

Abstract
We show here that infection of macrophages with murine γ-herpesvirus 68 (γHV68) actively induces H2AX phosphorylation (γH2AX), a key proximal step in the cellular DNA damage response pathway. Such infection-associated DNA damage signals have been considered as a host cell response to replicating viral DNA. However, we found that H2AX phosphorylation requires expression of the γHV68 encoded orf36 kinase, and is enhanced by ATM. γHV68 orf36 and its EBV homolog BGLF4 induce H2AX phosphorylation independent of other viral genes. Orf36-induced phosphorylation of H2AX in cells, and of recombinant H2AX in vitro, requires an intact kinase domain. Orf36 is important for γHV68 replication in infected animals, and orf36, H2AX, and ATM are all critical for efficient γHV68 replication in primary macrophages. Thus activation of proximal components of the DNA damage signaling response is an active viral kinase-driven strategy required for efficient γ-herpesvirus replication.
INTRODUCTION
Viruses have evolved elegant approaches to optimally use limited genetic capacity to manipulate the intracellular environment. One common viral strategy is to usurp cellular signaling pathways to create an optimal environment for viral replication. The DNA damage signaling system utilizes a series of ordered protein-protein interactions and modifications to detect and respond to lesions in the genetic material of the cell (Sancar et al., 2004; Rouse and Jackson, 2002). DNA damage can be generated during physiologic processes and by extracellular agents such as ionizing radiation and chemical mutagens. Initiation of the cellular DNA damage response results in the activation and rapid recruitment of proteins required for repair to the DNA damage site. In addition, cell cycle checkpoints are activated that prevent the cell from transitioning to the next stage of the cell cycle until the damage is repaired. Failure to repair the damaged DNA ultimately leads to death of the cell.
The ATM serine-threonine kinase is activated early in the response to DNA double strand breaks (McGowan and Russell, 2004; Barlow et al., 1996). One ATM substrate is the ubiquitously expressed histone protein H2AX. Phosphorylation of H2AX by ATM occurs exclusively at the site of the DNA double strand breaks (Burma et al., 2001). How phosphorylated H2AX (γH2AX) functions in the DNA damage response is not completely known, although it is thought to be involved in the recruitment and/or retention of repair factors at the site of DNA damage and possibly in the amplification of DNA damage signals (Celeste et al., 2003; Celeste et al., 2002; Paull et al., 2000; Bassing et al., 2002). The notion that H2AX is important for the DNA damage response is supported by the genomic instability and tumor predisposition associated with H2AX deficiency (Celeste et al., 2002; Bassing et al., 2002).
Murine γ-herpesvirus 68 (γHV68) is genetically and biologically related to human and simian γ-herpesviruses, such as Epstein-Barr virus (EBV), Kaposi Sarcoma-associated herpesvirus (KSHV), and Herpesvirus saimiri (Efstathiou et al., 1990; Virgin et al., 1997). While there are individual variations in the pathogenesis and cell tropism of these viruses, all γ-herpesviruses display tropism for lymphoid and/or myeloid cells of the host (Flano et al., 2000; Sunil-Chandra et al., 1992; Weck et al., 1999; Willer and Speck, 2003) and are associated with development of lymphoproliferative diseases (Damania, 2004; Jung et al., 1999; Young and Rickinson, 2004; Tarakanova et al., 2005). Study of γHV68 allows integrated in vivo and in vitro analysis of fundamental aspects of γ-herpesvirus biology in a facile experimental system.
Herpesvirus genomes are linear DNA molecules that circularize upon entry into the cell (Poffenberger and Roizman, 1985). The circular form of viral DNA can serve as a template for rolling circle type of replication with viral genome concatemers resolved by yet unidentified endonuclease(s) (Boehmer and Nimonkar, 2003). Replication of viral DNA is associated with increased levels of linear DNA molecules or abnormal DNA structures within the cell nucleus that could be recognized by cellular sensors of DNA damage and trigger DNA damage signaling. Indeed, DNA damage signaling is triggered upon infection with DNA viruses, including adenovirus, polyoma virus, and herpesviruses, including herpes simplex virus, human CMV, and Epstein-Barr virus (Gaspar and Shenk, 2006; Kudoh et al., 2005; Dahl et al., 2005; Shirata et al., 2005; Stracker et al., 2002). Whether this response requires the presence of viral DNA, viral DNA replication, or is an active viral strategy due to viral protein expression is unknown. Moreover, the impact of DNA damage responses on virus infection is not fully understood. DNA damage response is antiviral in case of human adenovirus infection, and is inhibited by adenovirus-encoded E4-orf6 and E1B-55K proteins (Stracker et al., 2002). For other viruses the consequences of DNA damage signaling are less well defined. For example, both beneficial and detrimental effects of DNA damage and repair proteins for the viral life cycle have been shown for HSV-1 (Lilley et al., 2005; Taylor and Knipe, 2004).
In this study we provide the mechanism of DNA damage signaling induction as well as its physiological relevance for murine γHV68 infection. We found that γHV68 infection leads to phosphorylation of H2AX in vitro and in vivo. Furthermore, using transposon and recombination-based mutagenesis of the viral genome, we identified and confirmed a requirement for a γHV68 kinase, encoded by orf36, for H2AX phosphorylation in γHV68-infected primary macrophages. The ability to increase H2AX phosphorylation is conserved between γHV68 and EBV kinases. Optimal orf36-mediated H2AX phosphorylation is dependent on ATM activity, demonstrating a critical synergistic interplay between viral and cellular encoded proteins in mediating this process. Proximal events of the DNA damage signaling are physiologically relevant since the DNA damage sensor H2AX, the DNA damage mediator kinase ATM, and orf36 kinase all facilitate efficient γHV68 replication in primary macrophages. Together these data define an active virus-driven mechanism of DNA damage response induction in infected cells that creates an optimal environment for viral replication in a physiologically relevant primary cell type.
RESULTS
γHV68 infection leads to H2AX phosphorylation in vivo
To determine whether proximal events in DNA damage signaling are initiated during γHV68 infection in vivo, H2AX phosphorylation was examined in spleens of γHV68-infected mice. To optimize the number of infected cells in histopathologic sections of organs, we infected STAT1 deficient mice as STAT1 deficiency allows for robust acute replication and improved detection of γHV68-infected cells. The profound immunodeficiency of these mice has been presumed to be due to decreased interferon-dependent viral resistance, although it has also been reported that DNA damage signaling downstream of H2AX phosphorylation may be altered in the absence of STAT1 (Townsend et al., 2005). STAT1 deficient mice were mock-infected or infected with 106 plaque forming units (PFU) of γHV68 and spleens were harvested three days later for immunohistochemical analysis. Spleens from infected mice contained numerous cells staining specifically with polyclonal anti γHV68 antibody (Fig 1A) indicating widespread γHV68 infection. Spleens from infected animals also had a marked increase in H2AX phosphorylation as compared to spleens from mock-infected animals (Fig. 1C, D), indicating that proximal events in DNA damage signaling are induced in vivo by infection with γHV68.
Figure 1. H2AX phosphorylation is increased upon γHV68 infection in vivo.
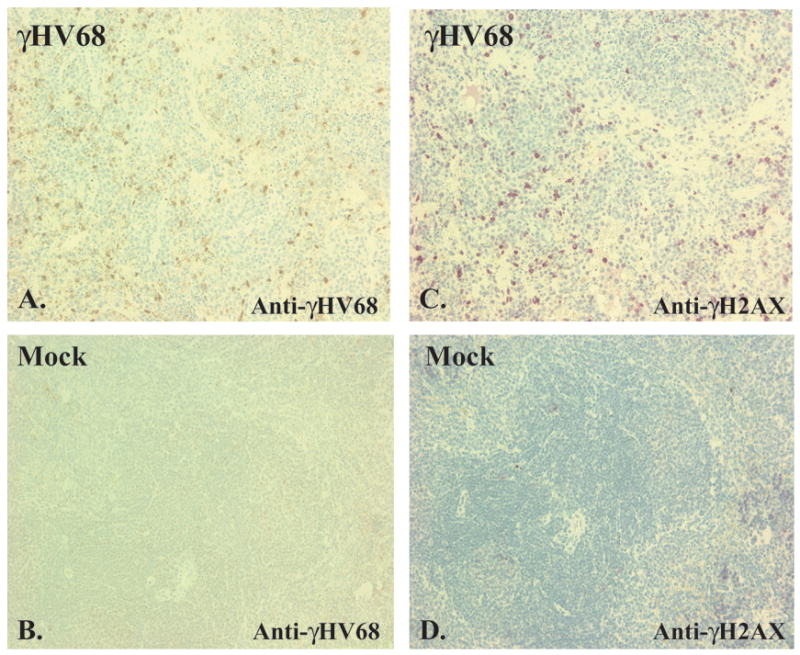
STAT1 deficient mice were inoculated intraperitoneally with 106 PFU of γHV68 or carrier (mock). At three days post infections, spleens were harvested, fixed, and subjected to immunohistochemistry using a polyclonal anti-γHV68 antibody (A, B) or anti-γH2AX antibody (C, D).
