

Abstract
The N-methyl-D-aspartate (NMDA) subtype of glutamate receptors plays an important role in brain physiology, but excessive receptor stimulation results in seizures and excitotoxic nerve cell death. NMDA receptor-mediated neuronal excitation and injury can be prevented by high, non-physiological concentrations of the neuroinhibitory tryptophan metabolite kynurenic acid (KYNA). Here we report that endogenous KYNA, which is formed in and released from astrocytes, controls NMDA receptors in vivo. This was revealed with the aid of the dopaminergic drugs d-amphetamine and apomorphine, which cause rapid, transient decreases in striatal KYNA levels in rats. Intrastriatal injections of the excitotoxins NMDA or quinolinate (but not the non-NMDA receptor agonist kainate) at the time of maximal KYNA reduction resulted in 2-3-fold increases in excitotoxic lesion size. Pre-treatment with kynurenine 3-hydroxylase inhibitors or dopamine receptor antagonists, two classes of pharmacological agents that prevented the reduction in brain KYNA caused by dopaminergic stimulation, abolished the potentiation of neurotoxicity. Thus, the present study identifies a previously unappreciated role of KYNA as a functional link between dopamine receptor stimulation and NMDA neurotoxicity in the striatum.
Keywords: Amphetamine, Astrocyte, Basal Ganglia, Dopamine, Kynurenine 3-hydroxylase, Quinolinic acid
Glutamatergic neurotransmission mediated by N-methyl-D-aspartate (NMDA) receptors plays important roles in striatal physiology. Because of the discrete pre- and postsynaptic, and intra-and extrasynaptic, localization of the various receptor subunits (Bernard and Bolam, 1998; Wang and Pickel, 2000; Dunah and Standaert, 2003; Galvan et al., 2006), striatal NMDA receptor stimulation has multiple and complex functional effects. Moderate activation of NMDA receptors controls, for example, monoaminergic, cholinergic, glutamatergic and peptidergic neurotransmission (Becquet et al., 1990; Young and Bradford, 1993; Knauber et al., 1999; Hathway et al., 2001; Radke et al., 2001; Marti et al., 2005) and, as a consequence, affects synaptic plasticity (Centonze et al., 2004; Dang et al., 2006), motor function (Hallett et al., 2005; Gardoni et al., 2006) and cellular bioenergetics (Moncada and Bolanos, 2006). Excessive stimulation of striatal NMDA receptors, on the other hand, triggers excitotoxicity, a mode of neuronal death that is believed to play a causative role in neurodegenerative diseases affecting the basal ganglia (Sonsalla et al., 1998; Waxman and Lynch, 2005; Fan and Raymond, 2006).
Because of the need to delicately balance receptor function, regulatory mechanisms have evolved to assure that NMDA receptors operate optimally and to safeguard against NMDA receptor malfunction (Schrattenholz and Soskic, 2006). Some of these modulatory influences involve small messenger molecules that originate from astrocytes situated in close apposition to NMDA receptor-bearing neurons. These glia-derived signals or “gliotransmitters” include, among others, D-serine (Schell, 2004; Nishikawa, 2005; Martineau et al., 2006) and the tryptophan metabolite kynurenic acid (KYNA; Kiss et al., 2003). The latter compound is especially interesting, because it not only inhibits the NMDA receptor directly by acting at the glycine co-agonist (“glycineB”) site (Kessler et al., 1989; Parsons et al., 1997) but also decreases extracellular glutamate levels by blocking presynaptic α7 nicotinic receptors (α7nAChRs) situated on glutamatergic nerve endings (Carpenedo et al., 2001; Rassoulpour et al., 2005). KYNA is therefore increasingly viewed as an important endogenous modulator of physiological and pathological events associated with glutamatergic neurotransmission (Schwarcz, 2004; Coyle, 2006).
In the striatum, removal of dopaminergic afferents confers neuroprotection against excitotoxic insults (Buisson et al., 1991; Meldrum et al., 2001). This and related evidence suggests that striatal excitotoxicity may be exacerbated by increased dopaminergic activity, and that timely anti-dopaminergic interventions may attenuate neuronal loss in Huntington’s disease and other neurodegenerative basal ganglia disorders (Jakel and Maragos, 2000). In line with this supposition, we demonstrated a few years ago that systemic administration of the psychostimulant d-amphetamine (d-Amph), which raises extracellular dopamine levels in the brain, is associated with an increased susceptibility of striatal neurons to the excitotoxic actions of NMDA (Poeggeler et al., 1998).
Through mechanisms that involve dopamine receptors but are otherwise not fully understood, d-Amph administration also causes an acute, transient reduction of cerebral KYNA levels. In rats, this effect can be seen at all ages but is especially prominent during the first postnatal weeks (Rassoulpour et al., 1998). Taken together with the ability of endogenous KYNA to control striatal vulnerability to excitotoxic injury (cf above; Harris et al., 1998; Sapko et al., 2006), it is therefore plausible that the potentiation of NMDA receptor-mediated excitotoxicity by d-Amph is caused by the synergistic effects of enhanced dopamine receptor activation and decreased KYNA formation.
With the availability of pharmacological agents that can selectively influence cerebral KYNA levels (Röver et al., 1997; Schwarcz and Pellicciari, 2002), it has now become possible to probe the relationships between dopamine, KYNA and NMDA receptor function in greater depth. In a first approach, we used immature rats to examine whether manipulations of KYNA by dopaminomimetic drugs [d-Amph and apomorphine (Apo)] predictably and causally influence neuronal vulnerability to excessive NMDA receptor activation. Our study confirmed the existence of such a connection and revealed a previously unappreciated, KYNA-sensitive component of striatal NMDA neurotoxicity.
EXPERIMENTAL PROCEDURES
Chemicals
KYNA, DL-3-hydroxykynurenine (3-HK), quinolinic acid (QUIN), NMDA, kainic acid, indole-3-propionic acid (IPA), d-Amph and Apo were purchased from Sigma (St. Louis, MO, USA). SCH 23390 and raclopride were obtained from Research Biochemicals International (Natick, MA, USA). CGP 40116 and Ro 61-8048 were generous gifts from Dr. Wolfgang Fröstl (Novartis, Basel, Switzerland). All other chemicals were purchased from various commercial suppliers and were of the highest purity available.
Animals
Female Sprague-Dawley rats with litters were purchased from Charles River Laboratories (Kingston, NY, USA). Pups and mothers were housed in an AAALAC-approved animal facility under a 12h/12h light/dark cycle, and had free access to food and water. One hour prior to experimentation, pups were placed in groups of six in cages that were kept warm with a heating lamp. All experiments were performed using 14 day-old male animals.
Drug administration
With the exception of Ro 61-8048, all drugs were dissolved in phosphate-buffered saline (PBS), pH 7.4, and injected either intraperitoneally (i.p.) or subcutaneously (s.c.). Ro 61-8048 was dissolved in 1% Tween and administered orally (p.o.). Control animals received appropriate vehicle treatments.
Intrastriatal excitotoxin injections
Rats were anesthetized with chloral hydrate (360 mg/kg, i.p.) and placed in a David Kopf strereotaxic frame (Tujunga, CA, USA). NMDA, QUIN or kainic acid were dissolved in 1 N NaOH, and the solutions were titrated to pH 7.4 with 0.1 M phosphate buffer. The excitotoxins were infused unilaterally into the striatum (1 μl over 10 min; coordinates: 0.8 mm anterior to bregma, 2.3 mm from the midline and 4.3 mm below the dura). Control rats received intrastriatal PBS (1 μl ). After surgery, the incision was closed with CrazyGlue®. The pups were kept under a heat lamp until they awakened from anesthesia and were then returned to their mother.
Apomorphine-induced rotations
Behavioral assessment was made 3 days after an intrastriatal excitotoxin injection (Schwarcz et al., 1979). To this end, animals were challenged with Apo (1 mg/kg, s.c.). Ten min later, the rats were placed into an acrylic bowl, and the net number of ipsilateral rotations/5 min was registered. Each rotation was defined as a complete 360° turn.
Quantitation of striatal lesion volume
Animals were killed by decapitation 4 days after an intrastriatal excitotoxin injection, and the brain was rapidly removed and frozen on dry ice. Cryostat sections (30 μm) were then cut coronally through the entire striatum, and one out of every four sections was collected on polylysinated slides and kept at −20°C. The sections were stained using a metal-enhanced cytochrome oxidase method replacing cobalt chloride with nickel ammonium sulfate, as described (Poeggeler et al., 1998). The slides were post-fixed in neutral buffered formaldehyde, dehydrated, and cover-slipped. Lesion volume was quantitated blindly using a video-based image analysis sytem with Loats Image Analyzer software (Silver Spring, MD, USA). Area measurements were multiplied by the intersection distance (120 μm) and summed to determine the lesion volume.
