leads to renal failure after a median time of 30 to 40 weeks.15,17 Renal failure (uremia) is the only significant cause of illness and death in this model.15 The studies were performed in syngeneic WAG/Rij/MCW rats that were bred and housed in a moderate-security barrier. The animals were free of Mycoplasma pulmonis, Pseudomonas and common rat viruses. No antibiotics or immunosuppressive drugs were used. The rats were maintained in the Biomedical Research Center of the Medical College of Wisconsin, which is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. The studies were approved by the College’s Animal Care and Use Committee.
Seven- to 8-week-old male rats underwent TBI with 18.8 Gy or 20.5 Gy, given in six fractions over 3 days, at a dose rate of 1.95 Gy/min. For the two daily treatments, the minimum interval was 4 hours and the maximum was 4.3 hours. Within 24 hours after TBI, the rats received a BMT from a syngeneic donor.15 The day of BMT was considered to be day zero for definition of time after irradiation.
Radiation dosimetry
A Pantak HF320 orthovoltage x-ray system was used for the TBI. It was operated at 300 kVp with a half value layer of 1.4 mm Cu. During the irradiation, each rat was confined in a separate chamber in a plastic jig; the jig consists of chambers, allowing irradiation of four rats simultaneously. The four chambers were placed on a plane perpendicular to the beam direction and were aligned in parallel with the x-ray tube. A collimator made of cerrobend was used to define a radiation field that was large enough to cover all four chambers with adequate (at least 2 cm) margin.
The absolute dose was measured using a Farmer-type ionization chamber (Wellhofer FC65-G) calibrated for these x-ray energies in a standard dosimetry laboratory. The ionization was measured in air and then converted to absolute dose in water following the American Association of Physicists in Medicine Task Group-61 protocol.18 The timer error of the x-ray machine was considered in the absolute dose determination. A two-dimensional radiation beam scanning system (Scanditronix-Wellhofer RA-200) with a RK-type ionization chamber was used to measure relative dose variation with depth in water for the field size and distance used. Subsequently, three-dimensional isodose distributions were generated; and based on this data, dose variation within and between the rat kidneys was estimated.
The dose at the centers of the four rat chambers varied by ±2%, and rats were randomly assigned to chambers to avoid any resulting bias. The detailed dosimetry done for this study revealed that renal doses were 11% greater than previously reported for this irradiation setup.
Monitoring the development of radiation nephropathy
Animals were monitored daily in all experiments. Development of severe nephropathy (BUN ≥ 120 mg/dl) was assessed for up to 62 weeks after TBI, and animals with symptomatic uremia were euthanized. Blood urea nitrogen (BUN), urine protein, and urine creatinine were determined with commercial kits; at a minimum these assays were done at 12, 17, 26, 35, 52 and 64 weeks after TBI. Systolic blood pressure (BP) was measured with a tail-cuff plethysmograph (IITC Life Sciences Instruments, Woodland Hills, CA) one week after the BUN and urine assays. Animals were conditioned to the BP apparatus and the reported BP was the average of readings on three successive days. Urine protein excretion is expressed as the ratio of urine protein to creatinine (UP/UC) in the same urine sample; this is done to account for the known urine concentrating defect that occurs in radiation nephropathy and to normalize for differences in animal size.
The NG108-15 cell line
NG108 cells were used to obtain a crude membrane fraction that was known to contain a high level of AT2 receptors.19 The NG108-15 cell line of mouse neuroblastoma-rat glioma hybrid cells was obtained from American Type Cell Culture (ATCC# HB-12317). NG108 cells were propagated in Dulbecco’s Modified Eagle’s Medium without sodium pyruvate, and with L-glutamine (4 mM), glucose (4.5 mg/L), pyridoxine hydrochloride (4.0 mg/L), hypoxanthine (0.1 mM), aminopterin (400 nM), thymidine (0.016 mM), sodium bicarbonate (3.7 g/L) and fetal bovine serum (10%). Incubator environment was maintained at 5% carbon dioxide in air at 37°C. Confluent cultures were treated with solution containing 0.25% trypsin and 0.3% EDTA to detach cells for subcultivation. Confluent cultures were used for preparation of crude membrane fractions.
AII blockers
The AT1 blocker (L158,809)20,21 was given in the drinking water (pH 7.5–7.8) at doses of 20, 40 or 80 mg/liter (20 mg/liter is the dose used in our previous studies5,12); this gives daily doses of 2, 4 or 8 mg/kg/day.13 Note that the pH of drinking water is critical as the AT1 blocker is not soluble at these concentrations if the pH drops below about 7.2. The AT2 blocker (PD-123319)22 was given subcutaneously at a dose of 15 mg/kg/day using Alzet infusion pumps.12 The Model 2ML4 pumps were loaded with PD-123319 at a concentration of 44–60 mg/ml in saline adjusted to pH 3.5; the exact concentration was based on the rat’s weight.
The timing of therapy with the AII blockers was based on previous studies that showed preferential efficacy of angiotensin converting enzyme (ACE) inhibitors or AT1 blockers when used during the first 4–12 weeks after irradiation.23,24 Thus, when the AT1 blocker was used alone in an experiment, therapy was started the day after completion of TBI and was continued for 12 weeks. The dose and duration of treatment for the AT2 blocker required further compromise between achieving maximum effect (long duration of treatment at high dose) and conserving a drug that is in short supply.12 The use of the AT2 blockers was also delayed until four weeks after irradiation because the surgical implantation of the minipumps was complicated by poor wound healing when these were placed earlier. Thus, when the AT2 blocker was used in an experiment, therapy with the blockers (both AT1 and AT2) was started at 4 weeks after TBI and continued for 8 weeks.
Under the terminology recently recommended by the U.S. National Cancer Institute25 these drug schedules are “mitigation” regimens, in that therapy starts after irradiation, but before there is symptomatic disease.
Crude membrane preparations (kidney)
Kidneys were minced in ice-cold buffer, then homogenized with a Dounce tissue grinder with 5 strokes at 160 μm clearance and then 5 strokes at 70 μm clearance. Membrane fractions were isolated and washed as described by Ernsberger et al26 In brief, the minced kidney tissue was homogenized in 10 ml ice-cold pH 7.4 HEPES “homogenization” buffer containing 265 mM sucrose, 100 μM 1,10-phenanthroline, and 50 μM phenylmethylsulfonyl fluoride (PMSF). Homogenates were centrifuged at 1,000 g for 5 min at 4°C to remove nuclei and debris. The pellets were resuspended in 7 ml of homogenization buffer and recentrifuged. The combined supernatants were centrifuged at 48,000 g for 18 min at 4°C, and the pellets were resuspended in 7 ml of 50 mM Tris-HCl buffer (pH 7.7) containing 5 mM EDTA. Membranes were recentrifuged and resuspended in Tris-HCl containing 25 mM NaCl, incubated for 30 min on ice, centrifuged a third time at 48,000 g. The resulting pellet was resuspended in “assay” buffer (50 mM Tris-HCl containing 120 mM NaCl, 2.0 mM MgCl2, 100 μM bacitracin, 50 μM PMSF, 10 μM phosphoramidon, and 10 μg/ml soybean trypsin inhibitor).
Aliquots were frozen in the assay buffer at −80°C, with one aliquot reserved for protein determination. Protein content was assayed using the Bicinchoninic acid protein assay (BCA-1, Sigma).
Crude membrane preparations (NG108)
Crude membrane fractions were prepared using an adaptation of the methods described above for renal membranes. Briefly, cells were harvested at 4°C by gentle scraping, suspended in ice-cold homogenization buffer and homogenized in a hand-held glass homogenizer. The homogenate was centrifuged at 1,000 g for 10 min at 4°C and the supernatant was collected. The pellet was resuspended in fresh buffer and centrifuged again. The supernatants were pooled and prepared as above.