Infection of primary bone marrow macrophages with γHV68 induces phosphorylation of H2AX
We next sought to determine whether γHV68 infection induces H2AX phosphorylation in cultured cells. Macrophages are a critical cell type for γHV68 infection and pathogenesis (Weck et al., 1999), and primary bone marrow derived macrophages are permissive for γHV68 infection in culture. We therefore used γHV68 infection of primary macrophages that have functional STAT1 to study the mechanism and significance of γHV68-induced initiation of DNA damage responses. For primary macrophage infections we utilized virus derived from the parental bacterial artificial chromosome used for signature tagged mutagenesis (STM BAC derived γHV68) as a wild type γHV68 control (Song et al., 2005).
γHV68 infection of primary bone marrow derived macrophages isolated from wild type mice induced a robust increase in H2AX phosphorylation starting 24 hours post infection and persisting for up to 72 hours (Fig. 2A). In contrast to the punctate pattern of γH2AX staining reported in irradiated cells (Paull et al., 2000), γH2AX staining in γHV68-infected macrophages had a diffuse nuclear pattern (Fig 2E). An increase in H2AX phosphorylation was also observed in mouse embryonic fibroblasts (MEFs) infected with γHV68 as early as 4 hours post infection, and infection with UV inactivated viral stock failed to increase γH2AX levels, demonstrating that viral gene expression was required for increased H2AX phosphorylation (Supplemental Fig. 1A). Interestingly, the increase in etoposide-induced H2AX phosphorylation in primary macrophages was lower than that observed in primary fibroblasts (data not shown). An increase in phosphorylation of serine 1981 of ATM was also observed in γHV68-infected macrophages indicating that DNA damage signaling was initiated by γHV68 (Fig. 2F). Together, these findings demonstrate that γHV68 infection increased H2AX phosphorylation and triggered proximal events of DNA damage signaling in primary bone marrow macrophages and fibroblasts in vitro.
Figure 2. γHV68 infection increases H2AX phosphorylation in primary bone marrow macrophages in a viral kinase-dependent manner.
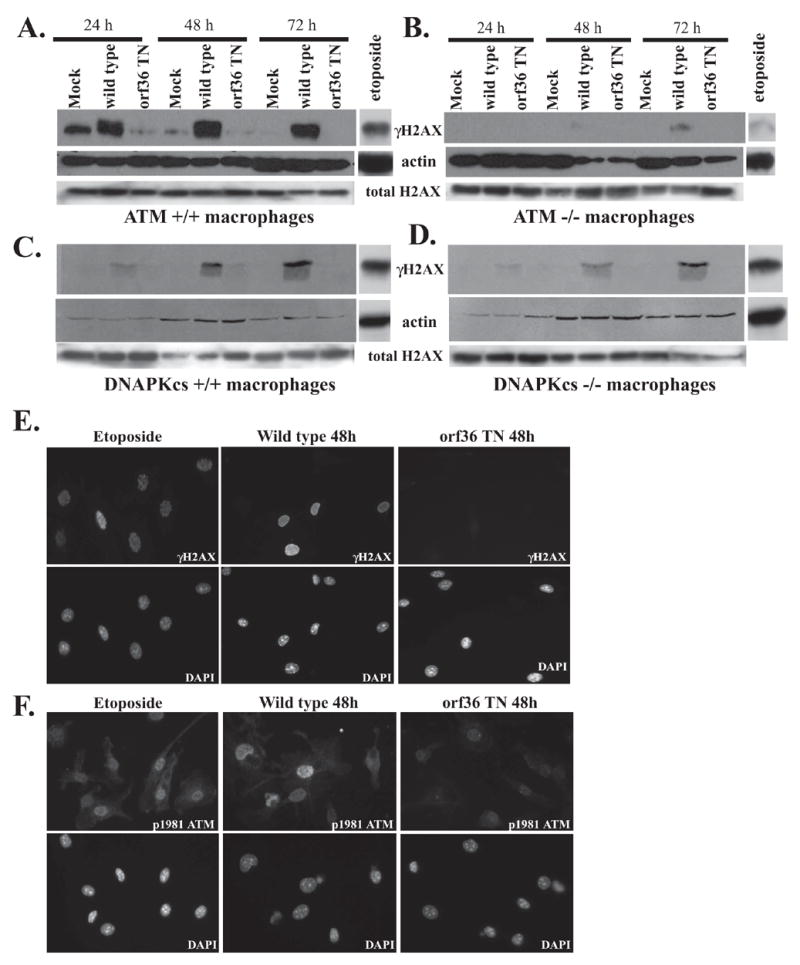
Primary bone marrow derived macrophages were infected at 10 PFU/cell with wild type γHV68 or a γHV68 mutant with disruption of the orf36 gene (orf36 TN). Lysates were collected at indicated times and analyzed by western using anti γH2AX, anti total H2AX, and anti-β-actin antibodies (A–D) or cells were subjected to immunoflourescence using anti-γH2AX (E) or anti-phospho serine 1981 of ATM (F) and counterstained with DAPI. As a positive control, macrophages were treated with 60 μM etoposide for 6 h prior to fixation or lysis.
H2AX phosphorylation is an active viral process requiring expression of a viral kinase
To answer the fundamental question of what initiates DNA damage responses in γHV68 infected cells, we used a library of γHV68 mutants generated by signature-tagged mutagenesis of the γHV68 viral genome as a bacterial artificial chromosome (Song et al., 2005) to test the hypothesis that a specific viral gene is responsible for increase of H2AX phosphorylation. A γHV68 mutant virus was identified that was incapable of increasing H2AX phosphorylation in primary macrophages. This mutant harbored a transposon insertion in viral orf36, a gene that is conserved throughout the γ-herpesvirus family and is predicted to encode a viral kinase (Virgin et al., 1997). Infection with the orf36 TN γHV68 mutant failed to increase H2AX phosphorylation in wild type primary macrophages (Fig 2A). Total H2AX levels remained unchanged throughout the infection (Fig. 2A–D; Supplemental Fig. 1B, C).
We considered that decreased H2AX phosphorylation observed after infection with orf36 TN was due to an inability of this virus to infect macrophages. However, when the expression of viral proteins was compared in cells infected with wild type versus orf36 TN virus, newly synthesized viral proteins were detected in both cases at comparable levels early after infection (Supplemental Fig. 1B, C). At later times after infection, orf36 was important for expression of some viral proteins. In addition, we confirmed that orf36 was required for efficient phosphorylation of H2AX in γHV68 infected MEFs (Supplemental Fig. 1D), where orf36 is not required for viral replication (Supplemental Fig. 3A, B). Together these data show that the transposon-mediated mutagenesis of γHV68 orf36 generates a virus that induces lower levels of H2AX phosphorylation than wild type virus, and that this decrease cannot be attributed to differences in infection or replication between wild-type and orf36 mutant viruses.
The inability of the orf36 TN mutant to increase H2AX phosphorylation in infected primary macrophages was recapitulated upon infection with a γHV68 mutant engineered to have three translational stops in the orf36 gene (orf36.Stop1, Supplemental Fig. 2A). Furthermore, other γHV68 mutants containing the STM transposon at different locations in the viral genome induced phosphorylation of H2AX normally (Supplemental Fig. 2C and data not shown). Together, these data demonstrate that the failure of the orf36 TN γHV68 mutant to induce phosphorylation of H2AX is due to the lack of orf36-encoded protein rather than a secondary effect of transposon-mediated mutagenesis.