Microscopy
Four days after an intrastriatal excitotoxin injection, animals were deeply anesthetized (chloral hydrate; 500 mg/kg, i.p.) and transcardially perfused with 4% paraformaldehyde in PBS for light microscopy; selected animals were perfused with 4% paraformaldehyde and 1% glutaraldehyde for electron microscopy. Brains used for light microscopy were cryoprotected and cut on a cryostat (30 μm). One series of sections was stained with cresyl violet, and an adjacent series was processed for the immunocytochemical localization of tyrosine hydroxylase (TH) using an antibody to TH (1:1000, Boehringer Mannheim, Germany) and reagents from the avidin biotin peroxidase kit (Vector, Burlingame, CA), using the recommended protocol.
Measurement of KYNA, 3-HK and QUIN
Animals were deeply anesthetized and decapitated, their brains were removed, and striata were dissected out on ice and stored at −80°C. For the determination of KYNA, 3-HK and QUIN, the tissue was thawed, homogenized in water, acidified and centrifuged. Details of this procedure, as well as the analysis of the three metabolites by HPLC with fluorimetric detection (KYNA, 3-HK) or gas chromatography with electron capture negative ionization mass spectrometry (QUIN) were identical to those described by Guidetti et al. (Guidetti et al., 2006). Protein was determined in aliquots of the original tissue homogenate (Lowry et al., 1951).
Data analysis
Data were expressed as the mean ± SEM. All comparisons were assessed using a one-way ANOVA followed by the Bonferroni test for multiple comparisons.
RESULTS
d-Amph administration reduces striat2al KYNA levels and increases neuronal vulnerability to NMDA
I.p. injection of d-Amph caused dose-dependent reductions of striatal KYNA levels within 1 h, reaching 50% of control levels at 5 mg/kg (Fig. 1A). This effect was transient, and KYNA levels returned to control values by 5 h (data not shown). Separate animals received a unilateral intrastriatal injection of NMDA (1.4 nmol/1 μl) 1 or 5 h after the d-Amph or vehicle injection. A subsequent challenge with Apo (1 mg/kg, s.c.) induced ipsilateral rotations in lesioned animals, a behavioral predictor of striatal lesion size (Schwarcz et al., 1979). The number of rotations increased significantly and dose-dependently in animals that had received d-Amph 1 h prior to NMDA (Fig. 1B). As illustrated in Fig. 1C, pre-treatment with d-Amph also caused a significant, dose-dependent increase in the size of NMDA-induced striatal lesions. Animals pre-treated with d-Amph 5 h before the NMDA injection showed no increase in Apo-induced rotations or striatal lesion volume compared to vehicle-pre-treated rats (p > 0.05; n = 6 per group, data not shown,).
Figure 1.
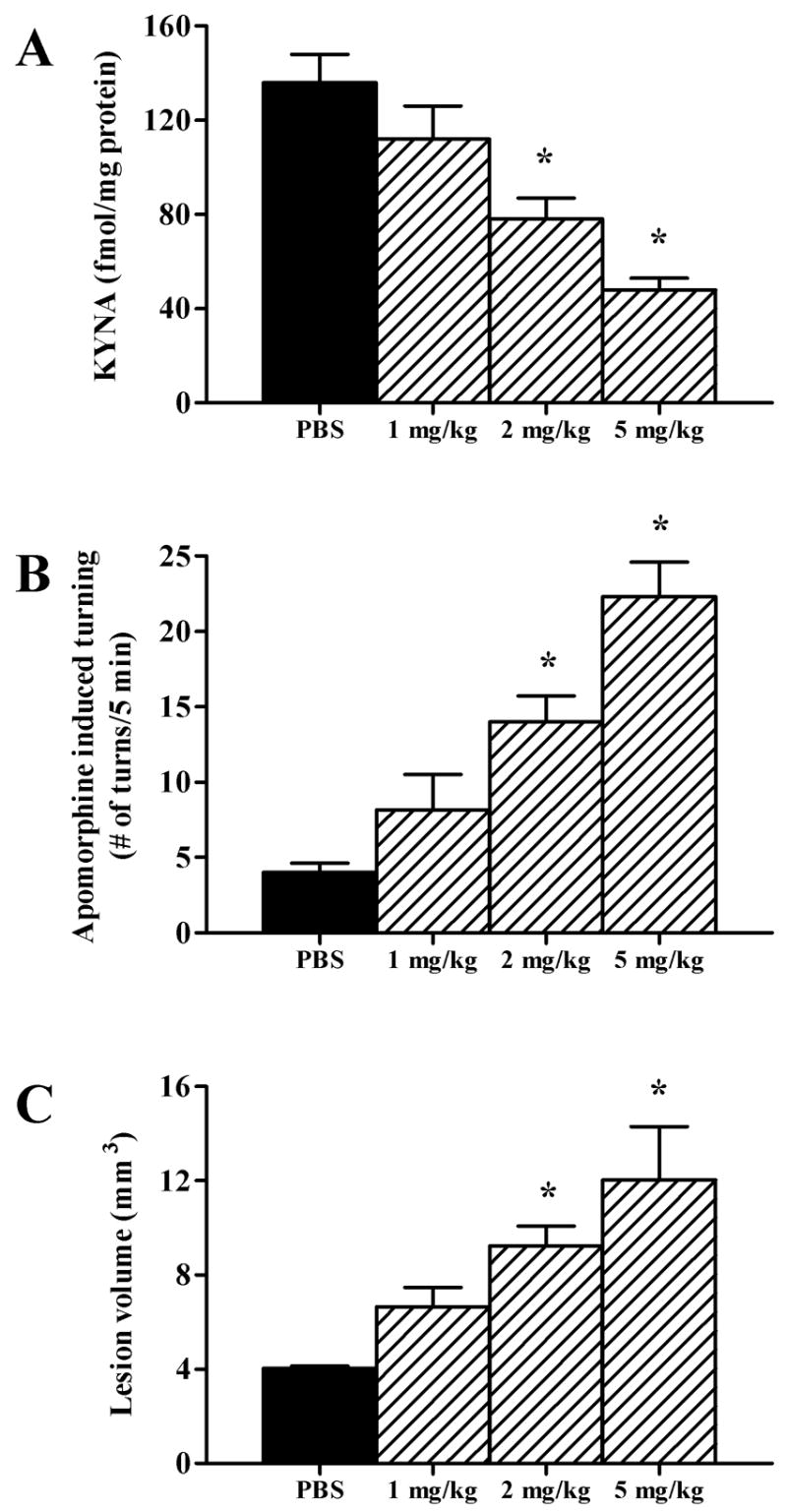
Pretreatment (1 h) with d-Amph dose-dependently decreases striatal KYNA levels and potentiates the striatal neurotoxicity of NMDA (1.4 nmol/1 μl). Controls received i.p. phosphate-buffered saline (PBS) instead of d-Amph. (A) Tissue KYNA content; (B) Behavioral assessment: Apo (1 mg/kg)-induced ipsilateral rotations; (C) Striatal lesion volume in animals tested in B. Data are the mean + SEM (n = 6 per group). *p < 0.05 vs. PBS.
The pro-excitotoxic effect of d-Amph is not associated with damage of dopaminergic afferents
Intrastriatal NMDA injections result in typical excitotoxic neuron loss, sparing axons of extrinsic origin (Guidetti and Schwarcz, 1999). Since d-Amph damages dopaminergic neurons at higher doses (Ryan et al., 1990), we wanted to ascertain that the enlarged NMDA-induced lesion seen in the striatum after d-Amph pre-treatment (Fig. 2A) was not accompanied by injury to afferent fibers originating in the substantia nigra. We therefore examined, in separate groups of animals, the degeneration of intrinsic striatal cells and the integrity of TH-immunoreactive nerve terminals by light and electron microscopy. Nissl-stained tissue sections from vehicle- and d-Amph-pretreated rats showed qualitatively identical features typical of NMDA-induced damage, i.e. a loss of neuronal somata, survival of a small population of large neurons, intact myelinated fiber bundles, and gliosis. Immunocytochemical analyses of adjacent sections revealed a dense network of surviving TH-positive fibers in both groups of animals, even in close vicinity of the track of the injection needle (Fig. 2B). Ultrastructural analysis confirmed the integrity of dopaminergic terminals in both vehicle- and d-Amph-pre-treated rats (micrographs not shown). These studies demonstrated that the characteristic “axon-sparing” nature of the NMDA-induced lesion was maintained in rats pre-treated with d-Amph.