AII binding assay
Thawed membrane preparations (125 μg/ml protein) were incubated for 45 min in fresh assay buffer containing 0.5 mg/ml BSA, I125 AII at 0.73 μM and 50–80 TBq/mmol (Perkin Elmer Life Science, Boston, MA), and AII receptor antagonists at various concentrations. Aliquots (200 μl each) of the incubation mix were placed in triplicate into a Millipore Multiscreen 96-well plate, incubated for 45 min, filtered and washed (x5) with Tris buffer. For determination of non-specific binding, cold AII at 700 nM was added to at least three aliquots of the incubation mix. The plate was dried overnight and 40 μl of Microscint-20 (Packard) was added to each well. The plate was counted on a Packard Top Count plate reader. Counts were corrected for nonspecific binding and then were converted to nanomolar AII concentrations using a quench correction curve that was created by adding I125 AII of known specific activity to 200 μl aliquots of assay buffer and quenching with graded amounts of carbon tetrachloride or food coloring. Binding levels were then converted to fmol AII per mg protein using the assayed protein concentration in the sample. Percent inhibition is the ratio of binding in presence and absence of an inhibitor. All total binding and nonspecific binding measurements for a specific inhibition determination were done with the same membrane preparation and on the same 96-well plate.
For the assay of AT1 receptors, L-158,809 was added to kidney cell membrane preparations at concentrations of 0.1 nM to 0.1 mM. For the assay of AT2 receptors, PD-123319 was added to a NG108 cell membrane preparation at concentrations of 0.01 nM to 3 μM; the AT1 blocker L-158,809 was added to the NG108 cell membrane preparation at 1 μM to block all AT1 receptors.
Assay of AII blockers in blood
The bioassay for the AT1 and AT2 blockers was adapted from that described by Macari et al27 The AT1 and AT2 blockers were extracted from blood using the methods previously described for extracting AII.17 In brief, one ml whole blood was collected from the retro-orbital sinus, added to 3 ml ice-cold methanol, and centrifuged for 10 min at 3000 rpm at 4°C. The supernatant was dried, resuspended in distilled water, extracted with a phenyl column (JT Baker, Phillipsburg, NJ), eluted with methanol, and then dried. The methanol extraction step precipitates protein and hence it removes protein-bound drug, so that this bioassay measures unbound (active) drug. The pellet was stored at −70°C. Samples were reconstituted in the buffer used for the AII binding assay.
To measure the extraction efficiency for the AT1 blocker (L-158,809), the blocker was added to whole blood of normal rats to final concentrations of 7–45 nM, then extracted as detailed above. The extracted sample was then added to renal membrane preparations and inhibition of AII binding was assessed as detailed above. Comparison to an inhibition curve for blocking of AII binding to renal crude membrane fractions by L-158,809 showed that extraction efficiency was 11.2±2.5% (mean ± standard deviation based on six separate extractions). An analogous sequence of procedures was done to calculate the extraction efficiency of the AT2 blocker (PD-123319) using the NG108 membrane preparation; the extraction efficiency for the AT2 blocker was 12.7±6.4% (mean ± standard deviation based on three separate extractions).
Blood samples from animals on the AII blockers were extracted in the same manner and compared, at a range of dilutions, with the relevant AII binding curve.
Statistical methods
Physiological data (e.g., BUN, BP, UP/UC) are shown as medians with 20–80% ranges. The two-group physiological data were compared by the Mann-Whitney test, and correlations were assessed with the Kendall rank correlation test. These non-parametric methods were used because physiological parameters in the groups showing abnormal renal function were often neither normally nor log-normally distributed. The incidence rates of renal failure were compared by an extension of the Kruskal-Wallis test,28 and the correlation of renal failure rates with drug doses was assessed with the Mantel-extension chi-square test. Where multiple comparison issues are relevant, they are discussed.
RESULTS
Efficacy of AT2 blockade
To confirm our previous report12 of the efficacy of the AT2 blocker in mitigation of radiation nephropathy, animals underwent 18.8 Gy TBI and were randomized to: the AT1 blocker at 2 mg/kg/day (n=5), the AT2 blocker at 15 mg/kg/day (n=6), the AT1 blocker at 2 mg/kg/day plus the AT2 blocker at 15 mg/kg/day (n=6), or no AII blockers (n=6). A fifth arm had 6 age-matched normal animals. All drugs were given from 4 to 12 weeks after TBI.
All three drug regimens protected the animals from long-term development of radiation-induced azotemia (P ≤ 0.018 at all time points, Figure 1). The two regimens that included the AT1 blocker gave better long-term control of radiation-induced azotemia than did the AT2 blocker alone (P ≤ 0.09 at 17 weeks, P < 0.01 at 26–52 weeks, Figure 1). The two regimens that included the AT1 blocker gave roughly equal control of radiation-induced azotemia (Figure 1).
Fig 1.
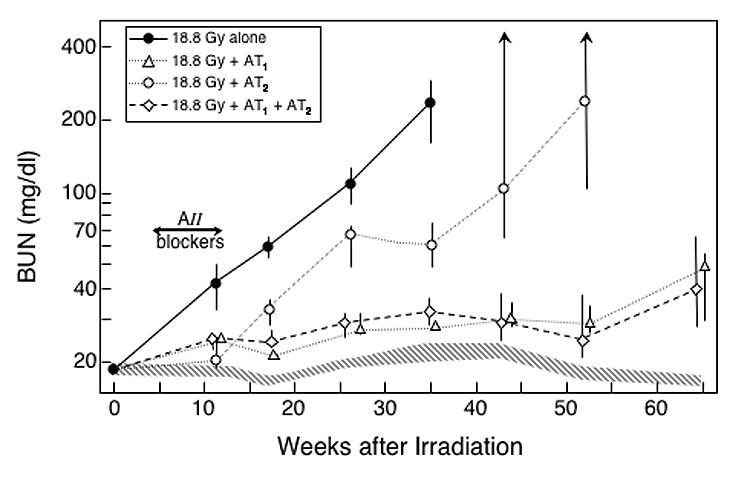
Effect of an 8-week treatment with AII blockers on the progression of azotemia (as BUN) induced by an 18.8 Gy radiation dose. Rats were given TBI in 6 fractions followed by a syngeneic BMT. Some animals received no further treatment (●), and others were treated from 4–12 weeks after irradiation with an AT1 blocker (L-158,809) at 2 mg/kg/day (△), with an AT2 blocker (PD-123319) at 15 mg/kg/day (○) or with a combination of the AT1 blocker and the AT2 blocker (●). Data is shown as medians with 20–80% ranges. Values for age-matched controls are shown by the hatched area.
The schedules with the AT1 blocker also yielded reductions in radiation-induced proteinuria and hypertension, both while the therapy was in progress and long-term (all P ≤ 0.025 compared to animals receiving irradiation alone, Tables I and II); but the AT2 blocker alone did not (Tables I and II). Finally, the actuarial risk of terminal radiation-induced uremia was significantly reduced by use of the AT2 blocker alone (P = 0.018, Figure 2), and completely eliminated in schedules that included the AT1 blocker (both P ≤ 0.002 versus radiation alone and P ≤ 0.018 versus the AT2 blocker, Figure 2).
Table I.