We next considered whether the decrease in H2AX phosphorylation observed in orf36 TN-infected macrophages was due to decreased viral DNA replication in cells infected with the mutant virus. To this end we examined H2AX phosphorylation in macrophages infected with wild type virus and treated with cidofovir, an inhibitor of viral DNA replication, at a concentration 125-fold higher than the inhibitory dose 50% for γHV68 replication (Neyts and De, 1998). As expected, addition of cidofovir decreased levels of γHV68 late proteins in both wild type and orf36 TN-infected cells (Supplemental Fig. 2B). Orf36 was required for induction of H2AX phosphorylation when viral DNA synthesis was inhibited by cidofovir. However, orf36-dependent H2AX phosphorylation in the presence of cidofovir was about 3.5 fold lower as compared to infected untreated cells (Supplemental Fig. 2B). Decreased H2AX phosphorylation seen in cidofovir treated macrophages may suggest that viral DNA synthesis contributes to H2AX phosphorylation in infected macrophages. It is also possible that lower level of H2AX phosphorylation in the presence of cidofovir is due to decreased expression of orf36, which has been identified as an early-late gene and whose full expression requires viral DNA synthesis (Martinez-Guzman et al., 2003).
ATM is required for efficient H2AX phosphorylation in γHV68-infected primary macrophages
ATM is one of the major kinases responsible for H2AX phosphorylation in response to DNA damage (Burma et al., 2001). To determine whether ATM is required for phosphorylation of H2AX upon γHV68 infection, we assayed for γH2AX in ATM deficient macrophages infected with wild type γHV68. Importantly, the orf36 TN γHV68 mutant was incapable of increasing H2AX phosphorylation in ATM−/− macrophages upon infection (Fig. 2B). H2AX phosphorylation was decreased in wild type γHV68-infected ATM −/− macrophages, indicating that ATM is required for efficient orf36-induced phosphorylation of H2AX throughout γHV68 infection (Fig. 2A and B). The catalytic subunit of the DNAPK complex, DNAPKcs, can also phosphorylate H2AX in response to DNA damage (Park et al., 2003). However, macrophages from SCID mice, which have a mutated non-functional DNAPKcs protein (Blunt et al., 1995), did not exhibit a decrease in H2AX phosphorylation until late time in infection (72 h, Fig. 2C and D, see quantitation in Supplemental Fig. 2D). Thus, efficient phosphorylation of H2AX throughout γHV68 infection requires the orf36 kinase, and is enhanced by ATM function.
γHV68 orf36 kinase is sufficient to increase H2AX phosphorylation independent of γHV68 infection
To determine whether γHV68 orf36 kinase is sufficient to elevate H2AX phosphorylation levels independent of other viral genes, a Flag-tagged version of γHV68 orf36 was transiently expressed in 3T12 mouse fibroblast cells. Transient expression of γHV68 orf36 kinase resulted in robust phosphorylation of H2AX (Fig. 3A), indicating that the expression of this viral gene alone is sufficient to induce phosphorylation of the DNA damage sensor H2AX, and confirming that viral DNA replication is not required. Generation of orf36-specific antibodies will be required to compare the efficiency of orf36-induced H2AX phosphorylation between infected and transfected cells.
Figure 3. γHV68 orf36 increases H2AX phosphorylation independent of viral infection.
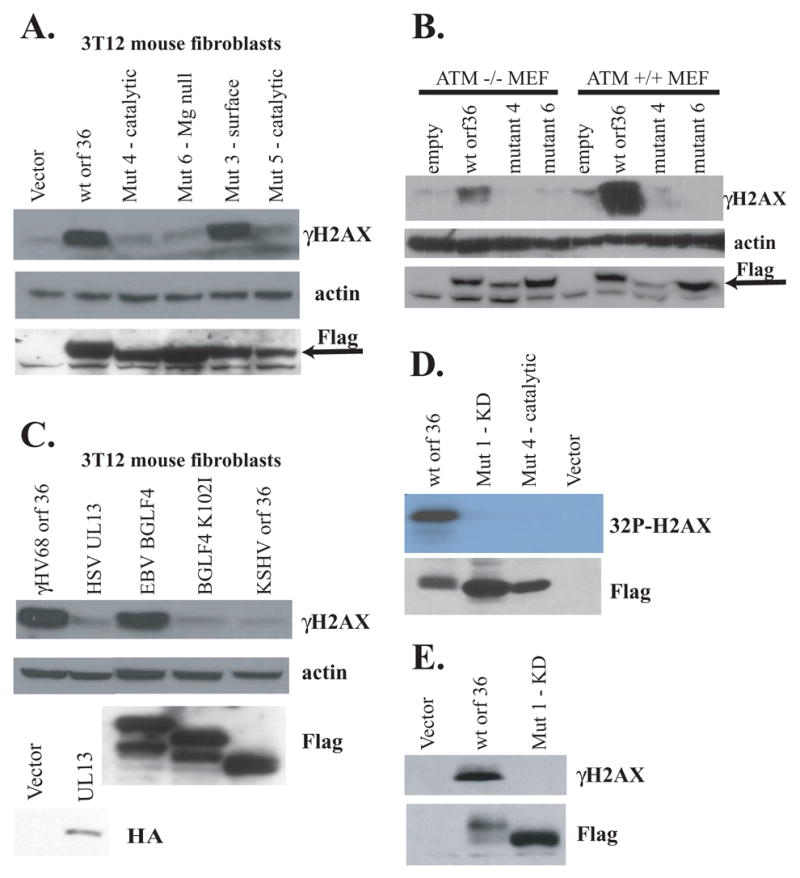
3T12 mouse fibroblasts (A, C) or primary MEFs (B) were transfected with indicated plasmids, and γH2AX levels were analyzed 36 h later. γHV68 orf36, its mutants, EBV BGLF4, and KSHV orf36, were detected with anti-flag antibody (specific band is denoted by an arrow). HSV-2 UL13 was detected with an anti-HA antibody. D, E. HeLa cells were transiently transfected with indicated constructs, expressed proteins were immunoprecipitated with anti-Flag, and incubated with recombinant H2AX in the presence of nucleotides plus γ32P ATP (D) or cold nucleotides (E). In vitro reactions were resolved by gel electrophoresis and exposed to film (D) or subjected to western analysis using anti-γH2AX antibody (E).
To test the requirement for orf36 kinase activity for H2AX phosphorylation, we engineered point mutants of γHV68 orf36. Several amino acids, including a conserved lysine residue, are essential for the kinase activity of serine/threonine type kinases (Krupa et al., 2004; Kim et al., 2005). These residues, important for ATP positioning, catalytic activity, and Mg+2 binding, are highly conserved among herpesvirus kinases and mouse protein kinase A, a serine/threonine protein kinase. Therefore, we created several point mutants affecting positional homologs of these critical amino acids in γHV68 orf36 (Table 1). Orf36 mutants harboring deleterious mutations in the kinase domain failed to significantly increase H2AX phosphorylation when transfected into fibroblast cells (Fig 3A). A control mutant with amino acid substitutions predicted to be on the external surface of the molecule and thus to be outside of the kinase active domain (Mutant 3) increased phosphorylation of H2AX similar to wild type orf36 (Fig. 3A). These data strongly suggest that the kinase activity of orf36 is important for H2AX phosphorylation in transfected fibroblasts.