Figure 2.
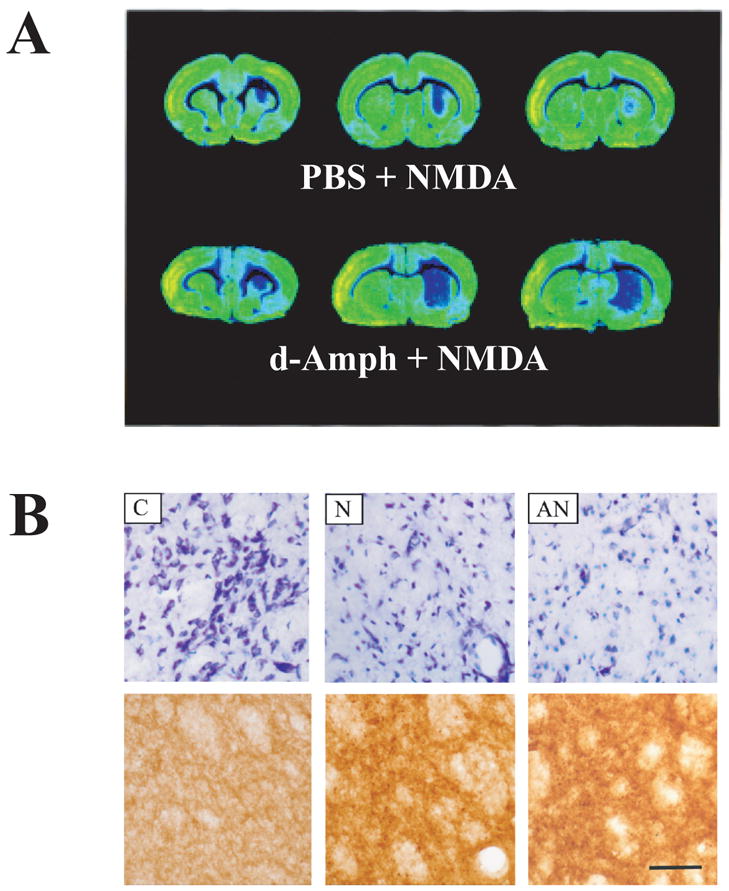
(A) Representative cytochrome oxidase-stained tissue sections from three rostro-caudal levels of the NMDA-lesioned striatum. Sections were taken from animals pretreated with PBS or d-Amph (5 mg/kg). Animals were killed 4 days after the NMDA infusion; (B) Microscopic appearance of the striatum 4 days after an intrastriatal NMDA injection without and with d-Amph pretreatment. Representative cresyl violet (Nissl)-stained (top row) and tyrosine hydroxylase (TH)-stained (bottom row) sections of control (C), NMDA-lesioned (N), and d-Amph + NMDA-lesioned (AN) striata. Photomicrographs of Nissl-stained and TH-immunoreactive tissue were taken from nearby sections. Scenes in N and AN were photographed at the center of the lesioned area. Scale bar: 0.5 μm.
The d-Amph-induced increase in NMDA neurotoxicity is causally related to reduced KYNA levels
The next set of experiments was designed to test whether the pro-excitotoxic effect of d-Amph was caused by the drug-induced reduction in striatal KYNA. To this end, we used 3,4-dimethoxy-N-[4-(3-nitrophenyl)thiazol-2-yl]benzenesulfonamide (Ro 61-8048), a potent inhibitor of kynurenine 3-hydroxylase, which shifts kynurenine pathway metabolism towards increased KYNA formation. Systemic administration of Ro 61-8048 to rats in vivo causes a steady increase in extracellular KYNA concentrations in the brain. KYNA levels peak around 5 h and gradually revert to normal over the next few hours (Röver et al., 1997). Administered at 2 mg/kg (p.o.), we found Ro 61-8048 to cause a modest 50% elevation in striatal KYNA levels by 5 h (Fig. 3A). This increase was insufficient to influence the neurotoxicity of intrastriatally injected NMDA (Figs. 3B,C). When given 5 h before d-Amph (5 mg/kg), however, the same dose of Ro 61-8048 not only counteracted the d-Amph-induced reduction in striatal KYNA (Fig. 3A) but also abolished the pro-excitotoxic effect of d-Amph (Figs. 3B,C). These findings demonstrated that the d-Amph-induced enhancement of NMDA neurotoxicity, i.e. its “pro-excitotoxic” effect, is directly related to the drug’s ability to reduce brain KYNA levels. Moreover, since the administration of Ro 61-8048 prevented only the potentiation of NMDA-induced neurotoxicity by d-Amph but failed to block neurodegeneration induced by NMDA alone, these experiments revealed a heretofore unrecognized, KYNA-dependent component of striatal excitotoxicity.
Figure 3.
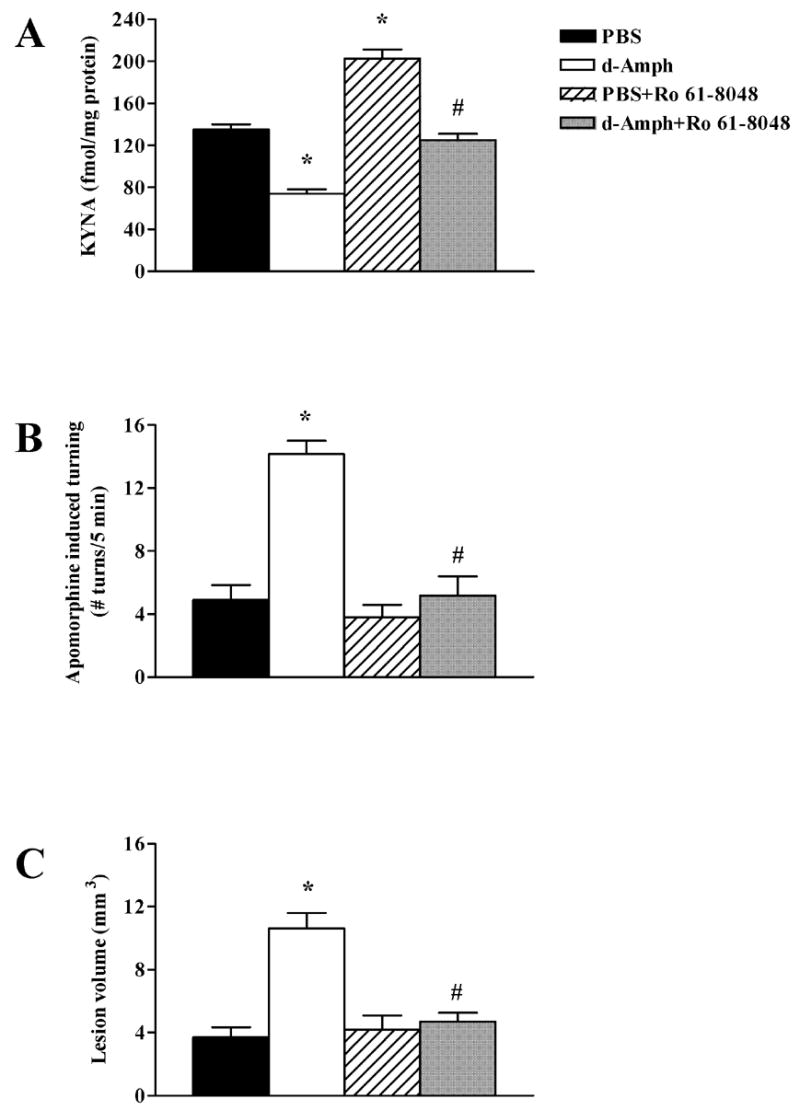
Prevention of d-Amph-induced effects on striatal KYNA levels and NMDA neurotoxicity by pre-treatment with the kynurenine 3-hydroxylase inhibitor Ro 61-8048. Ro 61-8048 (2 mg/kg) was administered 5 h before PBS or d-Amph (5 mg/kg). All animals received an intrastriatal injection of NMDA (1.4 nmol/1 μl) 1 h after PBS or d-Amph. (A) Ro 61-8048 increases striatal KYNA levels and reverses the effect of d-Amph on KYNA levels; (B) Ro 61-8048 prevents the pro-excitotoxic effect of d-Amph assessed behaviorally (Apo-induced rotations); (C) In the same animals tested in B, Ro 61-8048 prevents the pro-excitotoxic effect of d-Amph. Data are the mean + SEM (n = 6 per group). *p < 0.05 versus PBS, #p < 0.05 versus d-Amph.