Effect of AII Blockers on Radiation-Induced Nephropathy Assessed at 12 Weeks after Irradiation
| Initial Number of Animals | BUN(mg/dl)a | UP/UC(mg/mg)a | BP (mm Hg)a | |
|---|---|---|---|---|
| age-matched normal | 6 | 18 (17–18) | 0.3 (0.3–0.4) | 133 (132–138) |
| TBIb alone | 6 | 42 (33–50) | 15.1 (14.5–15.6) | 168 (167–173) |
| TBI + AT1 blockc | 5 | 25 (23–26) | 1.9 (1.4–2.2) | 146 (140–156) |
| TBI + AT2 blockd | 6 | 20 (19–24) | 11.5 (9.3–13.7) | 163 (157–165) |
| TBI + AT1 blockc + AT2 blockd | 6 | 25 (22–26) | 0.6 (0.5–1.4) | 142 (133–151) |
Median with 20–80% range
18.8 Gy in 6 fractions over 3 days
L158,809 in the drinking water at 2 mg/kg/day from 4–12 weeks after irradiation
PD123319 by s.c. osmotic pump at 15 mg/kg/day from 4–12 weeks after irradiation
Table II.
Effect of AII Blockers on Radiation-Induced Nephropathy Assessed at 26 Weeks after Irradiation
| BUN (mg/dl)a | UP/UC (mg/mg)a | BP (mm Hg)a | Time to develop renal failure (wks)b | |
|---|---|---|---|---|
| age-matched normal | 20 (18–20) | 1.0 (0.6–1.7) | 123 (116–124) | >61 |
| TBIc alone | 108 (90–126) | 10.9 (10.6–13.8) | 164 (152–171) | 38 (33, 50) |
| TBI + AT1 blockd | 27 (27–32) | 6.6 (6.4–6.6) | 135 (124–137) | >61 |
| TBI + AT2 blocke | 69 (48–71) | 14.7 (13.2–16.5) | 154 (140–157) | 54 (37, >61) |
| TBI + AT1 blockd + AT2 blocke | 29 (25–32) | 5.3 (3.7–5.9) | 133 (126–148) | >61 |
Median with 20–80% range
Median with range
18.8 Gy in 6 fractions over 3 days
L158,809 in the drinking water at 2 mg/kg/day from 4–12 weeks after irradiation
PD123319 by s.c. osmotic pump at 15 mg/kg/day from 4–12 weeks after irradiation
Fig 2.
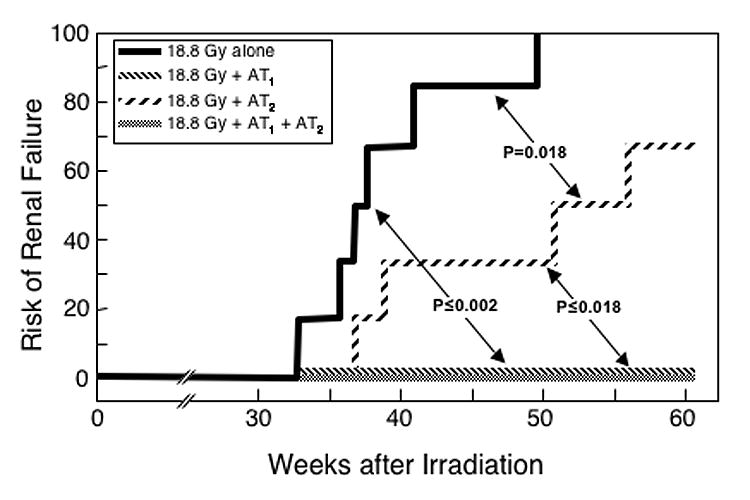
Effect of an 8-week treatment with AII blockers on the incidence of renal failure induced by an 18.8 Gy radiation dose. These are the same animals shown in Figure 1. There were no cases of renal failure in any groups treated with the AT1 blocker.
When combined with the AT1 blocker, the AT2 blocker did not significantly reduce the beneficial effect of the AT1 blocker on radiation-induced azotemia, proteinuria or hypertension; in fact, there was some indication that the combination improved control of radiation-induced proteinuria and hypertension, but the improvement was not statistically significant (Figure 1, Tables I and II).
Because the AT1 blocker was so effective after 18.8 Gy irradiation, studies at that dose provided little (Figure 1) or no (Figure 2) discrimination between the schedules that included the AT1 blocker. To provide better discrimination, the study was repeated with a higher radiation dose (20.5 Gy) where the AT1 blocker would be less effective. Again, the schedules using the AT1 blocker were effective in reducing the progression of radiation-induced azotemia (P ≤ 0.015 at all time points, Figure 3); but the schedule using the AT2 blocker alone was only marginally effective (Figure 3). The actuarial risk of terminal radiation-induced uremia was significantly reduced by use of the AT2 blocker alone (P = 0.033, Figure 4), and even further reduced in schedules that included the AT1 blocker (both P ≤ 0.002, Figure 4). When combined with the AT1 blocker, the AT2 blocker had no consistent effect on radiation-induced azotemia (Figure 3), proteinuria (data not shown) or hypertension (data not shown); but it significantly reduced actuarial risk of terminal radiation-induced uremia (P = 0.026, Figure 4).
Fig 3.
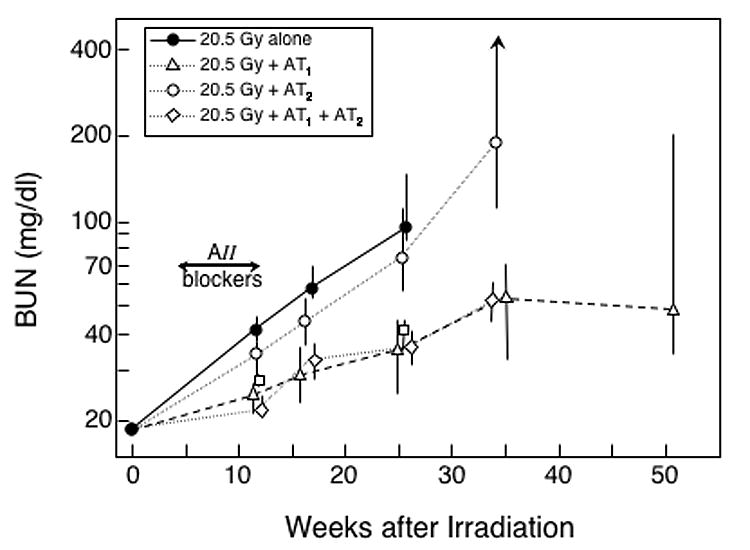
Effect of an 8-week treatment with AII blockers on the progression of azotemia (as BUN) induced by a 20.5 Gy radiation dose. The experimental design is identical to that shown in Figure 1, except that the radiation dose is higher.
Fig 4.
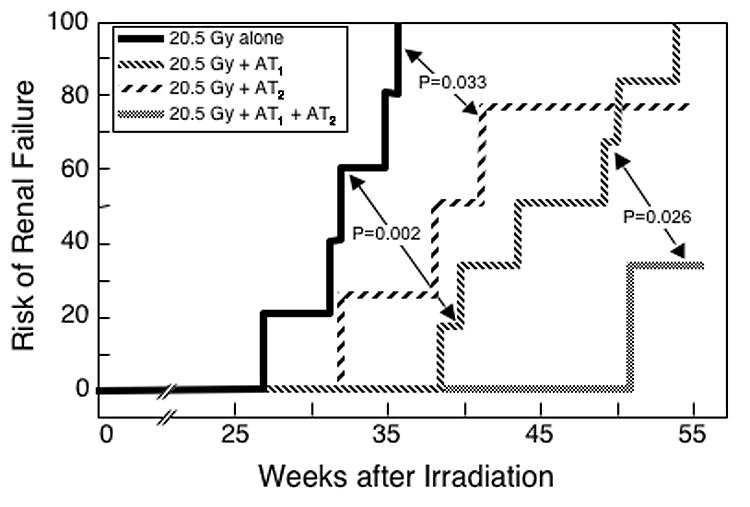
Effect of an 8-week treatment with AII blockers on the incidence of renal failure induced by a 20.5 Gy radiation dose. These are the same animals shown in Figure 3.