Table 1. orf36 mutants.
| Name of orf36 mutant | Amino acids mutated | Predicted location of mutation* |
|---|---|---|
| Mutant 1 | K108 to Q | ATP positioning |
| Mutant 3 | DGL to AAA (a.a.185–187) | External surface |
| Mutant 4 | D200 and V201 to A | Catalytic loop |
| Mutant 5 | N205 and I206 to A | Catalytic loop |
| Mutant 6 | D220 to A | Mg positioning |
Predictions were made based on alignments and information in references (Kim et al., 2005; Krupa et al., 2004)
To test the requirement for ATM in orf36-induced H2AX phosphorylation, wild type orf36 and the indicated orf36 mutants were transiently expressed in MEFs isolated from ATM −/− or littermate control mice (Fig. 3B). Similar to infection of ATM −/− macrophages, transiently expressed orf36 stimulated H2AX phosphorylation in a kinase domain-dependent manner in the absence of ATM (Fig. 3B). Expression of orf36 induced lower levels of H2AX phosphorylation in ATM−/− than in wild type cells, suggesting that while ATM is dispensable for H2AX phosphorylation in orf36-expressing cells, ATM functions to amplify orf36-dependent H2AX phosphorylation.
The ability of γHV68 orf36 to induce H2AX phosphorylation is shared by Epstein Barr virus kinase BGLF4
Since the orf36 gene is conserved among γ-herpesviruses, including human Epstein-Barr virus, we asked whether the homologous EBV kinase is capable of inducing H2AX phosphorylation. Upon transient transfection, the EBV viral kinase BGLF4 increased H2AX phosphorylation (Fig. 3C). Similar to γHV68 orf36, the kinase activity of EBV BGLF4 was required for H2AX phosphorylation since the kinase dead K102I mutant of BGLF4 failed to increase γH2AX levels above background (Fig. 3C). Interestingly, kinase active versions of viral kinases migrated at a higher molecular weight than their kinase dead counterparts, suggesting that these kinases may autophosphorylate. Expression of a phylogenetically more distant UL13 kinase of an alpha herpesvirus (HSV2) failed to induce phosphorylation of H2AX in mouse fibroblasts upon transient transfection (Fig. 3C). Expression of KSHV orf36 failed to induce H2AX phosphorylation (Fig. 3C). Together these data show that both human EBV and murine γ-herpesvirus encoded orf36 type kinases can induce phosphorylation of H2AX independent of other viral genes in a kinase domain-dependent fashion, but that this activity is not a property of all herpesvirus kinases.
Orf36 kinase induces H2AX phosphorylation in vitro
To test whether γHV68 orf36 can induce phosphorylation of H2AX in vitro, transiently expressed Flag-tagged wild type or kinase domain mutant orf36 was immunoprecipitated from HeLa cells. We used immunoprecipitated orf36 since we have been unable to express orf36 protein in soluble form in bacterial cells. Incubation of orf36 immune complexes with recombinant H2AX and γ-32P-labelled ATP induced robust phosphorylation of recombinant H2AX in vitro (Fig 3D). In contrast, orf36 mutants 1 and 4 (Table 1) assayed in a similar manner did not phosphorylate H2AX (Fig. 3D) above background levels, confirming the requirement for kinase activity for H2AX phosphorylation. The orf36 kinase assay does not define the specific activity of the viral protein for H2AX phosphorylation. These data are most consistent with H2AX being a direct substrate of orf36 kinase. We cannot rule out the alternative possibility that phosphorylation of H2AX in vitro is mediated by a cellular kinase that associates with or is activated by wild type, but not kinase dead, orf36.
To test the specificity of orf36-dependent phosphorylation of H2AX in vitro, immunoprecipitated wild type or mutant 1 orf36 kinase was incubated with recombinant H2AX in the presence of ATP. The resulting products were probed by western blot with a phosphoserine 139-specific H2AX (γH2AX) antibody. Serine 139 phosphorylated recombinant H2AX was present in reactions containing wild type but not orf36 mutant 1 (Fig. 3E), indicating that orf36-dependent in vitro phosphorylation of H2AX required both an active kinase domain and was directed to serine 139 of H2AX, a residue involved in H2AX-associated DNA damage responses (Lu et al., 2006; Burma et al., 2001; Park et al., 2003).
Orf36 is required for efficient replication of γHV68 in vivo and in primary macrophages in vitro
To determine the role of orf36 viral kinase in replication of γHV68 in vivo, wild type BL6 mice were infected with orf36 TN or wild type γHV68, and viral titers were measured in spleen, liver, and lung at 4 days post infection. Replication of the orf36 TN mutant was detected in spleens of infected animals at levels 1000-fold lower (p=0.0011) than those of control virus (Fig. 4A). In liver (Fig. 4B) and lung (data not shown) orf36 TN titers were below the level of detection, representing >30 fold (p=0.0112) and >11-fold (p=0.008) lower replication, respectively. Thus, disruption of the orf36 viral kinase gene significantly decreases γHV68 replication in vivo.
Figure 4. γHV68 orf36 is required for viral replication in vivo and γHV68 growth in primary macrophages in vitro.
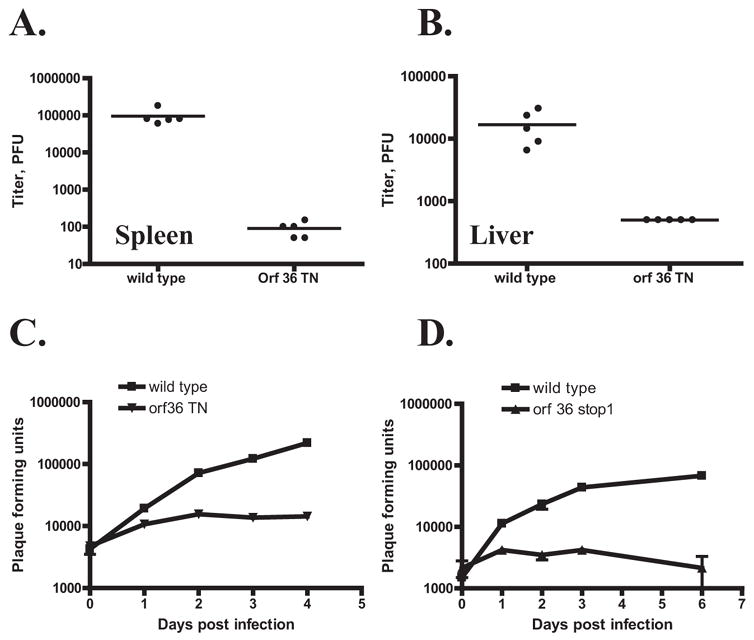
A, B. BL6 mice were intraperitoneally infected with 106 PFU of orf36 TN γHV68 mutant or a wild type γHV68. Acute titers were measured in spleens (A) and livers (B) at 4 days post infection. Each symbol represents an individual mouse. C, D. BL6 primary bone marrow macrophages were infected at 10 PFU/cell with wild type or orf36 TN mutant γHV68 (C). For D, the wild type control is the parental virus used to make orf36.stop1 γHV68 mutant. Viral yields were determined at indicated times. Each time point was measured in triplicate with average and standard error of measurement shown. A representative experiment of at least three repeats is shown in C, D.
We sought to correlate these striking deficiencies in replication in vivo with replication in primary cells. Replication of orf36 TN mutant in mouse embryonic fibroblasts was indistinguishable from replication of a wild type γHV68 at both high (10 PFU/cell, Supplemental fig. 3A) and low (0.01 PFU/cell, Supplemental fig. 3B) multiplicities of infection. However, orf36 TN mutant was attenuated for growth in primary macrophages over a range of multiplicities of infection from high (10 PFU/cell) to low (0.1 PFU/cell) (Fig 4C, data not shown). This same attenuation was observed upon infection of primary macrophages with γHV68 mutant engineered to have translational stops in orf36 gene (Fig 4D, orf36 stop 1), indicating that attenuation of the growth of the orf36 TN mutant in macrophages is due to a lack of the orf36-encoded viral kinase.
ATM and H2AX are required for efficient γHV68 replication in primary mouse macrophages
The above data are consistent with a model in which orf36 kinase-mediated phosphorylation of H2AX is important for optimal viral replication in primary macrophages but not fibroblasts. This predicts that ATM, which synergizes with orf36 in induction of H2AX phosphorylation, and H2AX itself, are important for γHV68 replication in physiologically relevant cells.