The pro-excitotoxic effect of d-Amph is unrelated to other kynurenine pathway metabolites
In the brain as in the periphery, KYNA is formed enzymatically by the irreversible transamination of the major tryptophan metabolite L-kynurenine. A separate, competing branch of the degradative kynurenine pathway produces two neurotoxic compounds, the free radical generator 3-HK (Okuda et al., 1996) and the NMDA receptor agonist QUIN (Stone and Perkins, 1981). We therefore examined whether the endogenous levels of 3-HK and/or QUIN in the striatum were affected by the drug regimens used to reveal the pro-excitotoxic effect of d-Amph. However, neither 3-HK nor QUIN levels were significantly influenced 1 h following 5 mg/kg d-Amph or 6 h after 2 mg/kg Ro 61-8048 (each p > 0.05 vs. controls; n = 6 per group, data not shown). Joint pre-treatment with Ro 61-8048 and d-Amph, using the same doses and treatment regimen, and euthanasia 1 h after the d-Amph administration, also left striatal 3-HK and QUIN levels unchanged (p > 0.05 vs. controls; n = 6, data not shown).
To examine the possible role of free radicals in the aggravation of excitotoxicity (Reynolds and Hastings, 1995; Guidetti and Schwarcz, 2003), we attempted to attenuate the pro-excitotoxic effect of d-Amph with the antioxidant IPA (80 mg/kg, i.p., given 15 min before d-Amph), a potent free radical scavenger (Poeggeler et al., 1999). However, IPA administration failed to influence the potentiation of NMDA neurotoxicity by d-Amph (Fig. 4A).
Figure 4.
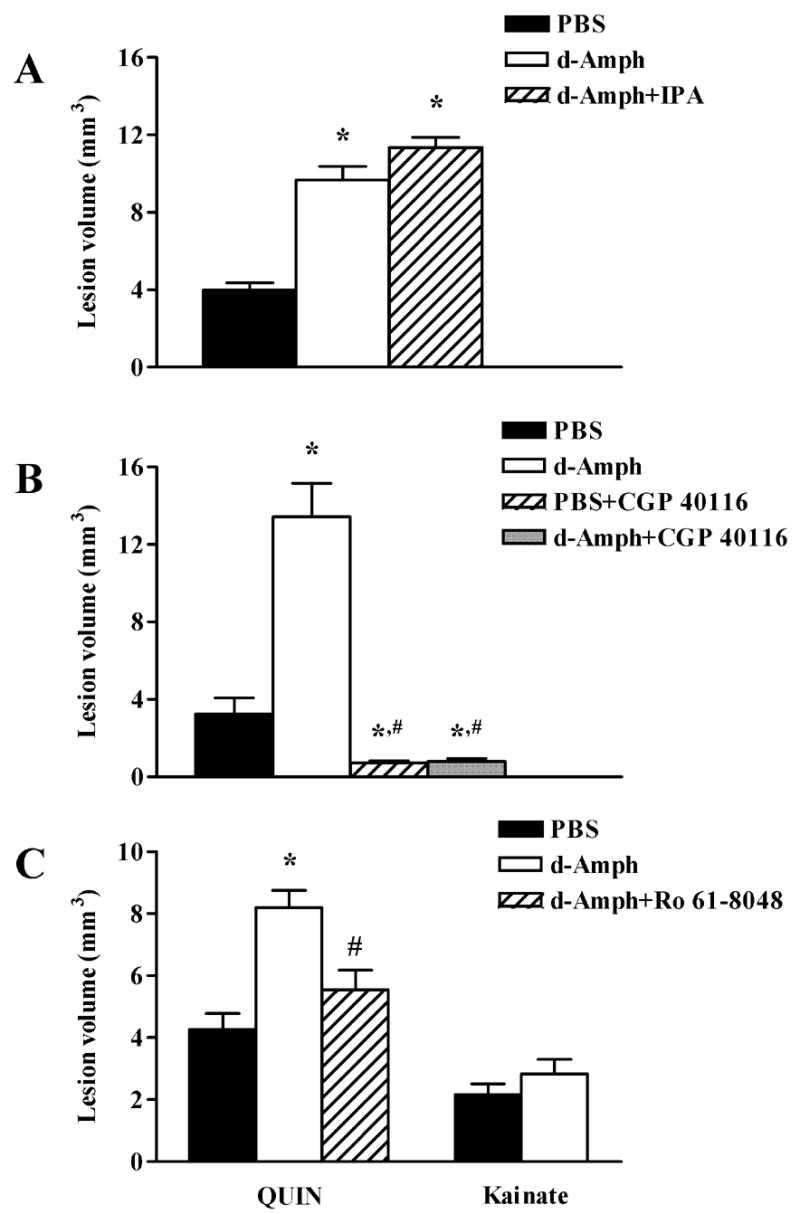
Role of free radicals and NMDA receptor activation in the pro-excitotoxic effect of d-Amph in the striatum. In all animals, excitotoxins (NMDA: 1.4 nmol; QUIN: 90 nmol; kainate: 5 nmol) were injected (1 μl) intrastriatally 1 h after PBS or d-Amph (5 mg/kg). (A) The free radical scavenger IPA (80 mg/kg, i.p., 15 min before d-Amph) fails to prevent the potentiation of NMDA neurotoxicity by d-Amph (p > 0.05 vs. d-Amph); (B) The NMDA receptor antagonist GCP 40116 (20 mg/kg, i.p., 15 min before NMDA) blocks both the neurotoxicity of NMDA alone and the potentiation of NMDA neurotoxicity by d-Amph; (C) d-Amph potentiates QUIN- but not kainate-induced neurotoxicity. Ro 61-8048 (2 mg/kg, 5 h before d-Amph) prevents the potentiation of QUIN neurotoxicity by d-Amph. Data are the mean + SEM (n = 6 per group). *p < 0.05 vs. PBS, #p < 0.05 vs. d-Amph.
Further pharma2cological evaluation of d-Amph’s pro-excitotoxic effect
The specificity of d-Amph’s pro-excitotoxic effect was tested with several distinct pharmacological tools. We first established that the potentiation of NMDA neurotoxicity by 5 mg/kg d-Amph was blocked by the competitive NMDA receptor antagonist CGP 40116 (20 mg/kg, i.p., given 15 min before NMDA; Fig. 4B). As expected (Sauer et al., 1993), CGP 40116 administration also prevented the neurotoxic effects of NMDA alone (Fig. 4B).
In order to test whether the pro-excitotoxic effect of d-Amph extends to NMDA receptor agonists other than NMDA itself, we administered d-Amph (5 mg/kg) 1 h prior to an intrastriatal injection of QUIN (90 nmol/1 μl). As illustrated in Fig. 4C, QUIN-induced neurotoxicity was also potentiated by d-Amph, and, as in the case of NMDA, this lesion enhancement was prevented by pre-treatment with Ro 61-8048 (2 mg/kg). However, no exacerbation by d-Amph was seen when the non-NMDA receptor agonist kainate (5 nmol/1 μl) was used as the toxic agent (Fig. 4C). Taken together, these findings provided evidence that a reduction in KYNA levels selectively enhances vulnerability to NMDA receptor agonists in the striatum.
In order to examine the role of dopamine receptor activation in the pro-excitotoxic effect of d-Amph, animals received an i.p. injection of either the dopamine D1 receptor antagonist SCH 23390 (1 mg/kg) or the D2 receptor antagonist raclopride (2 mg/kg) 15 min before d-Amph (5 mg/kg). As shown in Fig. 5A, both of these treatments prevented the reduction in striatal KYNA levels as well as the increase in NMDA-induced neurotoxicity caused by d-Amph.
Figure 5.