Escalation of AT1 blocker dose
The enhancement of the effects of AT1 blockade by the AT2 blocker (Moulder et al,12 and Figure 4) could be due to nonspecific binding of the AT2 blocker to the AT1 receptor.22,29 If so, then escalation of the dose of the AT1 blocker should also improve its efficacy. To assess this possibility, animals were given 20.5 Gy TBI and started on AT1 blocker the day after completion of TBI at doses of 2, 4 or 8 mg/kg/day; drug therapy was continued until 12 weeks after TBI. In the animals given TBI alone, azotemia, proteinuria and hypertension developed as expected, and this evolution was substantially attenuated by the AT1 blocker at all drug doses (Figure 5). Azotemia was significantly reduced at all drug doses at all follow-up times (all P ≤ 0.038, Figure 5); proteinuria and hypertension were attenuated during drug treatment (all P < 0.01, Figure 5), but the advantage gradually decreased after therapy stopped.
Fig 5.
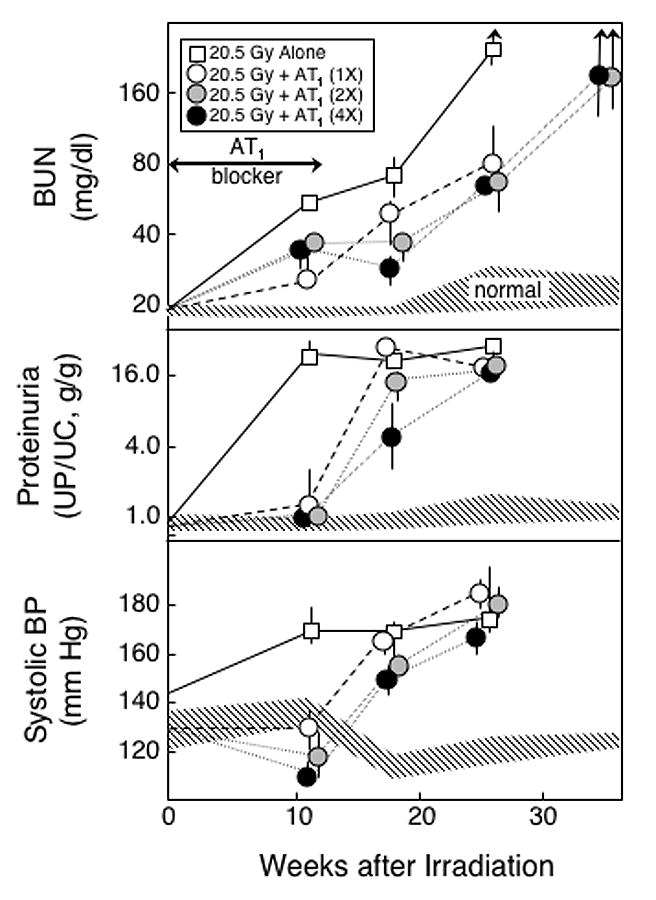
Effect of a 12-week treatment with different doses of an AT1 blocker on the progression of azotemia (as BUN), proteinuria (UP/UC), and hypertension (as systolic BP) induced by a 20.5 Gy radiation dose. Rats were given TBI in 6 fractions followed by a syngeneic BMT. Eleven animals received no further treatment (□), and others (n=6 per group) were treated for 12 weeks (starting immediately after the BMT) with an AT1 blocker (L-158,809) at 2 (○), 4 (●) or 8 (●) mg/kg/day. Data is shown as medians with 20–80% ranges. Values for age-matched controls are shown by the hatched area.
There was a small, but consistent, trend towards greater attenuation of radiation-induced physiological changes with escalating doses of the AT1 blocker. Radiation-induced proteinuria (Figure 5) was controlled after 11 wks of follow-up by all drug doses, and by 26 wks proteinuria had reached maximum values at all drug doses; however, at 17 wks there was a positive trend with drug dose (P < 0.001). Radiation-induced azotemia (Figure 5) was unrelated to drug dose after 11 wks of follow-up (P > 0.20), but there were positive trends with drug dose at 17 wks (P < 0.001) and 26 wks (P = 0.06) of follow-up. The reduction in radiation-induced hypertension (Figure 5) showed significant favorable trends at all follow-up times (all P ≤ 0.006).
The actuarial curves for freedom from terminal uremia (Figure 6) shows a clear-cut dose-response effect, with improving benefit at greater doses of the AT1 blocker (P = 0.01 for the trend in terminal uremia rates with drug dose).
Fig 6.
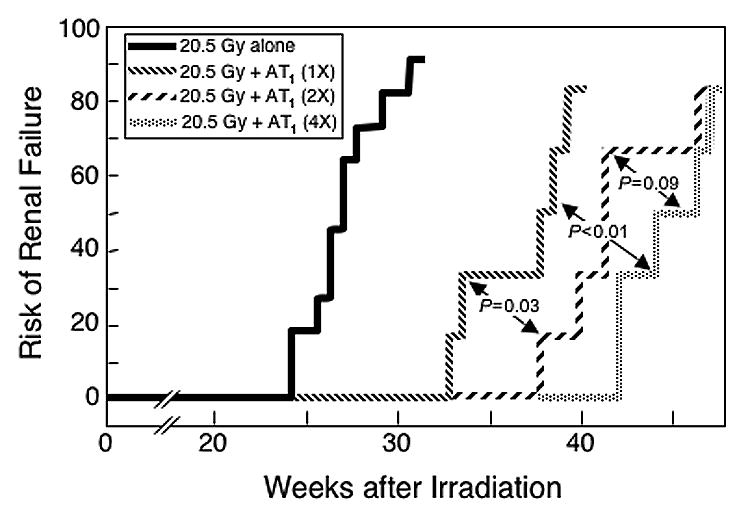
Effect of a 12-week treatment with different doses of an AT1 blocker on the incidence of renal failure induced by a 20.5 Gy radiation dose. These are the same animals shown in Figure 5.
Renal AII receptor binding
AII receptor binding studies were done with renal membrane preparations from irradiated rats and from age-matched normal rats. Inhibition of AII binding to renal membrane fractions was inhibited by the AT1 blocker at concentrations of 1 nM and above (Figure 7). There was a plateau in the binding inhibition at AT1 blocker concentrations of 100 nM or more; the maximum inhibition that could be achieved was 85% (95% confidence interval of 82–89%). There was no statistically significant difference between maximum inhibition levels in renal membrane fractions from irradiated rats and age-matched normal rats.
Fig 7.