To test this hypothesis, primary bone marrow macrophages derived from mice lacking H2AX (Bassing et al., 2002) or ATM (Barlow et al., 1996) and littermate controls were infected with γHV68. At a low multiplicity of infection, replication of wild type γHV68 was attenuated in H2AX −/− macrophages (Fig 5A), indicating that H2AX is required for efficient γHV68 replication in primary macrophages. Similarly, replication of wild type γHV68 in ATM deficient macrophages was significantly reduced compared to wild type cells (Fig. 5B). Attenuation of γHV68 growth at low multiplicities of infection was specific for ATM and H2AX deficiencies, as genetic deficiency of DNA-PK catalytic subunit in primary macrophages did not affect γHV68 replication at either low or high multiplicity of infection (Fig. 5C, supplemental figure 3C). Differences in titer at t=0 are attributable to batch-to-batch and mouse background variations in the growth of γHV68 in primary macrophages. These data indicate that H2AX and ATM but not the DNA-PK catalytic subunit are required for efficient viral growth when the viral inoculum is limiting.
Figure 5. ATM and H2AX are required for efficient γHV68 replication in primary macrophages.
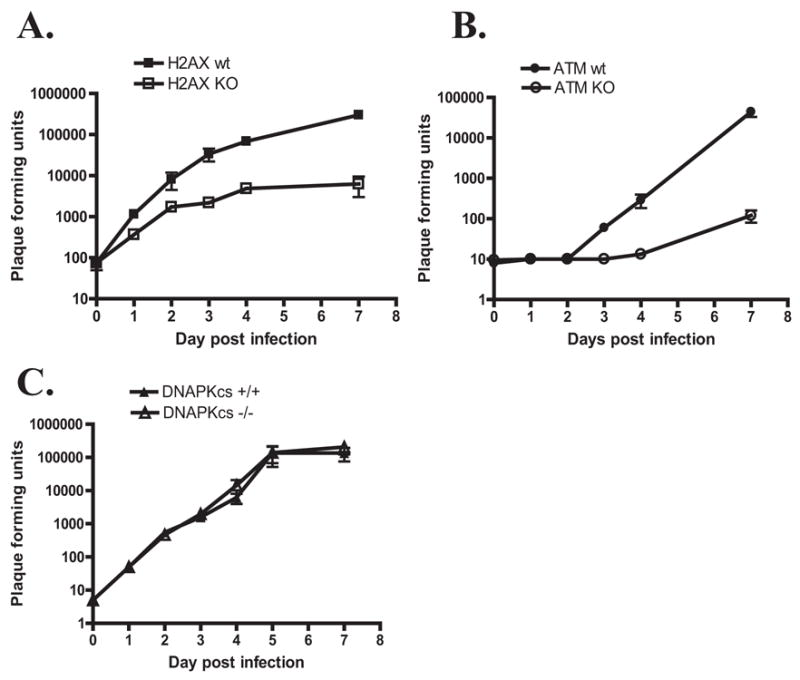
Primary macrophages were derived from mice with indicated genotypes, infected in vitro at 0.01 PFU/cell with wild type γHV68 and virus yield was measured on indicated days post infection. Each time point was measured in triplicate with average and standard error of measurement shown. A representative experiment of at least three repeats is shown.
Many herpesvirus mutants can show growth deficits after low MOI infection that can be compensated for by increasing the viral inoculum. We found that, neither ATM nor H2AX were required for efficient growth when a high MOI was used to infect macrophages (Supplemental figure 3D, E). These data show that the genetic defects in ATM or H2AX did not render macrophages incapable of supporting γHV68 replication. Furthermore, neither ATM nor H2AX nor orf36 kinase were required for γHV68 growth in MEFs regardless of whether a high MOI or a low MOI was used to infect the cells (data not shown). These data indicate that there are cell type-specific requirements for γHV68 replication and the function of the orf36 kinase, and raise the possibility that fibroblast cells, but not primary macrophages, contain factors that compensate for the absence of orf36, H2AX, or ATM.
If H2AX was the only physiologically meaningful substrate for the orf36-encoded viral kinase then one would predict that orf36 would be irrelevant for viral replication in cells lacking H2AX. We show that this is not the case since the orf36 TN virus replicates to lower levels than wild type γHV68 in H2AX deficient and ATM deficient macrophages (Supplemental fig. 3C, 3D). These data suggest that orf36 likely has additional substrates and/or functions that are important for growth in primary macrophages.
DISCUSSION
In this paper we show that γ-herpesviruses have an active mechanism for phosphorylation of a DNA damage sensor and induction of proximal events in DNA damage signaling. Phosphorylation of H2AX is induced by the expression of a viral kinase that is important for viral replication in vivo and in primary macrophages, but not fibroblasts. While the viral kinase is responsible for induction of H2AX phosphorylation, the host kinase ATM is important for amplification of this process. The genes involved in this pathway, including the viral kinase, ATM, and H2AX itself, are all important for growth of the virus in macrophages. Together these studies reveal a cell type-specific, active viral strategy in which the early steps of the DNA damage response are co-opted by a virus to create a cellular environment that is optimal for viral replication.
H2AX phosphorylation in virus infection: conserved event accomplished by diverse means
H2AX phosphorylation and ATM activation occur upon infection with numerous DNA viruses (Kudoh et al., 2005; Dahl et al., 2005; Shirata et al., 2005; Lilley et al., 2005). It has been thought, but not proven, that this is due to detection of viral DNA replication intermediates by cellular DNA damage sensor proteins. We found that γHV68 and EBV viral kinases orf36 and BGLF4 induce one aspect of the DNA damage response, phosphorylation of the important DNA damage sensor histone variant H2AX. This phosphorylation of H2AX also occurs upon transient transfection of orf36, indicating that viral DNA replication is not required for H2AX phosphorylation induction by orf36. This conclusion is further supported by experiments showing that orf36 has an important role in induction of phosphorylated H2AX even when viral DNA synthesis is inhibited by the antiviral drug cidofivir. Together these data make the important point that the initiation of the DNA damage response is not a passive result of viral DNA replication, but is rather an active viral strategy.
EBV BGLF4 is a viral kinase that accumulates by 8 hours post EBV reactivation and is incorporated into EBV virions (Asai et al., 2006; Gershburg et al., 2004). Interestingly, H2AX phosphorylation is induced by 12 hours post reactivation in EBV positive B95–8 cells (Kudoh et al., 2005), consistent with the timing of EBV BGLF4 expression. BGLF4 phosphorylates several viral proteins, including EBNA2 (Yue et al., 2005), EA-D (Chen et al., 2000), and BZLF1 (Asai et al., 2006), and cellular proteins, such as elongation factor 1delta (Kawaguchi et al., 2003), however the significance of these kinase substrates for the virus life cycle has not been completely defined.
Our study is the first that links a potential substrate of the mouse γ-herpesvirus viral kinase to its physiological role in viral replication. We demonstrate that both H2AX and ATM are important for replication of γHV68 in primary cells, raising the possibility that the same will be true for EBV. However, there are significant differences in the pathogenesis of these two viruses, and thus the physiologic effects observed in the murine system may not transfer directly to EBV biology. Our findings warrant future studies to define the potential interplay between BGLF4, DNA damage signaling, and EBV biology.
Importantly, we show that immunoprecipitated orf36 kinase is capable of inducing phosphorylation of recombinant H2AX on serine 139 in vitro in a kinase dependent manner. This observation is consistent with the hypothesis that H2AX is a direct substrate of orf36 kinase. We have not ruled out the possibility that immunoprecipitated active, but not kinase dead, orf36 is associated with another cellular kinase that phosphorylates H2AX in vitro. Defining the biochemistry of interactions between orf36 kinase and H2AX will require further experimentation.
While induction of H2AX phosphorylation by γHV68 infection requires the expression of the orf36-encoded viral kinase, it is likely that in the case of infection with other viruses distinct mechanisms initiate the DNA damage response. For example, polyoma virus does not encode a viral kinase, yet it is able to robustly induce DNA damage signaling upon infection (Dahl et al., 2005). In this study, the HSV kinase (UL13) and the human KSHV orf36 failed to induce H2AX phosphorylation in transfected cells, indicating that these viruses do not engage early steps of the DNA damage response in a manner similar to γHV68 or EBV, or alternatively that additional viral or cellular proteins may be required for H2AX phosphorylation by these viral kinases. It is interesting to speculate that a viral protein, perhaps one that interacts with host H2AX kinases is responsible for induction of DNA damage responses by these and other viruses.