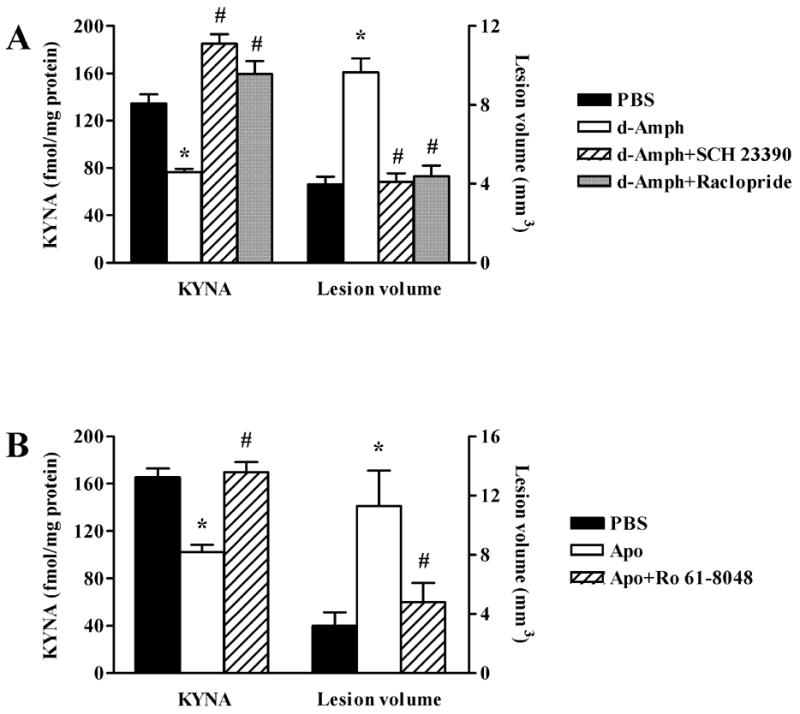
Dopamine receptors mediate the reduction of KYNA and the potentiation of striatal NMDA (1.4 nmol/1 μl) neurotoxicity. (A) Pretreatment with the D1 antagonist SCH 23390 (1 mg/kg) or the D2 antagonist raclopride (2 mg/kg), both administered 15 min before d-Amph (5 mg/kg), prevents the d-Amph-induced reduction of KYNA levels and the potentiation of NMDA neurotoxicity; (B) Dopamine receptor stimulation by Apo (1 mg/kg) causes a reduction in striatal KYNA after 1 h, and potentiates the striatal neurotoxicity of NMDA (injected 1 h after Apo). Ro 61-8048 (2 mg/kg, 5 h before Apo) prevents the potentiation of NMDA neurotoxicity by Apo. Data are the mean + SEM (n = 6 per group). *p < 0.05 versus PBS, #p < 0.05 vs. d-Amph or Apo.
Finally, to investigate the effects of a dopaminomimetic agent other than d-Amph, we performed a study using Apo (1 mg/kg, s.c.), a mixed D1 and D2 dopamine receptor agonist, in the same pre-treatment paradigm used to assess the effect of d-Amph. Apo administration caused a transient reduction in striatal KYNA levels, which was prevented by pre-treatment with Ro 61-8048 (2 mg/kg) (Fig. 5B). Similarly, Apo caused a 3-fold increase in NMDA-induced striatal neurotoxicity, and this pro-excitotoxic effect, like that of d-Amph, was abolished in animals pre-treated with 2 mg/kg Ro 61-8048 (Fig. 5B). These studies provided evidence that the KYNA-sensitive component of striatal NMDA toxicity is causally linked to a dopamine-mediated reduction in endogenous KYNA levels.
DISCUSSION
Using dopaminergic agents as pharmacological probes and assessing NMDA receptor-mediated striatal excitotoxicity as the experimental outcome measure, the present study revealed a novel component of NMDA receptor function, which is normally masked by endogenous KYNA. The relationship between a reduction in endogenous KYNA levels and enhanced vulnerability to an excitotoxic insult was ascertained using the kynurenine 3-hydroxylase inhibitor Ro 61-8048, which diverts kynurenine pathway metabolism towards enhanced KYNA formation. Administration of Ro 61-8048 not only offset the decrease in KYNA levels caused by dopamine receptor stimulation but also prevented the potentiation of neuronal vulnerability seen after d-Amph or Apo pre-treatment. These results indicate that acute fluctuations in KYNA levels in the striatum may be causally involved in NMDA receptor-mediated physiological and pathological events.
The mechanism underlying the reduction in cerebral KYNA levels following the peripheral application of dopaminomimetic drugs such as d-Amph or Apo has not been clarified so far. Using d-Amph as the model compound, we demonstrated previously that this effect is brain-specific, preventable by dopamine receptor antagonists, and especially pronounced in immature animals (Poeggeler et al., 1998; Rassoulpour et al., 1998; cf. Introduction). Another interesting feature of the phenomenon is that the decrease in KYNA levels caused by d-Amph can be prevented by dopamine D1 or D2 receptor antagonists, used here at doses that block the respective receptor subtype selectively (Andersen, 1988; Fig. 5A), and that, conversely, D1 and D2 receptor agonists can individually duplicate the effect of d-Amph (Poeggeler et al., 1998). These unusual pharmacological characteristics do not agree with the conventional view that D1 and D2 receptors have opposing functions in the brain (Greengard, 2001). Instead, they may be related, for example, to the existence of D1-D2 receptor heterooligomers, which exhibit unique functional cooperativity of the two receptor subtypes, albeit more so in older animals (So et al., 2005; Rashid et al., 2007).
The molecular and cellular events linking dopamine receptor activation and the decline in KYNA levels have not been elucidated so far. Since brain KYNA is formed in and released from astrocytes (Kiss et al., 2003), several mechanisms ought to be considered. Possibilities include a role of astrocytic D1 and D2 receptors (Khan et al., 2001; Reuss et al., 2001), intrastriatal neuron-astrocyte signalling or, to evoke a more complex hypothetical scenario, events secondary to extrastriatal dopamine receptor activation. These alternatives are currently being studied in our laboratory.
KYNA inhibits the glutamate binding site of the NMDA receptor competitively with an IC50 of approximately 200 μM (Ganong and Cotman, 1986) and has a similar, low affinity to non-NMDA receptors (Leeson et al., 1991). Inhibition of these sites accounts for the widely used classification of KYNA as a “broad spectrum” antagonist of excitatory amino acid receptors and explains why the local application of millimolar concentrations of KYNA effectively blocks both NMDA- and kainate-mediated excitotoxicity (Foster et al., 1984). As shown here in the case of NMDA toxicity, complete neuroprotection can also be achieved by peripherally administered drugs such as the potent and specific NMDA receptor antagonist CGP 40116 (Sauer et al., 1993; Fig. 4B).
Although superficially similar, these pharmacological effects are not particularly informative when considering the function and possible pathophysiological significance of endogenous KYNA in the brain. Thus, both the tissue and the extracellular concentration of KYNA in the mammalian brain range from low to high nanomolar (Moroni et al., 1988; Turski et al., 1988; Swartz et al., 1990), and even significant surges in these endogenous levels are insufficient to antagonize massive insults caused, for example, by focal injections of NMDA (Fig. 3; cf. also Obrenovitch and Urenjak, 2000).
The levels of KYNA normally found in the brain have long been assumed to preferentially affect the glycine co-agonist (“glycineB”) site of the NMDA receptor, which is inhibited by the metabolite with an IC50 of ~10 μM (Kessler et al., 1989; Parsons et al., 1997). In order to inhibit this site, however, KYNA must successfully compete with the more abundant and potent endogenous glycineB receptor agonists glycine and D-serine. This makes it more likely that another receptor, the α7nAChR, constitutes the primary target of KYNA in vivo. KYNA is a non-competitive α7nAChR antagonist and, importantly, shows significant inhibition of the receptor at nanomolar concentrations (Hilmas et al., 2001).
In the striatum as in other brain regions, α7nAChRs are preferentially localized presynaptically on glutamatergic nerve terminals to regulate glutamate release and function (Kaiser and Wonnacott, 2000; Marchi et al., 2002; Rousseau et al., 2005). Inhibition of these receptors by KYNA reduces extracellular glutamate levels (Carpenedo et al., 2001; Rassoulpour et al., 2005; Grilli et al., 2006). Preliminary evidence indicates that reductions in KYNA, conversely, enhance glutamatergic function by disinhibiting α7nAChRs (H.-Q. Wu and R. Schwarcz, unpublished observations). In other words, currently available data suggest that fluctuations in endogenous KYNA levels, via α7nAChRs, tonically regulate glutamate release.We assume that this mechanism underlies the novel, KYNA-sensitive component of striatal excitotoxicity described here. In particular, we propose that increased glutamate release, secondary to a dopamine-induced reduction in brain KYNA levels, accounts for the potentiation of NMDA receptor-mediated excitotoxicity. This hypothesis, which is currently being tested further using selective α7nAChR ligands, is in line with the observation that glutamate can exacerbate NMDA and QUIN neurotoxicity in the striatum (Orlando et al., 2001), and that mutant mice with a prolonged reduction in cerebral KYNA levels show increased susceptibility to an intrastriatal QUIN injection (Sapko et al., 2006). Notably, this proposition is also in agreement with the fact that the reduction in KYNA by d-Amph does not potentiate neuronal vulnerability to kainate (Fig. 4C). Thus, glutamate excitotoxicity is primarily mediated by NMDA, rather than non-NMDA, receptors (Choi et al., 1988; Monnerie et al., 2003), and KYNA-deficient mice fail to demonstrate enhanced striatal kainate toxicity (Sapko et al., 2006).