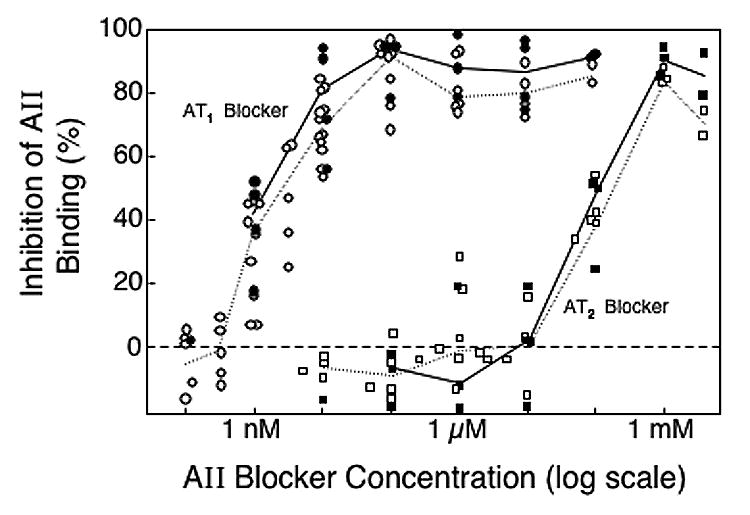
Inhibition of AII binding to renal membrane preparations from irradiated rats (solid symbols) and age-matched normal rats (open symbols) by the AT1 blocker L-158,809 (circles) or the AT2 blocker PD-123319 (squares). Irradiated rats had received 18.8 Gy TBI 4–9 weeks earlier. Each point represents a separate inhibition experiment study done with the AT1 blocker or the AT2 blocker. Solid lines connect the median inhibition levels for irradiated rats in each dose range; dotted lines connect the median inhibition levels for age-matched normal rats in each dose range.
Inhibition of AII binding by the AT2 blocker did not occur until the concentration exceeded 10 μM (Figure 7). At concentrations of 1000–4000 μM, where the AT2 blocker is reported to block AT1 receptors as well as AT2 receptors,22,29 AII binding was inhibited by 84% (78–90%). At concentrations of 0.1–10 μM, where the AT2 blocker should be blocking AT2 receptors,22,29 the upper 95% confidence interval on the level of inhibition was 3.9%; therefore, AT2 receptor levels of less than 4% would not have been detected.
Because the AT2 blocker (PD-123319) showed no activity at sub-mM levels in renal membrane fractions, NG108 cells, which are known to have AT2 receptors,19 were used to test the AT2 blocker and the AT2 receptor binding assay. Using membrane fractions from NG108 cells exposed to high levels (1 μM) of the AT1 blocker, there was progressive inhibition of residual AII binding by PD-123319 at concentrations ranging from 10 nM to 1000 nM (Figure 8). In renal membrane fractions, these concentrations of the AT2 blocker had no effect (Figure 7); thus, the lack of evidence for AT2 receptors in the rat renal membrane preparation was not due to problems with either the AT2 blocker or the AT2 receptor binding assay.
Fig 8.
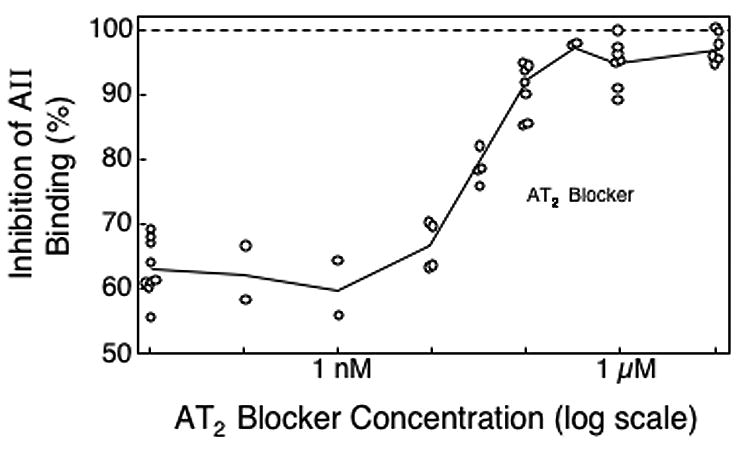
Inhibition of AII binding to crude membrane preparations from NG108 cells by an AT2 blocker (PD-123319) in the presence 1 μM of an AT1 blocker (L-158,809). Each point represents a separate inhibition experiment study. Solid lines connect the median inhibition levels in each dose range.
Adding the AT2 blocker at 100 nM to the AT1 blocker at 100 μM did not significantly increase the inhibition of AII binding to the renal membrane preparation; the maximum achieved with the combination of blockers was 87 (82–92)%. Thus there are AII receptors in the renal membrane preparation that do not appear to be blocked by either the AT1 blocker (L-158,809) or the AT2 blocker (PD-123319).
Bioassay of blood levels of the AII blockers
To determine the concentration of AII blockers in the blood of rats treated with these agents, blood samples were taken, extracted and assayed for inhibition of AII binding to the appropriate crude membrane fraction. The animals used for the bioassay studies were those shown in Tables I and II, and Figures 1 and 2; they were assayed 8 weeks after TBI (5 weeks after the start of therapy with the AII blockers).
When blood extracts from animals on the AT1 blocker at 2 or 8 mg/kg/day were assayed at full concentration (that is, the extract from 1 ml of blood suspended in 1 ml of assay buffer) they inhibited AII binding to renal crude membrane fractions by more than 70%, indicating that the L-158,809 concentration in the sample was greater than 4 nM (see Figure 7). Taking into account the extraction efficiency, this indicates blood concentrations of L-158,809 above 30 nM. To achieve a semiquantitative estimate of the actual blood concentrations of the AT1 blocker, the samples were diluted (1:3, 1:6 and/or 1:12) so that inhibition of AII binding would fall in the range (20–60%) where the dose-response curve was the steepest (Figure 7). Based on these dilutions, and with adjustment for extraction efficiency, the blood concentrations of the AT1 blocker was 100±25 nM for animals consuming 2 mg/kg/day, 235±75 nM for animals consuming 8 mg/kg/day, and 75±20 for animals consuming both the AT1 blocker at 2 mg/kg/day and the AT2 blocker at 15 mg/kg/day (means with standard deviation based on 4 animals). Thus, the dose of the AT1 blocker used in these studies yielded an unbound blood concentration that was more than sufficient (Figure 7) to block essentially all AT1 receptors.
Extracted blood samples from age-matched normal animals and animals given TBI only showed no detectable inhibition of AII binding to renal membrane fractions, even when assayed at 3X concentration; therefore, the blood of these animals had less capability to block AT1 receptors than L-158,809 at 0.3 nM. Animals on AT2 blocker (PD-123319) alone showed no detectable inhibition of AII binding in this assay when assayed at 3X concentration; thus, the AT2 blocker was used at a dose that did not achieve a blood concentration sufficient to bind AT1 receptors.
When blood extracts from animals on the AT2 blocker were assayed at full concentration in the presence of 1 μM of the AT1 blocker, they inhibited AII binding to NG108 membrane fractions by about 70%, indicating that the PD-123319 concentration in the sample was greater than 12 nM (Figure 8). Taking into account the extraction efficiency, this indicates blood concentrations of PD-123319 of 100 nM. To achieve a semiquantitative estimate of the actual blood concentrations of the AT2 blocker, the samples were concentrated (3:1) so that inhibition of AII binding would fall in the range (70–90%) where the dose-response curve was the steepest (Figure 8). Based on the assays of the concentrated blood extracts, and with adjustment for extraction efficiency, the blood concentrations of the AT2 blocker were 82±24 nM for animals on the AT2 blocker alone at 15 mg/kg/day, and 122±31 nM for animals on both the AT2 blocker at 15 mg/kg/day and the AT1 blocker at 2 mg/kg/day (means with standard deviations based on 3 animals). Thus, the dose of the AT2 blocker used in these studies yielded a blood concentration sufficient to achieve near-maximum blocking of AT2 receptors in NG108 cells (Figure 8), but which was 100-fold too low to block renal AT1 receptors (Figure 7).