H2AX phosphorylation was dramatically decreased in γHV68-infected macrophages derived from ATM −/− mice. While orf36 is capable of inducing phosphorylation of recombinant H2AX in vitro, it may not be very efficient at doing so in vivo, and therefore, ATM could act to amplify H2AX phosphorylation in a positive feedback loop. This idea is consistent with data demonstrating that transfection of orf36 induces more H2AX phosphorylation in wild type than in ATM−/− cells. ATM and H2AX are both important for γHV68 growth in macrophages, suggesting the possible physiologic relevance of this amplification to the virus. Interestingly, ATM is required for the efficient growth of polyoma virus after low MOI infection of primary MEFs (Dahl et al., 2005). Growth of HSV-1 is also attenuated in immortalized cells derived from ataxia telangiectasia patients with mutated ATM (Lilley et al., 2005).
DNA damage signaling downstream of ATM has diverse effects, including cell cycle arrest, sensitization to apoptosis, NFkB induction, and DNA repair (Wu et al., 2006; Sancar et al., 2004). Future studies will be needed to identify the specific processes downstream of H2AX phosphorylation and ATM activation that are important for completion of the viral life cycle. The striking importance of γHV68 encoded orf36 kinase demonstrated here for viral replication in vivo suggests that such studies may reveal pathways that are fundamentally significant for viral pathogenesis, and new targets for treatment of viral infection and the prevention of viral oncogenesis.
What does H2AX phosphorylation mean for γHV68 viral life cycle?
We found that H2AX phosphorylation is not merely a proximal step in the DNA damage signaling in virus-infected cells, but that H2AX itself is required for efficient γHV68 growth in primary macrophages. H2AX deficiency results in increased sensitivity to γ-irradiation and genomic instability (Celeste et al., 2002). While the absence of H2AX has a minimal effect on DNA damage-induced cell cycle arrest, H2AX is important for concentration and retention of DNA repair factors at the sites of DNA damage (Celeste et al., 2003; Celeste et al., 2002). An attractive hypothesis is that replication of viral DNA requires or is enhanced by the cellular DNA damage machinery. Factors that participate in DNA damage repair are recruited to viral replication compartment during both EBV and HSV infections (Daikoku et al., 2006; Lilley et al., 2005; Taylor and Knipe, 2004; Wilkinson and Weller, 2004), suggesting that γH2AX may serve as a scaffold to facilitate efficient viral DNA replication as well as viral DNA recombination that occurs in infected cells. H2AX also affects cell survival in response to UV irradiation (Lu et al., 2006), a function that may be modulated by γHV68. We found that the effects of H2AX and ATM on viral infection are observed in primary macrophages and not in primary fibroblasts. If the recruitment of DNA damage repair machinery to viral DNA is important for replication, then there must be multiple cell type-specific mechanisms to accomplish this.
An important remaining question is whether phosphorylated H2AX associates with viral DNA in infected cells. Mycobacteriophages Corndog and Omega encode a viral Ku70 homologue that associates with the ends of the linear viral genome and facilitates its circularization in bacterial host, a process critical for bacteriophage replication (Pitcher et al., 2006). It is tempting to speculate that H2AX phosphorylation may play a role in the circularization and/or processing of γHV68 genome in infected cells. Alternatively, other functions of H2AX outside of DNA damage response may also play a role in the virus life cycle.
The induction of H2AX phosphorylation and ATM activation, two of the key proximal steps in the fundamentally important DNA damage response, during γHV68 infection may have other consequences for viral pathogenesis. We show that H2AX phosphorylation is induced in infected tissues by γHV68 infection. DNA damage pathway activation upregulates ligands for the NKG2D receptor expressed by natural killer cells and activated CD8+ T cells (Gasser et al., 2005). The viral kinase BGLF4 was only detected in EBV infected cells undergoing lytic infection (Wang et al., 2005). Therefore, viral kinase-dependent induction of H2AX phosphorylation and ATM activation by γHV68 may allow the immune system to recognize the presence of lytic replication of the virus, while having very little effect on latently infected cells that likely lack viral kinase expression.
MATERIALS AND METHODS
Animals
C57BL/6J (BL6) mice obtained from Jackson Laboratories (Bar Harbor, Maine) were bred at Washington University, St. Louis, MO. STAT1 deficient mice were obtained from Dr. Schreiber. All mice were housed in a specific-pathogen-free barrier facility at Washington University in accordance with federal and institutional guidelines. Mice were infected between 8 and 10 weeks of age and organs from euthanized mice processed as previously described (Van Dyk et al., 2000).
Virus infections
γHV68 WUMS (ATCC VR1465) and BAC-derived γHV68 viruses were passaged and titered on NIH-3T12 cells (Weck et al., 1996). The orf36 TN and other mutants were a part of the signature transposon mutagenized library of γHV68 (Song et al., 2005). The orf36.stop1 γHV68 mutant was derived from a γHV68 BAC by homologous recombination to incorporate three translational stop codons in all three reading frames and a HindIII recognition site at a position corresponding to amino acid 26 of orf36 (S. Hwang and R. Sun, unpublished data). The BAC sequences were excised by passaging γHV68 BAC and BAC orf36.stop1 viruses through Cre-expressing cell line to generate orf36.stop 1 virus and its wild type control, loxP. Macrophages were infected for 1 h at 37C and 5% CO2 and washed three times with medium. To block viral DNA synthesis, macrophages were maintained in cidofovir (Gilead, Foster City, CA) at 1 μg/ml starting at 1 h post infection. Statistical analysis of the data was performed by students t-test.
Generation of bone marrow macrophages
Bone marrow was flushed from femurs and incubated in BM20 for the first 7 days of in vitro culture (10% FCS, 20% L-cell conditioned medium derived from GM-CSF producing fibroblast cell line, 5% horse serum in DMEM). On day 7 adherent cells were scraped and seeded in BM10 medium (10% FCS, 10% L-cell conditioned medium, 5% horse serum in DMEM). On day 10 cells were infected and maintained in DMEM10 medium (10% FCS). On day 7 of in vitro culture, 98% of adherent cells were F4/80 and CD11b positive as determined by flow cytometry (data not shown).
Plasmids
Wild type orf36 sequence was amplified from γHV68 viral DNA and cloned into pCMV2-Flag vector using following primers: GAATTCAATGGATTACCGACAGTTACCGC and GGATCCTCAAAAAAATCCAGAATAATCGACCCTTCC. Point mutants of γHV68 orf36 were generated using QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer’s instructions. The vector used to express HA-tagged UL13 kinase of HSV-2 was a kind gift of Dr. Lynda Morrison. EBV BGLF4 and the kinase dead K102I mutant were cloned into pTAG vector.
Western blot analysis
Macrophages were collected into laemmli buffer and analyzed as previously described (Lenschow et al., 2005). Antibodies used were anti-γH2AX and anti-total H2AX (Cell Signaling Technology, Danvers, MA), anti-β actin and anti-Flag (clone AC-74 and M2, Sigma, Saint Louis, MO), anti γHV68 (Dal Canto et al., 2000), and a secondary goat anti-mouse or anti-rabbit HRP-conjugated secondary antibody (Jackson Immunoresearch).
Immunofluorescence
At 48 h post infection, primary macrophages were fixed with 3.7% paraformaldehyde, permeabilized with methanol, and rehydrated with PBS. Coverslips were stained with anti-γH2AX (1:400) or anti-phospho serine 1981 of ATM (Rockland, Gilbertsville, PA) (1:200) followed by secondary Alexa-fluor 488 conjugated antibody (Invitrogen, Carlsbad, CA). Coverslips were mounted into DAPI containing medium (Vector labs, Burlingame, CA), and examined by fluorescent microscopy.
Transfections
3T12 mouse fibroblasts maintained in DMEM-10 medium were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) per manufacturer’s instructions. MEFs were electroporated using Amaxa kit T and program T-20 (Gaithersburg, MD) per manufacturer’s instructions.