Our study included additional experiments, which were in agreement with this hypothesis. Since high doses of d-Amph damage dopaminergic neurons (Ryan et al., 1990), we first tested the integrity of afferent dopaminergic fibers to ascertain that the “axon-sparing” feature characteristic of excitotoxic neurodegeneration (Olney, 1982) was preserved in the enlarged lesion. This was verified unequivocally using TH immunocytochemistry on the light and electron microscopic level (Fig. 2). We also examined if the d-Amph-induced reduction in KYNA levels was accompanied by an increased production of 3-HK and/or QUIN in the competing branch of the kynurenine pathway. By forming reactive free radicals and acting synergistically (Eastman and Guilarte, 1990; Guidetti and Schwarcz, 1999; Santamaria et al., 2001), a rapid up-regulation of 3-HK or QUIN synthesis could exacerbate NMDA receptor-mediated neurotoxicity, providing a simple alternative explanation for the potentiation of lesion size by d-Amph. However, both 3-HK and QUIN levels in the striatum were unaffected by d-Amph, and the free radical scavenger IPA failed to attenuate the enhanced neurotoxicity. Finally, we corroborated that Ro 61-8048, at a dose used to prevent the pro-excitotoxic effect of d-Amph, did not interfere with the ability of d-Amph to induce striatal dopamine release (H.-Q. Wu and R. Schwarcz, unpublished observations). The neuroprotective potency of Ro 61-8048, illustrated in Fig. 3, was therefore unrelated to the elevation of extracellular dopamine levels triggered by d-Amph.
The present study raises the possibility that (a reduction in) KYNA might be critically involved in the potentiation of neuronal vulnerability that occurs as a consequence of impaired cellular energy metabolism. Thus, chemicals that interfere with glycolysis or mitochondrial function, such as iodoacetate, malonate or 3-nitropropionic acid, substantially inhibit KYNA formation and also facilitate NMDA receptor-mediated excitotoxic neurodegeneration (Fiskum et al., 1999; Schwarcz et al., 1999; Massieu et al., 2000). The neurodegenerative properties of these metabolic inhibitors are especially pronounced in the basal ganglia, possibly due to the additional toxic effect of dopamine, which is highly concentrated in these brain areas. Based mostly on studies in animal models, dopaminergic activity is believed to be enhanced during the early stages of striatal injury in Huntington’s disease, hypoxia-ischemia and severe hypoglycemia (Jakel and Maragos, 2000). In these pathological conditions, the heightened vulnerability of NMDA receptor-bearing striatal neurons may therefore be a consequence of a reduction in striatal KYNA levels secondary to metabolic insults as well as excessive dopaminergic function.
In line with the emerging concept that astrocytes participate in the control of excitatory neurotransmission (Haydon, 2001; Schell, 2004), we demonstrated here that an acute reduction in the physiological levels of KYNA influences NMDA receptors in the striatum. By controlling NMDA receptors, endogenous KYNA may fulfill a previously unrecognized role in striatal pathology (Coyle, 2006). However, a decrease in brain KYNA levels is not necessarily harmful. Thus, a reduction in brain KYNA formation may well be physiologically and therapeutically advantageous when an enhancement of NMDA receptor function is desirable (Pittaluga et al., 1997; Carroll and Zukin, 2002; Coyle and Tsai, 2004; Nakazawa et al., 2004).
Acknowledgments
We thank Drs. C. Ceresoli-Borroni, P.S. Hodgkins and C. Kiss for useful discussions and Dr. J. Bartko for assistance with the statistical analyses. This work was supported by NIH grants HD16596 and MH66123.
Abbreviations
- Apo
Apomorphine
- d-Amph
d-Amphetamine
- 3-HK
3-hydroxykynurenine
- IPA
indole-3-propionic acid
- KYNA
Kynurenic acid
- QUIN
Quinolinic acid
- TH
Tyrosine hydroxylase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson PH. Comparison of the pharmacological characteristics of [3H]raclopride and [3H]SCH 23390 binding to dopamine receptors in vivo in mouse brain. Eur J Pharmacol. 1988;146:113–120. doi: 10.1016/0014-2999(88)90492-x. [DOI] [PubMed] [Google Scholar]
- Becquet D, Faudon M, Hery F. In vivo evidence for an inhibitory glutamatergic control of serotonin release in the cat caudate nucleus: involvement of GABA neurons. Brain Res. 1990;519:82–88. doi: 10.1016/0006-8993(90)90063-h. [DOI] [PubMed] [Google Scholar]
- Bernard V, Bolam JP. Subcellular and subsynaptic distribution of the NR1 subunit of the NMDA receptor in the neostriatum and globus pallidus of the rat: co-localization at synapses with the GluR2/3 subunit of the AMPA receptor. Eur J Neurosci. 1998;10:3721–3736. doi: 10.1046/j.1460-9568.1998.00380.x. [DOI] [PubMed] [Google Scholar]
- Buisson A, Pateau V, Plotkine M, Boulu RG. Nigrostriatal pathway modulates striatum vulnerability to quinolinic acid. Neurosci Lett. 1991;131:257–259. doi: 10.1016/0304-3940(91)90627-6. [DOI] [PubMed] [Google Scholar]
- Carpenedo R, Pittaluga A, Cozzi A, Attucci S, Galli A, Raiteri M, Moroni F. Presynaptic kynurenate-sensitive receptors inhibit glutamate release. Eur J Neurosci. 2001;13:2141–2147. doi: 10.1046/j.0953-816x.2001.01592.x. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Zukin RS. NMDA-receptor trafficking and targeting: implications for synaptic transmission and plasticity. Trends Neurosci. 2002;25:571–577. doi: 10.1016/s0166-2236(02)02272-5. [DOI] [PubMed] [Google Scholar]
- Centonze D, Usiello A, Costa C, Picconi B, Erbs E, Bernardi G, Borrelli E, Calabresi P. Chronic haloperidol promotes corticostriatal long-term potentiation by targeting dopamine D2L receptors. J Neurosci. 2004;24:8214–8222. doi: 10.1523/JNEUROSCI.1274-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Koh JY, Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;8:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle J, Tsai G. The NMDA receptor glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psychopharmacology (Berl) 2004;174:32–38. doi: 10.1007/s00213-003-1709-2. [DOI] [PubMed] [Google Scholar]
- Coyle JT. Glial metabolites of tryptophan and excitotoxicity: Coming unglued. Exp Neurol. 2006;197:4–7. doi: 10.1016/j.expneurol.2005.10.021. [DOI] [PubMed] [Google Scholar]
- Dang MT, Yokoi F, Yin HH, Lovinger DM, Wang Y, Li Y. Disrupted motor learning and long-term synaptic plasticity in mice lacking NMDAR1 in the striatum. Proc Natl Acad Sci U S A. 2006;103:15254–15259. doi: 10.1073/pnas.0601758103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, Standaert DG. Subcellular segregation of distinct heteromeric NMDA glutamate receptors in the striatum. J Neurochem. 2003;85:935–943. doi: 10.1046/j.1471-4159.2003.01744.x. [DOI] [PubMed] [Google Scholar]
- Eastman CL, Guilarte TR. The role of hydrogen peroxide in the in vitro cytotoxicity of 3-hydroxykynurenine. Neurochem Res. 1990;15:1101–1107. doi: 10.1007/BF01101711. [DOI] [PubMed] [Google Scholar]
- Fan MM, Raymond LA. Prog Neurobiol. 2006. N-Methyl-d-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab. 1999;19:351–369. doi: 10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- Foster AC, Vezzani A, French ED, Schwarcz R. Kynurenic acid blocks neurotoxicity and seizures induced in rats by the related brain metabolite quinolinic acid. Neurosci Lett. 1984;48:273–278. doi: 10.1016/0304-3940(84)90050-8. [DOI] [PubMed] [Google Scholar]
- Galvan A, Kuwajima M, Smith Y. Glutamate and GABA receptors and transporters in the basal ganglia: what does their subsynaptic localization reveal about their function? Neuroscience. 2006;143:351–375. doi: 10.1016/j.neuroscience.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganong AH, Cotman CW. Kynurenic acid and quinolinic acid act at N-methyl-D-aspartate receptors in the rat hippocampus. J Pharmacol Exp Ther. 1986;236:293–299. [PubMed] [Google Scholar]