DISCUSSION
These data confirm and extend our previous studies. The efficacy of AT1 blockade for mitigation of radiation nephropathy is clear (Figures 1–4, Tables I and II). It is also evident that the addition of the AT2 blocker did not reverse this benefit. Thus, we conclude that the role of the AT2 receptor is not necessarily in opposition to that of the AT1 receptor, as has been reported in other models.11 In addition, the use of the AT2 blocker alone yielded some benefit in mitigation of radiation nephropathy, as assessed by reduced azotemia (Figures 1 and 3) and a reduced actuarial risk of terminal uremia (Figures 2 and 4). This occurred despite a lack of major effect of the AT2 blocker on either proteinuria or blood pressure during the time of its administration (Tables I and II), and despite any evidence for the presence of detectable levels of AT2 receptor binding in renal membranes (Figure 7). Other studies have also shown that potentially adverse effects can be mediated by stimulation of AT2 receptors, including cardiac fibrosis and stimulation of renal tubular proliferation;30 this would suggest that in some circumstances blockade of the AT2 receptor could be beneficial.
Since some of our studies suggested that the AT2 blocker could enhance the effect of AT1 blockade, we explored the possibility that the added benefit of the AT2 blocker was achieved via enhanced AT1 blockade. If our standard 2 mg/kg/day dose of the AT1 blocker was already achieving maximal blockade of AT1 receptors, this would seem to rule out the possibility that the AT2 blocker was enhancing blockade of AT1 receptors. However, higher doses of the AT1 blocker (4 and 8 mg/kg/day) yielded an advantage over the lower dose, with reduced azotemia, proteinuria and hypertension (Figure 5), and a corresponding longer median time to develop terminal renal failure (Figure 6).
The added benefit of the higher doses of the AT1 blocker (Figures 5 and 6) was inconsistent with the observation that even the lowest dose gave a blood concentration (>50 nM) that was sufficient (Figure 7) to block essentially all AT1 receptors. However, this benefit of higher-dose AT1 blockade is consistent with the superior protection found by a ten-fold increase in the use of the AT1 blocker losartan in the remnant kidney model.31 In that study, glomerulosclerosis at 4 months after renal ablation was reduced by losartan at 50 mg/kg/day, but was reduced even more at 500 mg/kg/day. As in the radiation model, this benefit was accompanied by a modest, but significant reduction in blood pressure. Additional studies in the remnant model with the higher dose of losartan showed reduction of markers of inflammation, particularly interstitial macrophages. It is possible that the higher dose of L-158,809 in our model exerted benefit via such a mechanism. However, we doubt that interstitial inflammation is a critical mechanism of renal failure in radiation nephropathy, because histopathologically, radiation nephropathy does not have a major inflammatory component.
Renal membrane binding studies (Figure 7) showed blockade of AII binding by the AT1 blocker in concentrations ranging from nanomolar to micromolar, with an apparent plateau at 100 nM and beyond. The plateau was significantly below the 100% level, even when both the AT1 and the AT2 blocker were used, raising the question of the presence of receptors not blocked by either the AT1 or the AT2 antagonist. This could be the angiotensin IV (AT4) receptor, which has been found in rat kidney and which may act via AT1 receptor signaling.32,33
There was no inhibition of AII binding to the renal membrane fraction by the AT2 blocker until concentrations were reached at which the AT2 blocker has been reported to bind to the AT1 receptor.22,29,34 The lack of detectable inhibition of AII binding to renal membranes by the AT2 blocker (at 0.1–10 μM) occurred in both normal and irradiated animals, suggesting that the efficacy of the AT2 blocker in radiation nephropathy was not due to radiation-induced upregulation of AT2 receptors. The possibility that radiation-induced renal failure could lead to upregulation of AT2 receptors, as it does in other forms of renal failure,35 was not directly tested as the AT2 blocker was used in these studies before frank radiation injury had occurred.
The receptor binding data are consistent with other studies of AII binding in kidneys. Sechi et al34 for instance, show a Kd of 0.6 nM for the AT1 receptor in rat kidney, and that an AT2 antagonist had an inhibitory effect on binding only at micromolar or greater concentrations. The known nanomolar concentrations of AII in the renal tubular lumen36 are also consistent with these binding studies. Thus, the AT1 antagonist L-158,809 appears to be active in inhibiting binding to renal microsomes in concentrations that are stoichimetrically appropriate for known intra-renal concentrations of AII.
We next tested the levels of AII blocking activity in the blood of animals treated with the AT1 or AT2 blockers. Blood taken from animals treated with the AT1 blocker at 2 or 8 mg/kg/day yielded AII binding inhibition of ≥ 80% in the renal membrane preparations, a level consistent with blockade of essentially all AT1 receptors (Figure 7). In rats treated with the AT2 blocker, on the other hand, blood samples showed no effect on AT1 binding by renal membrane preparations. Bioassays using renal membrane preparations indicated that the unbound AT1 blocker blood concentrations were 100 mM and 235 mM in the animals treated at 2 and 8 mg/kg/day, respectively. In parallel experiments using an NG108 membrane binding bioassay, the unbound blood concentrations of the AT2 blocker were 80–120 nM. Although this concentration is sufficient to block AT2 receptors (Figure 8), it is orders of magnitude less than concentrations of the AT2 blocker required to inhibit AII binding in renal membrane preparations (Figure 7). Thus it is extremely unlikely that the benefit of the AT2 blocker that we have documented is due to blockade of AT1 receptors by the AT2 blocker.
Wong et al37 suggested that the AT2 antagonist PD-123177 could displace the AT1 antagonist losartan from its rat plasma protein binding sites, increasing the concentration of free losartan and enhancing AT1 blockade. This mechanism could help to explain the benefit of adding the AT2 blocker to the AT1 blocker in our model (if it also applied to L-158,809 and PD-123319), but it would not explain the benefit of the AT2 blocker when used by itself. In addition, our bioassay found no statistically significant effect on the blood concentration of the AT1 blocker when the AT2 blocker was also present.
Two studies of experimental radiation nephropathy have reported that an aldosterone antagonist exerts benefit in radiation nephropathy.38,39 It is possible that the added benefit of the AT2 blocker in the present studies is due to an effect on aldosterone secretion. This is unlikely, however, because the effect of AII on aldosterone secretion is mediated via the AT1 receptor, not the AT2 receptor.40
In our initial report of the benefit of AT2 blockade in radiation nephropathy, we speculated that this benefit could be related to attenuation of cell proliferation. We did not specifically test this possibility because concurrent studies in our laboratory have weakened the hypothesis that cell proliferation is a key mechanism of radiation nephropathy.9
Recent studies of intra-renal blood flow suggest mechanisms whereby AT2 blockade could be beneficial. In a 2 kidney 1 clip model, Duke et al41 showed that the AT2 blocker PD-123319 enhanced renal medullary blood flow by almost 20%, but not in control non-clipped rats; in contrast, an AT1 blocker (candesartan) enhanced cortical, but not medullary blood flow, in both clipped and non-clipped rats. The inability of the AT2 blockade to influence medullary blood flow in the non-clipped rats suggests that an activated renin-AII system is required to unmask the AT2 effect. This is supported by parallel experiments by Duke et al41 using arterial AII infusion to normal rats. In those studies, the AT2 blocker PD-123319 had a positive effect on medullary blood flow which was completely abolished by simultaneous use of an AT1 blocker (candesartan). Although we have found no evidence for enhanced levels of renin,17 AII,17 or AII receptor binding42 after renal irradiation, we have recently shown a trend towards enhanced renin-substrate, angiotensinogen, in radiation nephropathy.43 Thus, in radiation nephropathy, as in the 2 kidney 1 clip model and in AII-infused rats, there may be an overactivity of the renin-AII system, and by extension a beneficial medullary vasodilation by AT2 receptor blockade.