Immunohistochemistry
Formalin/paraffin sections of harvested mouse organs were deparaffinized in xylene and passed through descending alcohol concentrations to distilled water. Following quenching of endogenous peroxidase, antigen was retrieved in citrate buffer, pH 6.0. Slides were blocked and incubated with primary antibody overnight at 4C. Following rinses with PBS, secondary biotinylated antibody was applied followed by streptavidin-conjugated HRP. Chromogenic signal was detected with DAB, and sections were counterstained with hematoxylin.
In vitro orf36 kinase assay
1 × 106 HeLa cells were transfected with 5 μg of Flag-vector, Flag-orf36 wild-type, or Flag-orf36 mutant using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 24 hours, cells were lysed in mammalian cell lysis buffer (MCLB) (50 mM Tris-HCl [pH 8.0], 2 mM DTT, 5 mM EDTA, 0.5% Nonidet P-40, 100 mM NaCl, 1 μM microcystin, 1 mM sodium orthovanadate, 2 mM phenylmethylsulfonyl fluoride [PMSF], 10 μg of aprotinin/ml, 20 μM leupeptin). Lysates were pre-cleared with Protein A agarose (Pierce, Rockford, IL) and then incubated with anti-Flag agarose (Sigma, St. Louis, MO) for 2h at 4°C. For the hot kinase reactions the immunoprecipitates were washed 5 × with MCLB, 1 × with LiCl buffer (0.5 M LiCl, 50 mM Tris HCl [pH 8.0]) and 2 × with incomplete kinase buffer (50 mM Tris Cl [pH 7.4], 1 mM DTT, 10 mM MgCl2). Kinase reactions were prepared in 50 μl of incomplete kinase buffer with 20 μM ATP, 10 μCi of [γ-32P] ATP and 5 μg of purified recombinant H2AX (Alexis Biochemicals, San Diego, CA). Kinase reactions were allowed to proceed at 30°C for 30 min and resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and proteins were transferred to nitrocellulose membrane followed by visualization by autoradiography. For the cold kinase reactions, immunoprecipitates were washed 5 × with MCLB, 5 × with LiCl and 2 × with incomplete kinase buffer. The kinase reactions were prepared in 30 μl of incomplete kinase buffer with 20 μM ATP, 50 ng of purified H2AX, 0.4 μg/ml of 3×Flag peptide (Sigma, St. Louis, MO) and incubated at 30°C for 45 min with shaking. Reactions were resolved by SDS-PAGE, transferred to a nitrocellulose membrane and immunoblotted with anti-γH2AX.
Supplementary Material
Supplemental figure 1. A, D. MEFs were infected at 10 PFU/cell with indicated viruses. γH2AX levels and β-actin (D) were measured at indicated times post infection by western analysis. B, C. Primary macrophages were infected at 10 PFU/cell with indicated viruses, and γH2AX, viral antigen, or total H2AX levels were measured at times indicated.
Supplemental figure 2. A, B, C. Primary bone marrow macrophages were infected at 10 PFU/cell with indicated γHV68 mutants, and γH2AX and β-actin levels were measured at 48 h post infection (B, C) or as indicated (A). In B, cidofovir was added to some cultures at 1 h post infection and maintained through 48 h post infection. H. Quantitation of western data displayed in Figure 2C and D, representative experiment is shown.
Supplemental figure 3. A, B. MEFs were infected with wild type or orf36 TN γHV68 mutant at high (10 PFU/cell, A) or low (0.01 PFU/cell, B) multiplicities of infection and viral yield measured at indicated times. C, D, E. Primary bone marrow macrophages derived from mice with indicated genotypes were infected with with wild type or orf36 TN viruses at 10 PFU/cell viral yield was determined at indicated time points. Each time point was measured in triplicate with average and standard error of measurement shown.
Acknowledgments
We thank Darren Kreamalmayer for his outstanding expertise in animal breeding and members of the Virgin and Sleckman laboratories for helpful discussions. Dr. Lynda Morrison provided valuable reagents. H.W.V. was supported by National Institutes of Health (NIH) grant CA 74730. V.L.T. is a Leukemia and Lymphoma society fellow. This work was supported in part by NIH grant (GM47017) to H.P.-W. H.P.-W. is a Howard Hughes Medical Institute investigator. This work was supported in part by Digestive Disease Research Core Center, NIH Grant # P30 DK52514
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Asai R, Kato A, Kato K, Kanamori-Koyama M, Sugimoto K, Sairenji T, Nishiyama Y, Kawaguchi Y. Epstein-Barr virus protein kinase BGLF4 is a virion tegument protein that dissociates from virions in a phosphorylation-dependent process and phosphorylates the viral immediate-early protein BZLF1. J Virol. 2006;80:5125–5134. doi: 10.1128/JVI.02674-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, WynshawBoris A. Atm-deficient mice: A paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, Livingston DM, Ferguson DO, Scully R, Alt FW. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blunt T, Finnie NJ, Taccioli GE, Smith GCM, Demengeot J, Gottlieb TM, Mizuta R, Varghese AJ, Alt FW, Jeggo PA, Jackson SP. Defective Dna-Dependent Protein-Kinase Activity Is Linked to V(D)J Recombination and Dna-Repair Defects Associated with the Murine Scid Mutation. Cell. 1995;80:813–823. doi: 10.1016/0092-8674(95)90360-7. [DOI] [PubMed] [Google Scholar]
- Boehmer PE, Nimonkar AV. Herpes virus replication. Iubmb Life. 2003;55:13–22. doi: 10.1080/1521654031000070645. [DOI] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. Journal of Biological Chemistry. 2001;276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nature Cell Biology. 2003;5:675–U51. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MR, Chang SJ, Huang HW, Chen JY. A protein kinase activity associated with Epstein-Barr virus BGLF4 phosphorylates the viral early antigen EA-D in vitro. J Virol. 2000;74:3093–3104. doi: 10.1128/jvi.74.7.3093-3104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl J, You J, Benjamin TL. Induction and utilization of an ATM signaling pathway by polyomavirus. J Virol. 2005;79:13007–13017. doi: 10.1128/JVI.79.20.13007-13017.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daikoku T, Kudoh A, Sugaya Y, Iwahori S, Shirata N, Isomura H, Tsurumi T. Postreplicative mismatch repair factors are recruited to Epstein-Barr virus replication compartments. Journal of Biological Chemistry. 2006;281:11422–11430. doi: 10.1074/jbc.M510314200. [DOI] [PubMed] [Google Scholar]
- Dal Canto AJ, Virgin HW, Speck SH. Ongoing viral replication is required for gammaherpesvirus 68-induced vascular damage. J Virol. 2000;74:11304–11310. doi: 10.1128/jvi.74.23.11304-11310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damania B. Oncogenic gammaherpesviruses: comparison of viral proteins involved in tumorigenesis. Nat Rev Microbiol. 2004;2:656–668. doi: 10.1038/nrmicro958. [DOI] [PubMed] [Google Scholar]
- Efstathiou S, Ho YM, Hall S, Styles CJ, Scott SD, Gompels UA. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J Gen Virol. 1990;71:1365–1372. doi: 10.1099/0022-1317-71-6-1365. [DOI] [PubMed] [Google Scholar]
- Flano E, Husain SM, Sample JT, Woodland DL, Blackman MA. Latent murine gamma-herpesvirus infection is established in activated B cells, dendritic cells, and macrophages. J Immunol. 2000;165:1074–1081. doi: 10.4049/jimmunol.165.2.1074. [DOI] [PubMed] [Google Scholar]
- Gaspar M, Shenk T. Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:2821–2826. doi: 10.1073/pnas.0511148103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. 2005;436:1186–1190. doi: 10.1038/nature03884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershburg E, Marschall M, Hong K, Pagano JS. Expression and localization of the Epstein-Barr virus-encoded protein kinase. J Virol. 2004;78:12140–12146. doi: 10.1128/JVI.78.22.12140-12146.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JU, Choi JK, Ensser A, Biesinger B. Herpesvirus saimiri as a model for gammaherpesvirus oncogenesis. Semin Cancer Biol. 1999;9:231–239. doi: 10.1006/scbi.1998.0115. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Kato K, Tanaka M, Kanamori M, Nishiyama Y, Yamanashi Y. Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1 delta. J Virol. 2003;77:2359–2368. doi: 10.1128/JVI.77.4.2359-2368.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Xuong NH, Taylor SS. Crystal structure of a complex between the catalytic and regulatory (RI alpha) subunits of PKA. Science. 2005;307:690–696. doi: 10.1126/science.1104607. [DOI] [PubMed] [Google Scholar]