- Gardoni F, Picconi B, Ghiglieri V, Polli F, Bagetta V, Bernardi G, Cattabeni F, Di Luca M, Calabresi P. A critical interaction between NR2B and MAGUK in L-DOPA induced dyskinesia. J Neurosci. 2006;26:2914–2922. doi: 10.1523/JNEUROSCI.5326-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P. The neurobiology of dopamine signaling. Biosci Rep. 2001;21:247–269. doi: 10.1023/a:1013205230142. [DOI] [PubMed] [Google Scholar]
- Grilli M, Raiteri L, Patti L, Parodi M, Robino F, Raiteri M, Marchi M. Modulation of the function of presynaptic alpha7 and non-alpha7 nicotinic receptors by the tryptophan metabolites, 5-hydroxyindole and kynurenate in mouse brain. Br J Pharmacol. 2006;149:724–732. doi: 10.1038/sj.bjp.0706914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti P, Bates GP, Graham RK, Hayden MR, Leavitt BR, MacDonald ME, Slow EJ, Wheeler VC, Woodman B, Schwarcz R. Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol Dis. 2006;23:190–197. doi: 10.1016/j.nbd.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Schwarcz R. 3-Hydroxykynurenine potentiates quinolinate but not NMDA toxicity in the rat striatum. Eur J Neurosci. 1999;11:3857–3863. doi: 10.1046/j.1460-9568.1999.00806.x. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Schwarcz R. 3-Hydroxykynurenine and quinolinate: pathogenic synergism in early grade Huntington’s disease? In: Allegri G, et al., editors. Developments in tryptophan and serotonin metabolism. New York: Kluwer Academic Publishers; 2003. pp. 137–145. [DOI] [PubMed] [Google Scholar]
- Hallett PJ, Dunah AW, Ravenscroft P, Zhou S, Bezard E, Crossman AR, Brotchie JM, Standaert DG. Alterations of striatal NMDA receptor subunits associated with the development of dyskinesia in the MPTP-lesioned primate model of Parkinson’s disease. Neuropharmacology. 2005;48:503–516. doi: 10.1016/j.neuropharm.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Harris CA, Miranda AF, Tanguay JJ, Boegman RJ, Beninger RJ, Jhamandas K. Modulation of striatal quinolinate neurotoxicity by elevation of endogenous brain kynurenic acid. Br J Pharmacol. 1998;124:391–399. doi: 10.1038/sj.bjp.0701834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hathway GJ, Humphrey PP, Kendrick KM. Somatostatin release by glutamate in vivo is primarily regulated by AMPA receptors. Br J Pharmacol. 2001;134:1155–1158. doi: 10.1038/sj.bjp.0704362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon PG. Glia: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- Hilmas C, Pereira EF, Alkondon M, Rassoulpour A, Schwarcz R, Albuquerque EX. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: physiopathological implications. J Neurosci. 2001;21:7463–7473. doi: 10.1523/JNEUROSCI.21-19-07463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakel RJ, Maragos WF. Neuronal cell death in Huntington’s disease: a potential role for dopamine. Trends Neurosci. 2000;23:239–245. doi: 10.1016/s0166-2236(00)01568-x. [DOI] [PubMed] [Google Scholar]
- Kaiser S, Wonnacott S. alpha-bungarotoxin-sensitive nicotinic receptors indirectly modulate [(3)H]dopamine release in rat striatal slices via glutamate release. Mol Pharmacol. 2000;58:312–318. doi: 10.1124/mol.58.2.312. [DOI] [PubMed] [Google Scholar]
- Kessler M, Terramani T, Lynch G, Baudry M. A glycine site associated with N-methyl-D-aspartic acid receptors: characterization and identification of a new class of antagonists. J Neurochem. 1989;52:1319–1328. doi: 10.1111/j.1471-4159.1989.tb01881.x. [DOI] [PubMed] [Google Scholar]
- Khan ZU, Koulen P, Rubinstein M, Grandy DK, Goldman-Rakic PS. An astroglia-linked dopamine D2-receptor action in prefrontal cortex. Proc Natl Acad Sci U S A. 2001;98:1964–1969. doi: 10.1073/pnas.98.4.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss C, Ceresoli-Borroni G, Guidetti P, Zielke CL, Zielke HR, Schwarcz R. Kynurenate production by cultured human astrocytes. J Neural Transm. 2003;110:1–14. doi: 10.1007/s00702-002-0770-z. [DOI] [PubMed] [Google Scholar]
- Knauber J, Kischka U, Roth M, Schmidt WJ, Hennerici M, Fassbender K. Modulation of striatal acetylcholine concentrations by NMDA and the competitive NMDA receptor-antagonist AP-5: an in vivo microdialysis study. J Neural Transm. 1999;106:35–45. doi: 10.1007/s007020050139. [DOI] [PubMed] [Google Scholar]
- Leeson PD, Baker R, Carling RW, Curtis NR, Moore KW, Williams BJ, Foster AC, Donald AE, Kemp JA, Marshall GR. Kynurenic acid derivatives. Structure-activity relationships for excitatory amino acid antagonism and identification of potent and selective antagonists at the glycine site on the N-methyl-D-aspartate receptor. J Med Chem. 1991;34:1243–1252. doi: 10.1021/jm00108a002. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M. Direct evidence that release-stimulating alpha7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem. 2002;80:1071–1078. doi: 10.1046/j.0022-3042.2002.00805.x. [DOI] [PubMed] [Google Scholar]
- Marti M, Manzalini M, Fantin M, Bianchi C, Della Corte L, Morari M. Striatal glutamate release evoked in vivo by NMDA is dependent upon ongoing neuronal activity in the substantia nigra, endogenous striatal substance P and dopamine. J Neurochem. 2005;93:195–205. doi: 10.1111/j.1471-4159.2005.03015.x. [DOI] [PubMed] [Google Scholar]
- Martineau M, Baux G, Mothet JP. D-serine signalling in the brain: friend and foe. Trends Neurosci. 2006;29:481–491. doi: 10.1016/j.tins.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Massieu L, Gomez-Roman N, Montiel T. In vivo potentiation of glutamate-mediated neuronal damage after chronic administration of the glycolysis inhibitor iodoacetate. Exp Neurol. 2000;165:257–267. doi: 10.1006/exnr.2000.7481. [DOI] [PubMed] [Google Scholar]
- Meldrum A, Dunnett SB, Everitt BJ. Role of corticostriatal and nigrostriatal inputs in malonate-induced striatal toxicity. Neuroreport. 2001;12:89–93. doi: 10.1097/00001756-200101220-00025. [DOI] [PubMed] [Google Scholar]
- Moncada S, Bolanos JP. Nitric oxide, cell bioenergetics and neurodegeneration. J Neurochem. 2006;97:1676–1689. doi: 10.1111/j.1471-4159.2006.03988.x. [DOI] [PubMed] [Google Scholar]
- Monnerie H, Shashidhara S, Le Roux PD. Effect of excess extracellular glutamate on dendrite growth from cerebral cortical neurons at 3 days in vitro: Involvement of NMDA receptors. J Neurosci Res. 2003;74:688–700. doi: 10.1002/jnr.10797. [DOI] [PubMed] [Google Scholar]
- Moroni F, Russi P, Lombardi G, Beni M, Carla V. Presence of kynurenic acid in the mammalian brain. J Neurochem. 1988;51:177–180. doi: 10.1111/j.1471-4159.1988.tb04852.x. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci. 2004;5:361–372. doi: 10.1038/nrn1385. [DOI] [PubMed] [Google Scholar]
- Nishikawa T. A molecular pharmacological approach to the vulnerability to schizophrenia. Biol Psychiatry. 2005;57:145S. [Google Scholar]
- Obrenovitch TP, Urenjak J. In vivo assessment of kynurenate neuroprotective potency and quinolinate excitotoxicity. Amino Acids. 2000;19:299–309. doi: 10.1007/s007260070061. [DOI] [PubMed] [Google Scholar]
- Okuda S, Nishiyama N, Saito H, Katsuki H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc Natl Acad Sci U S A. 1996;93:12553–12558. doi: 10.1073/pnas.93.22.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW. The toxic effects of glutamate and related compounds in the retina and the brain. Retina. 1982;2:341–359. [PubMed] [Google Scholar]