It is noteworthy that there is an enhanced benefit of the higher dose of the AT1 blocker compared to the lower dose even though the blood level of the AT1 blocker is sufficient at the lower dose to provide maximal blocking of the AT1 receptor in a renal membrane preparation. It is possible that the higher dose of the AT1 blocker reaches renal tissue subdivisions that are not reached by the lower dose. A similar explanation is advanced by Fujihara et al31 to explain the benefit of very-high-dose losartan in their remnant kidney model.
In conclusion, we have confirmed that in our model of renal failure there is benefit, rather than a deleterious effect, of AT2 receptor antagonism. This is occurring despite a lack of change in renal AII binding after irradiation42 and a lack of evidence for AT2 receptor binding in renal membranes; and it does not depend on AT1 antagonism by the AT2 receptor blocker.
Acknowledgments
Natalya V. Morrow, Ph.D. assisted with the radiation dosimetry. Marylou Mäder provided expert technical assistance. Yvonne A. Morauski and Amt A. Irving assisted with preparation of the manuscript.
These studies were supported by NIH grant CA-24652 (JEM, EPC, MS). L-158,809 was supplied as a gift by Merck, Sharpe and Dohme, Rahway, NJ. PD-123319 was supplied as a gift by Parke-Davis Pharmaceutical Research, Ann Arbor, MI, and subsequently by the new parent company, Pfizer, Groton, CT.
Abbreviations
- AII
angiotensin II
- ACE
angiotensin converting enzyme
- AT1 receptor
angiotensin I type-1 receptor
- AT2 receptor
angiotensin II type-2 receptor
- BMT
bone marrow transplantation
- BSA
bovine serum albumin
- BP
blood pressure
- BUN
blood urea nitrogen
- EDTA
ethylenediaminetetraacetic acid
- HEPES
N-(2-hydroxyethyl)piperazine-N’-(2-ethanesulfonic acid)
- PMSF
Phenylmethylsulphonylfluoride
- TBI
total body irradiation
- UP/UC
urine protein to creatinine ratio
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kal HB, van Kempen-Harteveld ML. Renal dysfunction after total body irradiation: Dose-effect relationship. Int J Radiat Oncol Biol Phys. 2006;65:1228–32. doi: 10.1016/j.ijrobp.2006.02.021. [DOI] [PubMed] [Google Scholar]
- 2.Cohen EP, Robbins MEC. Radiation nephropathy. Semin Nephrol. 2003;23:486–99. doi: 10.1016/s0270-9295(03)00093-7. [DOI] [PubMed] [Google Scholar]
- 3.Zenz T, Schlenk RF, Glatting G, Neumaier B, Blumstein N, Buchmann I, et al. Bone marrow transplantation nephropathy after an intensified conditioning regimen with radioimmunotherapy and allogeneic stem cell transplantation. J Nuc Med. 2006;47:278–86. [PubMed] [Google Scholar]
- 4.Lambert B, Cybulla M, Weiner SM, Van De Wiele C, Ham H, Dierckx RA, et al. Renal toxicity after radionuclide therapy. Radiat Res. 2004;161:607–11. doi: 10.1667/rr3105. [DOI] [PubMed] [Google Scholar]
- 5.Moulder JE, Fish BL, Cohen EP. ACE inhibitors and AII receptor antagonists in the treatment and prevention of bone marrow transplant nephropathy. Curr Pharm Des. 2003;9:737–49. doi: 10.2174/1381612033455422. [DOI] [PubMed] [Google Scholar]
- 6.Oikawa T, Freeman M, Lo W, Vaughan DE, Fogo A. Modulation of plasminogen activator inhibitor-1 in vivo: A new mechanism for the anti-fibrotic effect of renin-angiotensin inhibition. Kidney Int. 1997;51:164–72. doi: 10.1038/ki.1997.20. [DOI] [PubMed] [Google Scholar]
- 7.Juncos LI, Carrasco Dueñas S, Cornejo JC, Broglia CA, Cejas H. Long-term enalapril and hydrochlorothiazide in radiation nephritis. Nephron. 1993;64:249–55. doi: 10.1159/000187322. [DOI] [PubMed] [Google Scholar]
- 8.Cohen EP, Hussain S, Moulder JE. Successful treatment of radiation nephropathy with angiotensin II blockade. Int J Radiat Oncol Biol Phys. 2003;55:190–3. doi: 10.1016/s0360-3016(02)03793-8. [DOI] [PubMed] [Google Scholar]
- 9.Moulder JE, Cohen EP. Future strategies for mitigation and treatment of chronic radiation-induced normal tissue injury. Semin Radiat Oncol. doi: 10.1016/j.semradonc.2006.11.010. in press. [DOI] [PubMed] [Google Scholar]
- 10.Cohen EP, Fish BL, Moulder JE. Angiotensin II infusion exacerbates radiation nephropathy. J Lab Clin Med. 1999;134:283–91. doi: 10.1016/s0022-2143(99)90209-3. [DOI] [PubMed] [Google Scholar]
- 11.Siragy HM. AT1 and AT2 receptor in the kidney: Role in health and disease. Semin Nephrol. 2004;24:93–100. doi: 10.1016/j.semnephrol.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Moulder JE, Fish BL, Cohen EP. Impact of angiotensin II type 2 receptor blockade on experimental radiation nephropathy. Radiat Res. 2004;161:312–7. doi: 10.1667/rr3129. [DOI] [PubMed] [Google Scholar]
- 13.Moulder JE, Fish BL, Cohen EP. Treatment of radiation nephropathy with ACE inhibitors and AII type-1 and type-2 receptor antagonists. Curr Pharm Des. 2007 doi: 10.2174/138161207780618821. in press. [DOI] [PubMed] [Google Scholar]
- 14.Cao Z, Bonnet F, Candido R, Nesteroff SP, Burns WC, Kawachi H, et al. Angiotensin type 2 receptor antagonism confers renal protection in a rat model of progressive renal injury. J Amer Soc Nephrol. 2002;13:1773–87. doi: 10.1097/01.asn.0000019409.17099.33. [DOI] [PubMed] [Google Scholar]
- 15.Moulder JE, Fish BL. Late toxicity of total body irradiation with bone marrow transplantation in a rat model. Int J Radiat Oncol Biol Phys. 1989;16:1501–9. doi: 10.1016/0360-3016(89)90955-3. [DOI] [PubMed] [Google Scholar]
- 16.Moulder JE, Cohen EP, Fish BL, Hill P. Prophylaxis of bone marrow transplant nephropathy with captopril, an inhibitor of angiotensin-converting enzyme. Radiat Res. 1993;136:404–7. [PubMed] [Google Scholar]
- 17.Cohen EP, Fish BL, Moulder JE. The renin-angiotensin system in experimental radiation nephropathy. J Lab Clin Med. 2002;139:251–7. doi: 10.1067/mlc.2002.122279. [DOI] [PubMed] [Google Scholar]
- 18.Ma CM, Coffey CW, DeWerd LA, Liu C, Nath R, Seltzer SM, et al. AAPM protocol for 40–300 kV x-ray beam dosimetry in radiotherapy and radiobiology. Med Phys. 2001;28:868–93. doi: 10.1118/1.1374247. [DOI] [PubMed] [Google Scholar]
- 19.Laflamme L, Gasparo M, Gallo JM, Payet MD, Gallo-Payet N. Angiotensin II induction of neurite outgrowth by AT2 receptors in NG108-15 cells. Effect counteracted by the AT1 receptors. J Biol Chem. 1996;271:22719–35. doi: 10.1074/jbc.271.37.22729. [DOI] [PubMed] [Google Scholar]