- Krupa A, Preethl G, Srinivasan N. Structural modes of stabilization of permissive phosphorylation sites in protein kinases: Distinct strategies in Ser/Thr and Tyr kinases. J Mol Bio. 2004;339:1025–1039. doi: 10.1016/j.jmb.2004.04.043. [DOI] [PubMed] [Google Scholar]
- Kudoh A, Fujita M, Zhang LM, Shirata N, Daikoku T, Sugaya Y, Isomura H, Nishiyama Y, Tsurumi T. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. Journal of Biological Chemistry. 2005;280:8156–8163. doi: 10.1074/jbc.M411405200. [DOI] [PubMed] [Google Scholar]
- Lenschow DJ, Giannakopoulos NV, Gunn LJ, Johnston C, O’Guin AK, Schmidt RE, Levine B, Virgin HW. Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo. J Virol. 2005;79:13974–13983. doi: 10.1128/JVI.79.22.13974-13983.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5844–5849. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CR, Zhu F, Cho YY, Tang FQ, Zykova T, Ma WY, Bode AM, Dong ZG. Cell apoptosis: Requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Molecular Cell. 2006;23:121–132. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Guzman D, Rickabaugh T, Wu TT, Brown H, Cole S, Song MJ, Tong L, Sun R. Transcription program of murine gammaherpesvirus 68. J Virol. 2003;77:10488–10503. doi: 10.1128/JVI.77.19.10488-10503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan CH, Russell P. The DNA damage response: sensing and signaling. Current Opinion in Cell Biology. 2004;16:629–633. doi: 10.1016/j.ceb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Neyts J, De CE. In vitro and in vivo inhibition of murine gamma herpesvirus 68 replication by selected antiviral agents. Antimicrob Agents Chemother. 1998;42:170–172. doi: 10.1128/aac.42.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EJ, Chan DW, Park JH, Oettinger MA, Kwon J. DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res. 2003;31:6819–6827. doi: 10.1093/nar/gkg921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Current Biology. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- Pitcher RS, Tonkin LM, Daley JM, Palmbos PL, Green AJ, Velting TL, Brzostek A, Korycka-Machala M, Cresawn S, Dziadek J, Hatfull GF, Wilson TE, Doherty AJ. Mycobacteriophage exploit NHEJ to facilitate genome circularization. Molecular Cell. 2006;23:743–748. doi: 10.1016/j.molcel.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Poffenberger KL, Roizman B. A noninverting genome of a viable herpes simplex virus 1: presence of head-to-tail linkages in packaged genomes and requirements for cirularization after infection. J Virol. 1985;53:587–595. doi: 10.1128/jvi.53.2.587-595.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annual Review of Biochemistry. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Shirata N, Kudoh A, Daikoku T, Tatsumi Y, Fujita M, Kiyono T, Sugaya Y, Isomura H, Ishizaki K, Tsurumi T. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. Journal of Biological Chemistry. 2005;280:30336–30341. doi: 10.1074/jbc.M500976200. [DOI] [PubMed] [Google Scholar]
- Song MJ, Hwang SM, Wong WH, Wu TT, Lee SM, Liao HI, Sun R. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3805–3810. doi: 10.1073/pnas.0404521102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature. 2002;418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- Sunil- Chandra NP, Efstathiou S, Nash AA. Murine gammaherpesvirus 68 establishes a latent infection in mouse B lymphocytes in vivo. J Gen Virol. 1992;73:3275–3279. doi: 10.1099/0022-1317-73-12-3275. [DOI] [PubMed] [Google Scholar]
- Tarakanova VL, Suarez FS, Tibbetts SA, Jacoby M, Weck KE, Hess JH, Speck SH, Virgin HW. Murine gammaherpesvirus 68 infection induces lymphoproliferative disease and lymphoma in BALB β2 microglobulin deficient mice. J Virol. 2005;79:14668–14679. doi: 10.1128/JVI.79.23.14668-14679.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor TJ, Knipe DM. Proteomics of herpes simplex virus replication compartments: Association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J Virol. 2004;78:5856–5866. doi: 10.1128/JVI.78.11.5856-5866.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend PA, Cragg MS, Davidson SM, McCormick J, Barry S, Lawrence KM, Knight RA, Hubank M, Chen PL, Latchman DS, Stephanou A. STAT-1 facilitates the ATM activated checkpoint pathway following DNA damage. Journal of Cell Science. 2005;118:1629–1639. doi: 10.1242/jcs.01728. [DOI] [PubMed] [Google Scholar]
- Van Dyk LF, Virgin HW, Speck SH. The murine gammaherpesvirus 68 v-cyclin is a critical regulator of reactivation from latency. J Virol. 2000;74:7451–7461. doi: 10.1128/jvi.74.16.7451-7461.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin HW, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J Virol. 1997;71:5894–5904. doi: 10.1128/jvi.71.8.5894-5904.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Yang PW, Lee CP, Han CH, Tsai CH, Chen MR. Detection of Epstein-Barr virus BGLF4 protein kinase in virus replication compartments and virus particles. J Gen Virol. 2005;86:3215–3225. doi: 10.1099/vir.0.81313-0. [DOI] [PubMed] [Google Scholar]
- Weck KE, Barkon ML, Yoo LI, Speck SH, Virgin HW. Mature B cells are required for acute splenic infection, but not for establishment of latency, by murine gammaherpesvirus 68. J Virol. 1996;70:6775–6780. doi: 10.1128/jvi.70.10.6775-6780.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weck KE, Kim SS, Virgin HW, Speck SH. Macrophages are the major reservoir of latent murine gammaherpesvirus 68 in peritoneal cells. J Virol. 1999;73:3273–3283. doi: 10.1128/jvi.73.4.3273-3283.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DE, Weller SK. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J Virol. 2004;78:4783–4796. doi: 10.1128/JVI.78.9.4783-4796.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer DO, Speck SH. Long-term latent murine Gammaherpesvirus 68 infection is preferentially found within the surface immunoglobulin D-negative subset of splenic B cells in vivo. J Virol. 2003;77:8310–8321. doi: 10.1128/JVI.77.15.8310-8321.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZH, Shi YL, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappa B signaling in response to genotoxic stimuli. Science. 2006;311:1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- Young LS, Rickinson A. Epstein-Barr Virus: 40 years on. Nature Reviews Immunology. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- Yue W, Gershburg E, Pagano JS. Hyperphosphorylation of EBNA2 by Epstein-Barr virus protein kinase suppresses transactivation of the LMP1 promoter. J Virol. 2005;79:5880–5885. doi: 10.1128/JVI.79.9.5880-5885.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental figure 1. A, D. MEFs were infected at 10 PFU/cell with indicated viruses. γH2AX levels and β-actin (D) were measured at indicated times post infection by western analysis. B, C. Primary macrophages were infected at 10 PFU/cell with indicated viruses, and γH2AX, viral antigen, or total H2AX levels were measured at times indicated.
Supplemental figure 2. A, B, C. Primary bone marrow macrophages were infected at 10 PFU/cell with indicated γHV68 mutants, and γH2AX and β-actin levels were measured at 48 h post infection (B, C) or as indicated (A). In B, cidofovir was added to some cultures at 1 h post infection and maintained through 48 h post infection. H. Quantitation of western data displayed in Figure 2C and D, representative experiment is shown.
Supplemental figure 3. A, B. MEFs were infected with wild type or orf36 TN γHV68 mutant at high (10 PFU/cell, A) or low (0.01 PFU/cell, B) multiplicities of infection and viral yield measured at indicated times. C, D, E. Primary bone marrow macrophages derived from mice with indicated genotypes were infected with with wild type or orf36 TN viruses at 10 PFU/cell viral yield was determined at indicated time points. Each time point was measured in triplicate with average and standard error of measurement shown.