- Orlando LR, Alsdorf SA, Penney JB, Jr, Young AB. The role of group I and group II metabotropic glutamate receptors in modulation of striatal NMDA and quinolinic acid toxicity. Exp Neurol. 2001;167:196–204. doi: 10.1006/exnr.2000.7542. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Danysz W, Quack G, Hartmann S, Lorenz B, Wollenburg C, Baran L, Przegalinski E, Kostowski W, Krzascik P, Chizh B, Headley PM. Novel systemically active antagonists of the glycine site of the N-methyl-D-aspartate receptor: electrophysiological, biochemical and behavioral characterization. J Pharmacol Exp Ther. 1997;283:1264–1275. [PubMed] [Google Scholar]
- Pittaluga A, Vaccari D, Raiteri M. The "kynurenate test", a biochemical assay for putative cognition enhancers. J Pharmacol Exp Ther. 1997;283:82–90. [PubMed] [Google Scholar]
- Poeggeler B, Pappolla MA, Hardeland R, Rassoulpour A, Hodgkins PS, Guidetti P, Schwarcz R. Indole-3-propionate: a potent hydroxyl radical scavenger in rat brain. Brain Res. 1999;815:382–388. doi: 10.1016/s0006-8993(98)01027-0. [DOI] [PubMed] [Google Scholar]
- Poeggeler B, Rassoulpour A, Guidetti P, Wu HQ, Schwarcz R. Dopaminergic control of kynurenate levels and N-methyl-D-aspartate toxicity in the developing rat striatum. Dev Neurosci. 1998;20:146–153. doi: 10.1159/000017309. [DOI] [PubMed] [Google Scholar]
- Radke JM, Owens MJ, Nemeroff CB. The effects of glutamate receptor agonists on neurotensin release using in vivo microdialysis. Eur J Pharmacol. 2001;411:129–134. doi: 10.1016/s0014-2999(00)00912-2. [DOI] [PubMed] [Google Scholar]
- Rashid AJ, So CH, Kong MM, Furtak T, El-Ghundi M, Cheng R, O’Dowd BF, George SR. D1–D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc Natl Acad Sci U S A. 2007;104:654–659. doi: 10.1073/pnas.0604049104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassoulpour A, Wu HQ, Ferré S, Schwarcz R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J Neurochem. 2005;93:762–765. doi: 10.1111/j.1471-4159.2005.03134.x. [DOI] [PubMed] [Google Scholar]
- Rassoulpour A, Wu HQ, Poeggeler B, Schwarcz R. Systemic d-amphetamine administration causes a reduction of kynurenic acid levels in rat brain. Brain Res. 1998;802:111–118. doi: 10.1016/s0006-8993(98)00577-0. [DOI] [PubMed] [Google Scholar]
- Reuss B, Lorenzen A, Unsicker K. Dopamine and epinephrine, but not serotonin, downregulate dopamine sensitivity in cultured cortical and striatal astroglial cells. Receptors Channels. 2001;7:441–451. [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau SJ, Jones IW, Pullar IA, Wonnacott S. Presynaptic alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate [3H]d-aspartate release from rat frontal cortex in vitro. Neuropharmacology. 2005;49:59–72. doi: 10.1016/j.neuropharm.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Röver S, Cesura AM, Huguenin P, Kettler R, Szente A. Synthesis and biochemical evaluation of N-(4-phenylthiazol-2-yl)benzenesulfonamides as high-affinity inhibitors of kynurenine 3-hydroxylase. J Med Chem. 1997;40:4378–4385. doi: 10.1021/jm970467t. [DOI] [PubMed] [Google Scholar]
- Ryan LJ, Linder JC, Martone ME, Groves PM. Histological and ultrastructural evidence that D-amphetamine causes degeneration in neostriatum and frontal cortex of rats. Brain Res. 1990;518:67–77. doi: 10.1016/0006-8993(90)90955-b. [DOI] [PubMed] [Google Scholar]
- Santamaria A, Jimenez-Capdeville ME, Camacho A, Rodriguez-Martinez E, Flores A, Galvan-Arzate S. In vivo hydroxyl radical formation after quinolinic acid infusion into rat corpus striatum. Neuroreport. 2001;12:2693–2696. doi: 10.1097/00001756-200108280-00020. [DOI] [PubMed] [Google Scholar]
- Sapko MT, Guidetti P, Yu P, Tagle DA, Pellicciari R, Schwarcz R. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: Implications for Huntington’s disease. Exp Neurol. 2006;197:31–40. doi: 10.1016/j.expneurol.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Sauer D, Allegrini PR, Cosenti A, Pataki A, Amacker H, Fagg GE. Characterization of the cerebroprotective efficacy of the competitive NMDA receptor antagonist CGP40116 in a rat model of focal cerebral ischemia: an in vivo magnetic resonance imaging study. J Cereb Blood Flow Metab. 1993;13:595–602. doi: 10.1038/jcbfm.1993.77. [DOI] [PubMed] [Google Scholar]
- Schell MJ. The N-methyl D-aspartate receptor glycine site and D-serine metabolism: an evolutionary perspective. Philos Trans R Soc Lond B Biol Sci. 2004;359:943–964. doi: 10.1098/rstb.2003.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrattenholz A, Soskic V. NMDA receptors are not alone: dynamic regulation of NMDA receptor structure and function by neuregulins and transient cholesterol-rich membrane domains leads to disease-specific nuances of glutamate-signalling. Curr Top Med Chem. 2006;6:663–686. doi: 10.2174/156802606776894519. [DOI] [PubMed] [Google Scholar]
- Schwarcz R. The kynurenine pathway of tryptophan degradation as a drug target. Curr Opin Pharmacol. 2004;4:12–17. doi: 10.1016/j.coph.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Ceresoli-Borroni G, Wu HQ, Rassoulpour A, Poeggeler B, Hodgkins PS, Guidetti P. Modulation and function of kynurenic acid in the immature rat brain. In: Huether G, et al., editors. Tryptophan, Serotonin, Melatonin - Basic Aspects and Applications. New York: Plenum; 1999. pp. 113–123. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Fuxe K, Agnati LF, Hokfelt T, Coyle JT. Rotational behaviour in rats with unilateral striatal kainic acid lesions: a behavioural model for studies on intact dopamine receptors. Brain Res. 1979;170:485–495. doi: 10.1016/0006-8993(79)90966-1. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Pellicciari R. Manipulation of brain kynurenines: glial targets, neuronal effects, and clinical opportunities. J Pharmacol Exp Ther. 2002;303:1–10. doi: 10.1124/jpet.102.034439. [DOI] [PubMed] [Google Scholar]
- So CH, Varghese G, Curley KJ, Kong MM, Alijaniaram M, Ji X, Nguyen T, O’Dowd BF, George SR. D1 and D2 dopamine receptors form heterooligomers and cointernalize after selective activation of either receptor. Mol Pharmacol. 2005;68:568–578. doi: 10.1124/mol.105.012229. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Albers DS, Zeevalk GD. Role of glutamate in neurodegeneration of dopamine neurons in several animal models of parkinsonism. Amino Acids. 1998;14:69–74. doi: 10.1007/BF01345245. [DOI] [PubMed] [Google Scholar]
- Stone TW, Perkins MN. Quinolinic acid: a potent endogenous excitant at amino acid receptors in CNS. Eur J Pharmacol. 1981;72:411–412. doi: 10.1016/0014-2999(81)90587-2. [DOI] [PubMed] [Google Scholar]
- Swartz KJ, Matson WR, MacGarvey U, Ryan EA, Beal MF. Measurement of kynurenic acid in mammalian brain extracts and cerebrospinal fluid by high-performance liquid chromatography with fluorometric and coulometric electrode array detection. Anal Biochem. 1990;185:363–376. doi: 10.1016/0003-2697(90)90309-w. [DOI] [PubMed] [Google Scholar]
- Turski WA, Nakamura M, Todd WP, Carpenter BK, Whetsell WO, Jr, Schwarcz R. Identification and quantification of kynurenic acid in human brain tissue. Brain Res. 1988;454:164–169. doi: 10.1016/0006-8993(88)90815-3. [DOI] [PubMed] [Google Scholar]
- Wang H, Pickel VM. Presence of NMDA-type glutamate receptors in cingulate corticostriatal terminals and their postsynaptic targets. Synapse. 2000;35:300–310. doi: 10.1002/(SICI)1098-2396(20000315)35:4<300::AID-SYN8>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Waxman EA, Lynch DR. N-methyl-D-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist. 2005;11:37–49. doi: 10.1177/1073858404269012. [DOI] [PubMed] [Google Scholar]
- Young AM, Bradford HF. N-methyl-D-aspartate releases gamma-aminobutyric acid from rat striatum in vivo: a microdialysis study using a novel preloading method. J Neurochem. 1993;60:487–492. doi: 10.1111/j.1471-4159.1993.tb03176.x. [DOI] [PubMed] [Google Scholar]