- 20.Siegl PKS, Chang RSL, Mantlo NB, Chakavarty PK, Ondeyka DL, Greenlee WJ, et al. In vivo pharmacology of L-158,809, a highly potent and selective non-peptide angiotensin II receptor blocker. J Pharmacol Exper Ther. 1992;262:139–44. [PubMed] [Google Scholar]
- 21.Chang RSL, Siegl PKS, Clineschmidt BV, Mantlo NB, Chakavarty PK, Greenlee WJ, et al. In vitro pharmacology of L-158,809, a new and highly potent and selective angiotensin II receptor antagonist. J Pharmacol Exper Ther. 1992;262:133–8. [PubMed] [Google Scholar]
- 22.Macari D, Bottari S, Whitebread S, De Gasparo M, Levens N. Renal actions of the selective angiotensin AT2 receptor ligands CGP 42112B and PD 123319 in the sodium-depleted rat. Europ J Pharm. 1993;249:85–93. doi: 10.1016/0014-2999(93)90665-5. [DOI] [PubMed] [Google Scholar]
- 23.Cohen EP, Fish BL, Moulder JE. Successful brief captopril treatment in radiation nephropathy. J Lab Clin Med. 1997;129:536–47. doi: 10.1016/s0022-2143(97)90008-1. [DOI] [PubMed] [Google Scholar]
- 24.Moulder JE, Fish BL, Cohen EP. Brief pharmacologic intervention in experimental radiation nephropathy. Radiat Res. 1998;150:535–41. [PubMed] [Google Scholar]
- 25.Stone HB, Moulder JE, Coleman CN, Ang KK, Anscher MS, Barcellos-Hoff MH, et al. Models for evaluating agents intended for the prophylaxis, mitigation and treatment of radiation injuries. Report of an NCI workshop, December 3–4, 2003. Radiat Res. 2004;162:711–28. doi: 10.1667/rr3276. [DOI] [PubMed] [Google Scholar]
- 26.Ernsberger P, Zhou J, Damon TH, Douglas JG. Angiotensin II receptor subtypes in cultured rat renal mesangial cells. Amer J Physiol. 1992;263:F411–F6. doi: 10.1152/ajprenal.1992.263.3.F411. [DOI] [PubMed] [Google Scholar]
- 27.Macari D, Whitebread S, Cumin F, DeGasparo M, Levens N. Renal actions of the angiotensin AT receptors ligands CGP-42112 and PD-123319 after blockade of the renin-angiotensin system. Europ J Pharm. 1994;259:27–36. doi: 10.1016/0014-2999(94)90153-8. [DOI] [PubMed] [Google Scholar]
- 28.Lee ET, Desu MM. A computer program for comparing K samples with right-censored data. Computer Programs in Biomedicine. 1972;2:315–21. doi: 10.1016/0010-468x(72)90019-0. [DOI] [PubMed] [Google Scholar]
- 29.Bottari SP, de Gasparo M, Steckelings UM, Levens NR. Angiotensin II receptor subtypes: characterization, signalling mechanisms, and possible physiological implications. Front Neuroendocrinol. 1993;14:123–71. doi: 10.1006/frne.1993.1005. [DOI] [PubMed] [Google Scholar]
- 30.Wolf G. ‘The road not taken’: role of angiotensin II type 2 receptor in pathophysiology. Nephrol Dial Trans. 2002;17:195–8. doi: 10.1093/ndt/17.2.195. [DOI] [PubMed] [Google Scholar]
- 31.Fujihara CK, Velho M, Malheiros DMAC, Zatz R. An extremely high dose of losartan affords superior renoprotection in the remnant model. Kidney Int. 2005;67:1913–24. doi: 10.1111/j.1523-1755.2005.00290.x. [DOI] [PubMed] [Google Scholar]
- 32.Li XC, Campbell DJ, Ohishi M, Yuan S, Zhuo JL. AT1 receptor-activated signaling mediates angiotensin IV-induced renal cortical vasoconstriction in rats. Amer J Physiol. 2006;290:F1024–F33. doi: 10.1152/ajprenal.00221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Handa RK, Krebs LT, Harding JW, Handa SE. Angiotensin IV AT4-receptor system in the rat kidney. Amer J Physiol. 1998;274:F290–F9. doi: 10.1152/ajprenal.1998.274.2.F290. [DOI] [PubMed] [Google Scholar]
- 34.Sechi LA, Grady EF, Griffin CA, Kalinyak JE, Schambelan M. Distribution of angiotensin II receptor subtypes in rat and human kidney. Amer J Physiol. 1992;262:F236–F40. doi: 10.1152/ajprenal.1992.262.2.F236. [DOI] [PubMed] [Google Scholar]
- 35.Bautista R, Sánchez A, Hernández J, Oyekan A, Escalante B. Angiotensin II type AT2 receptor mRNA expression and renal vasodilatation are increased in renal failure. Hypertension. 2001;38:669–73. doi: 10.1161/hy09t1.096186. [DOI] [PubMed] [Google Scholar]
- 36.Navar LG, Lewis L, Hymel A, Braam B, Mitchell KD. Tubular fluid concentrations and kidney contents of angiotensins I and II in anesthetized rats. J Amer Soc Nephrol. 1994;5:1153–8. doi: 10.1681/ASN.V541153. [DOI] [PubMed] [Google Scholar]
- 37.Wong PC, Christ DD, Timmermans PBMWM. Enhancement of losartan (DuP 753)-induced angiotensin II receptor antagonism by PD123177 in rats. Europ J Pharm. 1992;220:267–70. doi: 10.1016/0014-2999(92)90758-v. [DOI] [PubMed] [Google Scholar]
- 38.Jaggi JS, Seshan SV, McDevitt MR, Sgouros G, Hyjek E, Scheinberg DA. Mitigation of radiation nephropathy after internal α-particle irradiation of kidneys. Int J Radiat Oncol Biol Phys. 2006;64:1503–12. doi: 10.1016/j.ijrobp.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 39.Brown NJ, Nakamura S, Ma L, Nakamura I, Donnert E, Freeman M, et al. Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int. 2000;58:1219–27. doi: 10.1046/j.1523-1755.2000.00277.x. [DOI] [PubMed] [Google Scholar]
- 40.Tanabe A, Naruse M, Arai K, Naruse K, Yoshimoto T, Seki T, et al. Angiotensin II stimulates both aldosterone secretion and DNA synthesis via type 1 but not type 2 receptors in bovine adrenocortical cells. J Endocrinol Invest. 1998;21:668–72. doi: 10.1007/BF03350796. [DOI] [PubMed] [Google Scholar]
- 41.Duke LM, Widdop RE, Kett MM, Evans RG. AT2 receptors mediate toxic renal medullary vasoconstriction in renovascular hypertension. Br J Pharm. 2005;144:486–92. doi: 10.1038/sj.bjp.0706036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen EP, Joines MM, Moulder JE. Prevention and treatment of radiation injuries – The role of the renin-angiotensin system. In: Rubin P, Constine LS, Mark LB, Okunieff P, editors. Late Effects on Normal Tissues of Cancer Treatment. Heidelberg: Springer-Verlag; In Press. [Google Scholar]
- 43.Kobori H, Ozawa Y, Suzaki Y, Prieto-Carrasquero MC, Nishiyama A, Shoji T, et al. Intratubular angiotensinogen in hypertension and kidney diseases. Am J Hyperten. 2006;19:541–50. doi: 10.1016/j.amjhyper.2005.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]